
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAxially chiral racemic half-sandwich nickel(II) complexes by ring-closing metathesis†
Włodzimierz
Buchowicz
 *a,
Łukasz
Banach
a,
Radosław
Kamiński
b and
Piotr
Buchalski
a
*a,
Łukasz
Banach
a,
Radosław
Kamiński
b and
Piotr
Buchalski
a
aFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: wbuch@ch.pw.edu.pl; Fax: +48 22 234 5462; Tel: +48 22 234 5150
bDepartment of Chemistry, University of Warsaw, Żwirki i Wigury 101, 02-089 Warsaw, Poland
First published on 13th February 2017
Abstract
A remarkable nickelacycle has been synthesised via olefin metathesis of the α,ω-diene complex [Ni(η5-C5H4R)(Br)(NHC)] (R = C(CH3)2CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, NHC = 1-(6-hexenyl)-3-(2,4,6-trimethylphenyl)-imidazol-2-ylidene). Single-crystal X-ray analysis reveals a helical shape of the molecule and stretching of some interatomic distances in the Ni(II) coordination sphere. The Cp-NHC tethered complex shows catalytic activity resembling that of the parent complexes.
CH2, NHC = 1-(6-hexenyl)-3-(2,4,6-trimethylphenyl)-imidazol-2-ylidene). Single-crystal X-ray analysis reveals a helical shape of the molecule and stretching of some interatomic distances in the Ni(II) coordination sphere. The Cp-NHC tethered complex shows catalytic activity resembling that of the parent complexes.
Asymmetric catalysis with the chiral derivatives of the ubiquitous cyclopentadienyl (Cp) ligand remains an undeveloped area of research.1 The diamagnetic complexes [Ni(Cp)(X)(NHC)] (X = Cl, Br, I, alkyl; NHC = N-heterocyclic carbene), initially discovered by Abernethy et al.,2 have found numerous applications as pre-catalysts3 or robust substrates in the synthesis of novel species.4 We have presumed that a convenient synthesis of a Cp-NHC tethered nickel complex of this type could open up access to novel axially chiral molecular frameworks with applications in catalysis, chiral recognition, or separation.
Transition-metal complexes bearing chelating Cp-NHC ligands are usually prepared from bidentate pro-ligands and suitable metal sources (Scheme 1, path i).5 Despite the considerable variety of the so far reported complexes [Ni(Cp)(X)(NHC)], to the best of our knowledge, few examples of closely related indenyl-NHC Ni(II) complexes6 and C5Me4-NHC congeners7 have been synthesised by this route. We hypothesized that a Cp-NHC tether could be readily formed via ring-closing metathesis (RCM)8,9 in bis-alkenyl complexes (Scheme 1, path ii). Herein, we explore this novel route to axially chiral half-sandwich Ni(II) complexes.
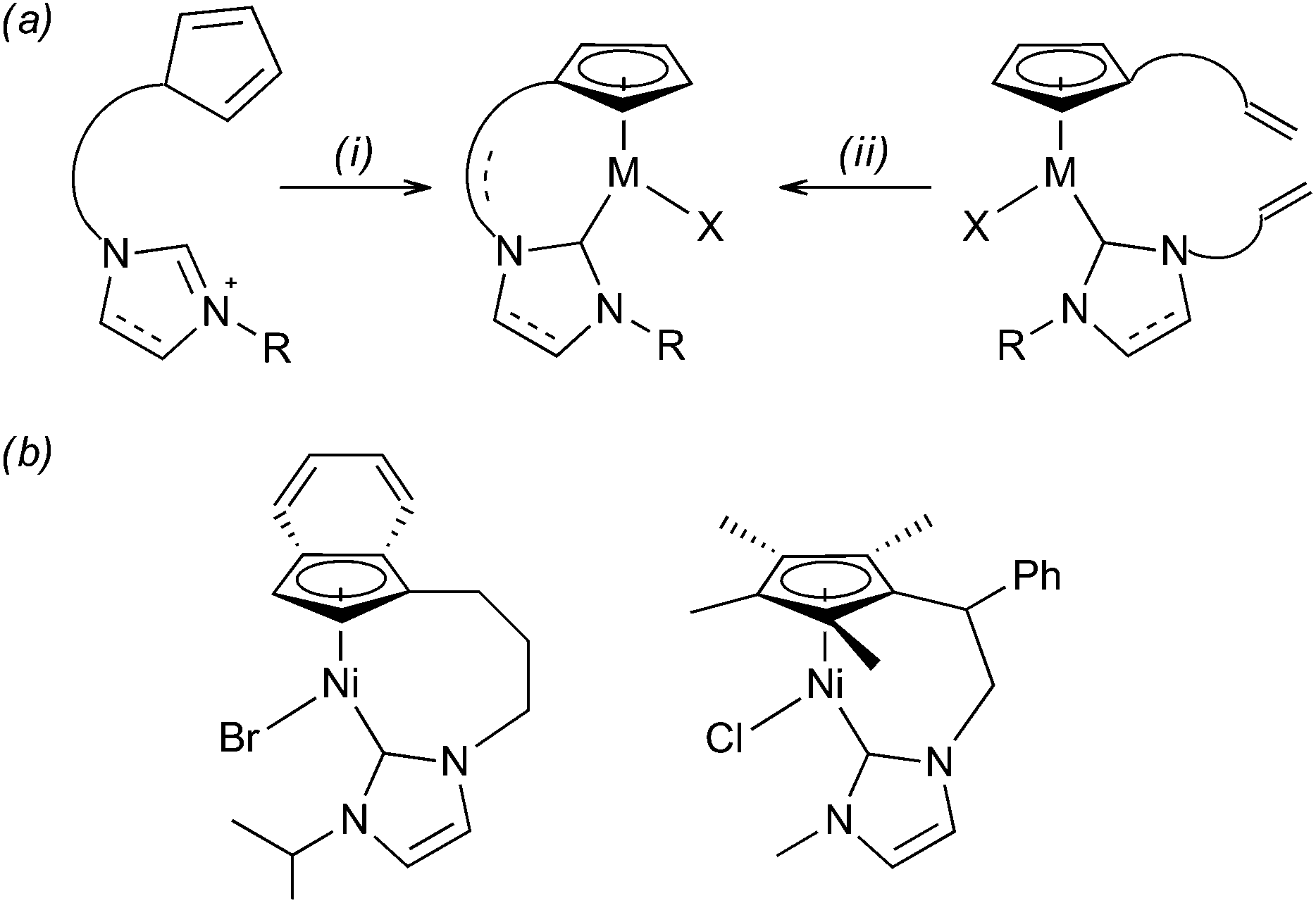 | ||
| Scheme 1 (a) General synthetic approaches to Cp-NHC complexes: (i) double deprotonation, then MX2 (ii) suitable metathesis catalyst, this work; (b) examples of Ni(II) complexes prepared by route i.6,7 | ||
Plausible RCM substrates, the α,ω-diene complexes 1 and 2, were prepared according to the standard procedure2 from the appropriate imidazolium salts and 1,1′-bis-(alkenyl)nickelocene (Scheme S1†).
Based on the precedence of complexes prepared from bidentate pro-ligands that are 6- or 7-membered metalacycles (Scheme 1b), we expected that complex 1 would readily undergo RCM to form a 10-membered ring. Thus, 1 was treated with 6 mol% of [Ru(CHPh)Cl2(PCy3)(SIMes)]10 under dilute conditions ([Ni] = 0.01 M) in refluxing toluene or CH2Cl2 (Scheme 2). Surprisingly, RCM in complex 1 proved to be ineffective.11 The presence of the expected product 3 could be determined only by means of mass spectrometry ([M]+ at m/z 496 (58Ni, 79Br)).
 | ||
| Scheme 2 RCM in complexes 1 and 2: (i) [Ru(CHPh)Cl2(PCy3)(SIMes)], 6 mol%, toluene or CH2Cl2, reflux. | ||
Fortunately, RCM in complex 2 catalysed by [Ru(CHPh)Cl2(PCy3)(SIMes)] proceeded smoothly in hot toluene to yield the 12-membered nickelacycle 4. The formation of the Cp-NHC tether renders all proton and carbon signals non-equivalent in the NMR spectra of 4, e.g. the Cp protons are represented by four multiplets at δ 4.96, 4.86, 4.61, and 4.02 ppm. Moreover, a detailed inspection of the 1H and 13C NMR spectra reveals the presence of two isomeric products. This finding is most clearly evidenced by two Ni–Ccarbene resonances at δ 165.3 ppm (major) and 165.8 ppm (minor). We identify these two products as, respectively, E and Z isomers of the CC double bond formed in the metathesis reaction (see below). The two isomers were obtained in a ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.12
:3 ratio.12
The X-ray diffraction studies allowed us to derive the crystal structures of complexes 1 and 4. Attempts to obtain a sufficient quality crystal structure of complex 2 were moderately successful (Fig. S1†). Nevertheless, the qualitative confirmation of the molecular structure of 2 shows its general similarity to that of 1. Consequently, a direct comparison of 1 and 4 allows one to gain structural insights into the differences and similarities of complexes before and after the intramolecular metathesis reaction.
Compounds 1 and 4 crystallise in the monoclinic P21/c and triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space groups, respectively. In both cases only one molecule is present in the asymmetric unit (Fig. 1). As both crystals are centrosymmetric, two enantiomers of each molecule are present in the analysed crystal structures. However, whereas in complex 1 rotation along the Ni1–C16 bond is possible, in 4 it is hindered due to the presence of the macrocyclic ring. This makes the molecules of 4 axially chiral, which has been confirmed in solution by NMR (Fig. 3).
space groups, respectively. In both cases only one molecule is present in the asymmetric unit (Fig. 1). As both crystals are centrosymmetric, two enantiomers of each molecule are present in the analysed crystal structures. However, whereas in complex 1 rotation along the Ni1–C16 bond is possible, in 4 it is hindered due to the presence of the macrocyclic ring. This makes the molecules of 4 axially chiral, which has been confirmed in solution by NMR (Fig. 3).
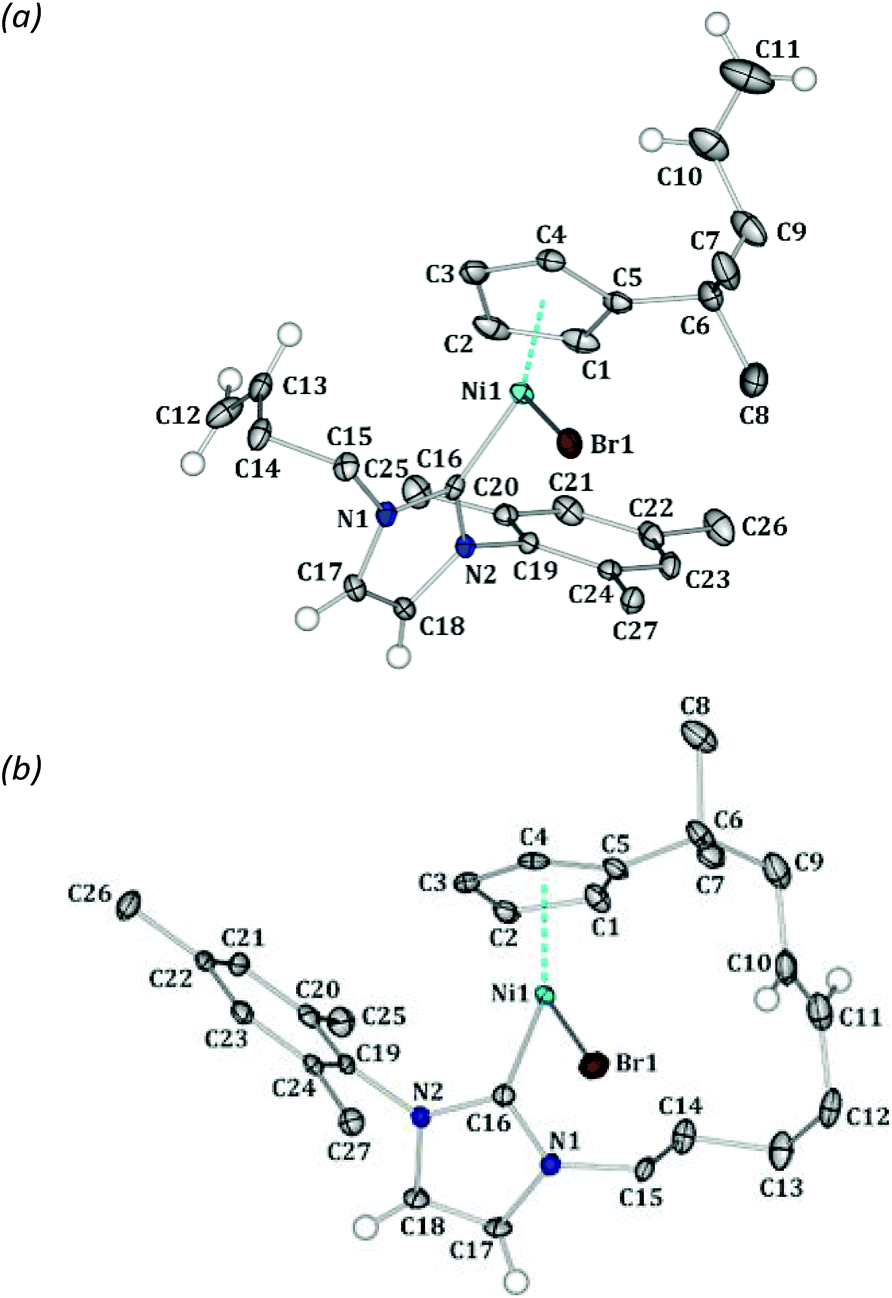 | ||
| Fig. 1 Molecular structures of complexes 1 (a) and 4 (E isomer) (b). Note that in 4 the C10–C11 (1.341(5) Å) bond is the double bond with E configuration. Atomic thermal motion is represented as ellipsoids (50% probability level), and some hydrogen atoms are removed for clarity. | ||
We have found a small fraction of the Z isomer (ca. 2–3%) in the crystal of 4, which is consistent with the NMR spectra. The olefin C10 and C11 atoms of the Z isomer are quite well visible on the residual density plot (Fig. S2†). Even though the full refinement of both isomers was not possible, it appears that the geometry of the CC double bond has no significant effect on the overall shape of the molecule.13
The coordination geometry at the nickel centre is comparable in both cases (Fig. 2). The most significant differences occur for the NHC ligand, which is oriented in the solid state in complex 1 in the opposite direction than in 4, as evidenced by the Br1–Ni1–C16–N2 torsion angles of −112.6(2)° and 114.5(2)° for 1 and 4, respectively. This suggests that 1 must adopt the less favourable conformation to facilitate the intramolecular metathesis.
 | ||
| Fig. 2 Overlay of 1 (red, labels in italics) and 4 (green, labels underlined). | ||
The formation of the macrocyclic ring also induces considerable distortions in the overall ligand arrangement. The large ring tends to push slightly the Cp and NHC ligands away. This is most clearly demonstrated by comparing the C16–Ni1–Cpcg angles (131.89(8)° for 1 and 132.14(8)° for 4; Cpcg = Cp ring centre of gravity). Moreover, the average Ni1–CCp bond lengths are approximately 0.02 Å larger for 4 (2.157 Å on average) than for 1 (2.140 Å on average) with the Ni1–C5 bond being the most significantly elongated (2.203(2) Å for 1 compared to 2.252(3) Å for 4). The same trend is observed for the Ni1–C16 bonds (1.877(2) Å in 1 and 1.884(3) Å in 4).
In order to probe the chirality of complex 4 in solution, its NMR spectra in the presence of an NMR chiral chemical shift reagent ((R)-(−)-1-(9-anthryl)-2,2,2-trifluoroethanol)14 (5) were recorded (Fig. 3). Doubling of the signals due to the formation of two diastereoisomers was clearly observed for the –NCH2 multiplet at δ 3.43 ppm, singlet of the o-CH3 group at δ 2.74 ppm (E isomer), and singlet of the C5H4C(CH3)2 group δ 1.26 ppm. This observation suggests that the chiral structure of 4 is also stable in this solution.15 Moreover, we conclude that the bromine ligand in 4 is the preferred site of its interaction with the polar reagent.
 | ||
| Fig. 3 (a) Selected sections of the 1H NMR (500 MHz, C6D6) spectrum of complex 4 (E and Z isomers), signals assigned to the –NCH2 proton, o-CH3 group and C5H4C(CH3)2 group (blue bottom line); (b) the same sample after addition of ca. an equimolar amount of 5 (brown top line). | ||
The catalytic activity of racemic 4 was tested in three C–C bond formation reactions (Table S1†). In the case of Suzuki coupling, the activity and selectivity to the desired cross-coupling product was comparable to those of the parent [Ni(Cp)(X)(NHC)] complexes (entry no. 1). Polymerization of styrene in the presence of 4 and methylalumoxane (MAO) yielded the expected atactic polystyrene (entries no. 2 and 3). Complex 4 with methyl methacrylate and MAO showed moderate activity at 50 °C (entry no. 4) and almost no activity at 20 °C (entry no. 5).
In summary, we have shown that a Cp-NHC tether can be readily formed via olefin metathesis in the Ni(II) coordination sphere. The length of the alkenyl substituents as well as the dynamics of the system in solution are both the key factors for determining the propensity of the intramolecular reaction. The helical shape of 4 opens up prospects for its applications in asymmetric catalysis after the resolution of enantiomers.
W. B., Ł. B. and P. B. would like to thank the National Science Centre for financial support (grant DEC-2011/01/B/ST5/06297).
Notes and references
- C. G. Newton, D. Kossler and N. Cramer, J. Am. Chem. Soc., 2016, 138, 3935 CrossRef CAS PubMed , and references therein.
- C. D. Abernethy, A. H. Cowley and R. A. Jones, J. Organomet. Chem., 2000, 596, 3 CrossRef CAS.
- Selected references: (a) R. A. Kelly III, N. M. Scott, S. Díez-González, E. D. Stevens and S. P. Nolan, Organometallics, 2005, 24, 3442 CrossRef; (b) W. Buchowicz, A. Kozioł, L. B. Jerzykiewicz, T. Lis, S. Pasynkiewicz, A. Pęcherzewska and A. Pietrzykowski, J. Mol. Catal. A: Chem., 2006, 257, 118 CrossRef CAS; (c) D. A. Malyshev, N. M. Scott, N. Marion, E. D. Stevens, V. P. Ananikov, I. P. Beletskaya and S. P. Nolan, Organometallics, 2006, 25, 4462 CrossRef CAS; (d) V. Ritleng, A. M. Oertel and M. J. Chetcuti, Dalton Trans., 2010, 39, 8153 RSC; (e) W. Buchowicz, W. Wojtczak, A. Pietrzykowski, A. Lupa, L. B. Jerzykiewicz, A. Makal and K. Woźniak, Eur. J. Inorg. Chem., 2010, 648 CrossRef CAS; (f) A. M. Oertel, V. Ritleng and M. J. Chetcuti, Organometallics, 2012, 31, 2829 CrossRef CAS; (g) L. P. Bheeter, M. Henrion, L. Brelot, C. Darcel, M. J. Chetcuti, J.-B. Sortais and V. Ritleng, Adv. Synth. Catal., 2012, 354, 2619 CrossRef CAS; (h) A. R. Martin, Y. Makida, S. Meiries, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2013, 32, 6265 CrossRef CAS; (i) Ł. Banach, P. A. Guńka, D. Górska, M. Podlewska, J. Zachara and W. Buchowicz, Eur. J. Inorg. Chem., 2015, 5677 CrossRef; (j) A. Włodarska, A. Kozioł, M. Dranka, J. Jurkowski and A. Pietrzykowski, J. Mol. Catal. A: Chem., 2014, 395, 481 CrossRef; (k) Ł. Banach, P. A. Guńka and W. Buchowicz, Dalton Trans., 2016, 45, 8688 RSC.
- Selected references: (a) C. Radloff, F. E. Hahn, T. Pape and R. Fröhlich, Dalton Trans., 2009, 7215 RSC; (b) A. M. Oertel, J. Freudenreich, J. Gein, V. Ritleng, L. F. Veiros and M. J. Chetcuti, Organometallics, 2011, 30, 3400 CrossRef CAS; (c) A. M. Oertel, V. Ritleng, L. Burr and M. J. Chetcuti, Organometallics, 2011, 30, 6685 CrossRef CAS; (d) W. Buchowicz, Ł. Banach, J. Conder, P. A. Guńka, D. Kubicki and P. Buchalski, Dalton Trans., 2014, 43, 5847 RSC; (e) S. Pelties, D. Herrmann, B. de Bruin, F. Hartl and R. Wolf, Chem. Commun., 2014, 50, 7014 RSC.
- (a) S. P. Downing and A. A. Danopoulos, Organometallics, 2006, 25, 1337 CrossRef CAS; (b) A. P. da Costa, M. Viciano, M. Sanaŭ, S. Merino, J. Tejeda, E. Persi and B. Royo, Organometallics, 2008, 27, 1305 CrossRef; (c) V. V. K. M. Kandepi, J. M. S. Cardoso, E. Peris and B. Royo, Organometallics, 2010, 29, 2777 CrossRef CAS; (d) J. M. S. Cardoso and B. Royo, Chem. Commun., 2012, 48, 4944 RSC; (e) B. Royo and E. Peris, Eur. J. Inorg. Chem., 2012, 1309 CrossRef CAS; (f) B. Royo, in Advances in Organometallic Chemistry and Catalysis: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, J. Wiley & Sons, 2014, vol. 10, pp. 133–143 Search PubMed.
- H.-M. Sun, D.-M. Hu, Y.-S. Wang, Q. Shen and Y. Zhang, J. Organomet. Chem., 2007, 692, 903 CrossRef CAS.
- (a) L. Postigo and B. Royo, Adv. Synth. Catal., 2012, 354, 2613 CrossRef CAS; (b) L. Postigo, R. Lopes and B. Royo, Dalton Trans., 2014, 43, 853 RSC.
- (a) E. B. Bauer and J. A. Gladysz, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, Germany, 2003, vol. 2, 2.11, pp. 403–431 Search PubMed; (b) A. Pietrzykowski and W. Buchowicz, in Advances in Organometallic Chemistry and Catalysis: The Silver/Gold Jubilee International Conference on Organometallic Chemistry Celebratory Book, ed. A. J. L. Pombeiro, J. Wiley & Sons, 2014, vol. 12, pp. 157–170 Search PubMed; (c) T. Fiedler and J. A. Gladysz, in Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, 2014, vol. 9, pp. 311–328 Search PubMed.
- Selected references: (a) M. Ogasawara, W. Y. Wu, S. Arae, K. Nakajima and T. Takahashi, Organometallics, 2013, 32, 6593 CrossRef CAS; (b) Y.-Y. Tseng, K. Kamikawa, Q. Wu, T. Takahashi and M. Ogasawara, Adv. Synth. Catal., 2015, 357, 2255 CrossRef CAS; (c) R. E. Andrew and A. B. Chaplin, Inorg. Chem., 2015, 54, 312 CrossRef CAS PubMed; (d) A. L. Estrada, T. Jia, N. Bhuvanesh, J. Blümel and J. A. Gladysz, Eur. J. Inorg. Chem., 2015, 5318 CrossRef CAS; (e) G. M. Lang, T. Shima, L. Wang, K. J. Cluff, K. Skopek, F. Hampel, J. Blümel and J. A. Gladysz, J. Am. Chem. Soc., 2016, 138, 7649 CrossRef CAS PubMed.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed.
- [Ru(CHPh)Cl2(PCy3)2] also did not afford efficient RCM in complex 1. Some related α,ω-diene complexes, e.g. [Ni(C5H4 CH2CHCH2)(Br)(allyl-IMes)], [Ni(C5H4 CH2CHCH2)(Cl)(allyl-IMes)], [Ni(C5H4(CH2)2CHCH2)(Br)(CH2CH(CH2)2IMes)] (see Fig. S3†) did not undergo RCM under conditions similar to those for 2.
- According to integration of doublets of the imidazole protons at δ 6.32 ppm (J = 1.9 Hz, E) and δ 6.30 ppm (J = 1.9 Hz, Z).
- A similar trend was observed for diansa-metallocenes: W. Buchowicz, A. Furmańczyk, J. Zachara and M. Majchrzak, Dalton Trans., 2014, 41, 9296–9271 Search PubMed.
- S. Braun, H.-O. Kalinowski and S. Berger, 100 and More Basic NMR Experiments: A Practical Course, VCH, Weinheim, 1996 Search PubMed.
- As noted by the referee, taking into account the chirality and E/Z isomers of the CC bond, four stereoisomers of 4 are formed in this reaction (Δ,Z; Δ,E; Λ,Z; Λ,E). Doubling of signals for the Z isomers in the presence of 5 was clearly observed for the NCH2– signal (see Fig. S4†). Preliminary attempts to hydrogenate the CC bond in 4 (H2/PtO2, toluene, room temp., 48 h) resulted in incomplete conversion of 4.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and figures. CCDC 1504275 and 1504276. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04811k |
| This journal is © The Royal Society of Chemistry 2017 |