DOI:
10.1039/C7DT00782E
(Paper)
Dalton Trans., 2017,
46, 6379-6391
The effects of introducing sterically demanding aryl substituents in [Cu(N^N)(P^P)]+ complexes†
Received
3rd March 2017
, Accepted 22nd April 2017
First published on 24th April 2017
Abstract
The syntheses and characterizations of six [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] compounds (POP = bis(2-(diphenylphosphino)phenyl)ether, xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene), in which N^N is a bpy ligand (1-Naphbpy, 2-Naphbpy, 1-Pyrbpy) bearing a sterically hindered 1-naphthyl, 2-naphthyl or 1-pyrenyl substituent in the 6-position, are reported. Single-crystal structure determinations of five complexes confirm a distorted tetrahedral environment for copper(I) and a preference for the N^N ligand to be oriented with the sterically-demanding aryl group being remote from the (C6H4)2O unit of POP or the xanthene ‘bowl’ of xantphos. The angle between the ring planes of the bpy range from 5.8 to 26.0° and this is associated with interactions between the aryl unit and the phenyl substituents of the P^P ligand. In solution at room temperature, the complexes undergo dynamic behaviour which has been investigated using variable temperature 2D NMR spectroscopy. The [Cu(N^N)(xantphos)]+ complexes exist as a mixture of conformers which interconvert through inversion of the xanthene bowl-shaped unit; the preference for one conformer over the other is significantly changed on going from N^N = Phbpy to 1-Pyrbpy (Phbpy = 6-phenyl-2,2′-bipyridine). The electrochemical and photophysical properties of the [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] compounds are presented; the compounds are orange emitters but the introduction of the 1-naphthyl, 2-naphthyl or 1-pyrenyl substituents result in poor photoluminescence quantum yields.
Introduction
In the last few years, there has been increased interest in harnessing the emissive properties of copper(I) complexes in low-cost, light-emitting electrochemical cells (LECs).1–3 Building upon the pioneering work of McMillin and coworkers,4,5 [Cu(N^N)(POP)]+ and [Cu(N^N)(xantphos)]+ complexes (POP = bis(2-(diphenylphosphino)phenyl)ether, xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene, N^N is an N,N′-chelating ligand) are established as favoured components in the emissive layers in LECs6–14 or organic light-emitting devices (OLEDs).15,16 Related complexes find application in oxygen sensing,17 CO2 reduction,18 and water reduction.19,20 The N^N ligand in [Cu(N^N)(POP)]+ and [Cu(N^N)(xantphos)]+ is usually a 2,2′-bipyridine (bpy) or 1,10-phenanthroline (phen) derivative, and the introduction of simple substituents into the 6- and 6′-positions of bpy or 2- and 9-positions of phen modulates the emission properties of the complexes.4,7,8
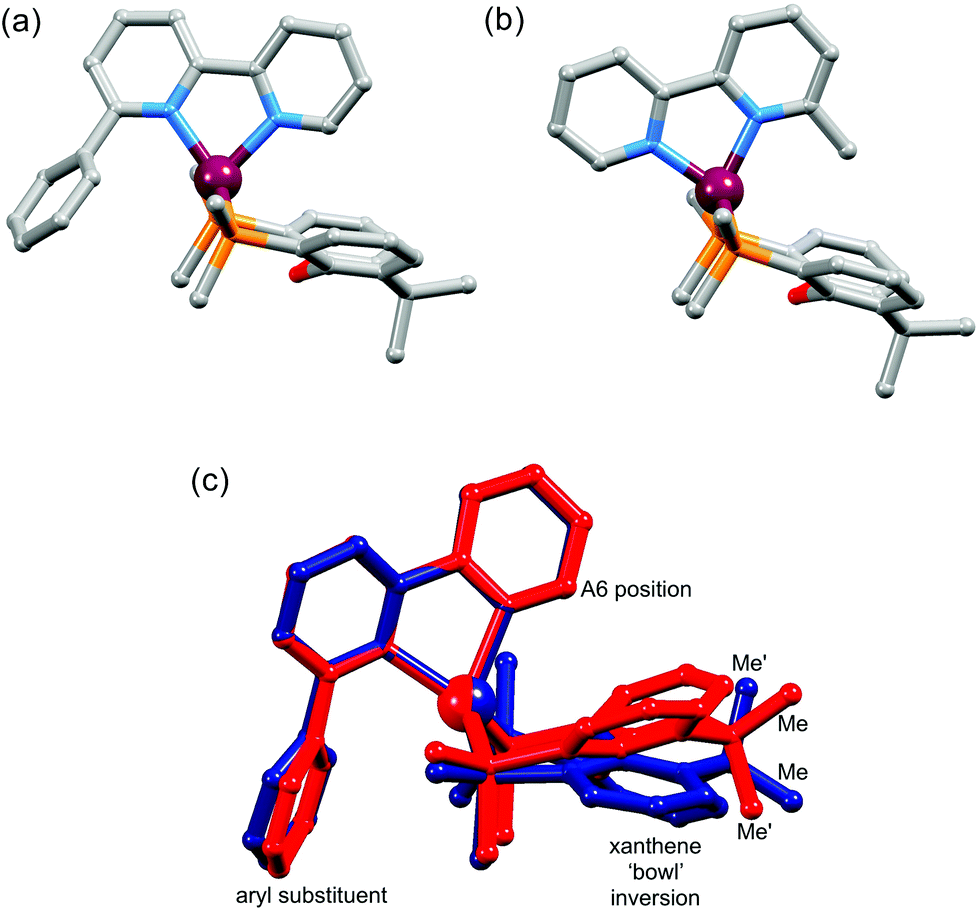
We recently demonstrated the remarkable performance of [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] (N^N = 6-methyl-2,2′-bipyridine, 6-ethyl-2,2′-bipyridine or 6,6′-dimethyl-2,2′-bipyridine) in LECs.8 However, achieving both high efficacy and long device lifetime remains challenging. A LEC with [Cu(Mebpy)(xantphos)][PF6] in the emissive layer had a lifetime of >15 h and an efficacy of 1.9 cd A−1. The latter increased to 3.0 cd A−1 on going to [Cu(Me2bpy)(xantphos)][PF6], but the lifetime decreased to 1 h. Introducing an ethyl group in place of the 6-methyl substituent led to a significant increase in lifetime (>40 h) but at the expense of luminance.8 Since POP and xantphos are both sterically demanding, there are significant constraints on the substituents that can be introduced into the 6- and 6′-positions of the bpy ligand. Ligand redistribution reactions which lead to the equilibrium mixtures of [Cu(N^N)(P^P)]+, [Cu(N^N)2]+ and [Cu(P^P)2]+ are a recognized problem.21 Nevertheless, the tetrahedral copper(I) coordination sphere in [Cu(N^N)(POP)]+ and [Cu(N^N)(xantphos)]+ is able to accommodate a 6-phenyl-2,2′-bipyridine (Phbpy) ligand giving complexes which are stable in CH2Cl2 solutions.8 While both [Cu(Phbpy)(POP)]+ and [Cu(Phbpy)(xantphos)]+ exhibit poor photoluminescence quantum yields,8 the compounds remain of interest from a structural viewpoint. In the solid-state, the orientation of the Phbpy ligand in [Cu(Phbpy)(POP)]+ and [Cu(Phbpy)(xantphos)]+ (Fig. 1a) is rotated ∼180° with respect to that in [Cu(Mebpy)(xantphos)]+ (Fig. 1b), [Cu(Etbpy)(POP)]+ and [Cu(Etbpy)(xantphos)]+, and in solution two conformers of [Cu(xantphos)(Phbpy)]+ exist with approximately equal populations.8 These conformers are related by the inversion of the xanthene unit which has a ‘bowl’-like structure (Fig. 1). We therefore decided to extend the investigation to larger aryl substituents to investigate the effect on solution dynamics and solid-state structure. The N^N ligands selected for the current study are shown in Scheme 1.
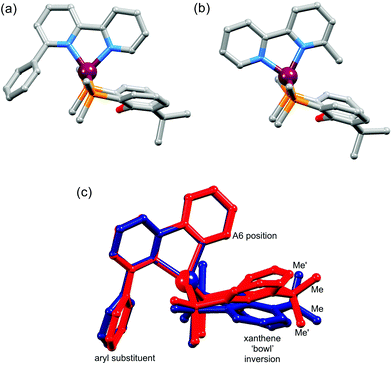 |
| | Fig. 1 Structures of (a) [Cu(Mebpy)(xantphos)]+ and (b) [Cu(Phbpy)(xantphos)]+ (c) overlay of the structures of two conformers of [Cu(xantphos)(Phbpy)]+ which are related by inversion of the xanthene unit. Only the ipso-C atoms of each PPh2 phenyl ring is shown and H atoms are omitted. The relevance of the ‘A6 position’ and the positions of the methyl groups (Me and Me′) are discussed later in the paper. Data for the figure are from ref. 8. | |
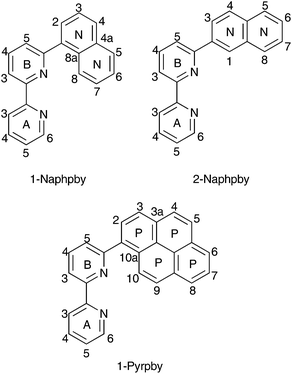 |
| | Scheme 1 Structures of the N^N ligands with ring and atom labelling for NMR spectroscopic assignments. | |
Experimental
General
Microwave reactions were performed in a Biotage Initiator 8 reactor. 1H, 13C and 31P NMR spectra were recorded using Bruker Avance III-400, 500 and 600 NMR spectrometers; spectra were recorded at ∼295 K unless otherwise stated. 1H and 13C NMR chemical shifts were referenced to the residual solvent peaks with respect to δ(TMS) = 0 ppm and 31P NMR chemical shifts with respect to δ(85% aqueous H3PO4) = 0 ppm. Solution absorption spectra were measured using an Agilent 8453 spectrophotometer. A Bruker esquire 3000 plus instrument was used to record electrospray ionization (ESI) mass spectra. Quantum yields were measured using a Hamamatsu absolute photoluminescence (PL) quantum yield spectrometer C11347 Quantaurus-QY. Emission lifetimes and powder emission spectra were measured with a Hamamatsu Compact Fluorescence lifetime Spectrometer C11367 Quantaurus-Tau, using an LED light source with λexc = 365 nm.
Electrochemical measurements were carried out using a CH Instruments 900B potentiostat with [nBu4N][PF6] (0.1 M) as supporting electrolyte and at a scan rate of 0.1 V s−1. The working electrode was glassy carbon, the reference electrode was a leakless Ag+/AgCl (eDAQ ET069-1) and the counter-electrode platinum wire. Final potentials were referenced with respect to the Fc/Fc+ couple.
6-Bromo-2,2′-bipyridine was prepared according to a literature procedure22 or by the more convenient method described below. POP and xantphos were purchased from Fluorochem and SPhosPd G2 from Sigma-Aldrich. [Cu(MeCN)4][PF6] was prepared by the published method.23
6-Bromo-2,2′-bipyridine
2,6-Dibromopyridine (1.27 g, 5.38 mmol) and [Pd(PPh3)4] (311 mg, 0.27 mmol) were placed in a 10–20 ml microwave vial. The vial was evacuated and refilled with N2 three times before dry THF (5 ml) was added. N2 was bubbled through the solution for 20 min before a solution of 2-pyridylzinc bromide (0.5 M in THF, 14.0 ml, 7.00 mmol) was added and the vial was sealed. The reaction was performed in a microwave reactor (90 °C, 2.5 h). Saturated aqueous NaHCO3 (30 ml) was added and the crude mixture was extracted with CH2Cl2 (3 × 70 ml). The combined organic layers were washed with water (3 × 180 ml) and dried over MgSO4. The solvent was removed in vacuo and the crude material was purified by column chromatography (Alox 90, hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 50:1) to give 6-bromo-2,2′-bipyridine (462 mg, 2.39 mmol, 44%) as a white powder. 1H NMR spectroscopic data were consistent with the literature data.1 ESI MS: m/z 234.7 [M + H]+ (base peak, calc. 235.0).
:ethyl acetate 50:1) to give 6-bromo-2,2′-bipyridine (462 mg, 2.39 mmol, 44%) as a white powder. 1H NMR spectroscopic data were consistent with the literature data.1 ESI MS: m/z 234.7 [M + H]+ (base peak, calc. 235.0).
6-(Naphthalen-1-yl)-2,2′-bipyridine (1-Naphbpy)
6-(Naphthalen-1-yl)-2,2′-bipyridine was prepared based on methodoloy described in the literature.24 A mixture of 6-bromo-2,2′-bipyridine (325 mg, 1.38 mmol), 1-naphthaleneboronic acid (261 mg, 1.51 mmol) and SPhosPd G2 (49.7 mg, 0.07 mmol) in toluene (15 ml), EtOH (7 ml) and aqueous Cs2CO3 (2 M, 1.5 ml) was heated at 80 °C under inert conditions for 23 h. The mixture was allowed to cool to room temperature before toluene (30 ml) was added. The organic layer was washed with water (3 × 50 ml) dried over MgSO4 and filtered. The solvent was removed in vacuo and the product was purified by column chromatography (Alox 90, cyclohexane:ethyl acetate 25:1) to give 6-(naphthalen-1-yl)-2,2′-bipyridine (259 mg, 0.92 mmol, 67%) as a white powder. 1H NMR (500 MHz, DMSO-d6) δ/ppm: 8.74 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H, HA6), 8.47 (dd, J = 7.9, 1.0 Hz, 1H, HB3), 8.37 (dt, J = 8.0, 1.1 Hz, 1H, HA3), 8.17 (m, 1H, HN8), 8.13 (t, J = 7.8 Hz, 1H, HB4), 8.07–8.03 (overlapping m, 2H, HN4+N5), 7.93 (ddd, J = 8.0, 7.5, 1.8 Hz, 1H, HA4), 7.74 (dd, J = 7.7, 1.0 Hz, 1H, HB5), 7.72 (dd, J = 7.1, 1.3 Hz, 1H, HN2), 7.64 (dd, J = 8.2, 7.1 Hz, 1H, HN3), 7.58 (ddd, J = 8.2, 6.8, 1.5 Hz, 1H, HN6), 7.54 (ddd, J = 8.2, 6.8, 1.6 Hz, 1H, HN7), 8.74 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H, HA5). 13C NMR (126 MHz, DMSO-d6) δ/ppm: 157.9 (CB6), 155.3 (CA2), 154.9 (CB2), 149.4 (CA6), 138.2 (CB4), 137.9 (CN1), 137.3 (CA4), 133.5 (CN4a), 130.6 (CN8a), 128.9 (CN4), 128.4 (CN5), 127.6 (CN2), 126.7 (CN7), 126.0 (CN6), 125.5 (CN3), 125.3 (CN8), 125.2 (CB5), 124.3 (CA5), 120.6 (CA3), 118.9 (CB3). MALDI-TOF MS: m/z 283.0 [M + H]+ (base peak, calc. 283.1). Found: C 84.78, H 5.37, N 9.73; C20H14N2 requires C 85.08, H 5.00, N 9.92%.
6-(Naphthalen-2-yl)-2,2′-bipyridine (2-Naphbpy)
6-(Naphthalen-2-yl)-2,2′-bipyridine was prepared using the same procedure as for 6-(naphthalen-1-yl)-2,2′-bipyridine but starting with 6-bromo-2,2′-bipyridine (350 mg, 1.49 mmol), 2-naphthaleneboronic acid (282 mg, 1.64 mmol) and SPhosPd G2 (53.6 mg, 0.07 mmol) in toluene (15 ml), EtOH (7 ml) and aqueous Cs2CO3 (2 M, 1.7 ml). 6-(Naphthalen-2-yl)-2,2′-bipyridine (288 mg, 1.02 mmol, 69%) was isolated as a white powder. 1H and 13C NMR data were consistent with the literature data.25 ESI MS: m/z 305.0 [M + Na]+ (base peak, calc. 305.1). Found: C 84.73, H 5.48, N 10.11; C20H14N2 requires C 85.08, H 5.00, N 9.92%.
6-(Pyrene-1-yl)-2,2′-bipyridine (1-Pyrbpy)
6-Bromo-2,2′-bipyridine (1.50 g, 4.89 mmol), pyrene-1-boronic acid (1.32 g, 5.38 mmol) and Na2CO3 (1.04 g, 9.78 mmol) were dissolved in toluene (200 ml) and water (50 ml). The mixture was degassed with N2 for 50 min before [Pd(PPh3)4] (282 mg, 0.24 mmol) was added. The reaction mixture was heated at reflux for 69 h under rigorous exclusion of light. The reaction mixture was cooled to room temperature and the aqueous phase was separated. The organic layer was washed with water (50 ml), dried over MgSO4 and evaporated to dryness. The crude product was purified by column chromatography (Alox 90, hexane:CH2Cl2 4:1 to pure CH2Cl2, then Silica 60, toluene:ethyl acetate 10:1 to 5:1, then Alox 90, pure CH2Cl2, the Silica 60 pure CH2Cl2 to CH2Cl2:ethyl acetate 10:1) to give 6-(pyrene-1-yl)-2,2′-bipyridine (1.25 g, 3.51 mmol, 72%) as a yellow powder. 1H NMR (500 MHz, acetone-d6) δ/ppm: 8.75 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H, HA6), 8.62 (overlapping dd, J = 7.9, 0.9 Hz, 1H, HB3), 8.62 (overlapping d, J = 9.1 Hz, 1H, HP10), 8.59 (d, J = 8.0 Hz, 1H, HA3), 8.42 (d, J = 7.9 Hz, 1H, HP3), 8.34 (d, J = 7.6 Hz, 1H, HP6), 8.33 (d, J = 7.9 Hz, 1H, HP2), 8.31 (d, J = 7.6 Hz, 1H, HP8), 8.26 (s, 2H, HP4+P5), 8.22 (d, J = 9.1 Hz, 1H, HP9), 8.18 (t, J = 7.8 Hz, 1H, HB4), 8.11 (t, J = 7.6 Hz, 1H, HP7), 7.93 (td, J = 7.8, 1.8 Hz, 1H, HA4), 7.91 (dd, J = 7.6, 0.9 Hz, 1H, HB5), 7.45 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H, HA5). 13C NMR (126 MHz, acetone-d6) δ/ppm: 159.8 (CB6), 156.9 (CA2), 156.7 (CB2), 150.2 (CA6), 138.7 (CB4), 137.9 (CA4), 132.4 (CP5a), 131.9 (CP8a), 129.6 (CP10c), 128.8 (CP9), 128.7 (CP2), 128.6 (CP4+P5), 128.4 (CP1), 127.2 (CP7), 126.6 (CB5), 126.4 (CP6), 126.1 (CP10a), 126.1 (CP8), 126.0 (CP10), 125.8 (CP10b), 125.7 (CP3), 125.6 (CP3a), 125.0 (CA5), 121.7 (CA3), 119.8 (CB3). ESI MS: m/z 357.1 [M + H]+ (base peak, calc. 357.1). Found: C 87.37, H 4.48, N 7.50; C26H16N2 requires C 87.62, H 4.52, N 7.86%.
[Cu(1-Naphbpy)(POP)][PF6]
[Cu(MeCN)4][PF6] (93.2 mg, 0.25 mmol) and POP (135 mg, 0.25 mmol) were dissolved in CH2Cl2 (30 ml) and the mixture was stirred for 2 h at room temperature. 1-Naphbpy (70.6 mg, 0.25 mmol) was added and stirring was continued for 2 h. The yellow solution was filtered and the solvent was removed from the filtrate under vacuum. The solid product was washed with hexane (2 × 30 ml) and dried under vacuum. [Cu(1-Naphbpy)(POP)][PF6] (244 mg, 0.21 mmol, 82%) was isolated as a yellow solid. 1H NMR (500 MHz, acetone-d6, 253 K) δ/ppm: 8.80 (dd, J = 8.1, 1.1 Hz, 1H, HB3), 8.72 (dt, J = 8.3, 1.0 Hz, 1H, HA3), 8.34 (t, J = 7.9 Hz, 1H, HB4), 8.17 (dd, J = 5.2, 0.9 Hz, 1H, HA6), 8.13–8.06 (overlapping m, 3H, HA4+N4+N6), 7.62 (dd, J = 7.7, 1.0 Hz, 1H, HB5), 7.54–7.49 (overlapping m, 2H, HC5a+N7), 7.45 (td, J = 7.4, 1.3 Hz, 1H, HD4a), 7.42–7.39 (overlapping m, 2H, HD4c+D4d), 7.36 (m, 1H, HC6a), 7.30–7.22 (overlapping m, 5H, HD3a+C5b+D3c), 7.20–7.14 (overlapping m, 4H, HN2+D3d+D4b), 7.14–7.08 (overlapping m, 4H, HA5+C4a+C6b+N5), 7.01–6.93 (overlapping m, 4H, HC4b+D2c+N8), 6.79–6.69 (overlapping m, 6H, HC3a+D2a+D3b+N3), 6.53 (m, 2H, HD2d), 6.20 (td, J = 7.8, 1.6 Hz, 1H, HC3b), 6.08 (m, 2H, HD2b). 13C NMR (126 MHz, acetone-d6, 253 K) δ/ppm: 159.5 (CB6), 157.8 (CC1a), 157.4 (CC1b), 154.1 (CB2), 153.0 (CA2), 149.3 (CA6), 139.2 (CA4), 139.0 (CB4), 138.8 (CN1), 135.5 (CC3a), 134.4 (CC3b), 134.4 (CD2a), 134.3 (CN4a), 133.6 (CD2c), 133.5 (CD2b), 132.9 (CD2d), 132.8 (CC5a), 132.5 (CC5b), 131.0 (CN8a), 130.6 (CD4a), 130.5 (CD4c+D4d), 130.4 (CD4b), 130.0 (CN4), 129.4 (CB5), 129.4 (CD3c), 129.3 (CD3a), 129.2 (CD3d), 129.0 (CN6), 128.9 (CD3b), 127.8 (CN5), 127.8 (CN8), 127.7 (CN2), 127.0 (CN7), 126.4 (CN3), 126.0 (CA5), 125.8 (CC2a), 125.6 (CC4b), 125.5 (CC4a), 124.1 (CC2b), 123.5 (CA3), 122.8 (CB3), 120.5 (CC6b), 119.9 (CC6a). 31P NMR (202 MHz, acetone-d6, T = 253 K) δ/ppm −12.7 (broad, FWHM = 200 Hz), −15.5 (br, FWHM = 220 Hz), −144.5 (septet, JPF = 710 Hz, [PF6]−). ESI MS: m/z 883.0 [M − PF6]+ (base peak, calc. 883.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 230 (57400), 287 (21900), 327sh (10500), 383 (2300). Found: C 64.94, H 4.49, N 3.05; C56H42CuF6N2OP3 requires C 65.34, H 4.11, N 2.72%.
[Cu(1-Naphbpy)(xantphos)][PF6]
[Cu(MeCN)4][PF6] (93.2 mg, 0.25 mmol) was dissolved in CH2Cl2 (15 ml). A solution of xantphos (148 mg, 0.25 mmol) and 1-Naphbpy (70.6 mg, 0.25 mmol) was added and the mixture turned red then yellow while it was stirred for 2 h at room temperature. The yellow solution was filtered and the solvent was removed from the filtrate. The solid material was washed with hexane (2 × 30 ml) and dried under vacuum. [Cu(1-Naphbpy)(xantphos)][PF6] (251 mg, 0.24 mmol, 94%) was isolated as a yellow solid. 1H NMR (500 MHz, acetone-d6, 223 K) δ/ppm: 8.87 (dd, J = 8.1, 1.1 Hz, 1H, HB3), 8.83 (d, J = 8.2 Hz, 1H, HA3), 8.41 (t, J = 7.9 Hz, 1H, HB4), 8.17 (d, J = 8.3 Hz, 1H, HN5), 8.30 (d, J = 8.3 Hz, 1H, HN4), 8.14 (td, J = 7.9, 1.7 Hz, 1H, HA4), 7.84 (dt, J = 8.0, 1.6 Hz, 2H, HC5a+C5b), 7.61 (dd, J = 7.7, 1.0 Hz, 1H, HB5), 7.57–7.49 (overlapping m, 2H, HD4a+N6), 7.46 (td, J = 7.4, 1.3 Hz, 1H, HD4b), 7.37–7.26 (overlapping m, 5H, HD3a+D3b+C4a), 7.25–7.13 (overlapping m, 4H, HC4b+N3+D4c+D4d), 7.08–7.03 (overlapping m, 2H, HA5+N8), 6.90–6.82 (overlapping m, 4H, HD3c+D3d), 6.69–6.53 (overlapping m, 7H, HA6+D2a+D2b+C3a+N7), 6.46 (d, J = 6.5 Hz, 1H, HN2), 6.30–6.20 (overlapping m, 3H, HC3b+D2c), 5.66 (broad m, 2H, HD2d), 2.06 (s, HMe, overlaps with acetone-d6 and assigned from 2D spectra), 1.32 (s, 3H, HMe′). 13C NMR (126 MHz, acetone-d6, 223 K) δ/ppm: 160.2 (CB6), 155.0 (CC1a), 154.4 (CC1b), 153.5 (CA2), 153.0 (CB2), 147.5 (CA6), 139.7 (CA4+B4), 139.0 (CN1), 134.6 (CN4a), 134.5 (CC6a+C6b), 134.4 (CD2a), 134.0 (CD2c), 133.3 (CD2b), 132.4 (CD2d), 131.3 (CC3b), 131.2 (CD4a), 131.1 (CN8a), 130.9 (CC3a), 130.7 (CD4b), 130.1 (CD4c), 130.1 (CN4), 129.7 (CD4d), 129.6 (CD1a), 129.5 (CD3a+D3b), 129.4 (CB5), 129.3 (CN5), 128.9 (CD1c+D1d), 128.7 (CD3c+D3d), 128.0 (CC5a+C5b), 127.5 (CN7), 127.4 (CN2), 127.3 (CN6), 126.8 (CN3), 126.1 (CC4b), 125.9 (CC4a), 125.8 (CN8+A5), 124.5 (CA3), 123.3 (CB3), 121.5 (CC2a), 120.2 (CC2b), 36.7 (Cq), 32.0 (CMe), 23.0 (CMe′). 31P NMR (202 MHz, acetone-d6, 223 K) δ/ppm −10.9 (broad d, J = 110 Hz), −13.4 (broad d, J = 110 Hz), −144.5 (septet, JPF = 710 Hz, [PF6]−). ESI MS: m/z 923.1 [M − PF6]+ (base peak, calc. 923.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 230 (60800), 289 (29900), 328sh (7800), 392 (2500). Found: C 66.29, H 4.78, N 2.86; C59H46CuF6N2OP3 requires C 66.26, H 4.34, N 2.62%.
[Cu(2-Naphbpy)(POP)][PF6]
[Cu(2-Naphbpy)(POP)][PF6] was prepared according to the procedure for [Cu(1-Naphbpy)(POP)][PF6] using 2-Naphbpy (70.6 mg, 0.25 mmol) in place of 1-Naphbpy. [Cu(2-Naphbpy)(POP)][PF6] was isolated as a yellow powder (251 mg, 0.24 mmol, 98%). 1H NMR (500 MHz, acetone-d6) δ/ppm: 8.67–8.62 (overlapping m, 2H, HA3+B3), 8.30 (t, J = 7.9 Hz, 1H, HB4), 8.12 (d, J = 5.1 Hz, 1H, HA6), 8.09 (td, J = 7.9, 1.7 Hz, 1H, HA4), 7.91 (m, 1H, HN5), 7.76 (dd, J = 7.7, 1.0 Hz, 1H, HB5), 7.65–7.61 (overlapping m, 2H, HN6+N8), 7.57–7.52 (overlapping m, 2H, HN1+N7), 7.44–7.33 (overlapping m, 6H, HN3+C5+D4a+N4), 7.31 (m, 2H, HD4b), 7.25 (m, 4H, HD3a), 7.16 (m, 2H, HC6), 7.13 (ddd, J = 7.6, 5.2, 1.1 Hz, 1H, HA5), 7.05–6.95 (overlapping m, 6H, HC4+D3b), 6.83 (m, 4H, HD2a), 6.64–6.54 (overlapping m, 6H, HC3+D2b). 13C NMR (126 MHz, acetone-d6) δ/ppm: 161.8 (CB6), 158.2 (t, J = 5.9 Hz, CC1), 154.4 (CB2), 153.8 (CA2), 149.7 (CA6), 140.3 (CB4), 139.7 (CA4), 139.0 (CN2), 135.3 (CC3), 134.6 (CD2b), 134.4 (CN4a), 133.5 (CN8a+D2a), 133.1 (CC5), 131.0 (CD4a), 130.8 (CD4b), 130.1 (CN4), 129.7 (CD3a), 129.5 (CN8), 129.3 (CD3b), 128.8 (CN1+N5), 128.1 (CN6), 128.0 (CB5), 127.9 (CN7), 126.4 (CA5+N3), 125.9 (t, J = 2.1 Hz, CC4), 124.6 (t, J = 14.2 Hz, CC2), 124.0 (CA3), 123.0 (CB3), 120.6 (CC6). 31P NMR (202 MHz, acetone-d6) δ/ppm −13.4 (broad, FWHM = 200 Hz), −144.3 (septet, JPF = 710 Hz, [PF6]−). ESI MS: m/z 883.1 [M − PF6]+ (base peak, calc. 883.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 232 (67600), 288 (28800), 389 (2600). Found: C 64.94, H 4.61, N 2.92; C56H42CuF6N2OP3 requires C 65.34, H 4.11, N 2.72%.
[Cu(2-Naphbpy)(xantphos)][PF6]
This compound was prepared by the same procedure as for [Cu(1-Naphbpy)(xantphos)][PF6] using 2-Naphbpy (70.6 mg, 0.25 mmol) instead of 1-Naphbpy. [Cu(2-Naphbpy)(xantphos)][PF6] was isolated as a yellow powder (256 mg, 0.24 mmol, 96%). 1H NMR (500 MHz, acetone-d6) δ/ppm: 8.63 (dt, J = 8.3, 1.0 Hz, 1H, HA3), 8.55 (dd, J = 8.0, 1.0 Hz, 1H, HB3), 8.24 (t, J = 7.9 Hz, 1H, HB4), 8.12 (m 1H, HA4), 8.00 (m, 1H, HN5), 7.77–7.73 (overlapping m, 4H, HC5+B5+N1), 7.67 (ddd, J = 8.3, 6.8, 1.3 Hz, 1H, HN6), 7.53 (ddd, J = 8.1, 6.7, 1.2 Hz, 1H, HN7), 7.50–7.42 (overlapping m Hz, 4H, HN3+N8+D4a), 7.28 (t, J = 7.7 Hz, 4H, HD3a), 7.28 (t, J = 7.7 Hz, 2H, HC4), 7.17–7.11 (overlapping m, 3H, HA5+D4b), 6.82 (m Hz, 4H, HD2a), 6.80–6.73 (overlapping m, 5H, HA6+D3b), 6.67–6.59 (overlapping m, 6H, HC3+D2b), 1.91 (broad s, 3H, HMe), 1.46 (s, 3H, HMe′). 13C NMR (126 MHz, acetone-d6) δ/ppm: 161.7 (CB6), 155.5 (t, J = 6.3 Hz, CC1), 154.3 (CA2), 154.0 (CB2), 148.9 (CA6), 140.3 (CB4), 140.1 (CA4), 139.2 (CN2), 135.6 (CD1a), 134.9 (CC6), 134.5 (CN4a), 134.1 (CN8a), 133.9 (t, J = 7.7 Hz, CD2a+D2b), 131.5 (CC3), 131.1 (CD4a), 130.8 (CD1b), 130.6 (CD4b), 129.8 (t, J = 4.5 Hz, CD3a), 129.5 (CN8), 129.1 (t, J = 4.7 Hz, CD3b), 128.9 (CN5), 128.7 (CN1), 128.3 (CC5), 128.2 (CN6), 127.9 (CB5), 126.4 (CA5), 126.2 (t, J = 2.2 Hz, CC4), 126.0 (CN3), 124.5 (CA3), 123.3 (CB3), 121.9 (CN7), 121.1 (t, J = 13.1 Hz, CC2), 36.8 (Cq), 30.8 (CMe′), 25.3 (CMe). 31P NMR (202 MHz, acetone-d6) δ/ppm −12.8 (broad, FWHM = 210 Hz), −144.3 (septet, JPF = 710 Hz, [PF6]−). ESI MS: m/z 923.2 [M − PF6]+ (base peak, calc. 923.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 230 (67000), 284 (32800), 388 (2500). Found: C 66.18, H 4.79, N 2.90; C59H46CuF6N2OP3 requires C 66.26, H 4.34, N 2.62%.
[Cu(1-Pyrbpy)(POP)][PF6]
[Cu(MeCN)4][PF6] (93.2 mg, 0.25 mmol) and POP (148 mg, 0.27 mmol) were dissolved in CH2Cl2 (30 ml) and the mixture was stirred for 2 h at room temperature. 1-Pyrbpy (89.6 mg, 0.25 mmol) was added and stirring was continued for 2 h. The yellow solution was filtered and the solvent was removed from the filtrate. The solid material was washed with hexane (2 × 30 ml), dried under vacuum and redissolved in a small amount of acetone. The solution was layered with Et2O and left to crystallize for 2 days. The resulting yellow crystals were ground to a powder and dried under vacuum to give [Cu(1-Pyrbpy)(POP)][PF6] (105 mg, 0.10 mmol, 38%) as a yellow powder. 1H NMR (600 MHz, acetone-d6, 238 K) δ/ppm: 8.89 (dd, J = 8.2, 1.0 Hz, 1H, HB3), 8.78 (d, J = 8.3 Hz, 1H, HA3), 8.47 (d, J = 7.6 Hz, 1H, HP6), 8.45 (t, J = 7.8 Hz, 1H, HB4), 8.42 (d, J = 8.9 Hz, 1H, HP5), 8.32 (d, J = 5.1 Hz, 1H, HA6), 8.23 (d, J = 7.6 Hz, 1H, HP8), 8.19 (d, J = 8.9 Hz, 1H, HP4), 8.14 (t, J = 7.6 Hz, 1H, HP7), 8.13 (t, J = 8.3 Hz, 1H, HA4), 7.80 (dd, J = 7.6, 1.0 Hz, 1H, HB5), 7.70 (d, J = 7.2 Hz, 1H, HP2), 7.69 (d, J = 9.4 Hz, 1H, HP9), 7.62 (t, J = 7.5 Hz, 1H, HD4a), 7.53 (t, J = 7.5 Hz, 1H, HD4b), 7.45–7.42 (overlapping m Hz, 2H, HD4c+C5b), 7.38–7.30 (overlapping m, 10H, HC6a+C6b+C5a+D3a+D3b+D3c+P3), 7.16 (ddd, J = 7.6, 5.1, 1.1 Hz, 1H, HA5), 7.09 (t, J = 8.2 Hz, 2H, HD2c), 7.04–7.01 (overlapping m, 2H, HC4b+P10), 6.95 (t, J = 7.7 Hz, 1H, HC4a), 6.70 (t, J = 7.7 Hz, 1H, HD4d), 6.66 (ddd, J = 7.8, 6.2, 1.7 Hz, 1H, HC3b), 6.52 (t, J = 8.1 Hz, 2H, HD2a), 6.39 (t, J = 8.2 Hz, 2H, HD2b), 6.13 (dd, J = 7.8, 6.2, 1.7 Hz, 1H, HC3a), 6.03 (m Hz, 2H, HD3d), 5.63 (m Hz, 2H, HD2d). 13C NMR (151 MHz, acetone-d6, 238 K) δ/ppm: 159.8 (CB6), 158.3 (CC1b), 156.7 (CC1a), 154.3 (CB2), 152.9 (CA2), 149.5 (CA6), 139.7 (CA4), 139.2 (CB4), 136.0 (CC3b), 135.5 (CP1), 134.7 (CC3a), 133.7 (CD2a), 133.4 (CD2c), 133.1 (CD2b), 132.8 (CC5b), 132.7 (CD2d), 132.4 (CC5a), 131.8 (CP3a), 131.6 (CP5a), 130.9 (CP8a), 130.5 (CD4b+D4c), 130.4 (CD4a), 129.8 (CB5+D4d), 129.4 (CD3b), 129.3 (CD3c), 129.2 (CP9), 129.1 (CD3a), 129.0 (CP5), 128.4 (CP10a), 128.0 (CD3d), 127.9 (CP4), 127.5 (CP2), 127.1 (CP7), 126.4 (CP6), 126.1 (CP8+C4a), 125.9 (CA5), 125.7 (CP3), 124.7 (CC4b+P10b), 124.5 (CP10c), 123.6 (CA3), 123.5 (CP10), 122.2 (CC6a), 117.7 (CC6b), 112.8 (CB3). 31P NMR (162 MHz, acetone-d6, 295 K) δ/ppm −13.7 (broad, FWHM = ∼350 Hz), −144.3 (septet, JPF = 707 Hz, [PF6]−); (243 MHz, acetone-d6, 238 K) δ/ppm −14.5 (broad, FWHM = 130 Hz). ESI MS: m/z 957.2 [M − PF6]+ (base peak, calc. 957.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 230 (58800), 245 (62000), 282 (38000), 354 (22500). Found: C 66.72, H 4.47, N 2.60; C62H44CuF6N2OP3 requires C 67.48, H 4.02, N 2.54%.
[Cu(1-Pyrbpy)(xantphos)][PF6]
[Cu(MeCN)4][PF6] (93.2 mg, 0.25 mmol) was dissolved in CH2Cl2 (15 ml). A solution of xantphos (148 mg, 0.25 mmol) and 1-Pyrbpy (89.6 mg, 0.25 mmol) was added and the mixture turned red then yellow while it was stirred for 2 h at room temperature. The yellow solution was filtered and the solvent was removed from the filtrate. The solid material was washed with hexane (2 × 30 ml), dried under vacuum and redissolved in a small amount of acetone. The solution was layered with Et2O and left to crystalize for 2 days. The resulting yellow crystals were ground to a powder and dried under vacuum to give [Cu(1-Pyrbpy)(xantphos)][PF6] (115 mg, 0.10 mmol, 40%) as a yellow powder. 1H NMR (600 MHz, acetone-d6, 238 K) δ/ppm: 8.89 (dd, J = 8.2, 1.1 Hz, 1H, HB3), 8.87 (d, J = 8.2 Hz, 1H, HA3), 8.61 (d, J = 7.6 Hz, 1H, HP6), 8.55 (d, J = 9.0 Hz, 1H, HP5), 8.50 (d, J = 7.8 Hz, 1H, HB4), 8.34 (d, J = 9.0 Hz, 1H, HP4), 8.21 (t, J = 7.6 Hz, 1H, HP7), 8.16 (td, J = 8.1, 1.1 Hz, 1H, HA4), 8.14 (d, J = 7.6 Hz, 1H, HP8), 7.95 (d, J = 7.8 Hz, 1H, HP3), 7.82 (dd, J = 7.9, 1.4 Hz, 2H, HC5a+C5b), 7.79 (dd, J = 7.5, 1.0 Hz, 1H, HB5), 7.66 (m, 1H, HD4d), 7.53 (m, 1H, HD4a), 7.43 (m, 2H, HD3d), 7.38 (m, 2H, HD3a), 7.37 (d, J = 9.2 Hz, 1H, HP10), 7.27 (td, J = 7.8, 1.1 Hz, 1H, HC4a), 7.20 (td, J = 7.8, 1.2 Hz, 1H, HC4b), 7.17 (dd, J = 9.2 Hz, 1H, HP9), 7.04 (ddd, J = 7.6, 5.1, 1.1 Hz, 1H, HA5), 7.00 (m, 1H, HD4b), 6.91 (d, J = 7.7 Hz, 1H, HP2), 6.71 (m, 2H, HD2a), 6.65 (d, J = 5.1 Hz, 1H, HA6), 6.64–6.58 (overlapping m, 3H, HD4c+D2d), 6.53 (m, 1H, HC3a), 6.43 (t, J = 7.0 Hz, 2H, HD3b), 6.21 (m, 1H, HC3b), 5.85 (m, 2H, HD2b), 5.53–5.41 (broad overlapping m, 4H, HD2c+D3c), 2.03 (s, 3H, HMe), 1.28 (s, 3H, HMe′). 13C NMR (151 MHz, acetone-d6, 238 K) δ/ppm: 160.3 (CB6), 154.9 (CC1b), 154.5 (CC1a), 153.2 (CA2), 152.9 (CB2), 147.6 (CA6), 139.7 (CB4), 139.4 (CA4), 135.9 (CP1), 134.4 (CD4c), 134.2 (CC6a+C6b), 133.5 (CD2b), 133.3 (CD2a), 132.3 (CP3a), 131.8 (CP5a), 131.7 (CD1d), 131.6 (CD1a), 131.5 (CD2c+D3c), 131.2 (CD4d), 131.1 (CC3b), 131.0 (CP8a), 130.8 (CC3a), 130.6 (CD4a), 129.8 (CD4b), 129.5 (CB5), 129.46 (CD3d), 129.4 (CD3a), 129.0 (CP5+D2d), 128.9 (CP9), 128.5 (CP10a), 128.3 (CD1b), 128.0 (CD3b+P4), 127.7 (CC5a+C5b), 127.4 (CP7), 127.1 (CP2), 126.6 (CP6), 126.4 (CP8), 126.0 (CP3), 125.8 (CC4b), 125.7 (CA5), 125.8 (CC4a), 124.8 (CP10b), 124.7 (CP10c), 124.6 (CP10), 124.3 (CA3), 123.1 (CB3), 121.1 (CC2a), 119.9 (CC2b), 36.5 (Cq), 31.6 (CMe), 22.6 (CMe′). 31P NMR (162 MHz, acetone-d6, 295 K) δ/ppm −12.0 (broad, FWHM = ∼550 Hz), −144.2 (septet, JPF = 707 Hz, [PF6]−); (243 MHz, acetone-d6, 238 K) δ/ppm −10.9 (broad d, J = 95 Hz), −13.5 (broad d, J = 95 Hz). ESI MS: m/z 997.3 [M − PF6]+ (base peak, calc. 997.2). UV-Vis (CH2Cl2, 2.5 × 10−5 mol dm−3): λ/nm (ε/dm3 mol−1 cm−1) 243 (68000), 280 (45000), 337sh (20000), 351 (24000). Found: C 67.51, H 4.81, N 2.59; C65H48CuF6N2OP3 requires C 68.27, H 4.23, N 2.45%.
Crystallography
Data were collected on a Bruker Kappa Apex2 diffractometer with data reduction, solution and refinement using the programs APEX26 and CRYSTALS.27 Structural analysis was carried out using Mercury v. 3.7.28,29 Crystallographic data are summarized in Table 1.
Table 1 Crystallographic data
| Compound |
[Cu(1-Naphbpy) (POP)][PF6]·0.25THF |
[Cu(2-Naphbpy) (POP)][PF6]·Me2CO |
[Cu(2-Naphbpy) (xantphos)][PF6] |
[Cu(1-Pyrbpy)(POP)] [PF6]·0.5Et2O·CH2Cl2 |
[Cu(1-Pyrbpy) (xantphos)][PF6]·0.5Et2O |
| Formula |
C57H44CuF6N2O1.25P3 |
C59H48CuF6N2O2P3 |
C59H46CuF6N2OP3 |
C65H51Cl2CuF6N2O1.5P3 |
C67H53CuF6N2O1.5P3 |
| Formula weight |
1047.44 |
1087.50 |
1069.48 |
1225.49 |
1180.63 |
| Crystal colour and habit |
Yellow block |
Yellow block |
Yellow plate |
Yellow block |
Yellow block |
| Crystal system |
Orthorhombic |
Monoclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
Pccn
|
P21/c |
Pn
|
C2/m |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a, b, c/Å |
22.6154(14) |
22.2781(11) |
11.4374(8) |
23.5648(17) |
13.0030(9) |
| 20.0116(12) |
14.7143(8) |
13.1660(9) |
43.959(3) |
14.4788(10) |
| 21.8373(14) |
15.4481(7) |
16.8565(12) |
12.3463(9) |
16.9143(11) |
|
α, β, γ/° |
90 |
90 |
90 |
90 |
72.110(3) |
| 90 |
99.249(3) |
98.647(3) |
94.788(3) |
84.932(4) |
| 90 |
90 |
90 |
90 |
75.976(4) |
|
U/Å3 |
9882.9(11) |
4998.1(4) |
2509.5(3) |
12744.7(16) |
2939.8(4) |
|
D
c/Mg m−3 |
1.408 |
1.445 |
1.415 |
1.277 |
1.334 |
|
Z
|
8 |
4 |
2 |
8 |
2 |
|
μ(Cu-Kα)/mm−1 |
2.103 |
2.112 |
2.079 |
2.464 |
1.834 |
|
T/K |
123 |
123 |
123 |
123 |
123 |
| Refln. collected (Rint) |
70584 (0.060) |
52879 (0.055) |
16429 (0.024) |
44681 (0.032) |
39266 (0.030) |
| Unique refln. |
9287 |
9320 |
7635 |
11839 |
10819 |
| Refln. for refinement |
7830 |
7470 |
7511 |
11247 |
9691 |
| Parameters |
645 |
661 |
650 |
257 |
748 |
| Threshold |
2σ |
2σ |
2σ |
2σ |
2σ |
|
R
1 (R1 all data) |
0.0605 (0.0713) |
0.0432 (0.0572) |
0.0472 (0.0476) |
0.0603 (0.0622) |
0.0698 (0.0761) |
| wR2 (wR2 all data) |
0.1485 (0.1530) |
0.0866 (0.1067) |
0.1120 (0.1121) |
0.1527 (0.1531) |
0.1719 (0.1737) |
| Goodness of fit |
1.0589 |
0.8433 |
0.7142 |
1.0572 |
1.0983 |
| CCDC deposition |
1528552
|
1528550
|
1528551
|
1528548
|
1528549
|
Results and discussion
Synthesis of N^N ligands and of the copper(I) complexes
The ligand 2-Naphbpy has previously been reported.25,30 However, we found that the palladium-catalysed coupling of 6-bromo-2,2′-bipyridine with 2-naphthaleneboronic acid (using the second generation Suzuki–Miyaura precatalyst SPhosPd G2) was a more convenient synthetic strategy than the literature methods. An analogous procedure was used to prepare 2-Naphbpy and 1-Pyrbpy. Each ligand was characterized by 1H and 13C NMR spectroscopies and mass spectrometry (see Experimental section).
Two different strategies are typically used to prepare the heteroleptic complexes [Cu(N^N)(P^P)][PF6] (P^P = POP or xantphos). The addition of POP to a solution of [Cu(MeCN)4][PF6] leads to the formation of three-coordinate complexes [Cu(POP)(MeCN)]+ or [Cu(POP-P,P′)(POP-P)]+. The steric demands of POP prevent the formation of the four-coordinate [Cu(POP-P,P′)2]+.31 The monodentate ligand in the three-coordinate intermediate is readily substituted by an N^N ligand such as bpy.6 With the less sterically demanding ligand xantphos, the formation of a homoleptic bis-chelate complex [Cu(xantphos-P,P′)2]+ is more likely. The optimized strategy for [Cu(N^N)(POP)][PF6] complexes is sequential addition of 1–1.2 equivalents of POP followed after ≈2 hours by a bpy ligand to a solution of [Cu(MeCN)4][PF6]. For [Cu(N^N)(xantphos)][PF6], a 1:1 solution of N^N and xantphos is added to a solution of [Cu(MeCN)4][PF6]. Immediately after mixing, the solution may appear red, indicating the presence of some [Cu(N^N)2]+ complex. However, after stirring for 2 hours, equilibration yields yellow [Cu(N^N)(xantphos)]+ as the preferred solution species.
The complexes [Cu(1-Naphbpy)(POP)][PF6], [Cu(2-Naphbpy)(POP)][PF6], [Cu(1-Pyrbpy)(POP)][PF6], [Cu(1-Naphbpy)(xantphos)][PF6], [Cu(2-Naphbpy)(xantphos)][PF6] and [Cu(1-Pyrbpy)(xantphos)][PF6] were isolated as yellow solids in yields of between 38 and 98%. The electrospray mass spectrum of each complex showed a peak corresponding to [M − PF6]+ with a characteristic isotope pattern. The presence of the [PF6]− counterion was confirmed by the appearance of a septet at δ −144.4 ppm in the 31P NMR spectrum, in addition to the signals for the POP or xantphos ligands (see later).
Single crystal structures
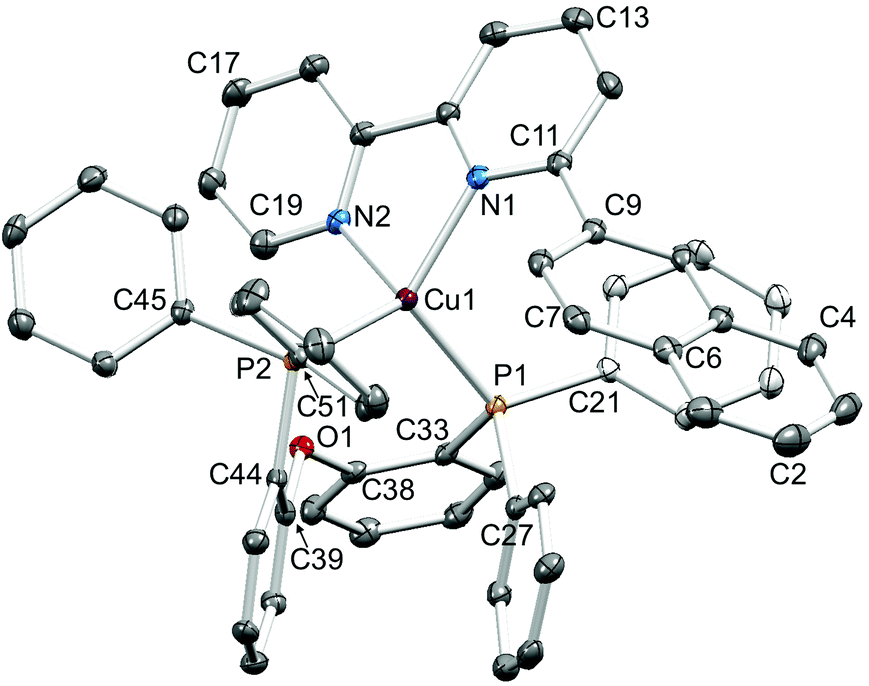
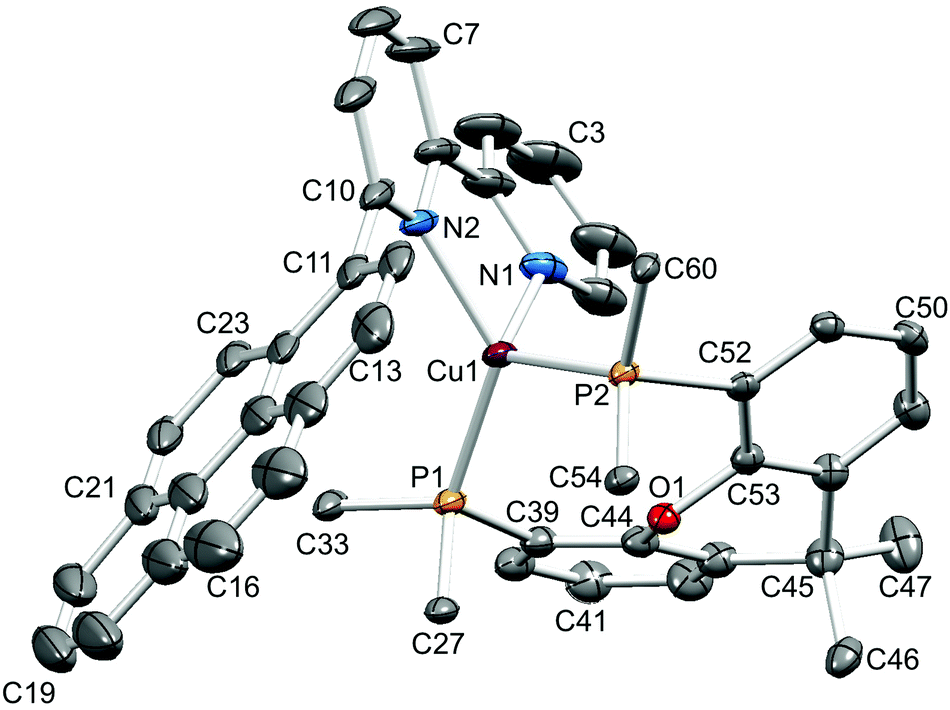
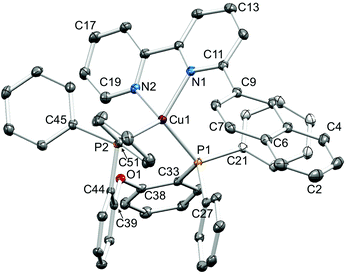
Single crystals of [Cu(1-Naphbpy)(POP)][PF6]·0.25THF, [Cu(2-Naphbpy)(POP)][PF6]·Me2CO, [Cu(1-Pyrbpy)(POP)][PF6]·0.5Et2O·CH2Cl2, [Cu(2-Naphbpy)(xantphos)][PF6] and [Cu(1-Pyrbpy)(xantphos)][PF6]·0.5Et2O were grown by diffusion of Et2O into solutions of the complexes in THF ([Cu(1-Naphbpy)(POP)][PF6]), Me2CO ([Cu(2-Naphbpy)(POP)][PF6]) or CH2Cl2 (the remaining complexes). The structures exhibit many similarities and it is appropriate to discuss them in a comparative manner. Fig. 2–6 show the structures of the [Cu(1-Naphbpy)(POP)]+, [Cu(2-Naphbpy)(POP)]+, [Cu(2-Naphbpy)(xantphos)]+, [Cu(1-Pyrbpy)(POP)]+ and [Cu(1-Pyrbpy)(xantphos)]+ cations, respectively, and parameters describing the copper(I) coordination environments are given in the figure captions. In each complex, the copper(I) atom is in a distorted tetrahedral environment with Cu–P and Cu–N bond lengths in the ranges 2.2453(10)–2.3101(11) and 2.085(4)–2.143(3) Å, respectively. The P–Cu–P angles fall in the range 109.97(3)–116.83(4)°, the largest being for the [Cu(2-Naphbpy)(xantphos)]+ cation. As expected, the N–Cu–N bond angles vary little across the series (79.16(14)–79.88(13)°). The asymmetry of the 6-substituted bpy ligand means that there are two possible orientations of the N^N ligand with respect to the P^P ligand, one with the 6-substituent lying over the ‘xanthene bowl’ and one with it positioned remote from the bowl as shown in Fig. 1. The two orientations are related by a 180° rotation of the bpy unit. We have previously reported the solid-state structures of [Cu(6-Mebpy)(xantphos)]+ and [Cu(6-Etbpy)(xantphos)]+ in which the 6-alkyl substituent lies over the ‘bowl’ of the xanthene unit (Fig. 1b); in contrast, the 6-phenyl substituent in [Cu(6-Phbpy)(xantphos)]+ is remote from the xanthene ‘bowl’ (Fig. 1a).8 Each of the cations shown in Fig. 2–6 exhibits the same N^N ligand orientation with the sterically-demanding naphthyl or pyrenyl group positioned away from the (C6H4)2O unit of POP or the xanthene ‘bowl’ of xantphos (Fig. 7a). We return to this point in the NMR spectroscopic discussion.
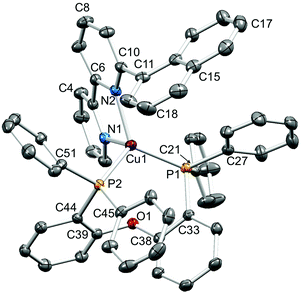 |
| | Fig. 2 Structure of the [Cu(1-Naphbpy)(POP)]+ cation in [Cu(1-Naphbpy)(POP)][PF6]·0.25THF with ellipsoids plotted at 40% probability level. H atoms and solvent molecules are omitted for clarity. Selected bond parameters: Cu1–P1 = 2.2723(8), Cu1–P2 = 2.2833(8), Cu1–N1 = 2.118(2), Cu1–N2 = 2.111(2) Å; P1–Cu1–P2 = 109.97(3), P1–Cu1–N1 = 106.14(7), P2–Cu1–N1 = 109.85(7), P1–Cu1–N2 = 128.14(7), P2–Cu1–N2 = 116.14(7), N1–Cu1–N2 = 79.68(9)°. | |
 |
| | Fig. 3 Structure of the [Cu(2-Naphbpy)(POP)]+ cation in [Cu(2-Naphbpy)(POP)][PF6]·Me2CO with ellipsoids plotted at 40% probability level. H atoms and solvent molecules are omitted for clarity. Selected bond parameters: Cu1–P1 = 2.2600(8), Cu1–P2 = 2.2716(8), Cu1–N1 = 2.105(2), Cu1–N2 = 2.099(2) Å; P1–Cu1–P2 = 112.01(3), P1–Cu1–N1 = 109.59(6), P2–Cu1–N1 = 131.94(6), P1–Cu1–N2 = 113.42(7), P2–Cu1–N2 = 104.26(7), N1–Cu1–N2 = 79.68(9)°. | |
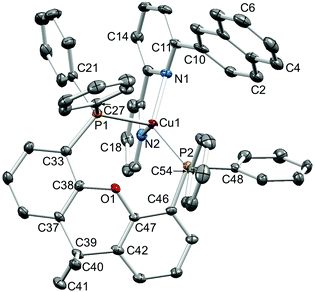 |
| | Fig. 4 Structure of the [Cu(2-Naphbpy)(xantphos)]+ cation in [Cu(2-Naphbpy)(xantphos)][PF6] with ellipsoids plotted at 30% probability level. H atoms omitted for clarity. Selected bond parameters: Cu1–P2 = 2.2453(10), Cu1–P1 = 2.3101(11), Cu1–N2 = 2.085(4), Cu1–N1 = 2.143(3) Å; P2–Cu1–P1 = 116.83(4), P2–Cu1–N2 = 113.47(10), P1–Cu1–N2 = 105.84(10), P2–Cu1–N1 = 130.15(10), P1–Cu1–N1 = 103.74(10), N2–Cu1–N1 = 79.16(14)°. | |
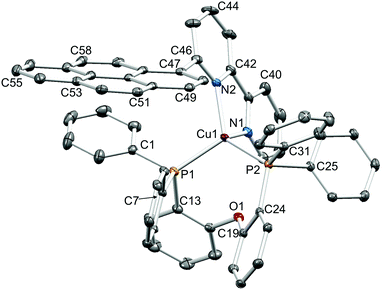 |
| | Fig. 5 Structure of the [Cu(1-Pyrbpy)(POP)]+ cation in [Cu(1-Pyrbpy)(POP)][PF6]·0.5Et2O·CH2Cl2 with ellipsoids plotted at 30% probability level. H atoms and solvent molecules omitted for clarity. Selected bond parameters: Cu1–P1 = 2.2543(7), Cu1–P2 = 2.2876(7), Cu1–N1 = 2.098(2), Cu1–N2 = 2.089(2) Å; P1–Cu1–P2 = 112.04(2), P1–Cu1–N1 = 111.02(6), P2–Cu1–N1 = 104.99(6), P1–Cu1–N2 = 114.25(6), P2–Cu1–N2 = 127.85(6), N1–Cu1–N2 = 79.52(8)°. | |
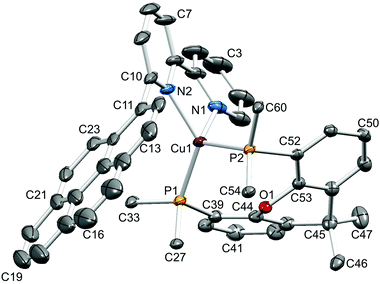 |
| | Fig. 6 Structure of the [Cu(1-Pyrbpy)(xantphos)]+ cation in [Cu(1-Pyrbpy)(xantphos)][PF6]·0.5Et2O with ellipsoids plotted at 30% probability level. H atoms and solvent molecules omitted for clarity, and only the ipso-C atom of each of the four P-attached phenyl rings is shown. Selected bond parameters: Cu1–P2 = 2.2576(7), Cu1–P1 = 2.2649(8), Cu1–N2 = 2.098(2), Cu1–N1 = 2.104(3) Å; P2–Cu1–P1 = 112.20(3), P2–Cu1–N2 = 110.70(8), P1–Cu–N2 = 130.64(7), P2–Cu1–N1 = 112.49(8), P1–Cu1–N1 = 104.67(8), N2–Cu1–N1 = 79.88(13)°. | |
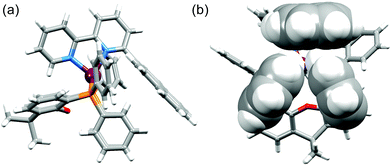 |
| | Fig. 7 (a) The [Cu(1-Pyrbpy)(xantphos)]+ cation showing the orientation of the 1-pyrenyl substituent (right) remote from the xanthene ‘bowl’ (left). (b) Edge-to-face positioning of the two phenyl rings of xantphos and the naphthyl unit in [Cu(2-Naphbpy)(xantphos)]+. | |
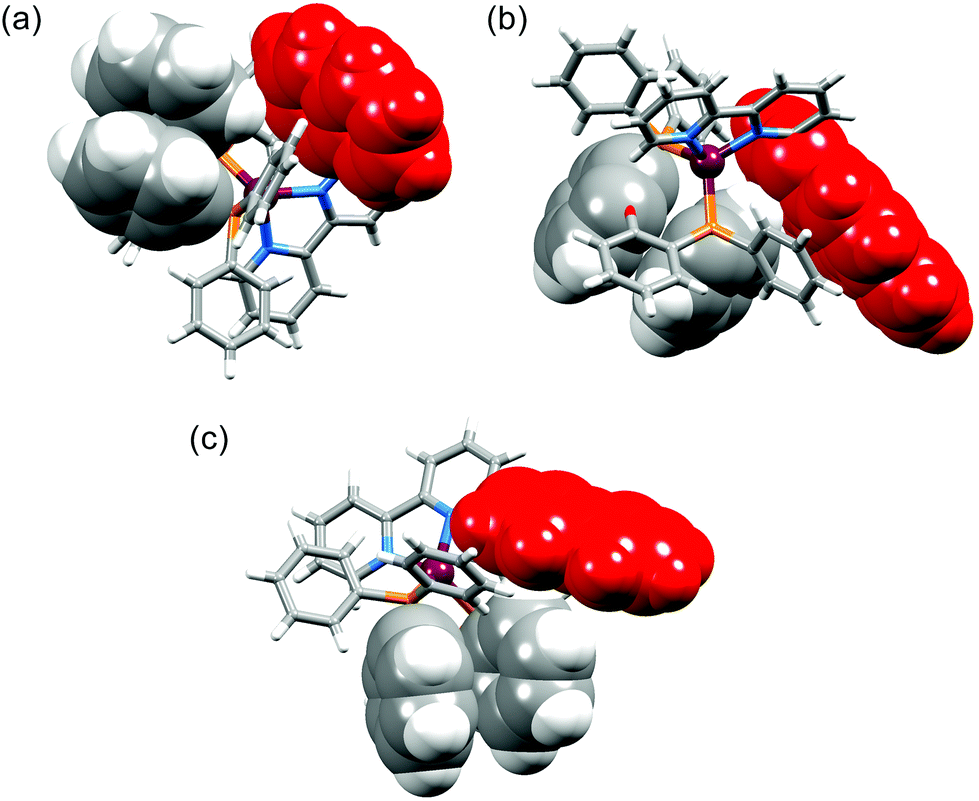
The interannular twist within the bpy unit varies significantly across the series of structures with the angle between the planes through the pyridine rings ranging from 5.8 to 26.0° (Table 2). This twist is coupled with the orientation of the pendant aryl unit with respect to the phenyl substituents of the POP or xantphos ligand. In [Cu(1-Pyrbpy)(xantphos)]+ and [Cu(2-Naphbpy)(xantphos)]+, one phenyl ring of each PPh2 group engages in an edge-to-face interaction with the pyrenyl or naphthyl group (Fig. 7). The closest CH⋯π contacts32 (measured to the centroid of the closest C6 ring) are 2.75 Å in [Cu(2-Naphbpy)(xantphos)]+ and 2.45 and 2.58 Å in [Cu(1-Pyrbpy)(xantphos)]+. Each POP-containing complex exhibits face-to-face π-stacking between one arene ring of the (C6H4)2O unit and one phenyl ring (Fig. 8). In [Cu(1-Naphbpy)(POP)]+ (Fig. 8a), this interaction is characterized by an angle between ring planes, centroid⋯centroid distance, and centroid⋯plane separation of 12.0°, 3.56 Å and 3.53 Å. The angles between the ring planes in [Cu(1-Pyrbpy)(POP)]+ and [Cu(2-Naphbpy)(POP)]+ are, however, 26.0 and 23.4°, respectively. This deviation from a coplanar orientation is associated with edge-to-face CH⋯π contacts involving the 6-substituted pyrenyl or naphthyl group (Fig. 8b and c).
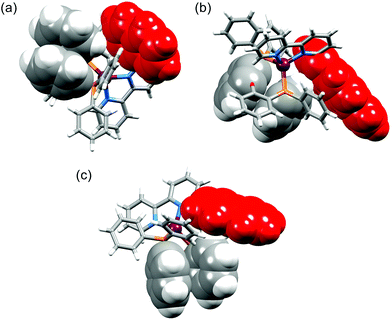 |
| | Fig. 8 Structures of (a) [Cu(1-Naphbpy)(POP)]+, (b) [Cu(1-Pyrbpy)(POP)]+ and (c) [Cu(2-Naphbpy)(POP)]+ showing π-stacking of arene rings within the POP ligand. See text discussion. The 6-substituted naphthyl or pyrenyl group is shown in red. | |
Table 2 Angular distortions of the N^N ligands in the [Cu(N^N)(P^P)]+ cations
| Complex |
Angle between planes of pyridine rings/° |
Angle between plane of pyridine ring and attached arene unit/° |
| [Cu(1-Naphbpy)(POP)]+ |
16.0 |
69.0 |
| [Cu(2-Naphbpy)(POP)]+ |
18.4 |
50.5 |
| [Cu(1-Pyrbpy)(POP)]+ |
5.8 |
66.6 |
| [Cu(2-Naphbpy)(xantphos)]+ |
22.8 |
44.0 |
| [Cu(1-Pyrbpy)(xantphos)]+ |
26.0 |
60.8 |
Interestingly, intermolecular face-to-face π-contacts play an insignificant role in packing in the solid state. Pyrenyl-units of adjacent cations in [Cu(1-Pyrbpy)(POP)][PF6]·0.5Et2O·CH2Cl2 approach closely (Fig. 9), but the orientation of the two arene moieties is not optimal for efficient stacking. The angle between the least squares planes through the two pyrenyl units is 21.8° and the centroid⋯centroid separation of the two closest C6-rings is 3.80 Å.
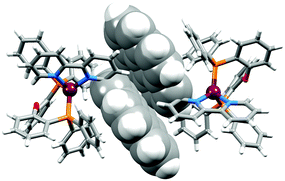 |
| | Fig. 9 Close approach of adjacent pyrenyl-units of adjacent [Cu(1-Pyrbpy)(POP)]+ cations (symmetry codes x, y, z and 1 − x, y, 1 − z). | |
NMR spectroscopy and dynamic behaviour of the [Cu(N^N)(P^P)][PF6] complexes
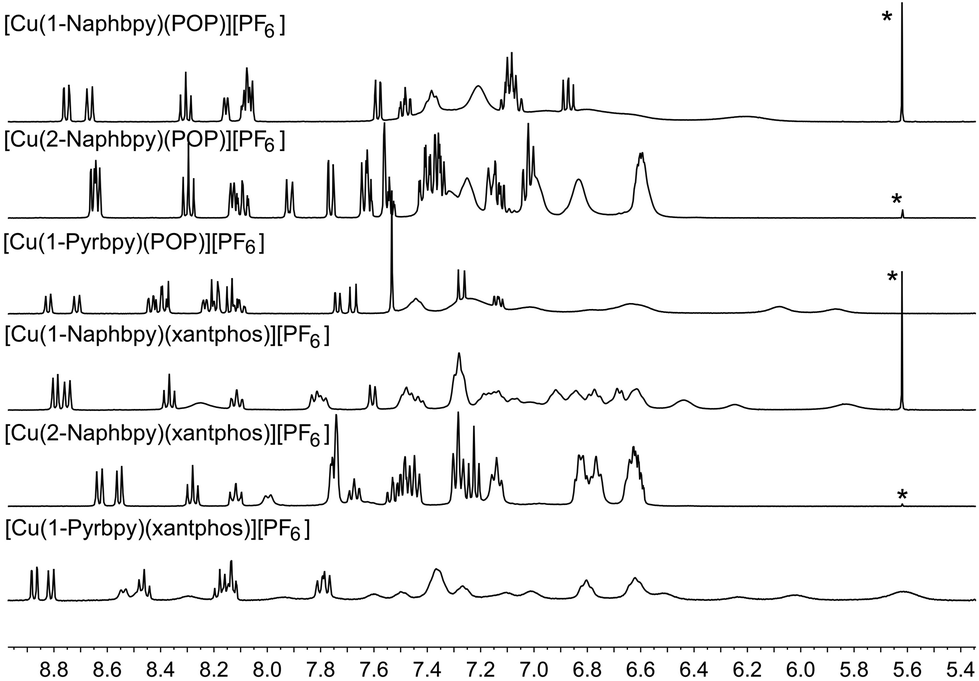
Acetone-d6 was chosen as the solvent for NMR spectroscopic studies because previous studies revealed that some [Cu(N^N)(P^P)]+ complexes are prone to ligand dissociation in CD2Cl2 after several days in solution.8,11Fig. 10 shows the room temperature 1H NMR spectra of the six complexes. Although the resonances arising from the bpy domain are sharp in all cases, those from the 6-aryl substituent are only sharp at 298 K for the 2-Naphthyl group. Signals from the POP and xantphos ligands are broadened in [Cu(2-Naphbpy)(POP)][PF6] and [Cu(2-Naphbpy)(xantphos)][PF6], and are very broad for the remaining complexes (Fig. 10). NMR spectroscopic characterization of [Cu(2-Naphbpy)(POP)][PF6] and [Cu(2-Naphbpy)(xantphos)][PF6] was carried out by recording COSY, NOESY, HMBC and HMQC spectra at 298 K. For the remaining complexes, ROESY, TOCSY, HSQC, HMBC and 1H–31P-HSQC (only for the complexes containing 1-Pyrbpy) spectra were measured at lower temperatures. Use of these 2D methods permitted full assignment of the 1H and 13C NMR spectra (see Experimental section) and atom labelling is given in Schemes 1 and 2.
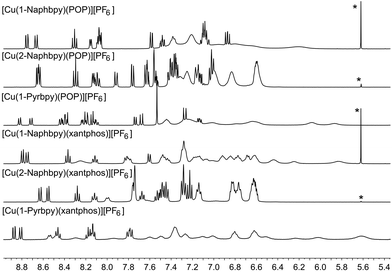 |
| | Fig. 10 Room temperature 400 MHz 1H NMR spectra of acetone-d6 solutions of the six complexes. The alkyl regions of the spectra are omitted. * = residual CH2Cl2. Chemical shifts in δ/ppm. | |
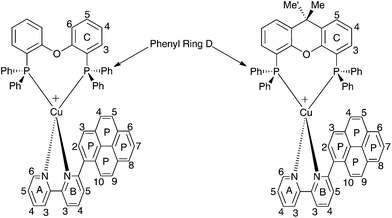 |
| | Scheme 2 Structures of [Cu(1-Pyrbpy)(POP)]+ (left) and [Cu(1-Pyrbpy)(xantphos)]+ with ring and atom labelling for NMR spectroscopic assignments. | |
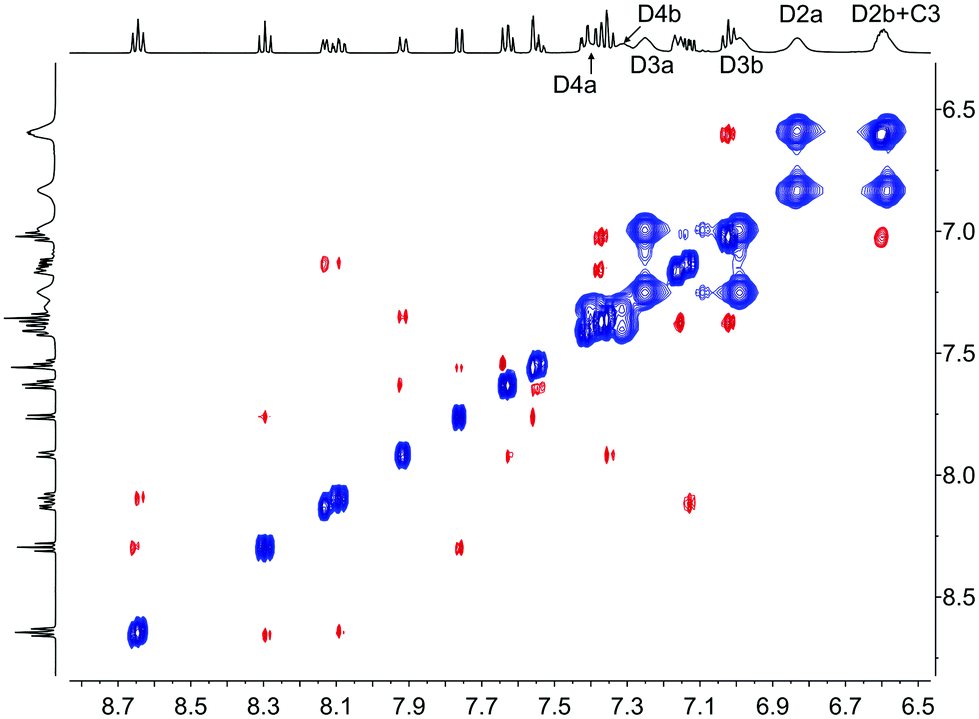
In the discussion below, we assume that dynamic behaviour arises from non-dissociative processes8 and initially consider the three complexes containing POP. In [Cu(2-Naphbpy)(POP)][PF6], one set of signals arising from the POP-backbone ring-C protons (correlating in the HMQC spectrum to one set of 13C NMR signals) is observed at 298 K. This is consistent with the [Cu(2-Naphbpy)(POP)]+ cation having C2 symmetry. Each PPh2 group of the POP ligand gives two sets of signals, one each for the phenyl rings (labelled Da and Db) pointing towards or away from the aryl group on the 6-position of the bpy. The conformational flexibility of the 8-membered chelate {CuPCCOCCP}-ring can exchange these positions as confirmed by exchange peaks (EXSY) in the ROESY spectrum between pairs HD2a/HD2b, HD3a/HD3b HD4a/HD4b (Fig. 11). Low temperature 13C and 1H NMR spectra of [Cu(1-Naphbpy)(POP)][PF6] and [Cu(1-Pyrbpy)(POP)][PF6] indicated loss of C2 symmetry and as an example, we focus on [Cu(1-Naphbpy)(POP)][PF6]. Two sets of signals for the C rings of the POP-backbone are observed (labelled Ca and Cb, see Experimental section) along with four sets of signals for the P-bonded phenyl rings (labelled Da, Db, Dc and Dd). The ROESY spectrum (at 253 K) is shown in Fig. S1,† and from this, EXSY cross-peaks the exchange of rings Ca/Cb, Da/Db and Dc/Dd at higher temperatures.
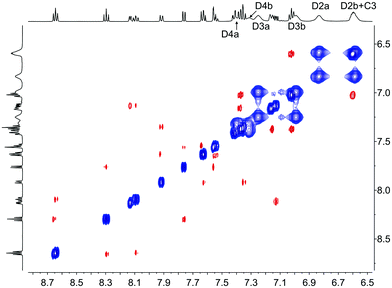 |
| | Fig. 11 500 MHz NOESY spectrum of [Cu(2-Naphbpy)(POP)][PF6] at 298 K in acetone-d6. Signals in phase with the diagonal (blue) correspond to EXSY peaks and signals with opposite phase relative to the diagonal (red) correspond to NOESY peaks. Chemical shifts in δ/ppm. | |
We now turn to the solution dynamic behaviour of [Cu(1-Naphbpy)(xantphos)]+, [Cu(2-Naphbpy)(xantphos)]+ and [Cu(1-Pyrbpy)(xantphos)]+ and Fig. 10 shows the room temperature 1H NMR spectra. As observed for the POP complexes, the spectra for [Cu(1-Naphbpy)(xantphos)][PF6] and [Cu(1-Pyrbpy)(xantphos)][PF6] are very broad at 298 K, indicating dynamic behaviour. Fig. 12 shows the effect of cooling an acetone-d6 solution of [Cu(1-Pyrbpy)(xantphos)][PF6]; similar changes are observed for [Cu(1-Naphbpy)(xantphos)][PF6]. The spectra at 238 K (N^N = 1-Pyrbpy) or 223 K (N^N = 1-Naphbpy) were fully assigned using 2D methods (see Experimental section).
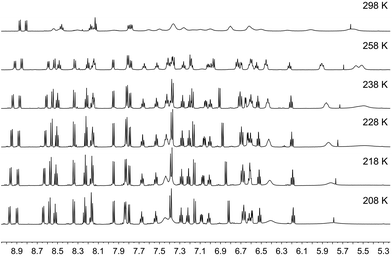 |
| | Fig. 12 Variable temperature 600 MHz 1H NMR spectra of an acetone-d6 solution of [Cu(1-Pyrbpy)(xantphos)][PF6]. The alkyl region of the spectrum is omitted. Chemical shifts in δ/ppm. | |
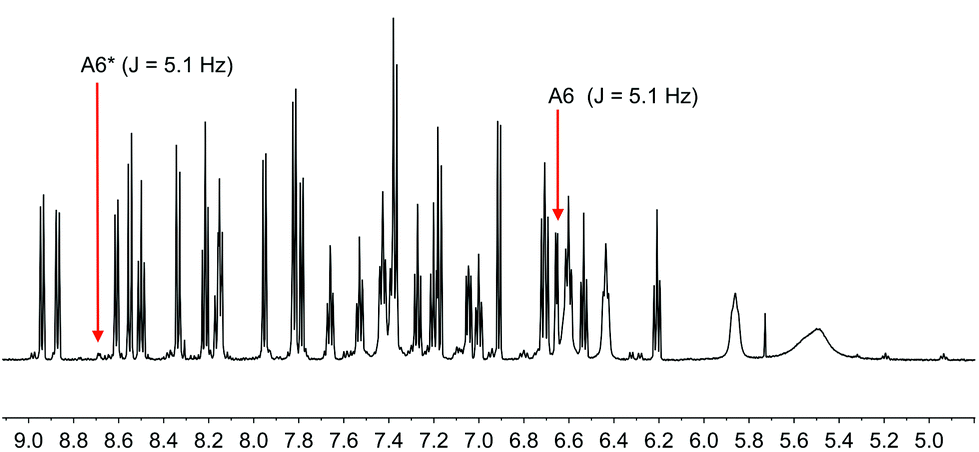
Although xantphos is more rigid than POP, the xanthene ‘bowl’ may undergo inversion.33 We previously reported that in solution, [Cu(Phbpy)(xantphos)]+ (Phbpy = 6-phenyl-2,2′-bipyridine) exists as two conformers which are related by the inversion of the xanthene ‘bowl’ (Fig. 1c). The relative population of the conformers of [Cu(Phbpy)(xantphos)]+ in CD2Cl2 was ∼1.0:0.9, and the calculated energy difference was 3.57 kcal mol−1. The characteristic 1H NMR signal distinguishing the two conformers is that for HA6 (see Scheme 2 and Fig. 1c for labelling) which appeared at δ 8.42 ppm in one conformer and δ 6.35 ppm in the other.8 In the low temperature 1H NMR spectra of [Cu(1-Naphbpy)(xantphos)][PF6] and [Cu(1-Pyrbpy)(xantphos)][PF6], a second set of signals with much lower intensity with respect to the dominant peaks (∼0.05:1.0) was observed. This is shown for [Cu(1-Pyrbpy)(xantphos)][PF6] in Fig. 13. For the major and minor components in the spectrum, the signal for HA6 (which exhibits a characteristic coupling constant of ∼5 Hz) appears at δ 6.65 and 8.69 ppm, respectively. These values are similar to those observed in [Cu(Phbpy)(xantphos)][PF6] (δ 6.35 and 8.42 ppm)8 (see above) and are consistent with the presence of two conformers of [Cu(1-Pyrbpy)(xantphos)]+ which interconvert through inversion of the xanthene ‘bowl’. EXSY cross-peaks observed in the ROESY spectrum (Fig. S2†) confirmed an exchange process. As we discussed in detail8 for [Cu(Phbpy)(xantphos)]+, the methyl region of the 1H NMR spectrum is very informative. The boat conformation (Fig. 4 and 6)8,34 of the xanthene unit forces one methyl group to be in the plane of the xantphos ligand backbone, whereas the second methyl is out-of-plane. This is clear in the projection of the [Cu(1-Pyrbpy)(xantphos)]+ cation in Fig. 7. Interconversion of the conformers (Fig. 1c) places the two methyl groups in different magnetic environments. In the dominant conformer, signals for the two Me groups are at δ 2.03 and 1.28 ppm, whereas in the second conformer, the corresponding resonances are δ 1.14 and 1.87 ppm, EXSY cross-peaks (δ 1.14 with 2.03 ppm, and δ 1.87 with 1.28 ppm) confirmed exchange. These observations are consistent with those described [Cu(Phbpy)(xantphos)][PF6].8 The significantly different populations of the conformers in the case of [Cu(Phbpy)(xantphos)][PF6] (∼1.0:0.9) versus [Cu(1-Pyrbpy)(xantphos)][PF6] (∼0.05:1.0) is consistent with the sterically more demanding pyrenyl group.
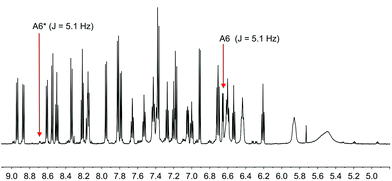 |
| | Fig. 13 Part of the solution 600 MHz 1H NMR spectrum of [Cu(1-Pyrbpy)(xantphos)][PF6] (acetone-d6) at 238 K. Chemical shifts in δ/ppm. See text for explanation of the subspectrum. | |
The room temperature 31P spectra of [Cu(2-Naphbpy)(POP)][PF6] and [Cu(2-Naphbpy)(xantphos)][PF6] show broad signals at δ −13.4 and −12.8 ppm, respectively. A similar broad signal at δ −14.5 ppm is observed for [Cu(1-Pyrbpy)(POP)][PF6] at 238 K. In contrast, [Cu(1-Pyrbpy)(xantphos)][PF6] shows two doublets at 238 K (2JPP = 95 Hz) at δ −10.9 and −13.5 ppm. A similar pattern is observed in the low temperature spectrum of [Cu(1-Naphbpy)(xantphos)][PF6] (2JPP = 110 Hz). Two 31P NMR signals (δ −12.7 and −15.5 ppm) are observed for [Cu(1-Naphbpy)(POP)][PF6] at 253 K, but the coupling could not be resolved. These observations are consistent with desymmetrization of the P^P ligand on the 1H NMR time-scale at low temperatures.
Electrochemistry
The [Cu(N^N)(P^P)][PF6] complexes are redox active and cyclic voltammetry was used to investigate the processes. Each complex undergoes a reversible copper-centred oxidation (Table 3), and the Eox1/2 values are similar to those of +0.82 and +0.81 V reported for [Cu(6,6′-Me2bpy)(POP)][BF4] and [Cu(6,6′-Me2bpy)(xantphos)][BF4] (MeCN solution, vs. Fc/Fc+).35 For each compound, reduction processes within the solvent accessible window were very poorly defined.
Table 3 Cyclic voltammetric data for [Cu(N^N)(P^P)][PF6] complexes referenced to internal Fc/Fc+ = 0 V; CH2Cl2 (freshly distilled) solutions with [nBu4N][PF6] as supporting electrolyte and scan rate of 0.1 V s−1
| Complex cation |
E
ox1/2/V (Epc − Epa/mV) |
| [Cu(1-Naphbpy)(POP)]+ |
+0.79 (97) |
| [Cu(1-Naphbpy)(xantphos)]+ |
+0.82 (101) |
| [Cu(2-Naphbpy)(POP)]+ |
+0.77 (96) |
| [Cu(2-Naphbpy)(xantphos)]+ |
+0.81 (101) |
| [Cu(1-Pyrbpy)(POP)]+ |
+0.77 (99) |
| [Cu(1-Pyrbpy)(xantphos)]+ |
+0.82 (88) |
Solution absorption and emission properties
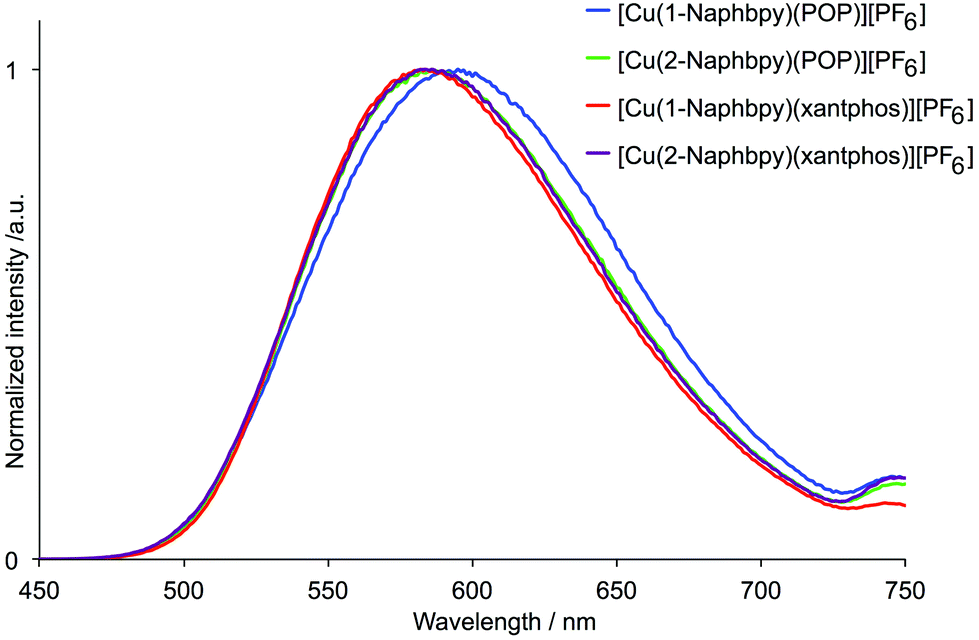
The solution absorption spectra of [Cu(N^N)(xantphos)][PF6] and [Cu(N^N)(POP)][PF6] are shown in Fig. 14. The intense absorption bands below 340 nm correspond to spin-allowed ligand-centred π* ← π transitions. For [Cu(1-Naphbpy)(P^P)][PF6] and [Cu(2-Naphbpy)(P^P)][PF6] (P^P = POP or xantphos), a broad absorption of low intensity appears at ∼390 nm arising from metal-to-ligand charge transfer (MLCT). For the complexes containing 1-Pyrbpy (dotted curves in Fig. 14), the profile of the bands between 225 and ∼335 nm reflects that of the absorption spectrum of the free 1-Pyrbpy ligand (Fig. S3†) which is turn is similar to, but broader than, that of pyrene. We assign the broad absorption around 350 nm to a charge-transfer from the pyrenyl to bpy unit.36,37
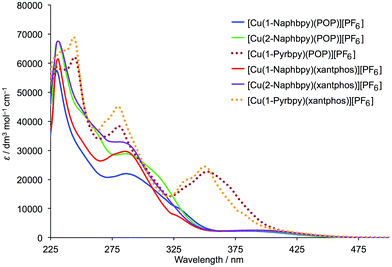 |
| | Fig. 14 Absorption spectra of the [Cu(N^N)(P^P)][PF6] complexes (CH2Cl2, 2.5 × 10−5 mol dm−3). | |
Dichloromethane solutions of the [Cu(N^N)(P^P)][PF6] complexes were poorly emissive. Emission data for powder samples are given in Table 4. The complexes are all orange emitters with values of λemmax in the range 582 to 617 nm. The emission spectra of the complexes containing 1-Naphby and 2-Naphbpy are shown in Fig. 15. Photoluminescence quantum yields (PLQY) were <6% and the emission lifetimes (τ1/2) were all ∼2 μs (Table 4). Both [Cu(1-Pyrbpy)(POP)][PF6] and [Cu(1-Pyrbpy)(xantphos)][PF6] are extremely weak emitters in the solid state. This may be due to a low lying triplet state which leads to quenching of the MLCT emission.
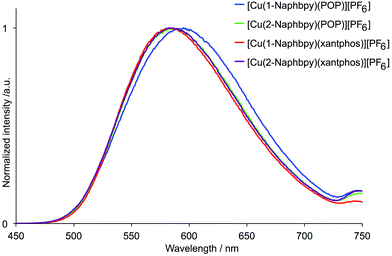 |
| | Fig. 15 Emission spectra of powder samples of [Cu(N^N)(P^P)][PF6] complexes containing 1-Naphby and 2-Naphbpy (λexc = 365 nm). | |
Table 4 Emission maxima and PLQY for powder samples of [Cu(P^P)(N^N)][PF6] complexes (λexc = 365 nm)
| Compound |
λ
maxem/nm |
PLQY/% |
τ
1/2(1)/μs (A1) |
τ
1/2(2)/μs (A2) |
τ
1/2(av)/μs |
|
Biexponential fit using the equation τ1/2(av) = ∑Aiτi/∑Ai where Ai is the pre-exponential factor for the lifetime.
Not measured.
|
| [Cu(1-Naphbpy)(POP)][PF6] |
595 |
4.0 |
2.9 (0.3812) |
1.1 (0.4664) |
1.9a |
| [Cu(2-Naphbpy)(POP)][PF6] |
586 |
4.6 |
2.5 (0.4636) |
0.8 (0.4101) |
1.7a |
| [Cu(1-Pyrbpy)(POP)][PF6] |
617 |
0.3 |
|
|
|
| [Cu(1-Naphbpy)(xantphos)][PF6] |
582 |
5.6 |
2.9 (0.4547) |
1.1 (0.3933) |
2.1a |
| [Cu(2-Naphbpy)(xantphos)][PF6] |
586 |
3.6 |
2.7 (0.4644) |
1.0 (0.3951) |
1.9a |
| [Cu(1-Pyrbpy)(xantphos)][PF6] |
576 |
0.3 |
|
|
|
Conclusions
We have prepared a series of [Cu(N^N)(POP)][PF6] and [Cu(N^N)(xantphos)][PF6] compounds in which N^N is a bpy ligand bearing a sterically hindered 1-naphthyl, 2-naphthyl or 1-pyrenyl substituent in the 6-position. Crystallographic data confirm that the copper(I) centre is in a distorted tetrahedral environment, and the N^N ligand is oriented with the sterically-demanding aryl substituent positioned away from the (C6H4)2O unit of POP or the xanthene ‘bowl’ of xantphos. Each POP-containing complex exhibits face-to-face π-stacking between one arene ring of the (C6H4)2O unit and one phenyl ring of a PPh2 unit. Despite the extended π-systems, intermolecular face-to-face π-contacts do not play a dominant role in the packing of the compounds in the solid state.
In solution, the complexes undergo dynamic behaviour, and the 1H NMR signals from the POP and xantphos ligands are broad to very broad at 298 K. At low temperatures, the phenyl rings in each PPh2 group in the POP-containing compounds gives two sets of signals (pointing towards or away from the 6-substituted aryl group on the bpy) but exchange associated with the conformationally flexible 8-membered chelate {CuPCCOCCP}-ring occurs at higher temperatures; this was confirmed using EXSY exchange spectroscopy. In acetone solution, the [Cu(N^N)(xantphos)]+ complexes exist as a mixture of conformers which interconvert through inversion of the xanthene ‘bowl’ (Fig. 1). Proton HA6 resides in very different magnetic environments in the two conformers, and the ratio of conformers in [Cu(1-Pyrbpy)(xantphos)][PF6] is ∼0.05:1.0. This compares to almost equal populations for the analogous conformers of [Cu(Phbpy)(xantphos)]+, demonstrating the significant steric influences of going from a phenyl to 1-pyrenyl 6-substituent.
Consistent with the trends previously observed on going from 6-alkyl to 6-phenyl substituents in the bpy ligand in [Cu(N^N)(POP)]+ and [Cu(N^N)(xantphos)]+ complexes,8 the introduction of the 1-Naphthyl or 2-Naphthyl substituents result in poor PLQY values. In the case of the 1-Pyrbpy ligand, a low lying triplet state may be responsible for quenching the MLCT emission.
Acknowledgements
We acknowledge the Swiss National Science Foundation (Grant number 162631) and the University of Basel for financial support. We thank Sarah Keller for invaluable suggestions.
Notes and references
- F. Dumur, Org. Electron., 2015, 21, 27 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- M. Magni, P. Biagini, A. Colombo, C. Dragonetti, D. Roberto and A. Valore, Coord. Chem. Rev., 2016, 322, 69 CrossRef CAS.
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6 CrossRef CAS PubMed.
- S.-M. Kuang, D. G. Cuttell, D. R. McMillin, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2002, 41, 3313 CrossRef CAS PubMed.
- R. D. Costa, D. Tordera, E. Ortí, H. J. Bolink, J. Schönle, S. Graber, C. E. Housecroft, E. C. Constable and J. A. Zampese, J. Mater. Chem., 2011, 21, 16108 RSC.
- S. Keller, E. C. Constable, C. E. Housecroft, M. Neuburger, A. Prescimone, G. Longo, A. Pertegás, M. Sessolo and H. J. Bolink, Dalton Trans., 2014, 43, 16593 RSC.
- S. Keller, A. Pertegás, G. Longo, L. Martinez, J. Cerdá, J. M. Junquera-Hernández, A. Prescimone, E. C. Constable, C. E. Housecroft, E. Ortí and H. J. Bolink, J. Mater. Chem. C, 2016, 4, 3857 RSC.
- C. Bizzarri, C. Strabler, J. Prock, B. Trettenbrein, M. Ruggenthaler, C.-H. Yang, F. Polo, A. Iordache, P. Brüggeller and L. De Cola, Inorg. Chem., 2014, 53, 10944 CrossRef CAS PubMed.
- M. D. Weber, C. Garino, G. Volpi, E. Casamassa, M. Milanesio, C. Barolo and R. D. Costa, Dalton Trans., 2016, 45, 8984 RSC.
- F. Brunner, L. Martínez-Sarti, S. Keller, A. Pertegás, A. Prescimone, E. C. Constable, H. J. Bolink and C. E. Housecroft, Dalton Trans., 2016, 45, 15180 RSC.
- R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322 CrossRef CAS PubMed.
- D. Asil, J. A. Foster, A. Patra, X. de Hatten, J. del Barrio, O. A. Scherman, J. R. Nitschke and R. H. Friend, Angew. Chem., Int. Ed., 2014, 53, 8388 CrossRef CAS PubMed.
- N. Armaroli, G. Accorsi, M. Holler, O. Moudam, J.-F. Nierengarten, Z. Zhou, R. T. Wegh and R. Welter, Adv. Mater., 2006, 18, 1313 CrossRef CAS.
- L. Zhang, B. Li and Z. Su, J. Phys. Chem. C, 2009, 113, 13968 CAS.
- C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854 CrossRef CAS PubMed.
- S. Medina-Rodríguez, F. J. Orriach-Fernández, C. Poole, P. Kumar, A. de al Torre-Vega, J. F. Fernández-Sánchez, E. Baranoff and A. Fernández-Gutiérrez, Chem. Commun., 2015, 51, 11401 RSC.
- H. Takeda, K. Ohashi, A. Sekine and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 4354 CrossRef CAS PubMed.
- A. J. J. Lennox, S. Fischer, M. Jurrat, S.-P. Luo, N. Rockstroh, H. Junge, R. Ludwig and M. Beller, Chem. – Eur. J., 2016, 22, 1233 CrossRef CAS PubMed.
- S.-P. Luo, N.-Y. Chen, Y.-Y. Sun, L.-M. Xia, Z.-C. Wu, H. Junge, M. Beller and Q.-A. Wu, Dyes Pigm., 2016, 134, 580 CrossRef CAS.
- A. Kaeser, M. Mohankumar, J. Mohanraj, F. Monti, M. Holler, J.-J. Cid, O. Moudam, I. Nierengarten, L. Karmazin-Brelot, C. Duhayon, B. Delavaux-Nicot, N. Armaroli and J.-F. Nierengarten, Inorg. Chem., 2013, 52, 12140 CrossRef CAS PubMed.
- T. Norrby, A. Börje, L. Zhang and B. Akermark, Acta Chem. Scand., 1998, 52, 77 CrossRef CAS.
- G. J. Kubas, Inorg. Synth., 1979, 19, 90 CrossRef CAS.
- Y. J. Pu, M. Miyamoto, K. Nakayama, T. Oyama, Y. Masaaki and J. Kido, Org. Electron., 2009, 10, 228 CrossRef CAS.
- G. E. Schneider, A. Pertegás, E. C. Constable, C. E. Housecroft, N. Hostettler, C. D. Morris, J. A. Zampese, H. J. Bolink, J. M. Junquera-Hernández, E. Ortí and M. Sessolo, J. Mater. Chem. C, 2014, 2, 7047 RSC.
-
APEX2, version 2 User Manual, M86-E01078, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2006 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389 CrossRef.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- J. G. Park and Y. Jahng, Bull. Korean Chem. Soc., 1998, 19, 436 CAS.
- J. Yuasa, M. Dan and T. Kawai, Dalton Trans., 2013, 42, 16096 RSC.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- See for example: G. Tárkányi, P. Király, G. Pálinkás and A. Deák, Magn. Reson. Chem., 2007, 45, 917 CrossRef CAS PubMed; A. Pintado-Alba, H. de la Riva, M. Nieuwhuyzen, D. Bautista, P. R. Raithby, H. A. Sparkes, S. J. Teat, J. M. López-de-Luzuriaga and M. C. Lagunas, Dalton Trans., 2004, 3459 RSC.
- S. Keller, F. Brunner, A. Prescimone, E. C. Constable and C. E. Housecroft, Inorg. Chem. Commun., 2015, 58, 64 CrossRef CAS and references cited therein.
- I. Andrés-Tomé, J. Fyson, F. Baiao Dias, A. P. Monkman, G. Iacobellis and P. Coppo, Dalton Trans., 2012, 41, 8669 RSC.
- E. C. Constable, C. E. Housecroft, M. Neuburger, P. Rösel, G. E. Schneider, J. A. Zampese, F. Monti, N. Armaroli, R. D. Costa and E. Ortí, Inorg. Chem., 2013, 52, 885 CrossRef CAS PubMed.
- A. Harriman and M. Hissler, Phys. Chem. Chem. Phys., 1999, 1, 4203 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2: additional NMR spectra; Fig. S3: absorption spectrum of 1-Pyrbpy. CCDC 1528548–1528552. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7dt00782e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Stefan
Graber
,
Yann
Baumgartner
,
Daniel
Häussinger
,
Stefan
Graber
,
Yann
Baumgartner
,
Daniel
Häussinger