
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring the solid state and solution structural chemistry of the utility amide potassium hexamethyldisilazide (KHMDS)†
Ana I.
Ojeda-Amador
,
Antonio J.
Martínez-Martínez
*,
Gemma M.
Robertson
,
Stuart D.
Robertson
,
Alan R.
Kennedy
 and
Charles T.
O'Hara
*
and
Charles T.
O'Hara
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: a.j.martinez@strath.ac.uk; charlie.ohara@strath.ac.uk
First published on 3rd May 2017
Abstract
The structural chemistry of eleven donor complexes of the important Brønsted base potassium 1,1,1,3,3,3-hexamethyldisilazide (KHMDS) has been studied. Depending on the donor, each complex adopted one of five general structural motifs. Specifically, in this study the donors employed were toluene (to give polymeric 1 and dimeric 2), THF (polymeric 3), N,N,N′,N′-tetramethylethylenediamine (TMEDA) (dimeric 4), (R,R)-N,N,N′,N′-tetramethyl-1,2-diaminocyclohexane [(R,R)-TMCDA] (dimeric 5), 12-crown-4 (dimeric 6), N,N,N′,N′-tetramethyldiaminoethyl ether (TMDAE) (tetranuclear dimeric 8 and monomeric 10), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (tetranuclear dimeric 7), tris[2-dimethyl(amino)ethyl]amine (Me6TREN) (tetranuclear dimeric 9) and tris{2-(2-methoxyethoxy)ethyl}amine (TMEEA) (monomeric 11). The complexes were also studied in solution by 1H and 13C NMR spectroscopy as well as DOSY NMR spectroscopy.
Introduction
Alkali metal complexes of 1,1,1,3,3,3-hexamethyldisilazide (HMDS) are commonly employed reagents in synthesis due to their non-nucleophilic but Brønsted basic nature.1,2 In comparison with related alkali metal secondary amide reagents such as diisopropylamide (DA) and 2,2,6,6-tetramethylpiperidide (TMP) salts, HMDS is weakly basic due to its inherent α-silyl stabilisation. The pK of LiHMDS in tetrahydrofuran (THF) solution is 24 whereas for LiDA and LiTMP are 35 and 36 respectively.3 Additionally, the absence of β-hydrogen atoms prevents β-hydride elimination, and their lipophilic character makes alkali metal derivatives of the HMDS group special candidates for many homogeneous reactions. Since lithium, sodium and potassium HMDS reagents are commercially available, they have become prominent transfer reagents for the synthesis of s-block metal,4 transition metal,5–7 lanthanide8 and actinide9,10 HMDS complexes via salt metathesis. In synthesis, they are used in a wide variety of chemical transformations ranging from drug synthesis to polymer production, such as in the formation of kinetic enolate anions,11–13 alkylations,14 arylations,15 isomerisations,16 polymerizations,17 ring closing reactions and Wittig reactions,18,19 and also, in the deprotonative metallation of acidic C–H bonds, like cyclopentadiene, indene and fluorene.20–22 Most recently, several organic transformations have been reported which use alkali metal HMDS salt complexes in combination with a Lewis basic donor molecule to induce catalysis. In particular, KHMDS has attracted special attention due to its slightly increased Brønsted basic character when compared with its lighter lithium or sodium congeners,3 but also because of its relatively low toxicity23 and as such can be used in medical-related fields which are incompatible with conventional transition metal-catalysed approaches. In this context, the seminal work by Kobayashi24 has recently shown that KHMDS in combination with chiral macrocyclic crown ethers (as Lewis basic donor molecules) acts as an effective catalytic system for the carbon–carbon bond-forming reaction of alkylnitriles and 1,5-dicarbonyl compounds with excellent diastereo- and enantioselectivities.25,26 Wilhelm reported that KHMDS exhibits catalytic activity in the presence of THF, at temperatures as low as −78 °C, for the [2 + 2] cycloaddition of ketenes with imines and aldehydes to produce biologically important β-lactam and β-lactone feedstocks.27 Zhang has shown that N-alkyl substituted carbamates undergo 5-exo-dig-cyclization to afford the corresponding functionalised oxazolidinone derivatives catalysed by KHMDS and 18-crown-6 in toluene solution.28 Panda and Carpentier have illustrated the use of KHMDS as a pre-catalyst with a higher activity than the lithium and sodium salts for cross-dehydrocoupling of boranes and silanes with amines for preparing aminoboranes and silazanes, respectively, with a high degree of conversion and chemoselectivity.29,30 The benefit of using potassium over lithium and sodium derivatives in synthesis could also be exemplified in the tert-butoxide congener of KHMDS (KOtBu). For instance, Shi31 reported the use of KOtBu in combination with the chelating Lewis base donor 1,10-phenanthroline as a transition metal-free catalytic system for constructing biaryl systems via radical cross-coupling between inert aromatic C–H bonds and aryl iodides or bromides. Murphy and Tuttle32 have studied in detail the mechanism of these transformations concluding that the greater basicity of KOtBu over its sodium and lithium counterparts is a crucial factor in allowing access to these electron transfer reactions.Since KHMDS is a widely used Brønsted base in synthesis and has recently shown excellent catalytic potential in THF or toluene solutions in the presence of Lewis basic donor molecules, it is important to understand the structural chemistry which is at play with these reagents in these solvents. The structural chemistry of LiHMDS and NaHMDS has been well studied; however, it is perhaps surprising that for the commercially available KHMDS (as toluene and THF solutions) it remains largely unexplored. To date, the solvent-free dimeric [(KHMDS)2],33 dimeric toluene-solvated [(KHMDS)2(toluene)2],34 dimeric ammonia-solvated [(KHMDS)2(NH3)4]35 complexes and the monomeric species [(KHMDS)(donor)] (donor = 1,4-dioxane36 and 18-crown-6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37,38) have been structurally characterized (Fig. 1a–d) alongside two examples of N-heterocyclic carbene (NHC) dimeric adducts of KHMDS, namely [(KHMDS)2(NHC)2], where NHC are 1,3-bis-2,6-di-isopropylphenyl-imidazol-2-ylidene and 1,3-diisopropyl-3,4,5,6-tetrahydropyrimid-2-ylidene (Fig. 1e and f).39,40 Additionally, one other related example containing a O,N-bidentate ligand derived from an ether-based backbone imidazolinium proligand [(KHMDS)2{(OCMe2CH2HC)NCH2CH2NiPr}2] has also been reported (Fig. 1g).41
37,38) have been structurally characterized (Fig. 1a–d) alongside two examples of N-heterocyclic carbene (NHC) dimeric adducts of KHMDS, namely [(KHMDS)2(NHC)2], where NHC are 1,3-bis-2,6-di-isopropylphenyl-imidazol-2-ylidene and 1,3-diisopropyl-3,4,5,6-tetrahydropyrimid-2-ylidene (Fig. 1e and f).39,40 Additionally, one other related example containing a O,N-bidentate ligand derived from an ether-based backbone imidazolinium proligand [(KHMDS)2{(OCMe2CH2HC)NCH2CH2NiPr}2] has also been reported (Fig. 1g).41
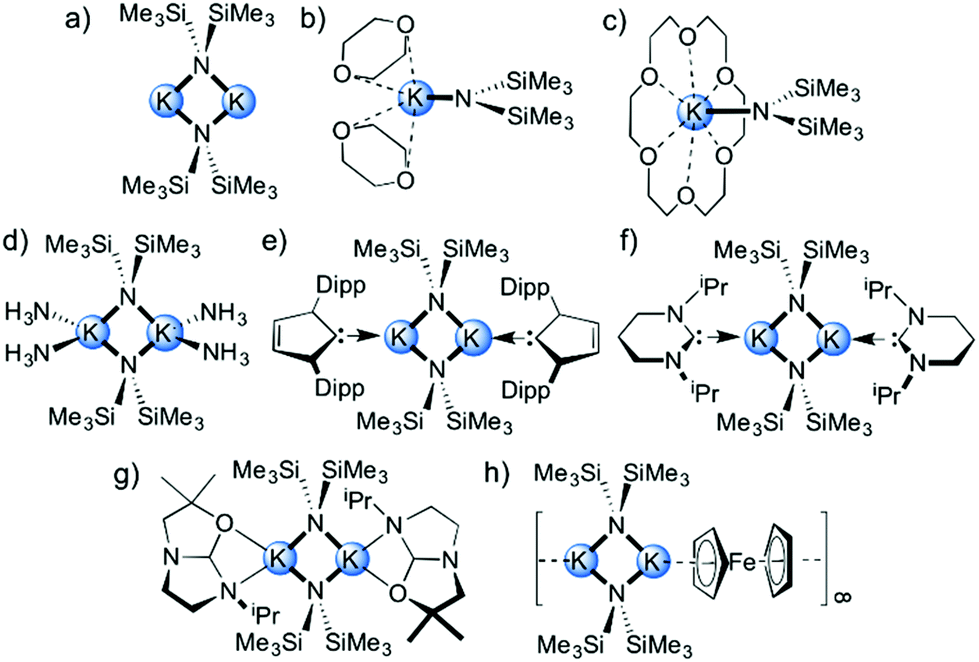 | ||
| Fig. 1 (a) Structurally known solvent-free dimeric (KHMDS)2 and (b–h) solvates of KHMDS. | ||
Most of the structurally known complexes contain an archetypal dinuclear (KNHMDS)2 core. This motif is also observed when ferrocene is added to KHMDS, resulting in the isolation of the unusual polymeric [(KHMDS)2(ferrocene)]∞ (Fig. 1h),42 where ferrocene π-coordinates to the potassium metal. When KHMDS is combined with alkaline earth metal amides forming mixed metallate species,43–45 it is possible to isolate inverse crown species such as hydrido-46 and oxo-variants47,48 as well as simpler binuclear heterobimetallic amide formulations49,50 also observed when the alkaline earth metal is replaced by lanthanides.51,52 As the structural chemistry of alkali metal reagents normally dictates and influences their reactivity, it was decided to isolate and characterise, (in the solid- and solution-state) a series of solvates of KHMDS containing key solubility- and reactivity-enhancing donor molecules. These molecules include toluene, THF, N,N,N′,N′-tetramethylethylenediamine (TMEDA), (R,R)-N,N,N′,N′-tetramethyl-1,2-diaminocyclohexane [(R,R)-TMCDA], 12-crown-4, N,N,N′,N′-tetramethyldiaminoethyl ether (TMDAE), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA), tris[2-dimethyl(amino)ethyl]amine (Me6TREN) and tris{2-(2-methoxyethoxy)ethyl}amine (TMEEA).
Results and discussion
Synthetic procedures
A total of eleven new solvates of KHMDS (1–11) have been prepared and structurally characterised, both in solution and in the solid state. All synthetic protocols were optimised for obtaining high-quality crystalline samples for single crystal X-ray diffraction studies.When toluene was employed as a donor, two distinct aggregates of KHMDS were obtained depending on the reaction conditions, a polymer [(KHMDS)2(toluene)]∞1 and a discrete dimer [(KHMDS)2(toluene)2] 2. The ‘toluene-deficient’ polymeric aggregate 1 was prepared by generating KHMDS in situ (via a salt metathesis reaction between equimolar quantities of LiHMDS and KOtBu). This reaction mixture results in a pale-yellow suspension in n-hexane from which 1 was successfully crystallised by adding toluene at ambient temperature (2:1 mixture of n-hexane:toluene) in 38% yield (Scheme 1). However, when the reaction mixture is stored at −27 °C the 1:1 toluene:KHMDS dimeric solvate 2 was isolated in 46% yield. THF-solvate [(KHMDS)2(THF)2]∞3 was obtained by adding two molar equivalents of THF to a slurry of KHMDS in n-hexane. The THF polymeric aggregate was isolated (in 34% yield) by storing the resultant solution at −27 °C (Scheme 1). To the best of our knowledge, this represents the first isolation and structural characterisation of a THF-containing derivative of KHMDS, despite this reagent-solvent combination being commonly used in synthetic chemistry. Next, two bidentate diamine donors were studied, namely TMEDA and its chiral analogue (R,R)-TMCDA. To achieve homogeneity, a single molar equivalent with respect to KHMDS was required. The corresponding dimeric complexes [(KHMDS)2(TMEDA)2] 4 and [(KHMDS)2{(R,R)-TMCDA}2] 5 were isolated in coincidentally identical yields of 46% at −27 °C (Scheme 1). In a similar reaction, the analogous 12-crown-4 dimeric solvate [(KHMDS)2(12-crown-4)2] 6 was isolated in almost quantitative yield (96%) from an n-hexane solution containing equimolar amounts of KHMDS and the crown ether molecule at −27 °C. Structurally distinct (vide infra) tetranuclear potassium aggregates, namely [(KHMDS)2(PMDETA·KHMDS)2] 7 and [(KHMDS)2(TMDAE·KHMDS)2] 8 (Scheme 2), were obtained by adding the corresponding tridentate donor PMDETA and TMDAE molecules to a suspension of KHMDS in n-hexane in 0.5:1 donor:KHMDS molar ratios, producing homogeneous solutions in both cases. Complex 7 was obtained at ambient temperature in 52% yield whilst 8 crystallised at −27 °C in 66% yield. Using a single molar equivalent of tetradentate Me6TREN produces a similar tetranuclear KHMDS complex, [(KHMDS)2(Me6TREN·KHMDS)2] 9; however, two molar equivalents of Me6TREN were required in n-hexane. Crystallisation at −35 °C yielded 9 in a moderate yield of 36%. Surprisingly, addition of two molar equivalents of Me6TREN did not produce full deaggregation (i.e. monomerisation) of KHMDS, but employing two molar equivalents of the N,O-containing tridentate TMDAE molecule with respect to KHMDS in n-hexane yielded the monomeric aggregate [KHMDS(TMDAE)2] 10 (Scheme 2) in good yield (62%). Finally, the N,O-containing potentially heptadentate TMEEA was added to a toluene solution of KHMDS in a 1:1 donor:KHMDS molar ratio from which [KHMDS(TMEEA)] 11 was obtained in 69% yield upon cooling down the resulting solution to −27 °C. Complexes 10 and 11 represent rare examples of monomeric KHMDS complexes (Scheme 2).
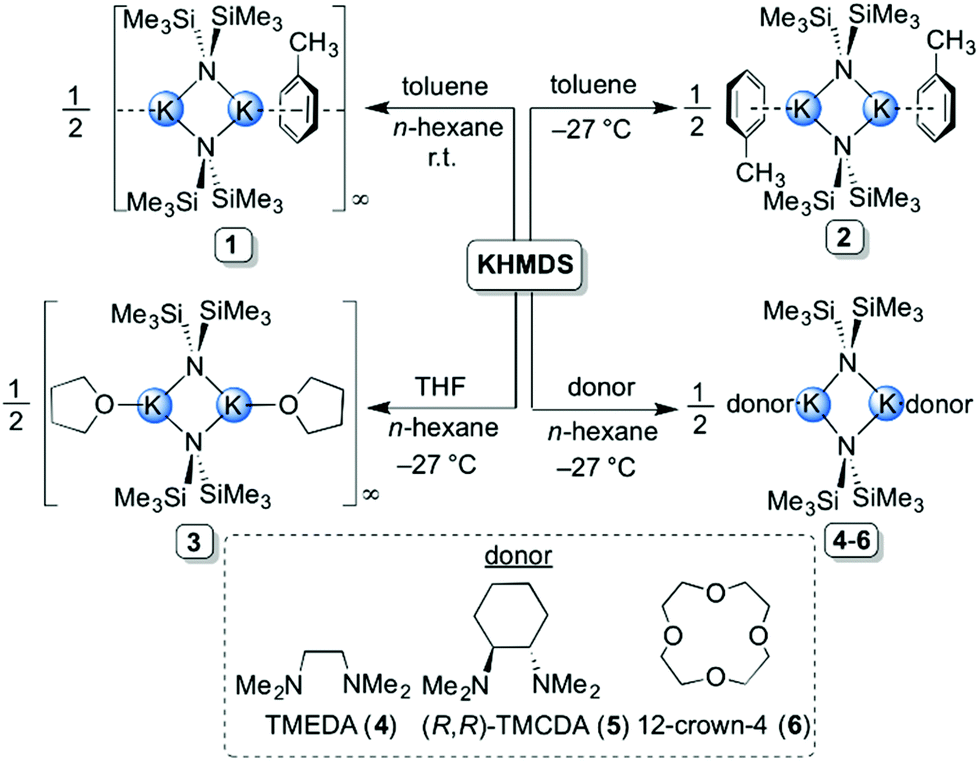 | ||
| Scheme 1 Synthesis of polymeric [(KHMDS)2(toluene)]∞1, dimeric [(KHMDS)2(toluene)2] 2, polymeric [(KHMDS)2(THF)2]∞3 and dimeric [(KHMDS)2(TMEDA)2] 4, [(KHMDS)2{(R,R)-TMCDA}2] 5 and [(KHMDS)2(12-crown-4)2] 6. | ||
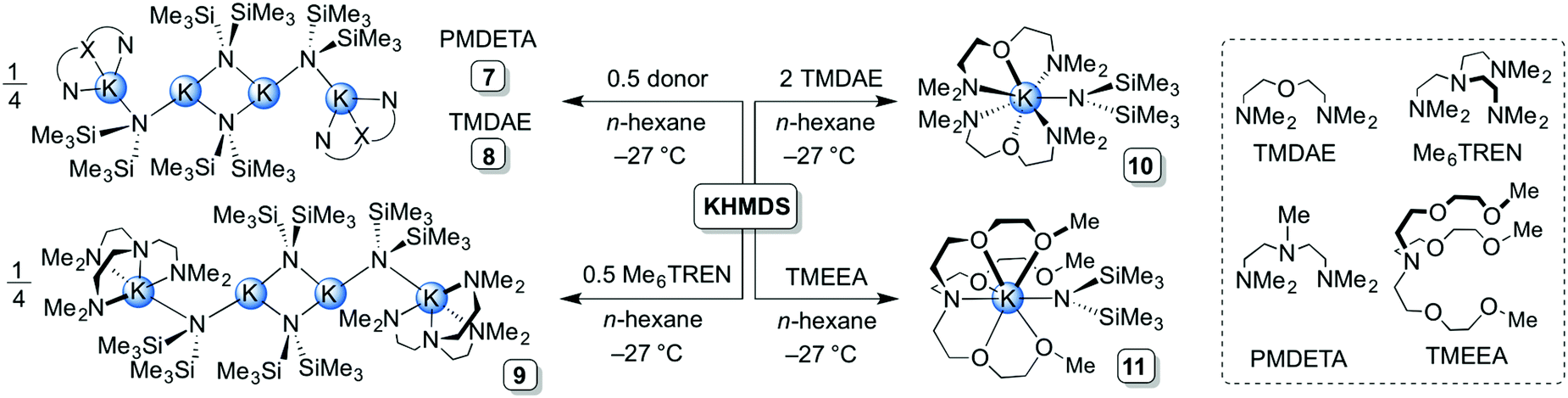 | ||
| Scheme 2 Synthesis of tetranuclear [(KHMDS)2(PMDETA·KHMDS)2] 7, [(KHMDS)2(TMDAE·KHMDS)2] 8 and [(KHMDS)2(Me6TREN·KHMDS)2] 9, and monomeric [KHMDS(TMDAE)2] 10 and [KHMDS(TMEEA)] 11. | ||
X-ray diffraction studies
As discussed earlier, depending on the crystallisation temperature, it is possible to isolate two distinct toluene solvates of KHMDS, namely polymeric [(KHMDS)2(toluene)]∞1 (Fig. 2a and b) and dimeric [(KHMDS)2(toluene)2] 2 (Fig. 2c). These crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and in the monoclinic space group C2/c, respectively. The asymmetric unit cell of 1 is composed of a dimeric (KHMDS)2 arrangement where one molecule of toluene is coordinated to the K metal centre in a η6-manner via π–arene interactions [K1⋯arene(centroid) 3.045 Å]. The structure of 1 propagates through the crystallographic a-axis via π–arene interactions of this toluene molecule with a second K metal centre from a neighbouring (KHMDS)2 unit, ultimately forming a linear polymeric chain arrangement [K2⋯arene(centroid) 3.015 Å] (Fig. 2b). Complex 2 resembles the lattice parameters previously reported by Williard, although in this initial report the toluene solvent molecules were described as not coordinating to the K metal centres.34 Here we have included our interpretation of the structural description of 2 by considering it as a four-membered [K–N–K–N] cyclodimeric unit, where each K atom is solvated by a molecule of toluene in an η6-manner [K⋯arene(centroid) 2.967 Å]. Each arene molecule binds approximately in a perpendicular array to the K2N2 plane (87.5°) resulting in a discrete dimeric arrangement (Fig. 2c). Comparing 1 and 2, in the former the toluene molecules are disposed in a cisoid manner in the polymeric chain (Fig. 2b), whereas in 2, the toluene molecules are transoidal. This η6-bonding mode of an arene to a metal is a common structural feature in heavy alkali metal organometallic complexes and these K⋯arene(centroid) distances are similar to those found in related benzene- or toluene-solvated potassium amide complexes (range, 2.862–3.108 Å).23,45,53–67 The K2N2 units in both the non-centrosymmetric 1 and centrosymmetric 2 resemble those of (KHMDS)2 reported by Tesh and Hanusa.33 Both K atoms are connected through bridging μ-HMDS ligands where the N atoms lie in approximately the same plane as the K metal centres [torsion angle K1–N1–K2–N2 of 1.5°, for 1; coplanar in 2]. The inclusion of an additional molecule of toluene per potassium atom, does not have a pronounced effect on the respective K–N bond distances found in 1 and 2 [K1–N 2.7726(15) and 2.7619(15) Å and K2–N 2.7506(15) and 2.7593(15) Å in 1; and 2.739(2) and 2.800(2) Å for 2, respectively; see Table 1]. These K–N bond distances are similar to those found for the solvent-free (KHMDS)2 [2.770(3) and 2.803(3) Å].33 The distinct K–N bond lengths for K1 and K2 observed in 1 result in two slightly distinct N–K–N angles [N–K1–N 94.58(4) and N–K2–N 95.13(4)°], whilst the K–N–K angles are identical [K1–N1–K2 85.14(4) and K1–N2–K2 85.11(4)°]. These bond angles are similar to those found in 2 [N1–K1–N1′ 94.17(7) and K1–N1–K1, 85.83(7)°]. In addition, each K atom exhibits long K⋯C interactions with Me groups from the HMDS ligands in 1 and 2 [K1⋯C range 3.237(2)–3.2951(19) and 3.303(4)–3.402(4) Å for 1 and 2, respectively].
and in the monoclinic space group C2/c, respectively. The asymmetric unit cell of 1 is composed of a dimeric (KHMDS)2 arrangement where one molecule of toluene is coordinated to the K metal centre in a η6-manner via π–arene interactions [K1⋯arene(centroid) 3.045 Å]. The structure of 1 propagates through the crystallographic a-axis via π–arene interactions of this toluene molecule with a second K metal centre from a neighbouring (KHMDS)2 unit, ultimately forming a linear polymeric chain arrangement [K2⋯arene(centroid) 3.015 Å] (Fig. 2b). Complex 2 resembles the lattice parameters previously reported by Williard, although in this initial report the toluene solvent molecules were described as not coordinating to the K metal centres.34 Here we have included our interpretation of the structural description of 2 by considering it as a four-membered [K–N–K–N] cyclodimeric unit, where each K atom is solvated by a molecule of toluene in an η6-manner [K⋯arene(centroid) 2.967 Å]. Each arene molecule binds approximately in a perpendicular array to the K2N2 plane (87.5°) resulting in a discrete dimeric arrangement (Fig. 2c). Comparing 1 and 2, in the former the toluene molecules are disposed in a cisoid manner in the polymeric chain (Fig. 2b), whereas in 2, the toluene molecules are transoidal. This η6-bonding mode of an arene to a metal is a common structural feature in heavy alkali metal organometallic complexes and these K⋯arene(centroid) distances are similar to those found in related benzene- or toluene-solvated potassium amide complexes (range, 2.862–3.108 Å).23,45,53–67 The K2N2 units in both the non-centrosymmetric 1 and centrosymmetric 2 resemble those of (KHMDS)2 reported by Tesh and Hanusa.33 Both K atoms are connected through bridging μ-HMDS ligands where the N atoms lie in approximately the same plane as the K metal centres [torsion angle K1–N1–K2–N2 of 1.5°, for 1; coplanar in 2]. The inclusion of an additional molecule of toluene per potassium atom, does not have a pronounced effect on the respective K–N bond distances found in 1 and 2 [K1–N 2.7726(15) and 2.7619(15) Å and K2–N 2.7506(15) and 2.7593(15) Å in 1; and 2.739(2) and 2.800(2) Å for 2, respectively; see Table 1]. These K–N bond distances are similar to those found for the solvent-free (KHMDS)2 [2.770(3) and 2.803(3) Å].33 The distinct K–N bond lengths for K1 and K2 observed in 1 result in two slightly distinct N–K–N angles [N–K1–N 94.58(4) and N–K2–N 95.13(4)°], whilst the K–N–K angles are identical [K1–N1–K2 85.14(4) and K1–N2–K2 85.11(4)°]. These bond angles are similar to those found in 2 [N1–K1–N1′ 94.17(7) and K1–N1–K1, 85.83(7)°]. In addition, each K atom exhibits long K⋯C interactions with Me groups from the HMDS ligands in 1 and 2 [K1⋯C range 3.237(2)–3.2951(19) and 3.303(4)–3.402(4) Å for 1 and 2, respectively].
 | ||
| Fig. 2 (a) Molecular structure of [(KHMDS)2(toluene)]∞1, showing the contents of the asymmetric unit. (b) Section of the extended linear polymeric structural framework of 1 showing atomic connectivity between K and arene(centroids). (c) Molecular structure of [(KHMDS)2(toluene)2] 2. Hydrogen atoms are omitted for simplicity and displacement ellipsoids are displayed at 35% probability. The arene(centroids) are pictured as translucent black spheres. The dashed lines illustrate both K⋯C contacts and K⋯arene(centroid) interactions. Selected bond distances (Å) and angles (°): for 1, K1–N1 2.7619(15), K1–N2 2.7726(15), K2–N2 2.7506(15), K2–N1 2.7593(15), K1⋯arene(centroid) 3.045, K2⋯arene(centroid) 3.015, K1⋯C1 3.237(2), K1⋯C10 3.2610(19), K2⋯C6 3.2951(19), K2⋯C8 3.283(2), N1–K1–N2 94.58(4), N2–K2–N1 95.13(4), K2–N1–K1 85.14(4), K2–N2–K1 85.11(4); for 2, K1–N1 2.739(2), K1–N1′ 2.800(2), K1–C1 3.384(4), K1–C3′ 3.402(4), K1–C4 3.303(4), K1–C6′ 3.385(4), K1⋯arene(centroid) 2.967, K1⋯C1 3.384(4), K1⋯C4 3.303(4), K1⋯C6′ 3.385(4), N1–K1–N1′ 94.17(7), K1–N1–K1′ 85.83(7), N1–K1⋯arene(centroid) 129.04, N1′–K1⋯arene(centroid) 136.76. The symmetry operations used to generate the equivalent atoms numbered with ′ and ′′ for 1 are x + 1, y, z and x − 1, y, z, respectively; and ′ for 2 is −x + 1/2, −y + 1/2, −z + 1. | ||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Toluene | Toluene | THF | TMEDA | (R,R)-TMCDA | 12-c-4 | PMDETA | TMDAE | Me6TREN | TMDAE | TMEEA | |
| Polymer | Dimer | Polymer | Dimer | Dimer | Dimer | Tetranuclear dimer | Tetranuclear dimer | Tetranuclear dimer | Monomer | Monomer | |
| a K–N–K/N–K–N bond angles involving the terminal K (donor) atoms in 7–8. | |||||||||||
| K–NHMDS | 2.7629(15) | 2.739(2) | 2.7442(13) | 2.8365(16) | 2.884(2) | 2.913(2) | 2.8236(13) | 2.8288(11) | 2.846(2) | 2.794(3) | 2.7889(9) |
| 2.7726(15) | 2.800(2) | 2.8534(12) | 2.7942(17) | 2.908(2) | 3.073(2) | 2.8452(13) | 2.8660(11) | 2.848(2) | |||
| 2.7506(15) | 2.7876(17) | 2.785(2) | 2.8808(11) | 2.9254(11) | 2.906(2) | ||||||
| 2.7593(15) | 2.8765(17) | 2.764(2) | 2.7536(12) | 2.8367(11) | 2.913(2) | ||||||
| K⋯CMe | 3.237(2) | 3.303(4) | 3.2226(15) | 3.313(2) | 3.185(3) | 3.313(3) | 3.3223(17) | 3.3147(14) | 3.239(4) | — | — |
| 3.2610(19) | 3.180(3) | ||||||||||
| 3.223(3) | |||||||||||
| K–N–K | 85.14(4) | 85.83(7) | 83.73(3) | 80.99(4) | 84.78(6) | 83.08(5) | 84.08(3) | 84.35(3) | 83.42(6) | — | — |
| 85.11(4) | 81.81(4) | 85.62(6) | 106.43(4)a | 108.36(3)a | 117.64(7)a | ||||||
| N–K–N | 94.58(4) | 94.17(7) | 96.27(3) | 98.99(5) | 92.04(6) | 96.92(5) | 95.93(3) | 95.65(3) | 96.58(6) | — | — |
| 95.13(4) | 98.19(5) | 97.37(7) | 141.54(4)a | 140.39(3) a | 132.40(6) a | ||||||
| 122.13(4)a | 121.12(3) a | 126.95(6) a | |||||||||
Complex 3 crystallises in the monoclinic space group P21/c. Like most known KHMDS structures,34,36,39,41,49,56,68,693 crystallises as a dimeric (KHMDS)2 unit, and it contains two molecules of THF, one coordinating to each K atom (Fig. 3a). The key features and metrics resemble those found for 1 and 2, a four membered [K–N–K–N] planar ring, two bridging μ-HMDS ligands and for 2, two distinct K–N bond lengths [2.7442(13) and 2.8534(12) Å]. The two THF ligands are coordinated to the K atoms in a transoidal manner [K–O bond length is 2.7199(13) Å]. Reflecting the fact that the K atoms are only partially saturated by the donor molecule in 3 (in comparison with 2), there is an intermolecular interaction with an HMDS methyl group of a neighbouring (KHMDS)2 unit. This results in the formation of a two-dimensional layer arrangement through K⋯Me interactions [Fig. 3b and c, K1⋯C6′′ 3.2226(15) Å]. Each [(KHMDS)2(THF)2] unit represents a branch point towards four distinct directions in the polymeric two-dimensional sheet, two through its K atoms and two via two methyl groups from each HMDS ligand.
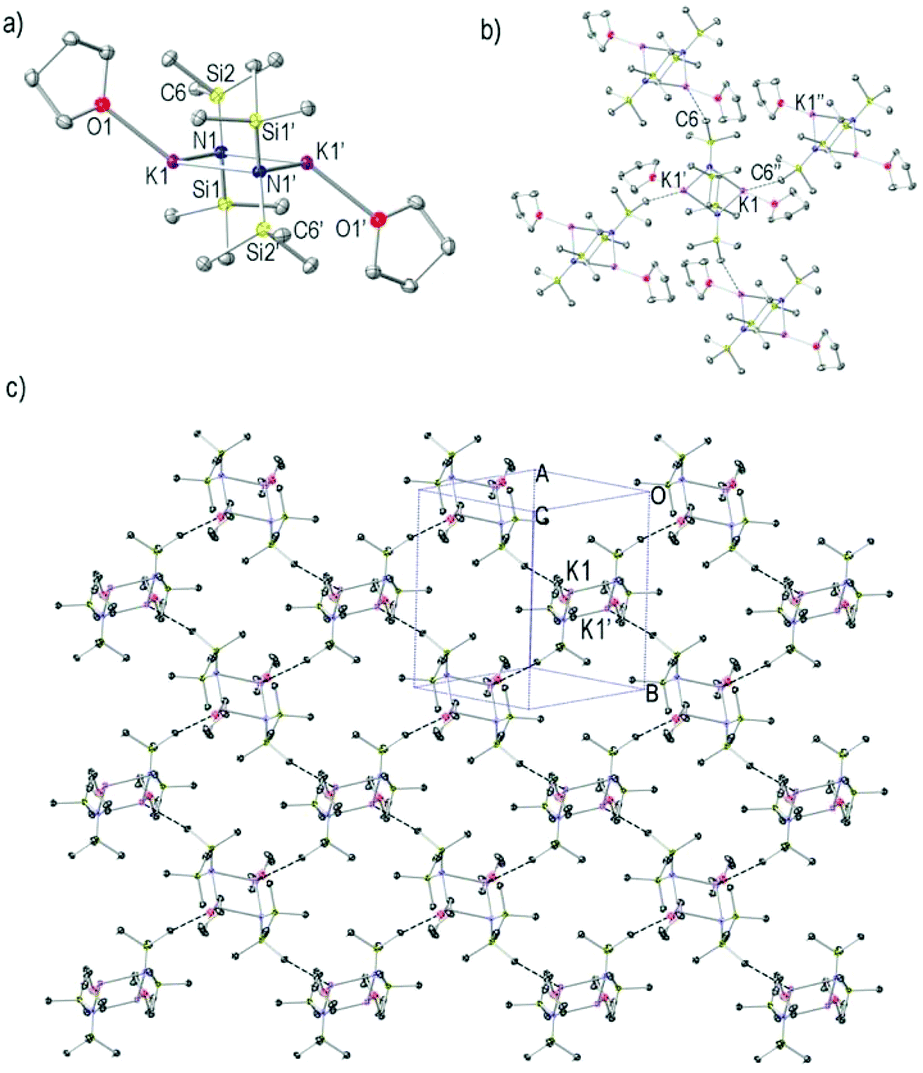 | ||
| Fig. 3 (a) Molecular structure of [(KHMDS)2(THF)2]∞3. Hydrogen atoms and one disordered component for the CH2 framework in the THF ligand are omitted for simplicity. Displacement ellipsoids are displayed at 35% probability. The dashed lines illustrate K⋯C interactions. (b) Section of the extended framework structure showing K⋯C atom connectivity between K and Me and (c) the resulting two-dimensional monolayer packing. Selected bond distances (Å) and angles (°): K1–O1 2.7199(13), K1–N1 2.7442(13), K1–N1′ 2.8534(12), K1⋯C6′′ 3.2226(15), O1–K1–N1 129.65(4), O1–K1–N1′ 114.86(4), N1–K1–N1′ 96.27(3), K1–N1–K1′ 83.73(3), O1–K1–C6′′ 85.98(4), N1–K1–C6′′ 99.32(4), N1′–K1–C6′′ 135.03(4). The symmetry operations used to generate the equivalent atoms numbered with ′ and ′′ are −x + 1, −y + 2, −z and −x + 3/2, y + 1/2, −z + 1/2, respectively. | ||
Derivatives 4–6 crystallised as discrete dimers (Fig. 4a–c). Whilst 4 and 6 are both in the monoclinic space group P21/c, 5 is in the orthorhombic Sohncke space group P212121. TMEDA and its chiral analogue (R,R)-TMCDA are widely utilised bidentate Lewis donor ligands in the chemistry of alkali metals;70–72 thus surprisingly, 4 and 5 represent the first structural examples of a KHMDS species solvated by these important diamine ligands. The asymmetric unit of 4 contains three distinct molecules of [(KHMDS)2(TMEDA)2]; two of them present some degree of disorder, which hampers their structural discussion. The third is discussed below. Complexes 4–6 resemble the archetypical planar four-membered K2N2 metalla-cyclo-dimer [sum of endocyclic angles, 359° for 4 and 357° for 5] previously seen in 1–3 and in the solvent-free [(KHMDS)2]∞.33,34 In 4 and 5, each K atom is coordinated to two bridging μ-HMDS amido groups and to the chelating TMEDA and (R,R)-TMCDA ligands, respectively, resulting in the metals being four coordinate in a distorted tetrahedral geometry [N–K–N range 63.27(5)–135.20(5) and 62.86(5)–133.38(5)° for K5 and K6 in 4, and 55.22(6)–130.71(7) and 60.14(6)–138.81(7)° for K1 and K2 in 5; Fig. 4a and b, respectively]. However, each K in the centrosymmetric 6 binds a tetradentate 12-crown-4 ligand resulting in six-coordinated metal centres. In 4, the K–NHMDS bond distances are identical [2.7942(17) and 2.7876(17) Å], whilst for 5 there is a short and long set of K–NHMDS bonds [short K2–N1 and K2–N2 lengths of 2.785(2) and 2.764(2) Å, and long K1–N1 and K1–N1 lengths of 2.908(2) K1–N2 2.884(2) Å, respectively]. As expected, these are shorter than the K–NTMEDA lone-pair dative interactions with either TMEDA or (R,R)-TMCDA ligands in 4 and 5, respectively [K–NTMEDA range 2.8529(19)–2.9489(19) Å in 4 and K–N(R,R)-TMCDA range 2.878(2)–3.035(2) Å in 5]. The coordination modes of the K atoms in 4 and 5 closely resemble those found in related dimeric [(KDA)2(TMEDA)2]73 and [(KTMP)2(TMEDA)2].74 Turning to 6 (Fig. 4c), the K–NHMDS bond distances are noticeably longer [2.913(2) and 3.073(2) Å] than those for 4 and 5, and as expected longer than that in the donor-free reagent [mean 2.787 Å].33 This is presumably the result of the different nature and higher denticity of the 12-crown-4 ligand resulting in six coordinate K atoms. The 12-crown-4 ligand coordinates the K metal centres being slightly distorted, thus resulting in four distinct K–O bond lengths. Three of them are in the range 2.773(2)–2.912(2) Å; however, the fourth K–O distance (K1–O4) is considerably longer at 3.121(2) Å. The K atoms are located approximately 2.1 Å above the mean plane of O atoms within the 12-crown-4 molecules. The internal angles at the K atoms in the centrosymmetric K2N2 ring are approximately 14° wider than the angles at the N atoms [N–K–N 96.92(5), K–N–K 83.08(5)°], where the latter present an intermediate value between those found in 4 and 5. The mean K–N bond distance of the central (KHMDS)2 ring of 6 is 2.99 Å. In keeping with the other complexes discussed herein, the coordination spheres of both K atoms in 4–6 are completed by a series of long intramolecular K⋯CHMDS interactions [range 3.313(3)–3.397(2) and 3.180(3)–3.369(3) Å in 4 and 5, respectively] while a single K⋯CHMDS interaction can be found in 6 for each potassium atom [3.313(3) Å]. Complex 6 is a rare example of a potassium amide 12-crown-4 dimer, only two other examples have been reported with different amides, (2-phenylamido)pyridine75 and (trimethylsilylamido)pyridine.76
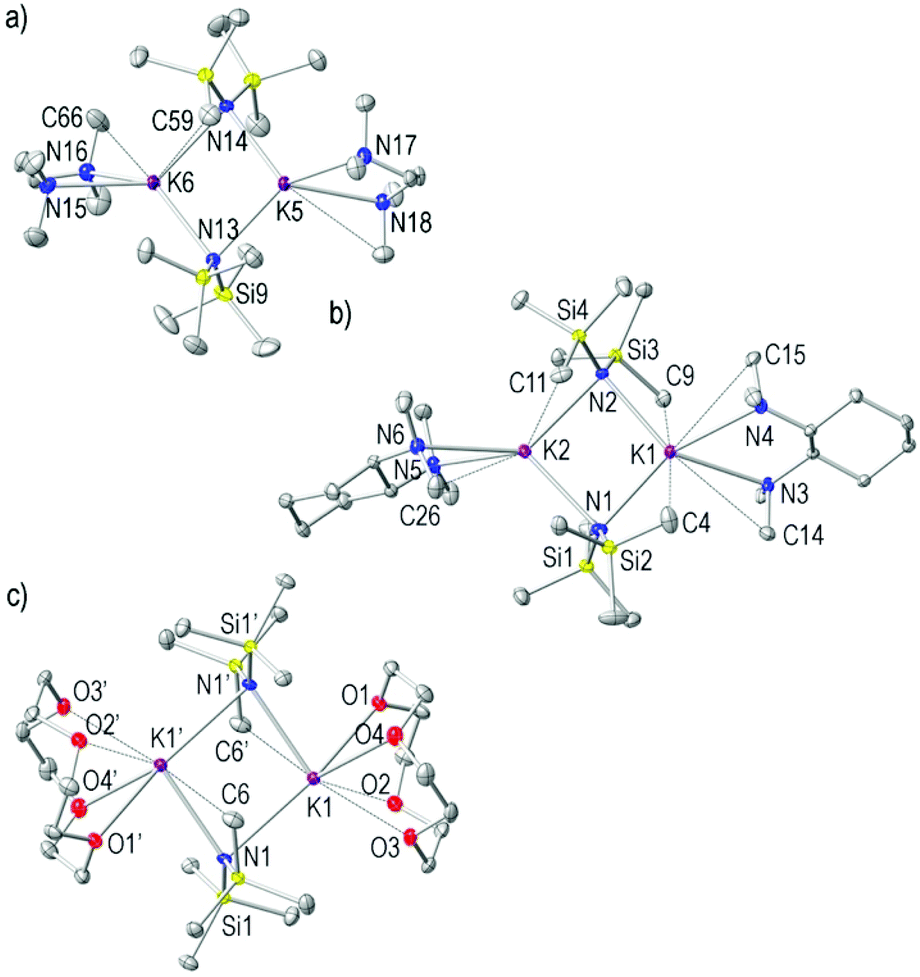 | ||
| Fig. 4 (a) Molecular structure of [(KHMDS)2(TMEDA)2] 4, showing one of the three crystallographically independent molecules within the asymmetric unit. (b) Molecular structure of [(KHMDS)2{(R,R)-TMCDA}2] 5. (c) Molecular structure of the centrosymmetric [(KHMDS)2(12-crown-4)2] 6. Hydrogen atoms, one disordered component of one SiMe3 group of 4 and one disordered molecule of toluene of crystallisation of 6 are omitted for simplicity. Displacement ellipsoids are displayed at 35% probability. The dashed lines illustrate K⋯C and K⋯O long interactions. Selected bond distances (Å) and angles (°): for 4, K5–N13 2.8365(16), K5–N14 2.7942(17), K5–N17 2.9244(17), K5–N18 2.8762(17), K6–N13 2.7876(17), K6–N14 2.8765(17), K6–N15 2.9489(19), K6–N16 2.8529(19), K5⋯C71 3.373(3), K5⋯C59 3.397(3), K6⋯C66 3.313(2), N13–K5–N14 98.99(5), N13–K5–N17 135.20(5), N13–K5–N18 120.17(5), N14–K5–N17 115.45(5), N14–K5–N18 123.69(5), N17–K5–N18 63.27(5), N13–K6–N16 133.38(5), N13–K6–N14 98.19(5), N16–K6–N14 111.74(5), N13–K6–N15 118.70(5), N15–K6–N16 62.86(5), N14–K6–N15 133.61(5), K5–N14–K6 80.99(4), K5–N13–K6 81.81(4); for 5, K1–N2 2.884(2), K1–N1 2.908(2), K1–N5 2.959(2), K1–N6 3.035(2), K2–N1 2.785(2), K2–N3 2.878(2), K2–N2 2.764(2), K2–N4 2.878(2), K1–C11 3.369(3), K1–C26 3.185(3), K2–C4 3.223(3), K2–C9 3.180(3), K2–C14 3.346(3), K2–C15 3.299(3), N1–K1–N2 92.04(6), N2–K1–N5 128.12(7), N1–K1–N5 125.61(7), N2–K1–N6 130.71(7), N1–K1–N6 125.71(7), N5–K1–N6 57.22(6), K1–N1–K2 84.78(6), N1–K2–N2 97.37(7), N2–K2–N3 138.81(7), N1–K2–N3 115.26(7), N2–K2–N4 106.44(7), N1–K2–N4 144.64(7), N3–K2–N4 60.14(6), K1–N2–K2 85.62(6); for 6, K1–O1 2.9124(19), K1–O2 2.773(2), K1–O3 2.8024(18), K1–O4 3.121(2), K1–N1 3.073(2), K1–N1′ 2.913(2), K1′–N1 2.913(2), K1⋯C6′ 3.313(3), O2–K1–O3 61.01(6), O2–K1–O1 59.05(6), O3–K1–O1 84.81(5), O2–K1–N1′ 91.72(6), O3–K1–N1′ 79.98(6), O1–K1–N1′ 150.78(6), O2–K1–N1 138.32(6), O3–K1–N1 160.67(6), O1–K1–N1 105.29(6), K1–N1–K1′ 83.08(5), N1′–K1–N1 96.92(5), O2–K1–O4 88.27(6), O3–K1–O4 55.82(5), O1–K1–O4 56.26(6), N1′–K1–O4 128.82(6), N1–K1–O4 116.04(5). The symmetry operation used to generate the equivalent atoms numbered with ′ in 6 is −x + 1, −y, −z. | ||
Complexes 7 and 8 both crystallised in the monoclinic system space group P21/n, whilst 9 crystallised in the triclinic system P. Complexes 7–9 (Fig. 5a–c) are essentially isostructural and can be considered as being composed of two [(donor)K–N–K–N] chains which are linked together though two K–NHMDS bonds forming a 1:2 donor:KHMDS tetranuclear discrete arrangement with a central cyclo-dimer (KHMDS)2 unit. The central K2N2 ring is planar in 7–9 (sum of endocyclic angles, 360° for 7–9). Two distinct types of K atoms are found in 7–9 occupying ‘internal’ and ‘external’ positions. Tridentate PMDETA and TMDAE, and the tetradentate Me6TREN ligand respectively coordinate the external K atoms in an anti-disposition. The terminal metals are in a distorted tetrahedral environment in 7 and distorted square planar environment in 8. In 9, the tetradentate amine Me6TREN coordinates the external K centres in a η4-manner, rendering these metals five-coordinated [N–K–N range 62.42(7)–165.97(6)°]. The three N-donor arms emerging from the central N donor in the tripodal Me6TREN ligand are disposed in a plane in which the K atoms occupy a position approximately 1.41 Å above it. The internal K atoms bind to two bridging NHMDS atoms forming the cyclic (KHMDS)2 motif (mean K–NHMDS 2.83, 2.85 and 2.88 Å for 7, 8 and 9, respectively) and additionally to a bridging NHMDS atom from a [(KHMDS)(donor)] unit [K1–N2 2.8808(11) for 7, K2–N3 2.9254(11) for 8 and K2–N5 2.906(2) Å for 9]. The K atoms in the central cyclo-dimer present a distorted trigonal planar geometry in the three examples [range N–K–N 95.93(3)–141.54(4) for 7, N4′–K2–N3 95.65(3)–140.39(3) for 8 and 96.58(6)–132.40(6)° for 9]. The K–NHMDS bond distances are similar in 7–9 and as alluded to previously it is they are shorter than the corresponding K–Ndonor bond length [mean K–NPMDETA distance 2.90 Å for 7, mean K–NTMDAE and K–OTMDAE distances of 2.98 and 2.7362(10), respectively, for 8, and mean K–NMe6TREN distance 3.00 Å for 9]. Additionally, the K atoms in 7, 8 and 9 show stabilizing long K⋯C contacts with methyl groups from the HMDS and donor ligands [K⋯C range 3.3223(17)–3.366(3) for 7, 3.3147(14)–3.3353(15) for 8, 3.239(4)–3.402(3) Å for 9]. A search of the Cambridge Crystallographic Database77 reveals only four solid state structures with Me6TREN coordinating to a K metal centre, including benzyl potassium complexes [PhCH2K(Me6TREN)] and 3,5-dimethylbenzyl potassium [3,5-Me2C6H3CH2K].55,78
 | ||
| Fig. 5 (a) Molecular structures of [(KHMDS)2{(PMDETA·KHMDS)2}] 7, (b) [(KHMDS)2{(TMDAE·KHMDS)2}] 8 and (c) [(KHMDS)2{(Me6TREN·KHMDS)2}] 9. Hydrogen atoms and one toluene molecule of crystallisation for 9 are omitted for simplicity. Displacement ellipsoids are displayed at 35% probability. Selected bond distances (Å) and angles (°): for 7, K1–N1 2.8236(13), K1–N1′ 2.8452(13), K1–N2 2.8808(11), N1–K1′ 2.8451(13), K2–N2 2.7536(12), K2–N4 2.8525(13), K2–N3 2.9035(15), K2–N5 2.9528(13), K2⋯C9 3.3223(17), K2⋯C15 3.366(3), K1–N1–K1′ 84.08(3), K1–N2–K2 106.43(4), N1–K1–N1′ 95.93(3), N1–K1–N2 141.54(4), N1′–K1–N2 122.13(4), N2–K2–N4 120.54(4), N2–K2–N3 131.85(4), N4–K2–N3 63.47(4), N2–K2–N5 123.17(4), N4–K2–N5 64.90(4), N3–K2–N5 102.46(4), the symmetry operation used to generate the equivalent atoms numbered with ′ is −x + 1, −y + 1, −z + 1; for 8, K1–O1 2.7362(10), K1–N3 2.8367(11), K1–N2 2.9579(12), K1–N1 2.9972(13), K2–N4′ 2.8288(11), K2–N4 2.8660(11), K2–N3 2.9254(11), O1–K1–N3 173.68(3), O1–K1–N2 59.83(3), K1⋯C18′ 3.3147(14), K2⋯C18′ 3.3353(15), K2–N4–K2′ 84.35(3), K2–N3–K1 108.36(3), N3–K1–N2 126.25(3), O1–K1–N1 60.04(4), N3–K1–N1 113.95(4), N2–K1–N1 119.79(4), N4′–K2–N4 95.65(3), N4′–K2–N3 121.12(3), N4–K2–N3 140.39(3), the symmetry operation used to generate the equivalent atoms numbered with ′ is −x + 2, −y + 1, −z + 1; for 9, K1–N5 2.913(2), K1–N2 2.919(2), K1–N1 3.003(3), K1–N4 3.018(2), K1–N3 3.048(2), N5–K2 2.906(2), K2–N6′ 2.846(2), K2–N6 2.848(2), N6–K2′ 2.846(2), K1⋯C23 3.402(3), K2⋯C30′ 3.239(4), K2–N6–K2′ 83.42(6), K2–N5–K1′ 117.64(7), N5–K1–N2 119.91(6), N5–K1–N1 126.90(7), N2–K1–N1 101.57(7), N5–K1–N4 165.97(6), N2–K1–N4 62.44(6), N1–K1–N4 62.42(7), N5–K1–N3 105.29(7), N2–K1–N3 96.24(7), N1–K1–N3 101.16(8), N4–K1–N3 60.96(6), K2–N5–K1 117.64(7), N6′–K2–N6 96.58(6), N6′–K2–N5 132.40(6), N6–K2–N5 126.95(6), the symmetry operation used to generate the equivalent atoms labelled with ′ is −x + 3, −y + 1, −z + 1. | ||
In an effort to prepare and characterise the solid state structures of monomeric KHMDS species we used TMDAE and the heptadentate TMEEA donor molecules in 2:1 and 1:1 donor:HMDS molar ratios yielding 10 and 11, respectively. X-ray crystallographic analysis reveals that 10 crystallises in the monoclinic space group P121/a1 whilst 11 crystallises in the triclinic space group P. The structures of 10 and 11 (Fig. 6a and b, respectively) consist of discrete solvated monomeric species of KHMDS containing two molecules of TMDAE and one molecule of the multidentate TMEEA molecule coordinating the K metal centres, respectively. Two chemically identical but crystallographically distinct molecules of 10 are observed within its asymmetric cell; however, for simplicity and due to disorder in one of the molecules, the structural parameters of only one molecule will be discussed. The K metal centre is seven-coordinate bound to the TMDAE and HMDS ligands in 10, increasing its coordination number to eight in 11. The salient structural feature of 10 and 11 is that these compounds are monomeric. In keeping with the tendency observed in the previous examples, the K–NHMDS bond lengths found in 10 are shorter than the K–Ndonor bond distances [K2–N6HMDS 2.794(3), mean K–Ndonor 3.208 Å] with K–O bond distances of 2.814(2) and 2.824(2) for K2–O3 and K2–O4, respectively. Also, in 11, the K–NHMDS bond length [K1–N2 2.7889(9) Å] is shorter than the K–Ndonor [K1–N1 3.1364(9) Å], and noticeably one of the K–O bond lengths is shorter than K–NHMDS bond distance [K1–O1 2.7613(8) Å] while the rest of the K–O bond lengths are in the range 2.8131(9)–3.3145(10) Å. One example of a solvated monomeric species [KHMDS(18-crown-6)] was previously crystallographically characterised79 where the K atom is hepta-coordinated to the chelating crown ligand and HMDS group. Both compounds, 10 and 11, crystallise from toluene solutions. The affinity of heavy alkali metals to engage π-interactions with arenes is well known;42,80–86 however, in the presence of the corresponding multidentate ligands TMDAE and TMEEA in 10 and 11, respectively, there is no interaction with the arene in the solid-state structures.
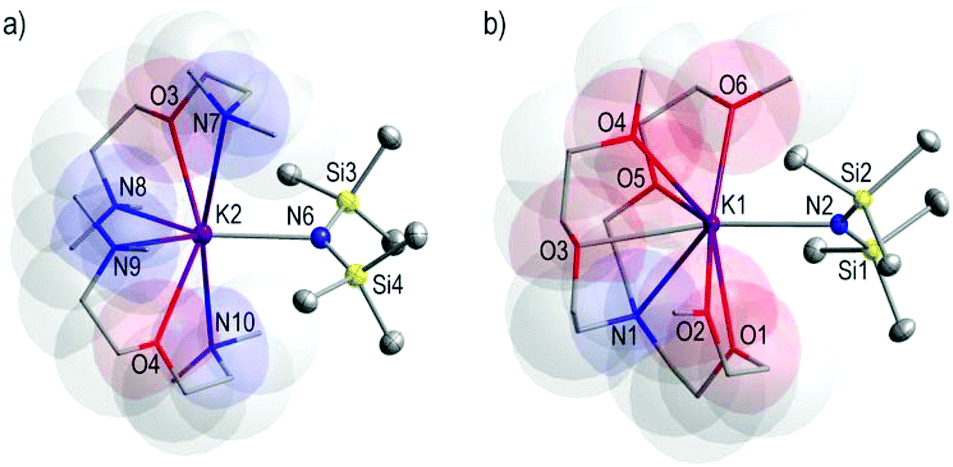 | ||
| Fig. 6 (a) Molecular structure of [KHMDS(TMDAE)2] 10, showing one of the two crystallographically independent molecules within the asymmetric unit. (b) Molecular structure of [KHMDS(TMEEA)] 11. Hydrogen atoms are omitted for simplicity. Displacement ellipsoids are displayed at 35% probability. The TMDAE and TMEEA ligands are pictured in 10 and 11 as capped sticks with translucent space-filling van der Waals surfaces for a probe of 1.5 Å radius. Selected bond distances (Å) and angles (°): for 10, K2–N6 2.794(3), K2–O3 2.814(2), K2–O4 2.824(2), K2–N9 2.960(3), K2⋯N10 3.559(3), K2–N8 2.975(3), K2⋯N7 3.337(3), N6–K2–O3 111.44(8), N6–K2–O4 106.42(9), O3–K2–O4 141.91(8), N6–K2–N9 132.63(8), O3–K2–N9 95.50(9), O4–K2–N9 60.13(9), N6–K2–N8 134.58(9), O3–K2–N8 59.22(8), O4–K2–N8 90.93(9), N9–K2–N8 92.54(9), N6–K2–N7 83.46(9), O3–K2–N7 55.33(8), O4–K2–N7 135.52(8), N9–K2–N7 81.01(9), N8–K2–N7 113.00(9), N6–K2–N10 83.97(8), N10–K2–N7 166.99(8); for 11, K1–O1 2.7613(8), K1–N2 2.7889(9), K1–O6 2.8131(8), K1–O4 2.8736(8), K1–O5 2.9078(8), K1–O3 2.9652(8), K1–N1 3.1364(9), K1⋯O2 3.3145(10), O1–K1–N2 80.00(3), O1–K1–O6 150.12(3), N2–K1–O6 83.93(3), O1–K1–O4 137.21(3), N2–K1–O4 121.03(3), O6–K1–O4 72.63(2), O1–K1–O5 111.53(2), N2–K1–O5 122.61(2), O6–K1–O5 57.96(2), O4–K1–O5 88.90(2), O1–K1–O3 90.45(2), N2–K1–O3 161.50(2), O6–K1–O3 111.11(2), O4–K1–O3 57.56(2), O5–K1–O3 75.61(2), O1–K1–N1 57.01(2), N2–K1–N1 126.60(2), O6–K1–N1 117.35(2), O4–K1–N1 112.19(2), O5–K1–N1 59.71(2), O3–K1–N1 57.04(2), O1–K1–O2 56.12(2), N2–K1–O2 85.08(3), O6–K1–O2 147.03(2), O4–K1–O2 86.86(2), O5–K1–O2 149.24(2), O3–K1–O2 76.45(2), N1–K1–O2 94.02(2). | ||
NMR spectroscopic studies
Crystalline compounds 1–11 were studied by 1H, 13C and DOSY NMR spectroscopies in C6D6 solution. The 1H and 13C NMR spectra of 1–11 showed two distinct set of signals corresponding to the Lewis base donor of choice and the HMDS group. In general, the Lewis donor ligand:HMDS ratio found in the 1H NMR spectra is in agreement with the proportions found in the crystalline structures of complexes. However, the observed toluene:HMDS ratios in the 1H NMR spectra of 1 and 2 were slightly smaller than expected (1:2 and 1:1, respectively) presumably due to the partial removal of the labile arene ligand during their isolation. The 1H NMR chemical shifts for the HMDS ligand in the toluene-containing compounds 1 and 2, and in the PMDETA-solvate 7 are practically identical (0.13 ppm for 1 and 2, and 0.14 ppm for 7), whilst it is marginally shifted downfield for the THF-solvate 3 (0.16 ppm). For the dimeric complexes containing the bidentate N-donor ligands TMEDA and (R,R)-TMCDA, 4 and 5, the HMDS singlet appears only slightly shifted downfield (0.23 ppm in both) with respect to 1–3, and is similar to that found for the TMEDA species 4 (0.24 ppm). However, the 1H NMR resonance for the HMDS ligand for the 12-crown-4, Me6TREN and TMDAE derivatives 6, 9 and 10 appears markedly further downfield (0.35, 0.30 and 0.37 ppm, respectively), being even farther shifted for the monomeric TMEEA species 11 (0.49 ppm). The HMDS resonance in the 13C NMR spectra for the complexes 1–11 appears in the range 7.2–7.5 ppm.
Conclusions
The solid and solution structural chemistry of KHMDS has been developed by exploring its coordination with a series of Lewis donor molecules. In the solid state, four different aggregation forms of KHMDS have been found, namely, a linear polymeric chain array (in the presence of toluene), two-dimensional polymeric layer arrangement (with THF), dimeric arrangements (with toluene, TMEDA, R,R-TMCDA and 12-crown-4), tetranuclear aggregates (with PMDETA, TMDAE and Me6TREN), and discrete monomeric species (with TMDAE and TMEEA). Solution studies by DOSY NMR spectroscopy in [D8]THF and arene (C6D6 and D8-toluene) reveal that monomerisation of KHMDS and displacement of the Lewis donor by these solvents occur in all the examples studied at ambient temperature.Experimental
General procedures
All manipulation and reactions were performed under an atmosphere of dry pure argon gas using standard high vacuum Schlenk and glove box techniques. n-Hexane, toluene and tetrahydrofuran (THF) were distilled under reflux with sodium metal and benzophenone under a nitrogen atmosphere. Toluene was additionally stored over activated 4 Å molecular sieves. C6D6, [D8]toluene and [D12]cyclohexane were degassed and dried over activated 4 Å molecular sieves. [D8]THF was stored over a potassium metal mirror. KHMDS was purchased from Aldrich and used as received. N,N,N′,N′-Tetramethylethylenediamine (TMEDA), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) and N,N,N′,N′-tetramethyldiaminoethyl ether (TMDAE) were purchased from Aldrich, distilled with CaH2 under a nitrogen atmosphere and stored over activated 4 Å molecular sieves. Tris{2-(2-methoxyethoxy)ethyl}amine (TMEEA) and 12-crown-4 were purchased from Aldrich and stored over activated 4 Å. Tris[2-dimethyl(amino)ethyl]amine was prepared by a previously reported method87 and stored over activated 4 Å molecular sieves. NMR spectra were collected on a Bruker AV400 MHz spectrometer operating at 400.1 and 100.6 MHz for 1H and 13C, respectively. 1H and 13C{1H} NMR chemical shifts are expressed in parts per million (δ, ppm) and referenced to residual solvent peaks. Microanalyses were obtained using a PerkinElmer 2400 elemental analyser.X-ray crystallography
Crystallographic data were collected at 123(2) K on Oxford Diffraction Xcalibur (Mo-Kα radiation, λ = 0.71073 Å, for 3, 4, 6, and 11) and Gemini (Mo-Kα radiation, λ = 0.71073 Å, for 9; Cu-Kα radiation, λ = 1.5418 Å, for 1, 2, 5, 7, 8 and 10) diffractometers. The structures were solved and refined to convergence on F2 and against all independent reflections by full-matrix least-squares using SHELXS and SHELXL programs,88 respectively. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were geometrically placed and allowed to ride on their parent atoms. The THF ligand in 3, SiMe3 groups and a TMEDA ligand in 4, the chelate group of 10, and toluene molecules of crystallisation in 6 and 9 were modelled as disordered over two sites with the geometry and displacement parameters of these groups restrained to approximate typical values. Selected crystallographic and refinement details are provided in Tables S1 and S2.† CCDC 1537847 to 1537857 (1–11) contain the supplementary crystallographic data for this paper.:1 mixture of n-hexane/toluene (9 mL) in a hot water bath (24 h). The crystalline material was filtered, washed with cold n-hexane (5 mL) and dried under vacuum for 10 min. Yield: 0.44 g, 0.94 mmol, 38%. In agreement with 1H NMR and microelemental analyses toluene is partially removed (approximately 25%) during isolation of 1. 1H NMR (400.1 MHz, C6D6, 300 K): δ 0.13 (s, 36 H, Me3Si), 2.11 (s, 3 H, Me-toluene), 7.03 (m, 3 H, toluene), 7.12 (d, 2 H, 3JHH = 8.0 Hz, toluene). 13C{1H} NMR (100.6 MHz, C6D6, 300 K): δ 7.2 (Me3Si), 21.4 (Me-toluene), 125.7 (para-CH, toluene), 128.6 (meta-CH, toluene), 129.3 (ortho-CH, toluene), 137.9 (ipso-C, toluene). Anal. calcd (found) for C12H36K2N2Si4·C5.25H6: C, 44.26 (45.44); H, 9.04 (9.25); N, 5.98% (5.69%).
:5 mixture of n-hexane/toluene (6 mL) at −27 °C (12 h). The crystalline material was filtered, washed with cold n-hexane (10 mL) and dried under vacuum for 15 min. Yield: 0.72 g, 1.38 mmol, 69%. 1H NMR (400.1 MHz, C6D6, 300 K): δ 0.49 (s, 18 H, Me3Si), 2.25 (t, 6 H, 3JHH = 5.0 Hz, CH2–N), 3.16 (t, 6 H, 3JHH = 5.0 Hz, N–CH2–CH2–O), 3.22 (s, 9 H, MeO), 3.31 (m, 6 H, CH2–OMe), 3.36 (m, 6 H, O–CH2–CH2–OMe). 13C{1H} NMR (100.6 MHz, C6D6, 300 K): δ 7.4 (Me3Si), 55.2 (CH2–N), 58.9 (MeO), 68.6 (N–CH2–CH2–O), 70.2 (O–CH2–CH2–OMe), 72.1 (CH2–OMe). Anal. calcd (found) for C21H51KN2O6Si2: C, 48.24 (48.42); H, 9.83 (10.18); N, 5.36% (5.63%).
Acknowledgements
We would like to thank Prof. Robert Mulvey and Prof. Eva Hevia for useful and insightful discussions and the financial support of the Engineering and Physical Science Research Council (EPSRC) (Career Acceleration Fellowship, EP/J001872/1 and EP/L001497/1). The research data associated with this paper are openly available at 10.15129/6e4353f6-8046-4cfd-82b3-a9c7d8f465e9.References
- R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470 CrossRef CAS PubMed.
- M. P. Coles, Coord. Chem. Rev., 2015, 297–298, 2 CrossRef CAS.
- A. Streitwieser, A. Facchetti, L. Xie, X. Zhang and E. C. Wu, J. Org. Chem., 2012, 77, 985 CrossRef CAS PubMed.
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O'Hara, Inorg. Chem., 2016, 55, 5719 CrossRef CAS PubMed.
- D. C. Bradley, R. G. Copperthwaite, M. W. Extine, W. W. Reichert and M. H. Chisholm, in Inorganic Syntheses, John Wiley & Sons, Inc., 1978, p. 112 Search PubMed.
- H. Bürger and U. Wannagat, Monatsh. Chem., 1963, 94, 1007 CrossRef.
- H. Bürger and U. Wannagat, Monatsh. Chem., 1964, 95, 1099 CrossRef.
- S. A. Schuetz, V. W. Day, R. D. Sommer, A. L. Rheingold and J. A. Belot, Inorg. Chem., 2001, 40, 5292 CrossRef CAS PubMed.
- L. R. Avens, S. G. Bott, D. L. Clark, A. P. Sattelberger, J. G. Watkin and B. D. Zwick, Inorg. Chem., 1994, 33, 2248 CrossRef CAS.
- R. A. Andersen, Inorg. Chem., 1979, 18, 1507 CrossRef CAS.
- A. Huang, J. J. Kodanko and L. E. Overman, J. Am. Chem. Soc., 2004, 126, 14043 CrossRef CAS PubMed.
- D. A. Evans and T. C. Britton, J. Am. Chem. Soc., 1987, 109, 6881 CrossRef CAS.
- P. F. Godenschwager and D. B. Collum, J. Am. Chem. Soc., 2008, 130, 8726 CrossRef CAS PubMed.
- A. J. McNeil and D. B. Collum, J. Am. Chem. Soc., 2005, 127, 5655 CrossRef CAS PubMed.
- B. C. Hamann and J. F. Hartwig, J. Am. Chem. Soc., 1997, 119, 12382 CrossRef CAS.
- F. A. Davis and K. R. Prasad, J. Org. Chem., 2003, 68, 7249 CrossRef CAS PubMed.
- T. Yokozawa and Y. Ohta, Chem. Commun., 2013, 49, 8281 RSC.
- J. Åhman, T. Jarevång and P. Somfai, J. Org. Chem., 1996, 61, 8148 CrossRef.
- R. C. Sawant, Y.-H. Lih, S.-A. Yang, C.-H. Yeh, H.-J. Tai, C.-L. Huang, H.-S. Lin, S. S. Badsara and S.-Y. Luo, RSC Adv., 2014, 4, 26524 RSC.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354 RSC.
- R. Michel, T. Nack, R. Neufeld, J. M. Dieterich, R. A. Mata and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 734 CrossRef CAS PubMed.
- T. J. Katz, A. Sudhakar, M. F. Teasley, A. M. Gilbert, W. E. Geiger, M. P. Robben, M. Wuensch and M. D. Ward, J. Am. Chem. Soc., 1993, 115, 3182 CrossRef CAS.
- K. Searles, A. K. Das, R. W. Buell, M. Pink, C.-H. Chen, K. Pal, D. G. Morgan, D. J. Mindiola and K. G. Caulton, Inorg. Chem., 2013, 52, 5611 CrossRef CAS PubMed.
- Y. Yamashita and S. Kobayashi, Chem. – Eur. J., 2013, 19, 9420 CrossRef CAS PubMed.
- Y. Yamashita, I. Sato, H. Suzuki and S. Kobayashi, Chem. – Asian J., 2015, 10, 2143 CrossRef CAS PubMed.
- H. Suzuki, I. Sato, Y. Yamashita and S. Kobayashi, J. Am. Chem. Soc., 2015, 137, 4336 CrossRef CAS PubMed.
- S. Tabassum, O. Sereda, P. V. G. Reddy and R. Wilhelm, Org. Biomol. Chem., 2009, 7, 4009 CAS.
- W.-Z. Zhang, X. Ren and X.-B. Lu, Chin. J. Chem., 2015, 33, 610 CrossRef CAS.
- A. Harinath, S. Anga and T. K. Panda, RSC Adv., 2016, 6, 35648 RSC.
- S. Anga, Y. Sarazin, J.-F. Carpentier and T. K. Panda, ChemCatChem, 2016, 8, 1373 CrossRef CAS.
- C.-L. Sun, H. Li, D.-G. Yu, M. Yu, X. Zhou, X.-Y. Lu, K. Huang, S.-F. Zheng, B.-J. Li and Z.-J. Shi, Nat. Chem., 2010, 2, 1044 CrossRef CAS PubMed.
- J. P. Barham, G. Coulthard, K. J. Emery, E. Doni, F. Cumine, G. Nocera, M. P. John, L. E. A. Berlouis, T. McGuire, T. Tuttle and J. A. Murphy, J. Am. Chem. Soc., 2016, 138, 7402 CrossRef CAS PubMed.
- K. F. Tesh, T. P. Hanusa and J. C. Huffman, Inorg. Chem., 1990, 29, 1584 CrossRef CAS.
- P. G. Williard, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 270 CrossRef.
- R. Neufeld, R. Michel, R. Herbst-Irmer, R. Schöne and D. Stalke, Chem. – Eur. J., 2016, 22, 12340 CrossRef CAS PubMed.
- A. M. Domingos and G. M. Sheldrick, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 517 CrossRef CAS.
- W. J. Evans, D. B. Rego and J. W. Ziller, Inorg. Chem., 2006, 45, 3437 CrossRef CAS PubMed.
- C. G. Werncke, E. Suturina, P. C. Bunting, L. Vendier, J. R. Long, M. Atanasov, F. Neese, S. Sabo-Etienne and S. Bontemps, Chem. – Eur. J., 2016, 22, 1668 CrossRef CAS PubMed.
- R. W. Alder, M. E. Blake, C. Bortolotti, S. Bufali, C. P. Butts, E. Linehan, J. M. Oliva, A. G. Orpen and M. J. Quayle, Chem. Commun., 1999, 241 RSC.
- M. S. Hill, G. Kociok-Köhn and D. J. MacDougall, Inorg. Chem., 2011, 50, 5234 CrossRef CAS PubMed.
- P. L. Arnold, I. J. Casely, Z. R. Turner, R. Bellabarba and R. B. Tooze, Dalton Trans., 2009, 7236 RSC.
- J. J. Morris, B. C. Noll, G. W. Honeyman, C. T. O'Hara, A. R. Kennedy, R. E. Mulvey and K. W. Henderson, Chem. – Eur. J., 2007, 13, 4418 CrossRef CAS PubMed.
- X. He, B. C. Noll, A. Beatty, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444 CrossRef CAS PubMed.
- B. Conway, P. García-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, New J. Chem., 2010, 34, 1707 RSC.
- W. Clegg, G. C. Forbes, A. R. Kennedy, R. E. Mulvey and S. T. Liddle, Chem. Commun., 2003, 406 RSC.
- D. J. Liptrot, M. S. Hill and M. F. Mahon, Chem. – Eur. J., 2014, 20, 9871 CrossRef CAS PubMed.
- G. C. Forbes, A. R. Kennedy, R. E. Mulvey, R. B. Rowlings, W. Clegg, S. T. Liddle and C. C. Wilson, Chem. Commun., 2000, 1759 RSC.
- A. R. Kennedy, R. E. Mulvey, C. L. Raston, B. A. Roberts and R. B. Rowlings, Chem. Commun., 1999, 353 RSC.
- J. J. Morris, B. C. Noll and K. W. Henderson, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2477 CAS.
- P. G. Williard and M. A. Nichols, J. Am. Chem. Soc., 1991, 113, 9671 CrossRef CAS.
- K. R. Meihaus, J. F. Corbey, M. Fang, J. W. Ziller, J. R. Long and W. J. Evans, Inorg. Chem., 2014, 53, 3099 CrossRef CAS PubMed.
- W. J. Evans, M. Fang, G. Zucchi, F. Furche, J. W. Ziller, R. M. Hoekstra and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 11195 CrossRef CAS PubMed.
- P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and R. B. Rowlings, Eur. J. Inorg. Chem., 2003, 3354 CrossRef CAS.
- L. T. J. Evans, F. G. N. Cloke, M. P. Coles and P. B. Hitchcock, Inorg. Chim. Acta, 2007, 360, 1258 CrossRef CAS.
- D. R. Armstrong, M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Inorg. Chem., 2013, 52, 12023 CrossRef CAS PubMed.
- C. Glock, F. M. Younis, S. Ziemann, H. Görls, W. Imhof, S. Krieck and M. Westerhausen, Organometallics, 2013, 32, 2649 CrossRef CAS.
- D. Hoffmann, W. Bauer, P. v. R. Schleyer, U. Pieper and D. Stalke, Organometallics, 1993, 12, 1193 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and R. Sablong, Dalton Trans., 2006, 4146 RSC.
- M. Zhao, L. Wang, P. Li, J. Ma and W. Zheng, Dalton Trans., 2016, 45, 11172 RSC.
- G. Bai, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2002, 21, 2789 CrossRef CAS.
- I. Korobkov, G. Aharonian, S. Gambarotta and G. P. A. Yap, Organometallics, 2002, 21, 4899 CrossRef CAS.
- K. L. Hull, I. Carmichael, B. C. Noll and K. W. Henderson, Chem. – Eur. J., 2008, 14, 3939 CrossRef CAS PubMed.
- A. Arunachalampillai, P. Crewdson, I. Korobkov and S. Gambarotta, Organometallics, 2006, 25, 3856 CrossRef CAS.
- G. Bai, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, J. Chem. Soc., Dalton Trans., 2002, 2437 RSC.
- A. A. Danopoulos, P. Braunstein, K. Y. Monakhov, J. van Leusen, P. Kögerler, M. Clémancey, J.-M. Latour, A. Benayad, M. Tromp, E. Rezabal and G. Frison, Dalton Trans., 2017, 46, 1163 RSC.
- K. C. Mullane, A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2014, 53, 9129 CrossRef CAS PubMed.
- P. L. Arnold, J. B. Love, A. J. Blake and C. Wilson, Recent Adv. Actinide. Sci., 2006, 305, 228 CAS.
- K. Naktode, J. Bhattacharjee, H. P. Nayek and T. K. Panda, Dalton Trans., 2015, 44, 7458 RSC.
- W. J. Evans, M. A. Johnston, R. D. Clark, R. Anwander and J. W. Ziller, Polyhedron, 2001, 20, 2483 CrossRef CAS.
- A. I. Ojeda Amador, A. J. Martinez-Martinez, A. R. Kennedy, D. R. Armstrong and C. T. O'Hara, Chem. Commun., 2017, 53, 324 RSC.
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2007, 129, 8952 CrossRef CAS PubMed.
- C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2008, 130, 11719 CrossRef CAS PubMed.
- W. Clegg, S. Kleditzsch, R. E. Mulvey and P. O'Shaughnessy, J. Organomet. Chem., 1998, 558, 193 CrossRef CAS.
- D. R. Armstrong, D. V. Graham, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Chem. – Eur. J., 2008, 14, 8025 CrossRef CAS PubMed.
- S. T. Liddle, W. Clegg and C. A. Morrison, Dalton Trans., 2004, 2514 RSC.
- S. T. Liddle and W. Clegg, J. Chem. Soc., Dalton Trans., 2001, 402 RSC.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364 CrossRef CAS PubMed.
- W. J. Evans, D. B. Rego and J. W. Ziller, Inorg. Chem., 2006, 45, 3437 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles and C. M. Fitchett, Organometallics, 2015, 34, 2500 CrossRef CAS.
- E. Hevia, G. W. Honeyman, A. R. Kennedy, R. E. Mulvey and D. C. Sherrington, Angew. Chem., Int. Ed., 2005, 44, 68 CrossRef CAS PubMed.
- C. Glock, H. Görls and M. Westerhausen, Inorg. Chem., 2009, 48, 394 CrossRef CAS PubMed.
- P. C. Andrews, V. L. Blair, E. C. Border, A. C. Peatt, J. G. MacLellan and C. D. Thompson, Organometallics, 2013, 32, 7509 CrossRef CAS.
- P. C. Andrews, A. R. Kennedy, R. E. Mulvey, C. L. Raston, B. A. Roberts and R. B. Rowlings, Angew. Chem., Int. Ed., 2000, 39, 1960 CrossRef CAS PubMed.
- M. L. Cole, A. J. Davies, C. Jones and P. C. Junk, J. Organomet. Chem., 2007, 692, 2508 CrossRef CAS.
- F. Antolini, P. B. Hitchcock, A. V. Khvostov and M. F. Lappert, Eur. J. Inorg. Chem., 2003, 2003, 3391 CrossRef.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C and DOSY NMR spectroscopic analyses, selected crystallographic and refinement details for all new compounds. CCDC 1537847–1537857. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt01118k |
| This journal is © The Royal Society of Chemistry 2017 |