
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and characterization of metastable trigonal layered MSb2O6 phases (M = Co, Ni, Cu, Zn, and Mg) and considerations on FeSb2O6†
A. Yu.
Nikulin
a,
E. A.
Zvereva
b,
V. B.
Nalbandyan
 *a,
I. L.
Shukaev
a,
A. I.
Kurbakov
cd,
M. D.
Kuchugura
cd,
G. V.
Raganyan
b,
Yu. V.
Popov
e,
V. D.
Ivanchenko
a and
A. N.
Vasiliev
bfg
*a,
I. L.
Shukaev
a,
A. I.
Kurbakov
cd,
M. D.
Kuchugura
cd,
G. V.
Raganyan
b,
Yu. V.
Popov
e,
V. D.
Ivanchenko
a and
A. N.
Vasiliev
bfg
aChemistry Faculty, Southern Federal University, 7, ul. Zorge, 344090 Rostov-on-Don, Russia. E-mail: vbn@sfedu.ru
bFaculty of Physics, Moscow State University, 119991 Moscow, Russia
cPetersburg Nuclear Physics Institute – NRC Kurchatov Institute, 188300 Gatchina, Russia
dFaculty of Physics, St.Petersburg State University, 198504 St. Petersburg, Russia
eInstitute for Earth Sciences, Southern Federal University, 40, ul. Zorge, 344090 Rostov-on-Don, Russia
fUral Federal University, 620002 Ekaterinburg, Russia
gNational University of Science and Technology “MISiS”, Moscow 119049, Russia
First published on 28th March 2017
Abstract
MSb2O6 compounds (M = Mg, Co, Ni, Cu, Zn) are known in the tetragonal trirutile forms, slightly distorted monoclinically with M = Cu due to the Jahn–Teller effect. In this study, using a low-temperature exchange reaction between ilmenite-type NaSbO3 and molten MSO4–KCl (or MgCl2–KCl) mixtures, these five compositions were prepared for the first time as trigonal layered rosiaite (PbSb2O6)-type phases. Upon heating, they irreversibly transform to the known phases via amorphous intermediates, in contrast to previously studied isostructural MnSb2O6, where the stable phase is structurally related to the metastable phase. The same method was found to be applicable for preparing stable rosiaite-type CdSb2O6. The formula volumes of the new phases show an excellent correlation with the ionic radii (except for M = Cu, for which a Jahn–Teller distortion is suspected) and are 2–3% larger than those for the known forms although all coordination numbers are the same. The crystal structure of CoSb2O6 was refined via the Rietveld method: P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) 1m, a = 5.1318(3) Å, and c = 4.5520(3) Å. Compounds with M = Co and Ni antiferromagnetically order at 11 and 15 K, respectively, whereas the copper compound does not show long-range magnetic order down to 1.5 K. A comparison between the magnetic behavior of the metastable and stable polymorphs was carried out. FeSb2O6 could not be prepared because of the 2Fe2+ + Sb5+ = 2Fe3+ + Sb3+ redox reaction. This electron transfer produces an additional 5s2 shell for Sb and results in a volume increase. A comparison of the formula volume for the stable mixture FeSbO4 + 0.5Sb2O4 with that extrapolated for FeSb2O6 predicted that the trirutile-type FeSb2O6 can be stabilized at high pressures.
1m, a = 5.1318(3) Å, and c = 4.5520(3) Å. Compounds with M = Co and Ni antiferromagnetically order at 11 and 15 K, respectively, whereas the copper compound does not show long-range magnetic order down to 1.5 K. A comparison between the magnetic behavior of the metastable and stable polymorphs was carried out. FeSb2O6 could not be prepared because of the 2Fe2+ + Sb5+ = 2Fe3+ + Sb3+ redox reaction. This electron transfer produces an additional 5s2 shell for Sb and results in a volume increase. A comparison of the formula volume for the stable mixture FeSbO4 + 0.5Sb2O4 with that extrapolated for FeSb2O6 predicted that the trirutile-type FeSb2O6 can be stabilized at high pressures.
1. Introduction
Preparation of a new crystal form for a well-known composition is challenging and very interesting because it can provide new properties and improve our understanding of the composition-structure–property relations. Recently, using a low-temperature ion-exchange reaction, a new form of MnSb2O6, isostructural with rosiaite PbSb2O6, was prepared and its magnetic properties and thermal transformations were studied.1 In this structure, magnetic cations are arranged in trigonal layers that antiferromagnetically order at low temperatures, forming spin-frustrated triangles.In this study, the same preparative approach was expanded to the entire MSb2O6 family (M = Mg, Fe, Co, Ni, Cu, Zn, and Cd) and the magnetic properties of the new phases with M = Ni, Co, and Cu were studied. A total of five of the seven listed compositions, with M = Mg, Co, Ni, Cu, and Zn, are known to crystallize in the tetragonal trirutile-structure type,2 which is slightly monoclinically distorted for M = Cu due to the Jahn–Teller effect, whereas CdSb2O6 is found in the rosiaite form.3 MSb2O6 compounds with a partially filled d subshell (M = Co, Ni, and Cu) have attracted attention due to their magnetic properties,4–13 whereas antimonates with nonmagnetic ions (M = Mg, Zn, and Cd) may be interesting as photocatalysts,14,15 transparent conductors,16 and/or sensors.17–19 Initially, FeSb2O6 was also reported to exist in the trirutile form; however, later studies showed that the mineral known as FeSb2O6 is actually FeSbO4 (Fe1/2Sb1/2O2) with disordered rutile-type structure.20 An attempt to prepare FeSb2O6via solid-state reactions of Sb2O3 and FeSO4·7H2O at 1000 °C in air was made and a mixture of FeSbO4 and Sb2O4 was obtained.20 This result is explained by the oxidation of Fe2+ in air and does not preclude an attempted preparation in an inert atmosphere at much lower temperatures.
2. Experimental
2.1. Sample preparation and analysis
Only reagent-grade starting materials were used. NaSbO3 was prepared at 550–850 °C from a stoichiometric mixture of dried Sb2O3, Na2CO3, and NaNO3, as previously reported,1,21 and its phase purity was confirmed via powder X-ray diffraction (XRD). MSO4·xH2O (M = Fe, Co, Ni, Cu, Zn, and Cd) was dried at 400 °C (in flowing argon atmosphere for FeSO4). Subsequently, NaSbO3, MSO4, and KCl were carefully mixed using a mortar and pestle at the molar ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:5, providing a five-fold excess of MSO4 against the stoichiometric amount. For M = Cd, a double excess was also tested, with a 1:1:2 ratio of NaSbO3, CdSO4, and KCl. In some instances, half of the KCl was substituted with KBr to further lower the solidus temperature.
:2.5:5, providing a five-fold excess of MSO4 against the stoichiometric amount. For M = Cd, a double excess was also tested, with a 1:1:2 ratio of NaSbO3, CdSO4, and KCl. In some instances, half of the KCl was substituted with KBr to further lower the solidus temperature.
For M = Mg, this approach was inapplicable due to the excessively high eutectic temperature of the MgSO4–KCl join, 665 °C.22 Therefore, a MgCl2–KCl mixture was used instead.
The mixtures were heated for various periods at various temperatures in air (except for M = Fe, for which a CO2 atmosphere was used), cooled, washed with warm water, dried, and examined via XRD.
The elemental composition of the samples was estimated using an electron microprobe (INCA ENERGY 450/XT) equipped with an X-Act ADD detector based on an electron microscope VEGA II LMU (Tescan) operated at the accelerating voltage of 20 kV. No potassium was detected; however, some samples showed a non-zero sodium content. The molar ratios M/Sb and Na/Sb were averaged on 8–11 points in each sample.
2.2. Diffraction studies
XRD was performed using Cu Kα radiation by an ARL X'TRA diffractometer equipped with a solid-state Si(Li) detector eliminating Co and Fe fluorescence and other undesired wavelengths (although sometimes very weak reflections from CuKβ and WLα radiations were observed). Rietveld refinements were performed using the GSAS + EXPGUI suite.23,24Neutron diffraction experiments were carried out via the cold neutron two-axis diffractometer G4.1 located at ORPHEE reactor, Laboratory Léon Brillouin, Saclay, France. The wavelength of the incident monochromatic neutrons was 2.426 Å. Neutron diffraction data were obtained in the 2θ range from 6.0 to 85.9° with the step of 0.1°. The measurements were carried out using the special Orange cryostat for neutron research. The sample was placed in a cylindrical vanadium container having a diameter of 6 mm. The diffraction pattern was treated by the Rietveld method using the FullProf suite.25
2.3. Magnetic measurements
The magnetic properties (temperature dependencies of the magnetic susceptibility and magnetization isotherms) of the MSb2O6 (M = Co, Ni, and Cu) samples in the temperature range from 2 to 350 K under a magnetic field up to 9 T were measured using Quantum Design MPMS-7T and VSM option of the Quantum Design PPMS-9T devices. The electron spin resonance spectra at room temperature were obtained using an X-band ESR spectrometer CMS 8400 (ADANI) (f ≈ 9.4 GHz, B ≤ 0.7 T). The effective g-factors of the samples were calculated with respect to an external reference for the resonance field. BDPA (a,g-bisdiphenyline-b-phenylallyl), gref = 2.00359, was used as the reference material.3. Results and discussion
3.1. Preparation, identification, and phase transformations
As expected, reactions of NaSbO3 with low-melting mixtures of MSO4 (M = Co, Ni, Cu, and Zn) or MgCl2 with KCl (or KCl–KBr) resulted in formation of five new trigonal layered phases of the rosiaite type (Table 1, Fig. 1). However, this required careful selection of the temperature and duration, typically in the ranges of 420–500 °C and 40–80 minutes. The lower limits were determined by the solidus temperature of the salt mixture and incomplete reaction, whereas the higher limits were determined by the MSb2O6 transformation to the stable form. Moreover, the results were also dependent on the total mass of the reaction mixture. Because the furnace temperature was set only slightly higher than the solidus temperature, complete melting of the salt mixture required considerable time, especially long with large batches. To prepare 1–2 g of the desired product in one stage, a 5–10 g batch was required, which usually resulted in either incomplete exchange or (at least partial) conversion to the stable trirutile-type polymorph.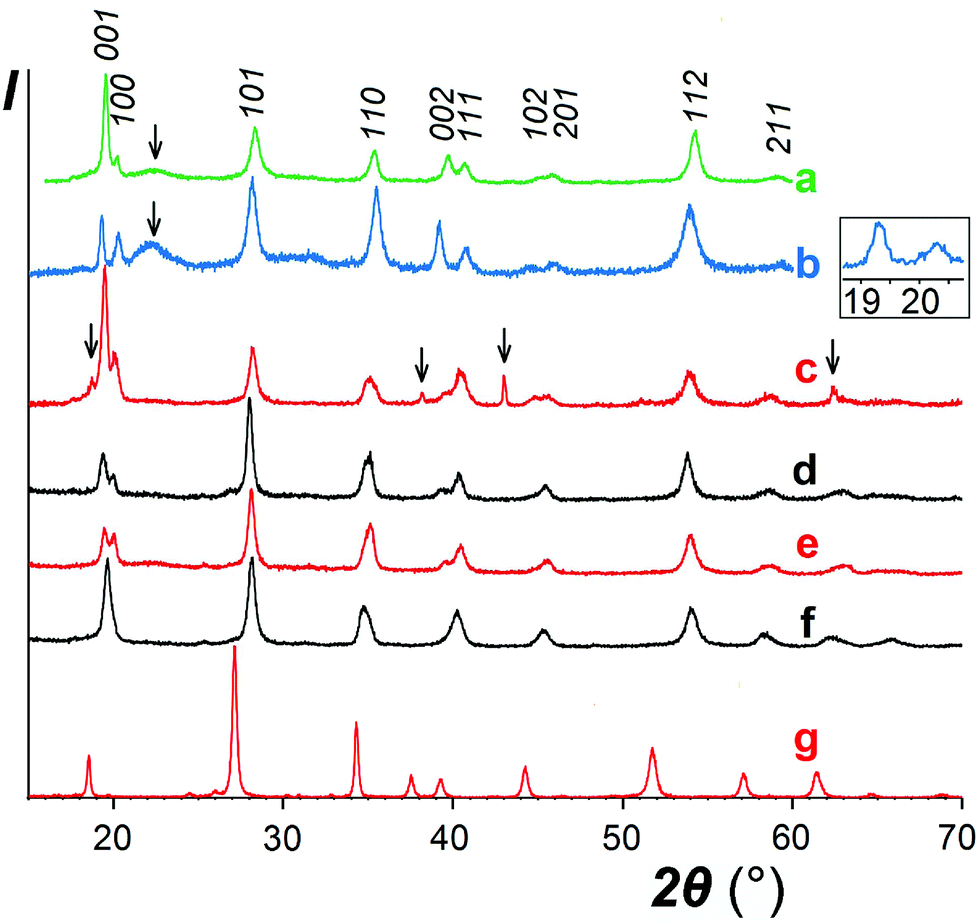 | ||
| Fig. 1 XRD patterns of the MSb2O6 samples prepared via ion exchange. (a) NiSb2O6, (b) CuSb2O6, (c) MgSb2O6, (d) ZnSb2O6, (e) CoSb2O6 (sample I), (f) CoSb2O6 (sample II), and (g) CdSb2O6 (sample I). Inset shows the expanded low-angle part for CuSb2O6. The arrows point to the reflections from foreign phases. | ||
| M |
VI
R, Å26 |
Metastable trigonal layered polymorphs (stable for M = Cd) | Trirutile27 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| a, Å | c, Å | c/a | V/Z | M/2Sb | Na/2Sb | V/Z | ΔV, % | ||
| a Treated with aqueous acetic acid to remove free MgO before analysis. b Sodium content could not be estimated due to overlap of its strongest line with the line from Zn. c Extremely inhomogeneous distribution of Na: only found in 2–4 points of 10 or 11. | |||||||||
| Ni | 0.83 | 5.076(4) | 4.5329(16) | 0.893 | 101.2 | 1.02(6) | 0 | 99.35 | 1.9 |
| Cu | 0.87 | 5.054(4) | 4.5881(10) | 0.908 | 101.5 | 1.00(12) | 0 | 99.81 | 1.9 |
| Mga | 0.86 | 5.116(5) | 4.538(8) | 0.887 | 102.8 | 1.07(15) | 0.11(3) | 99.86 | 3.0 |
| Zn | 0.88 | 5.114(3) | 4.5679(16) | 0.893 | 103.4 | 0.93(4) | ?b | 100.88 | 2.5 |
| Co(I) | 0.885 | 5.1318(3) | 4.5520(3) | 0.887 | 103.8 | 0.99(5) | 0 | 100.53 | 3.3 |
| Same, calcined at 950° | 1.00(5) | 0.02(5)c | |||||||
| Co(II) | 5.164(2) | 4.503(2) | 0.872 | 104.0 | 0.96(3) | 0.04(8)c | |||
| Same, calcined at 950° | 0.99(3) | 0.06(8)c | |||||||
| Cd(I) | 1.09 | 5.2405(20) | 4.796(3) | 0.915 | 114.1 | 1.03(3) | 0.06(3) | ||
| Same, calcined at 950° | 1.02(2) | 0.03(3) | |||||||
| Cd(II) | 5.2403(16) | 4.803(3) | 0.917 | 114.2 | 0.99(2) | 0.08(3) | |||
| Cd3 | 5.2399(2) | 4.8045(4) | 0.917 | 114.2 | |||||
The same approach was applied to the preparation of CdSb2O6, for which a rosiate-type structure is the stable form. Thus, the limitations of reaction temperature and time were unnecessary, and the exchange was performed at 500 °C for 2 h. This resulted in the formation of single-phase products with both 2- and 5-fold excess of CdSO4, marked I and II, respectively. Both have much better crystallinity than others (Fig. 1g) and the lattice parameters are in good agreement with literature data (Table 1).
In contrast to the samples prepared from sulfates, our MgSb2O6 contained admixtures of foreign crystalline phases including MgO (Fig. 1c). This is because we failed to avoid hydrolysis while drying the starting hydrous MgCl2.
The analytical data, as shown in Table 1, despite considerable scattering from point to point, confirm that obtained NiSb2O6 and CuSb2O6 are sufficiently pure: the M/Sb ratio is stoichiometric within the experimental accuracy and the Na content is essentially zero. On the other hand, the MgSb2O6 contained foreign phase(s) even after the removal of MgO via acid leaching. In addition to XRD, this is evident from the microprobe data showing the elevated average Mg/Sb ratio and especially high scatter.
With other three compounds, the situation was less obvious. The single analyzed sample of ZnSb2O6 and one of the two CoSb2O6 samples (labelled II) showed somewhat reduced M/Sb ratio (although within the experimental accuracy) associated with a non-zero sodium content in both Co samples (in the presence of Zn, determination of Na was unreliable). This suggests the possibility of sodium substitution for M2+ and correlates with variations in the XRD data. A comparison of the XRD patterns of CoSb2O6 I (Fig. 1e) and CoSb2O6 II (Fig. 1f) shows that the less pure sample CoSb2O6 II, in contrast to CoSb2O6 I, does not show splitting of the pairs 001 + 100 and 002 + 111, indicating a considerably lower c/a ratio. Similar variations were observed with the ZnSb2O6 samples, but those with unresolved 001 + 100 reflections have not been reported herein. The different behaviours of Zn and Co compounds compared to those of Ni and Cu might be tentatively attributed to the somewhat larger ionic radii of the former pair of cations, which is favourable for sodium substitution. However, ionic radius of Cd2+ is much larger and closer to that of Na+, but no Cd deficiency indicating sodium substitution can be seen in Table 1, especially with sample I, where a lower excess of the Cd salt might favour incomplete ion exchange. Therefore, we hypothesized that the elevated sodium content in the CdSb2O6 samples might be due to the adsorbed or occluded salts rather than ionic substitutions. Note that the sodium content in CdSb2O6(I) diminishes on calcination, probably due to volatilization of the salt impurity. The role of Na in MSb2O6 has been further discussed in Section 3.2.
It was noticed that the transformation to the stable polymorph was markedly enhanced in the presence of a salt melt, obviously due to the dissolution-precipitation mechanism. As illustrated in the XRD patterns in Fig. 2, for ZnSb2O6, taken as a typical example, prolonged treatment with the molten salt at 420–500 °C resulted in the conversion of the metastable trigonal layered rosiaite-type phase into the stable trirutile-type polymorph. On the other hand, this conversion was not observed at considerably higher temperature of 750 °C if ZnSb2O6 was free of the salt mixture. Note that exchange reaction of NaSbO3 with ZnCl2 under hydrothermal conditions yielded stable ZnSb2O6 at even lower temperature of 230 °C (for 4 days), whereas phase-pure products with M = Pb, Cu, Ni, and Mn could not be obtained via this method.14
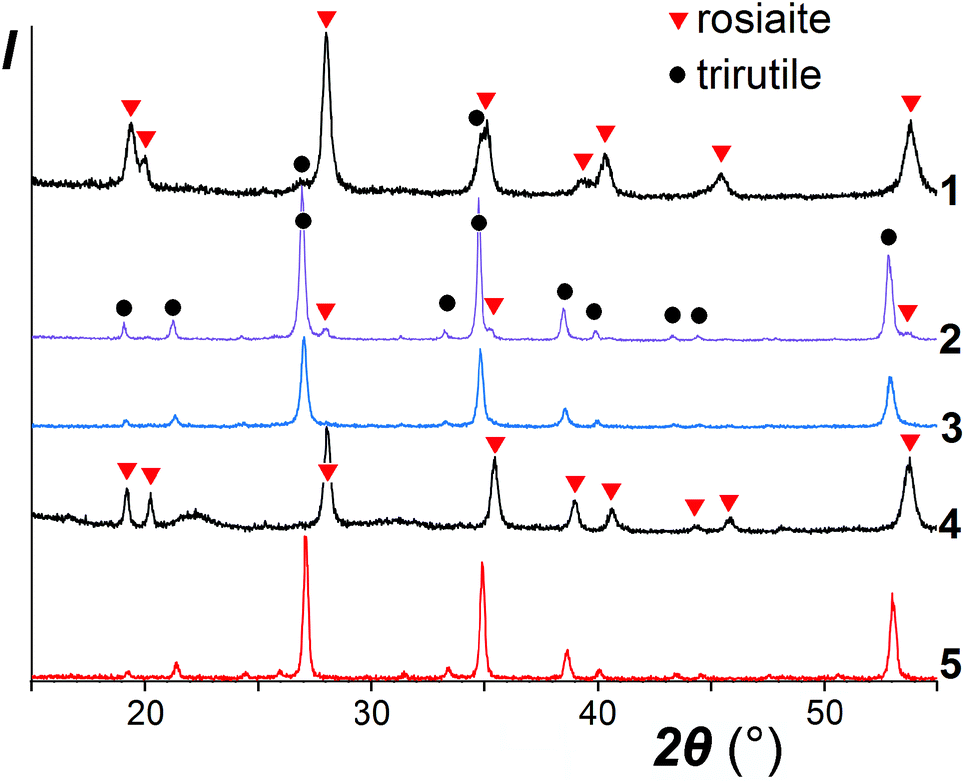 | ||
| Fig. 2 XRD patterns of ZnSb2O6 samples with different thermal history: 1, prepared by ion exchange for 6 h at 420 °C: rosiaite type with only a trace of the stable trirutile-type phase; 2, same after an additional 10 h treatment with the salt mixture at 420 °C: trirutile type with only trace of the starting rosiaite; 3, prepared for 5 h at 500 °C: only trirutile type; 4, rosiaite-type phase prepared under the optimal conditions, washed, and annealed for 2 h at 750 °C: no trirutile; 5, same annealed for an hour at 950 °C: only trirutile type. Red triangles, reflections from rosiaite; black circles, reflections from trirutile. | ||
A comparison of the patterns 1 and 4 depicted in Fig. 2 shows that annealing of ZnSb2O6 at 750 °C resulted in an increased axial ratio c/a and the appearance of large broad halos in the XRD pattern, which are characteristic of an amorphous phase. Similar halos are also visible in the patterns of CuSb2O6 and NiSb2O6 in Fig. 1a and b. Annealing at 950 °C resulted in complete transformation of trigonal MSb2O6 (M = Co, Ni, Cu, and Zn) to stable trirutile-type polymorphs (Fig. 3 and pattern 5 in Fig. 2). No additional phases were found within the sensitivity of the XRD. This means that the amorphous phases have the same composition as the crystalline products, being intermediates in the transformation between the trigonal and tetragonal phases. This is different from the previously studied case of MnSb2O6, where the stable phase was structurally related to the metastable phase and the transformation might have proceeded within an intact crystal.1
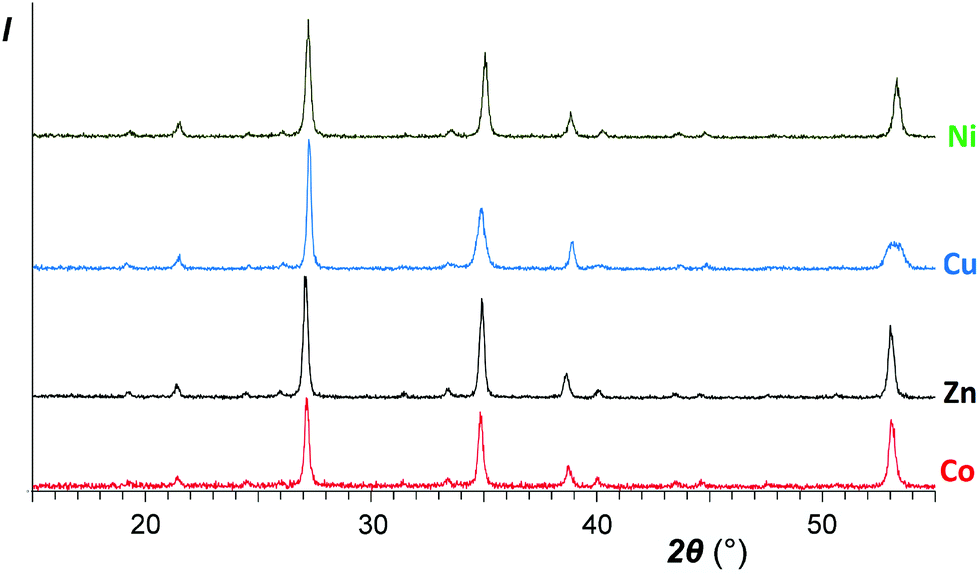 | ||
| Fig. 3 XRD patterns of the same samples as abovementioned in Fig. 1 after calcinations for an hour at 950 °C. Top to bottom: NiSb2O6, CuSb2O6, ZnSb2O6, and CoSb2O6 (sample I). | ||
3.2. Structural characterization
As shown in Table 1 and Fig. 4, the formula volumes of trigonal MSb2O6 (except for M = Cu) have an excellent correlation with the ionic radii of M2+, and for the five new phases, they are 2–3% larger than those for the stable trirutile-type phases although all coordination numbers are the same. Fig. 4 presents an additional example of a morphotropic series where substitution of a larger cation (herein, Co2+ in CoSb2O6) by an even larger cation (herein, Mn2+ or Cd2+) leads to less dense structure types,28 opposite to the widely assumed homology rule.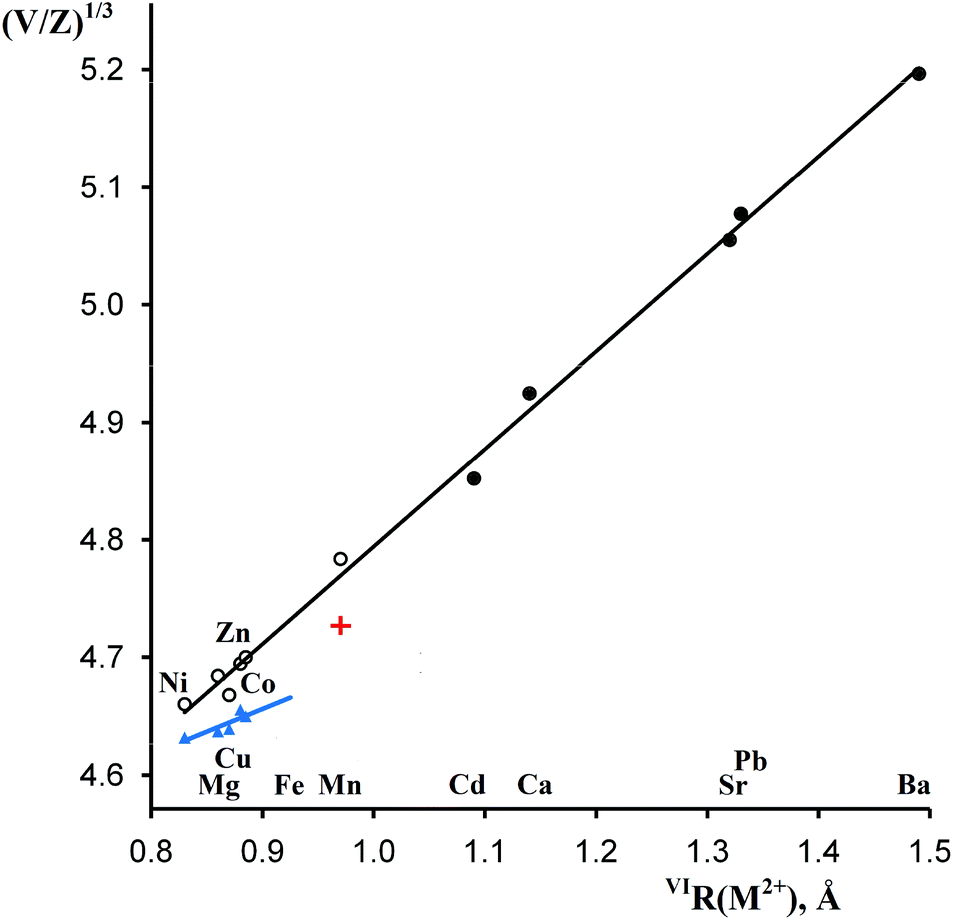 | ||
| Fig. 4 Correlations between the octahedral ionic radii26 of M2+ and reduced cell parameters (V/Z)1/3 of MSb2O6. Empty and filled circles, rosiaite type, metastable and stable phases, respectively; blue triangles, stable phases of the trirutile type; red cross, stable MnSb2O6, P321. | ||
As discussed earlier,1 the ilmenite-type structure of NaSbO3 and rosiaite-type structure of MSb2O6 (M = Ca, Cd, Sr, Pb, and Ba) are based on the same octahedral SbO6/2 layers. Therefore, M2+ substitution for 2Na+ may occur by mere gliding of the layers to avoid the face sharing of MO6 and SbO6 octahedra. This gliding may result in stacking faults. Therefore, it is not surprising that XRD peaks in the powder patterns (Fig. 1) are considerably broader than those of starting NaSbO3. Largest broadening is observed with M = Cu. This, together with the deviation seen in Fig. 4, may be an indication of lowered symmetry due to the Jahn–Teller effect characteristic of Cu2+. Inset in Fig. 1 shows that the reflection 100 that may split upon transition from the trigonal to any lower crystal system is markedly broader than 001, which does not split in any case. However, the quality of the powder pattern precludes a detailed structural investigation.
Only the crystal structure of CoSb2O6 (the sample I with minimum sodium impurity, if any, see Table 1) was refined using the Rietveld method. The main results are listed in Tables 2–4 and in the ESI† (cif). Fig. 5 compares the experimental and calculated XRD profiles, whereas Table 4 and Fig. 6 compare the new and previously known structures of CoSb2O6.
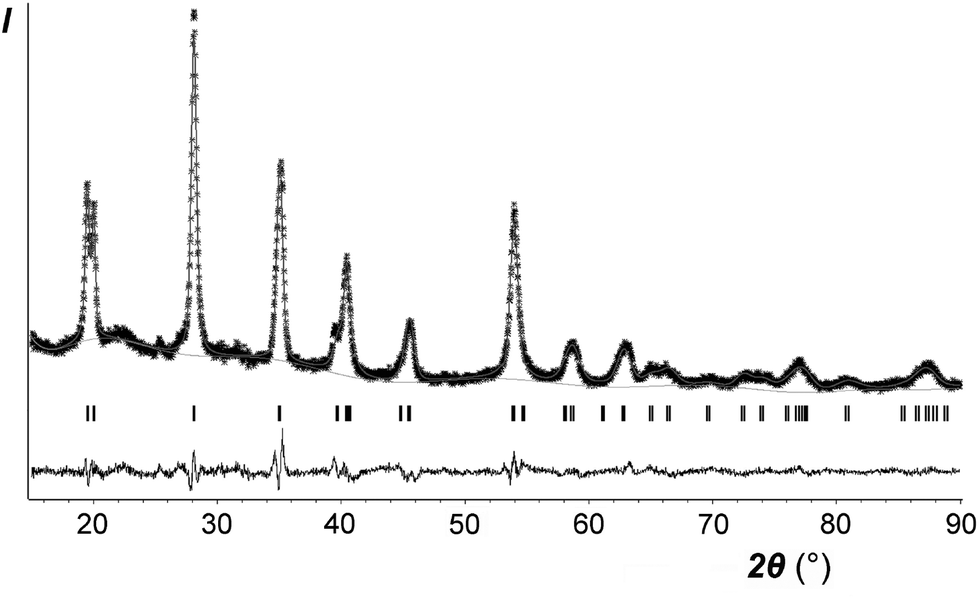 | ||
| Fig. 5 Rietveld refinement results for CoSb2O6. Stars, experimental data; line, calculated profile; line at the bottom, difference profile; vertical bars, calculated positions of the Bragg reflections. | ||
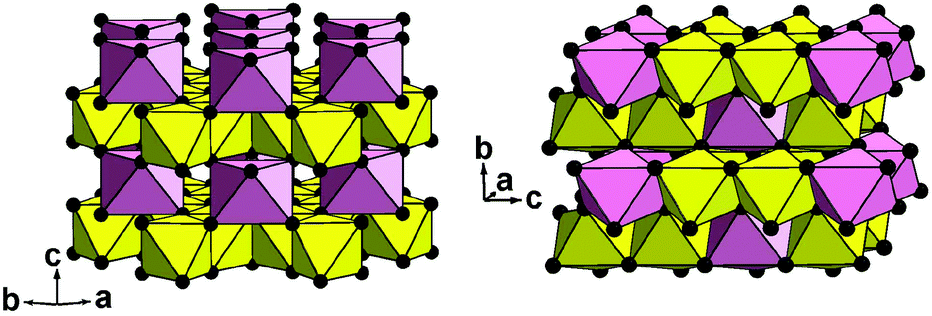 | ||
| Fig. 6 Two crystal structures of CoSb2O6 in a polyhedral presentation: new trigonal form (left) and known tetragonal form5 (right). Yellow octahedra, SbO6; pink octahedra, CoO6; and black balls, oxygen anions. | ||
| Crystal system | Trigonal | Density (calc.), g cm−3 | 6.372 | ||
|---|---|---|---|---|---|
| Space group |
P1m (no. 162) |
Texture parameters (March–Dollase) | Axis 001 | ||
| Ratio 0.9873 | |||||
| Lattice constants, Å | a | 5.13183(25) | 2Θ range, ° | 15.00–89.96 | |
| c | 4.55202(34) | Step width, ° | 0.02 | ||
| Cell volume, Å3 | 103.820(11) | No. of data points | 3748 | ||
| Formula weight | 398.4 | No. of reflections | |||
| Calc. (α1 only) | 39 | ||||
| Z | 1 | No. of variables | 44 (6 – of structure) | ||
| Wavelengths, Å | α 1 | 1.54056 | Agreement factors | R(F2) | 0.05085 |
| α 2 | 1.54439 | R p | 0.0611 | ||
| Ratio | 0.5 | R wp | 0.0776 | ||
| χ 2 | 2.166 | ||||
| Atom | Wyckoff position | Site symmetry | Occup. | x/a | y/b | z/c | U iso |
|---|---|---|---|---|---|---|---|
| Mn | 1a | m |
1 | 0 | 0 | 0 | 0.01 fixed |
| Sb | 2d | 32 | 1 | 1/3 | 2/3 | 1/2 | 0.01958 equi. |
| O | 6k | m | 1 | 0.3721(18) | 0 | 0.2518(13) | 0.03 fixed |
| U 11 | U 22 = U11 | U 33 | U 12 = U11/2 | U 13 | U 23 | |
|---|---|---|---|---|---|---|
| Sb | 0.0156(4) | 0.0156 | 0.0276(8) | 0.00779 | 0 | 0 |
| Bonds, angles | Rosiaite type, P1m |
Trirutile type, P42/mnm | Sum of radii |
|---|---|---|---|
| Co–O | 2.227(8) × 6 | 2.054 (average of six) | 2.105 |
| Sb–O | 1.975(4) × 6 | 1.985 (average of six) | 1.96 |
| O–Co–O | 84.1–95.9 | 77.7–102.3 | |
| O–Sb–O | 82.8–97.0 | 79.2–99.6 | |
| Co–O1–Sb | 125.40 × 2 | 129.62 × 2 | |
| Sb–O1–Sb | 97.17 | 100.75 | |
| Sum of the three | 348.0 | 360.0 | |
| Co–O2–Sb | 126.36 × 2 | ||
| Sb–O2–Sb | 133.29 | ||
| Sum of the three | 360.0 |
A comparison of the bond lengths and bond angles in the two polymorphs (Table 4) shows that both the CoO6 and SbO6 octahedra, especially the former, are much more regular in the new phase than in the stable trirutile-type counterpart. The same was noticed for MnSb2O61 although its stable form is not of the trirutile type.
The trigonal structure of CoSb2O6 is based on the distorted hexagonal close packing (better to say, 2H eutaxy) of oxygen anions, where cations fill half octahedral voids. Each oxygen atom has 10 oxygen neighbours at the distances of 2.61–2.98 Å plus two oxygen neighbours more distant at 3.31 Å. Although the rutile and trirutile structure types are also often described as 2H derivatives, it is actually incorrect.2 Indeed, the two independent anions in tetragonal CoSb2O65 have oxygen environments of 11 + 2 and 10 + 1 + 2; thus, the tetragonal close packing with 11 (rather than 12) neighbours is a more adequate term in this case.2 The cation arrangements in the two polymorphs also significantly differ. In the trirutile-structure type, each metal–oxygen octahedron shares two edges with other octahedra, and higher-valence cations have only one common edge. In the rosiaite-structure type, the number of shared edges is the same only as an arithmetic mean. Octahedra populated with lower-valence cations have no shared edges at all, whereas those populated with higher-valence cations share three edges each (Fig. 6). In addition, the trigonal form, despite the larger formula volume, exhibits shorter Sb–Sb distances across the shared edges: 2.96 Å in contrast to 3.05 and 3.11 Å for Sb–Sb and Sb–Co, respectively, in the tetragonal form. All this leads to stronger cation–cation repulsion in the rosiaite type and explains its instability with respect to the trirutile type. For large M2+ (Cd⋯Ba), size disparity with Sb5+ precludes the formation of the trirutile type; then, the rosiate type becomes stable.
The crystal-chemistry role of the sodium impurity remains unclear. Previously,1 we suggested the existence of small non-exchanged ilmenite-type fragments as stacking faults in the main MnSb2O6 phase. However, this disagrees with the lattice parameter changes. Pure NaSbO3 has a c/a ratio of 3.011 for the three-layered rhombohedral packing.29 For the primitive one-layered packing, it should be 3.011/3 = 1.004. These admixtures must increase the c/a ratio of our products. However, a comparison of the samples I and II of CoSb2O6 in Table 1 shows the opposite.
As discussed in Section 3.1, sodium substitution for M2+ was most probable with M = Cd but could not be supported by the analytical data and lattice parameters (Table 1). In addition, the introduction of large Na+ cations in the interlayer is also expected to increase the c/a. Moreover, to provide charge balance, there should be either additional Na+ ions (for which we do not see available sites) or holes that can be easily interpreted as Co3+; however, this is hardly applicable for zinc or cadmium. The problem could not be solved by powder XRD owing to the very small content of sodium, its relatively small X-ray scattering factor, and diffuse nature of the diffraction patterns. The crystal structure refinement reported herein is based on the ideal composition CoSb2O6.
3.3. Special case of FeSb2O6
Our low-temperature preparation of FeSb2O6 in an inert atmosphere differs from the preceding high-temperature preparation in air:20 instead of the mixture FeSbO4 + 0.5Sb2O4, we obtained FeSbO4 + 0.5Sb2O3 (Fig. 7), due to the reducing power of the excess FeSO4. In any case, FeSb2O6 could not be prepared, due to incompatible oxidation states, reacting according to the following scheme: 2Fe2+ + Sb5+ → 2Fe3+ + Sb3+.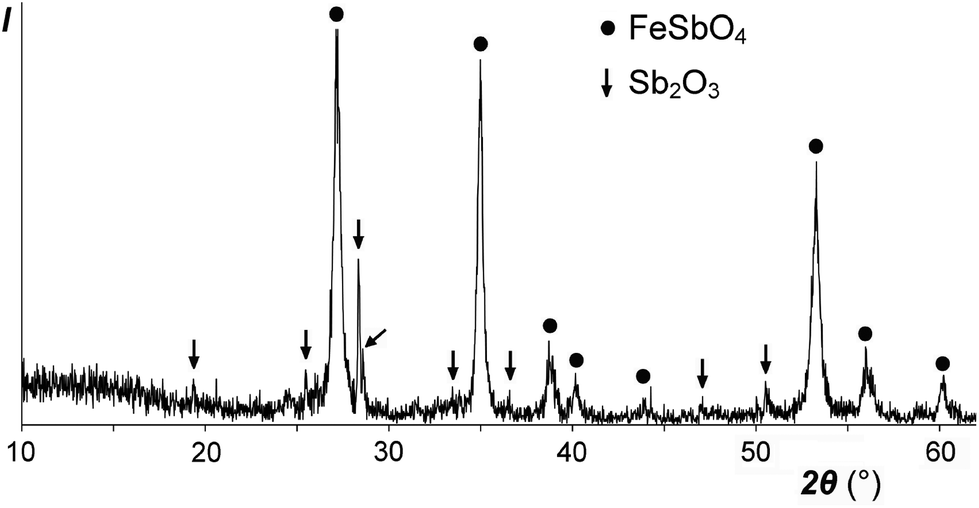 | ||
| Fig. 7 XRD pattern of the reaction product between a NaSbO3 and FeSO4–KCl melt in a CO2 atmosphere at 500 °C. Filled circles, reflections from FeSbO4 (rutile type); arrows, reflections from Sb2O3 (valentinite). | ||
This electron transfer from Fe2+ (3d6) to Sb5+ (4d10) creates an additional 5s2 shell for Sb and thus should result in an increase in the total volume. The formula volume of the hypothetical trirutile-type FeSb2O6 was estimated to be approximately 101.4 Å3 by extrapolation using the plot in Fig. 4 and Shannon's ionic radius for high-spin Fe2+. The volume of the equivalent stable mixture FeSbO4 + 0.5Sb2O4 is considerably higher, 103.9 or 104.4 Å3, obtained using the data for monoclinic or orthorhombic Sb2O4,30 respectively. Therefore, trirutile-type FeSb2O6, which is unstable at normal pressure, may be stabilized at high pressures although this may be complicated by high-spin-to-low-spin transitions.
3.4. Magnetic properties
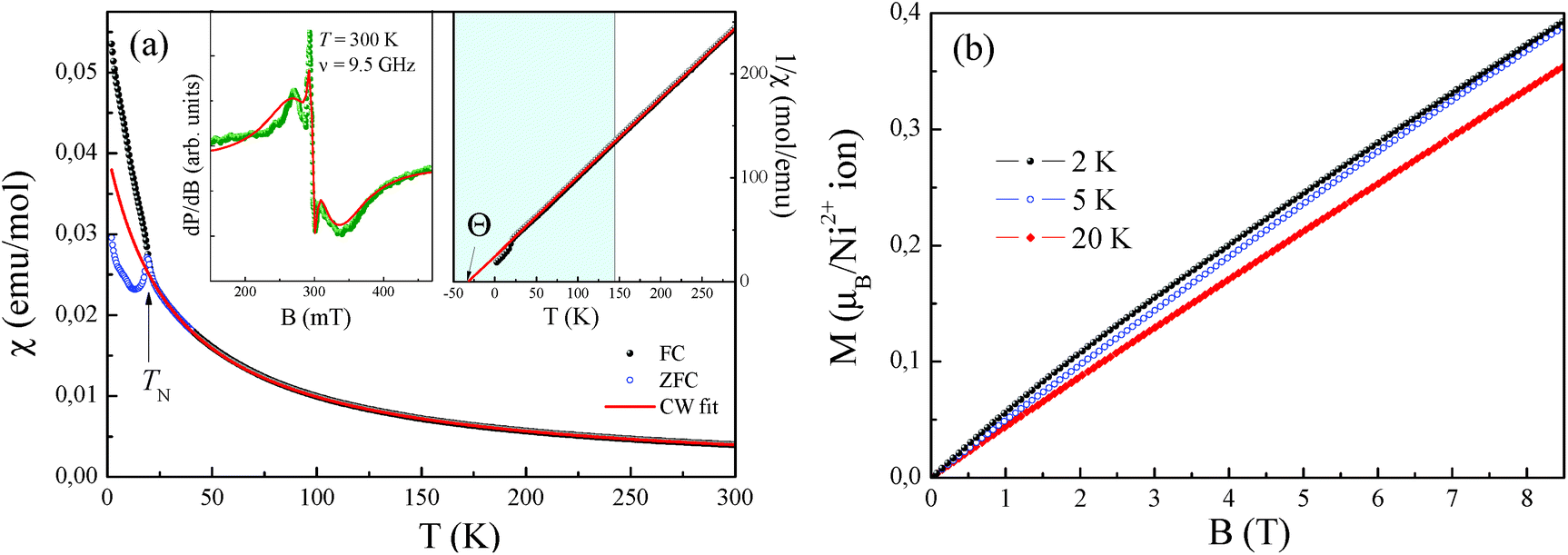 | ||
| Fig. 8 (a) Temperature-dependence of the magnetic susceptibility of NiSb2O6 measured at B = 0.1 T. (b) Magnetization isotherms of NiSb2O6 at various temperatures. Insets in (a): ESR spectrum at room temperature (left side) and inverse value of the magnetic susceptibility 1/χ, emphasizing the predominance of the antiferromagnetic correlations (right side). | ||
The effective magnetic moment calculated from the obtained Curie constant takes μeff = 3.2μB/Ni2+. This value is in reasonable agreement with the theoretical estimation according to 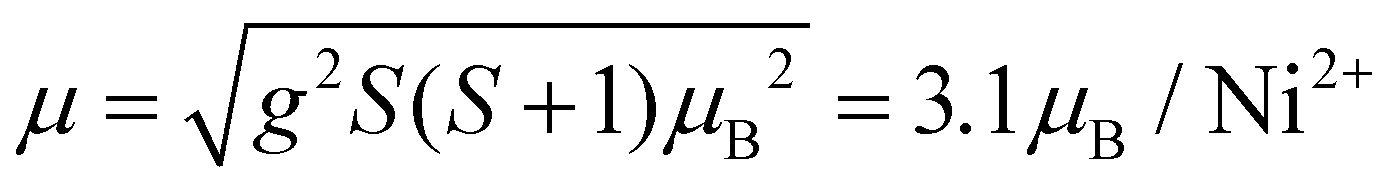 , where the average effective g-factor was directly measured via electron spin resonance (ESR) g = 2.21, which is well consistent with typical for Ni2+ values.
, where the average effective g-factor was directly measured via electron spin resonance (ESR) g = 2.21, which is well consistent with typical for Ni2+ values.
The negative sign of the Weiss temperature indicates the predominance of antiferromagnetic exchange interactions in the system, in agreement with the long-range antiferromagnetic ordering. The field dependence of the magnetization of NiSb2O6 (Fig. 8b) demonstrates characteristic S-type behaviour at low temperatures, which indicates the competing exchange interactions of both the ferro- and antiferromagnetic nature.
Interestingly, note that the magnetic properties observed herein for this novel phase possessing the trigonal layered rosiaite-type structure are essentially different from those reported for its stable polymorph with the tetragonal trirutile-type structure (space group P42/mnm). The magnetic behaviour of all the trirutile-type compounds MSb2O6 (M = Ni, Co, and Cu) is quite similar. Their low-dimensional magnetic behaviour was confirmed from the magnetic susceptibility, specific heat and thermal expansion measurements, ESR and NMR/NQR techniques, etc. as well as by electronic structure calculations. A characteristic feature is the presence of a wide temperature range, where all MSb2O6 with a trirutile structure exhibit short-range antiferromagnetic order. For the case of trirutile NiSb2O6, it was found that the χ(T) demonstrates a clear low-dimensional broad maximum at Tmax ∼ 40 K followed by long-range antiferromagnetic order below TN = 2.5 K.4 Despite the almost perfect 2D square Ni2+ sublattice, the data were well described within the formalism of antiferromagnetic S = 1 1D spin chain via Ni–O–O–Ni superexchange pathways with the main exchange parameter J/kB ∼ −45 K.
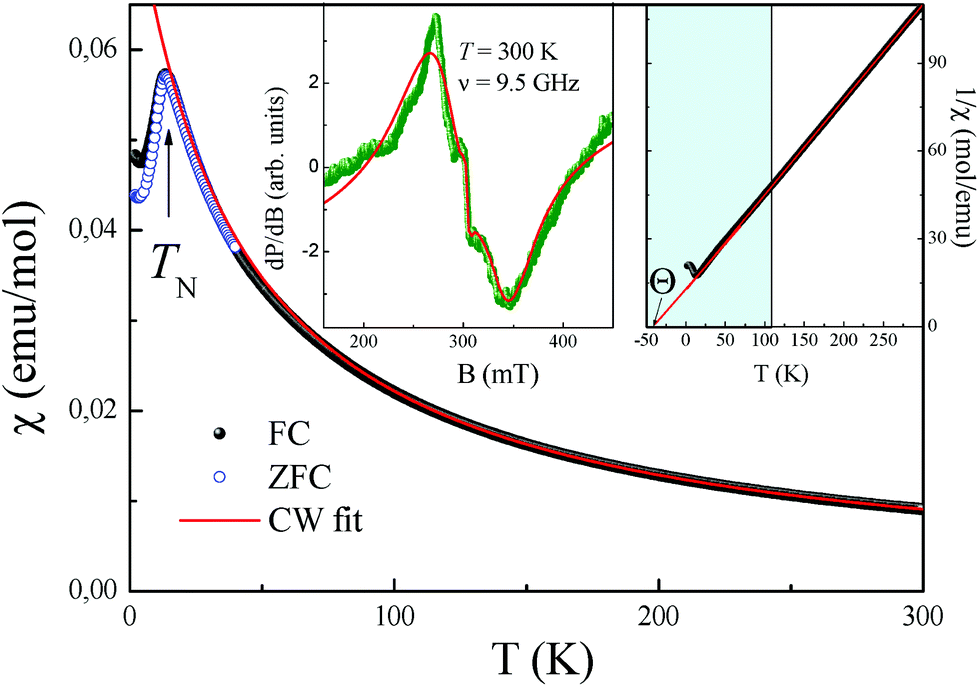 | ||
| Fig. 9 Temperature-dependence of the magnetic susceptibility of CoSb2O6-II measured at B = 0.1 T. Insets: ESR spectrum at room temperature (left side) and inverse value of magnetic susceptibility 1/χ, emphasizing the predominance of the antiferromagnetic correlations (right side). | ||
Moreover, the latter low-temperature phase transition has attracted significant interest because it was suggested to be possible spin-Peierls-like phase transition.9,12 The octahedral oxygen environment of the magnetic Cu2+ ions gives rise to the dominant Cu–O–O–Cu superexchange pathway, which forms magnetic chains along the (a + b) direction at z = 0, and (a − b) direction at z = 0.513 with the main intrachain exchange integral J∥/kB ∼ −94 K (ref. 13) and remarkably small ratio J∥/J⊥ = 120.6
The temperature-dependence of the magnetic susceptibility χ = M/B of new rosiaite-type CuSb2O6, measured at B = 0.1 T, is shown in Fig. 10(a). This follows the Curie–Weiss behaviour at elevated temperatures and does not show clear signature of any long-range order transition down to 2 K. However, left-side bending at low temperatures and a clear deviation from the Curie–Weiss law at T < 100 K can be observed, indicating rather strong short-range exchange interactions in this compound. The dependencies of magnetic susceptibility measured in the field-cooled (FC) and zero-field cooled (ZFC) regimes do not demonstrate any divergence, confirming the absence of any spin disorder effects. The fit of the experimental data in the high-temperature range with the function χ = χ0 + C/(T − Θ) allows an estimation of the temperature-independent term χ0 = 1.46 × 10−5 emu mol−1, Curie constant C = 0.447 emu K mol−1, and Weiss temperature Θ = −4 K.
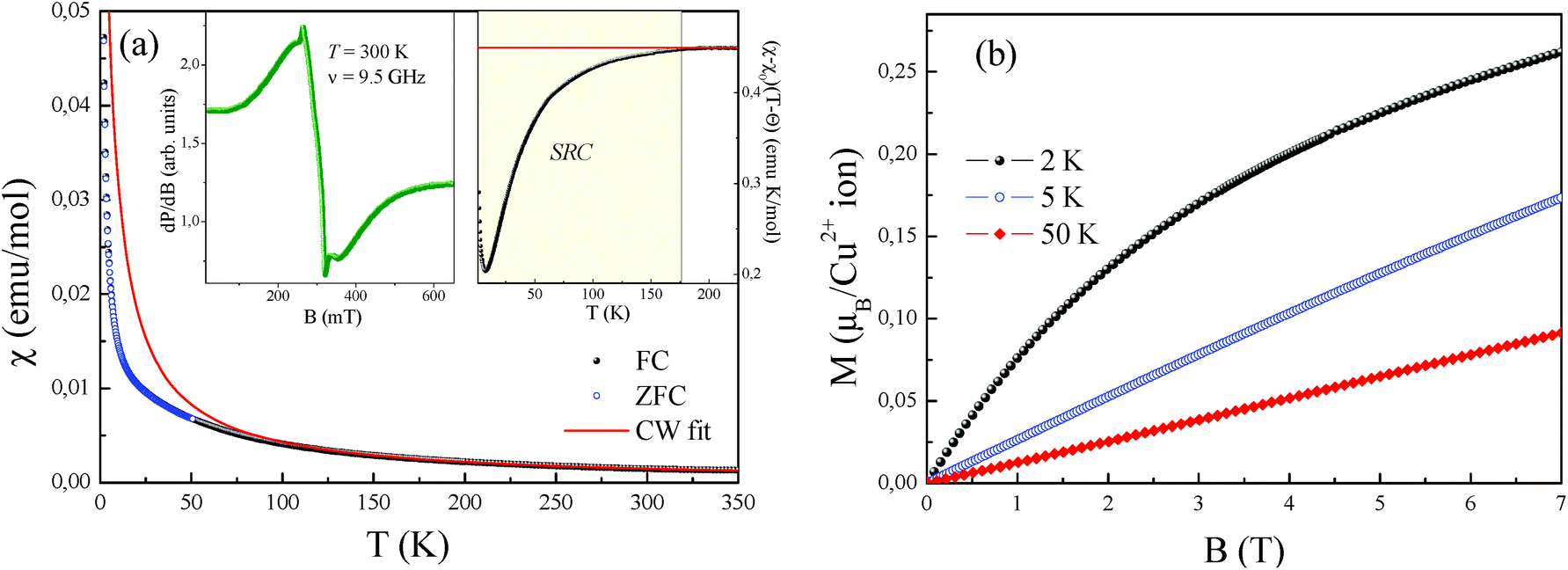 | ||
| Fig. 10 (a) Temperature-dependence of the magnetic susceptibility of CuSb2O6 measured at B = 0.1 T. (b) Magnetization isotherms of CuSb2O6 at various temperatures. Insets: ESR spectrum at room temperature (left side) and special scale of magnetic susceptibility (χ − χ0)(T − Θ), emphasizing the nature of the magnetic exchange interactions (right side). | ||
Similar to the NiSb2O6 and CoSb2O6 samples, the temperature-independent term χ0 comprises diamagnetic Pascal and paramagnetic van Vleck contributions. The positive value of χ0 indicates the predominance of the van Vleck term χvV = 7.18 × 10−4 emu mol−1 over the sum of the Pascal constant of individual ions χdia = −1.12 × 10−4 emu mol−1.31 The van Vleck contribution is defined by the splitting of the d-shell of Cu2+ ions in a distorted octahedral environment.
The effective magnetic moment calculated from the obtained Curie constant takes μeff = 1.9μB/Cu2+. This value is in excellent agreement with the theoretical estimation according to 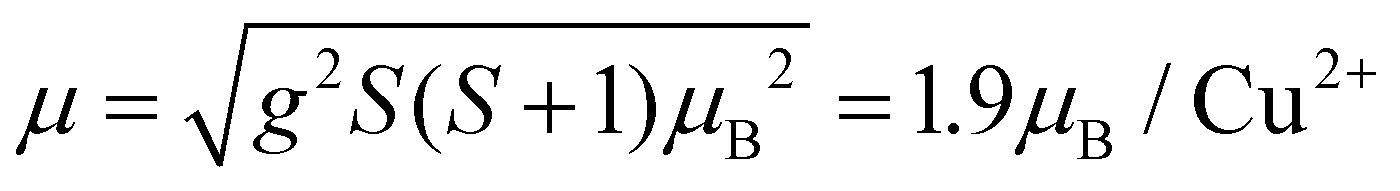 , where the average effective g-factor was directly measured by electron spin resonance (ESR). As depicted in the left inset in Fig. 10(a), the ESR spectrum shows relatively broad anisotropic absorption line, which is typical of the powder pattern of the Cu2+ compounds. The estimated values of the principal components of g-tensor are g∥ = 2.20 and g⊥ = 2.19, providing an average gav = 2.196.
, where the average effective g-factor was directly measured by electron spin resonance (ESR). As depicted in the left inset in Fig. 10(a), the ESR spectrum shows relatively broad anisotropic absorption line, which is typical of the powder pattern of the Cu2+ compounds. The estimated values of the principal components of g-tensor are g∥ = 2.20 and g⊥ = 2.19, providing an average gav = 2.196.
The negative sign of the Weiss temperature indicated the predominance of antiferromagnetic exchange interactions in the system. This is also confirmed by the temperature-dependence of the scaled product (χ − χ0)(T − Θ) shown in the right inset of Fig. 10(a). At high temperatures, this product follows a horizontal line, obeying the Curie–Weiss law, whereas below approximately 175 K, it deviates downward, indicating the predominance of the antiferromagnetic short-range correlations. Despite the wide range of correlations, the system does not reach ordered state, which probably relates to frustration and competing exchange interactions. The presence of exchange interactions of both the ferro- and antiferromagnetic nature follows from the field dependence of the magnetization (Fig. 10(b)), which demonstrates characteristic S-type behaviour at 2 K, which is similar to those for the isostructural compound with Ni2+. However, at higher temperature, magnetization shows ordinary linear trend, typical of antiferro- or paramagnets. This behaviour is completely different from the trirutile-type CuSb2O6, which demonstrated clear spin-flop transition with HSF ∼ 1.3 T at 5 K below the Néel temperature TN = 8.5 K.6,9 Interestingly, the relatively high Néel temperature for trirutile-type CuSb2O6 has not been confirmed from theoretical calculations due to the tiny balance in the energy levels of dx2−y2 and d3z2−r2 electronic shells of Cu2+ ions in the octahedral oxygen environments and hence is related to large intrinsic magnetic anisotropy.11 The value predicted from this theoretical analysis is less than 1.5 K.11 Consequently, the structural changes for the new polymorph lead to more regular octahedral parameters, reducing the magnetic anisotropy for rosiaite-type compound, which in turn results in an essential decrease in the ordering temperature.
The parallel and independent low-temperature neutron diffraction study of CuSb2O6 confirmed the absence of long-range magnetic order down to 1.5 K. Fig. 11 only shows the same reflections that were observed by XRD at room temperature although their relative intensities are entirely different. In particular, the first pair of reflections 001 + 100 is practically invisible. Good agreement between the experimental and calculated profiles confirmed the rosiaite-type structure; however, full-scale structure refinement was not attempted due to the scarce experimental data.
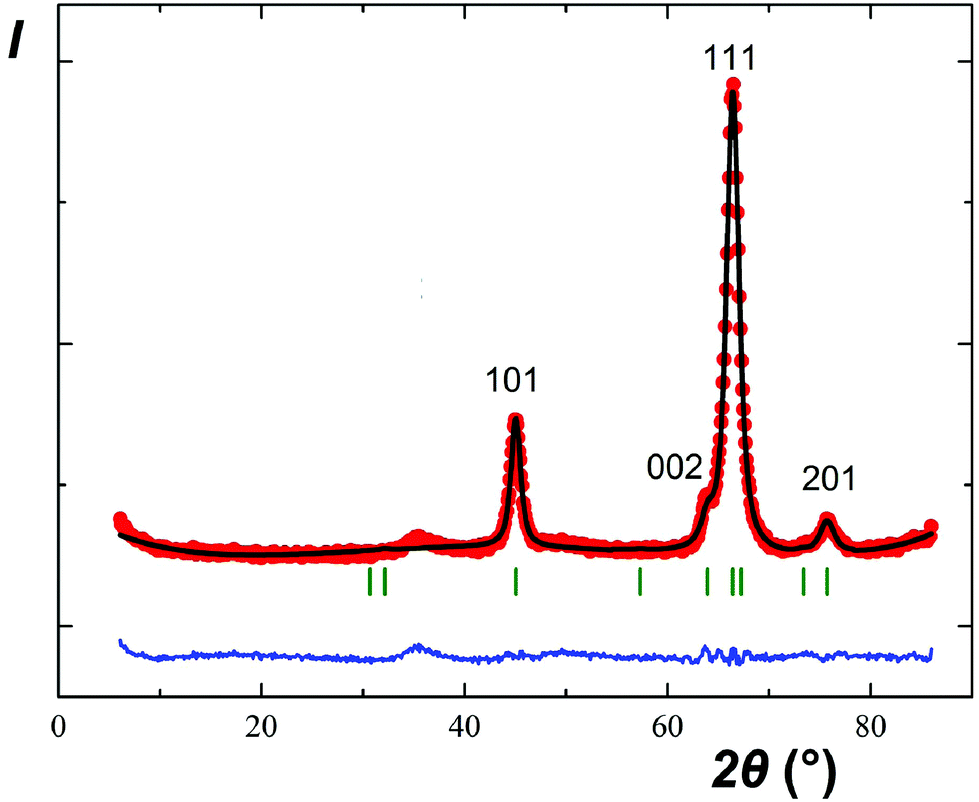 | ||
| Fig. 11 Rietveld refinement results of the powder neutron diffraction pattern of CuSb2O6 at 1.5 K. Red filled circles, experimental data; black line, calculated profile; blue line at the bottom, difference profile; and green vertical bars, calculated positions of the Bragg reflections. | ||
The diffuse maximum between 33 and 39°, as shown in Fig. 11, coincides, in terms of the interplanar spacings d, with that observed in the XRD pattern of Fig. 1b between 21 and 24°. Therefore, it is not related to short-range magnetic ordering.
4. Conclusions
Herein, five new metastable trigonal layered MSb2O6 phases (M = Co, Ni, Cu, Zn, and Mg) were prepared by the low-temperature ion-exchange reactions, together with previously known CdSb2O6. FeSb2O6 could not be prepared due to the incompatible oxidation states but is predicted to stabilize at high pressures. CoSb2O6 and NiSb2O6 antiferromagnetically order at 11 and 15 K, respectively. In contrast to their stable trirutile-type polymorphs, they do not show any signs of low-dimensional magnetic behaviour at higher temperatures. CuSb2O6 does not exhibit long-range magnetic order down to 1.5 K but shows antiferromagnetic short-range correlations.Acknowledgements
The work was supported by the Russian Foundation for Basic Research under the grant 14-03-01122. A. N. V. acknowledges the support in part from the Ministry of Education and Science of the Russian Federation in the framework of Increase Competitiveness Program of NUST <MISiS> (no. K2-2016-066) and by Act 211 of the Government of Russian Federation, contract no. 02.A03.21.0006.References
- V. B. Nalbandyan, E. A. Zvereva, A.Yu. Nikulin, I. L. Shukaev, M.-H. Whangbo, H.-J. Koo, M. Abdel-Hafiez, X.-J. Chen, C. Koo, A. N. Vasiliev and R. Klingeler, Inorg. Chem., 2015, 54, 1705–1711 CrossRef CAS PubMed.
- W. H. Baur, Crystallogr. Rev., 2007, 13, 65–113 CrossRef CAS.
- A. Castro, I. Rasines, M. C. Sanchez-Martos and P. Garcia-Casado, Powder Diffr., 1988, 3, 219–221 CrossRef CAS.
- E. Ramos, F. Fernandez, A. Jerez, C. Pico, J. Rodriguez-Carvajal, R. Saez-Puche and M. L. Veiga, Mater. Res. Bull., 1992, 27, 1041–1047 CrossRef CAS.
- J. N. Reimers, J. E. Greedan, C. V. Stager and R. Kremer, J. Solid State Chem., 1989, 83, 20–30 CrossRef CAS.
- A. B. Christian, S. H. Masunaga, A. T. Schye, A. Rebello and J. J. Neumeier, Phys. Rev. B: Condens. Matter, 2014, 90, 224423 CrossRef.
- A. B. Christian, A. Rebello, M. G. Smith and J. J. Neumeier, Phys. Rev. B: Condens. Matter, 2015, 92, 174425 CrossRef.
- E.-O. Giere, A. Brahimi, H. J. Deiseroth and D. Reinen, J. Solid State Chem., 1997, 131, 263–274 CrossRef CAS.
- A. Rebello, M. G. Smith, J. J. Neumeier, B. D. White and Yi.-K. Yu, Phys. Rev. B: Condens. Matter, 2013, 87, 224427 CrossRef.
- A. M. Nakua and J. E. Greedan, J. Solid State Chem., 1995, 118, 199–201 CrossRef CAS.
- D. Kasinathan, K. Koepernik and H. Rosner, Phys. Rev. Lett., 2008, 100, 237202 CrossRef PubMed.
- M. Kato, A. Hatazaki, K. Yoshimura and K. Kosuge, Physica B, 2000, 281&282, 663–664 CAS.
- M. Herak, D. Žilić, D. Matković-Calogović and H. Berger, Phys. Rev. B: Condens. Matter, 2015, 91, 174436 CrossRef.
- J. Singh, N. Bhardwaj and S. Uma, Bull. Mater. Sci., 2013, 36, 287–291 CrossRef CAS.
- D. P. Dutta, A. Ballal, A. Singh, M. H. Fulekarc and A. K. Tyagi, Dalton Trans., 2013, 42, 16887–16897 RSC.
- H. Mizoguchi and P. M. Woodward, Chem. Mater., 2004, 16, 5233–5248 CrossRef CAS.
- J. Tamaki, Y. Yamada, Y. Yamamoto, M. Matsuoka and I. Ota, Sens. Actuators, B, 2000, 66, 70–73 CrossRef CAS.
- C. R. Michel, N. L. L. Contreras, M. A. López-Alvarez and A. H. Martinez-Preciado, Sens. Actuators, B, 2012, 171-172, 686–690 CrossRef CAS.
- H. Guillen-Bonilla, V.-M. Rodriguez-Betancourtt, J.-T. Guillen-Bonilla, J. Reyes-Gomez, L. Gildo-Ortiz, M. Flores-Martinez, M. de la L. Olvera-Amador and J. Santoyo-Salazar, J. Nanomater., 2015, 979543 Search PubMed.
- A. R. Basso, R. Cabella, G. Lucchetti, P. Marescotti and A. Martinelli, Neues Jahrb. Mineral., Monatsh., 2003, 9, 407–420 CrossRef.
- V. B. Nalbandyan, M. Avdeev and A. A. Pospelov, Solid State Sci., 2006, 8, 1430–1437 CrossRef CAS.
- A. G. Bergman and M. S. Golubeva, Dokl. Akad. Nauk SSSR, 1953, 89, 471–473 CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR 86-748, Los Alamos National Laboratory, Los Alamos, NM, 2004 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- J. Rodrigues-Carvajal, FullProf_suite. http://www.ill.eu/sites/fullprof/.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- Powder Diffraction File. International Centre for Diffraction Data, Newtown Square, Pennsylvania, USA, 2006. Cards 00-17-284, 00-18-403, 00-37-1470, 00-38-453, 00-38-1083 Search PubMed.
- A. A. Novikova and V. B. Nalbandyan, Crystallogr. Rev., 2013, 19, 125–148 CrossRef CAS.
- B. Wang, S. C. Chen and M. Greenblatt, J. Solid State Chem., 1994, 108, 184–188 CrossRef CAS.
- J. Amador, E. G. Puebla, M. A. Monge, I. Rasines and C. R. Valero, Inorg. Chem., 1988, 27, 1367–1370 CrossRef CAS.
- G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt04859e |
| This journal is © The Royal Society of Chemistry 2017 |