
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of bidentate ligand donor types on the formation and stability in 2 + 1 fac-[MI(CO)3]+ (M = Re, 99mTc) complexes†
Thomas R.
Hayes
a,
Shalina C.
Bottorff
a,
Winston S.
Slocumb
a,
Charles L.
Barnes
b,
Aurora E.
Clark
a and
Paul D.
Benny
 *a
*a
aWashington State University, Department of Chemistry, Pullman, WA 99164, USA. E-mail: bennyp@wsu.edu
bDepartment of Chemistry, University of Missouri, Columbia, MO 65211, USA
First published on 20th December 2016
Abstract
In the last two decades, a number of chelate strategies have been proposed for the fac-[MI(CO)3]+ (M = Re, 99mTc) core in radiopharmaceutical applications. However, the development of new ligands/complexes with improved function and in vivo performance has been limited in recent years. Expanding on our previous studies using the 2 + 1 labeling strategy, a series of bidentate ligands (neutral vs. anionic) containing an aromatic amine in combination with monodentate pyridine analogs or imidazole were explored to determine the influence of the bidentate and monodentate ligands on the formation and stability of the respective complexes. The 2 + 1 complexes with Re and 99mTc were synthesized in two steps and characterized by standard radio/chemical methods. X-ray characterization and density functional theory analysis of the Re 2 + 1 complexes with the complete bidentate series with 4-dimethylaminopyridine were conducted, indicating enhanced ligand binding energies of the neutral over anionic ligands. In the 99mTc studies, anionic bidentate ligands had significantly higher formation yields of the 2 + 1 product, but neutral ligands appear to have increased stability in an amino acid challenge assay. Both bidentate series exhibited improved stability by increasing the basicity of the pyridine ligands.
Introduction
Pioneered by Alberto, the organometallic fac-[99mMI(OH2)3(CO)3]+ (M = Re, Tc) complex has provided a unique platform for bioinorganic medicinal applications.1,2 In nuclear medicine, the 99mTc analog has ideal nuclear decay characteristics (t1/2 = 6.0 h, γ = 140 keV (89%)) appropriate for single photon emission computed tomography (SPECT) imaging, while the radioactive 186/188Re analogs provide isostructural theranostics by combined gamma and beta (β−) particle emissions (1.07 and 2.12 MeV), respectively.3–5 In the last decades, the development of fac-[99mTcI(OH2)3(CO)3]+ based radiopharmaceuticals has expanded due the quantitative preparation with the Isolink kit and the ability to accommodate a wide range of ligand types and denticity on the fac-[MI(CO)3]+ core.6–8 The flexibility of the fac-[MI(CO)3]+ core has led to the examination of several ligands strategies (e.g., mono-, bi-, tridentate or a combination) to displace the coordinated waters and saturate the coordination sphere. A variety of donors have been reported for the low spin d6 center of the fac-[MI(CO)3]+ core, but it typically favors soft ligands (e.g., amines, pyridines, imidazoles, triazoles, pyrazoles) over harder ligands. Tridentate systems containing at least one aromatic amine (e.g., dipicolylamine, histidine, click to chelate) have garnered the highest attention due to radiochemical yields, ease of synthesis, and stability (in vitro and in vivo) with the fac-[MI(CO)3]+ core.6,7 While providing a unique combinatorial ability, monodentate and 2 + 1 (mono- and bidentate) systems have not exhibited the same degree of stability obtained by tridentate ligands, which may be due to the substitution lability and absence of the chelate effect for the monodentate donors.Current trends in fac-[MI(CO)3]+ radiopharmaceutical design continue to utilize small molecule or peptide targeting agents with established chelate systems, without significant improvement on the chelates themselves. However, several groups have started to examine the influence of functionalization on donors to tailor the pharmacological behavior and in vivo stability.9–11 In our previous study, the behavior of substitutions on monodentate pyridine ligands with electron withdrawing and activating groups in a 2 + 1 system were examined with fac-[MI(CO)3]+.10 The direct behavior of pyridine monodentate ligands was explored using picolinic acid as a bidentate ligand, to yield neutral 2 + 1 complexes. The basicity of the pyridine ligands had profound impact on the synthesis and in vitro stability. Pyridine ligands functionalized in the 4 position with electron activating groups (e.g., –OMe, –NH2, –NMe2) with higher pKa's than pyridine demonstrated higher synthetic yields and stability than electron withdrawing groups (e.g., –NO2) with lower pKa's. While this study conclusively determined the influence of monodentate pyridine basicity, the influence of the donor type in the bidentate ligand and complex charge on the overall system was not explored.
In this paper, a series of bidentate ligands was explored using a 2 + 1 strategy to examine the influence of the ligands on radiochemical preparation and stability of the respective fac-[MI(CO)3]+ (M = Re, 99mTc) complexes. Two general classes of bidentate ligands were selected for evaluation, neutral and anionic ligands. In each class of bidentate ligands, one donor remained constant (neutral: pyridine, anionic: carboxylate) while being paired with a series of aromatic nitrogen donors (e.g., pyridine, imidazole, triazole) (Fig. 1). These combinations permitted the evaluation of both the overall charge of the system as well as the type of aromatic nitrogen donor. Rhenium analogs were prepared for chemical evaluation. X-ray crystallography data of the 2 + 1 series containing 4-dimethylaminopyridine (DMAP) were collected and used in conjunction with density functional theory (DFT) calculations to determine the relative ligand binding energies. Radioactive 99mTc complexes were also synthesized for comparison with the Re analogs and evaluation of the effect the bidentate ligands exhibited on the radiochemical labeling and stability of 2 + 1 complexes.
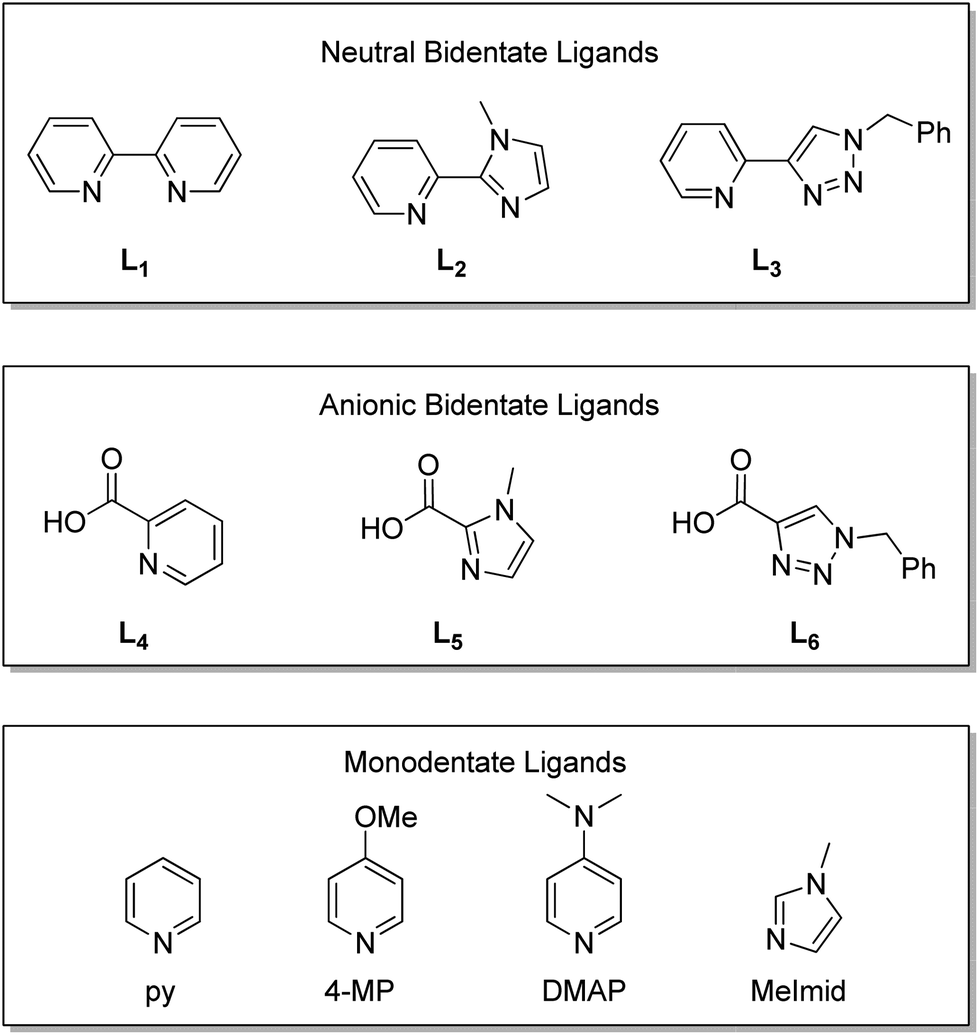 | ||
| Fig. 1 Neutral, anionic and monodentate ligands investigated. | ||
Experimental
Materials and methods
All reagents and organic solvents of reagent grade or better were used as purchased from Alfa Aesar, Acros, Chem-Impex, or Fluka without further purification. Compounds 2-(1-methyl-1H-imidazol-2-yl)pyridine, L2, 2-(1-benzyl-1H-1,2,3-triazol-4-yl)pyridine, L3, 1-benzyl-1H-1,2,3-triazole-4-carboxylic acid, L6, and rhenium complexes fac-[ReI(CO)3(L1)(py)](SO3CF3), 1a, fac-[ReI(CO)3(L1)(DMAP)](SO3CF3), 1c, fac-[ReI(CO)3(L1)(MeImid)](SO3CF3), 1d, fac-[ReI(CO)3(L2)(MeImid)](SO3CF3) 2d, fac-[ReI(CO)3(L3)(py)](SO3CF3), 3a, fac-[ReI(CO)3(L3)(DMAP)](SO3CF3), 3c, fac-[ReI(OH2)(CO)3(L4)], 4, fac-[ReI(CO)3(L4)(py)], 4a, fac-[ReI(CO)3(L4)(4-MP)], 4b, fac-[ReI(CO)3(L4)(DMAP)], 4c, and fac-[ReI(OH2)(CO)3(L6)], 6 were prepared as previously described.6,11–18 The rhenium starting materials [Re(CO)5OTf] and fac-[ReI(OH2)3(CO)3](SO3CF3) were prepared by literature methods from Re2(CO)10 purchased from Strem.19,2099mTc was obtained in the form of Na[99mTcO4] from Cardinal Health (Spokane, WA) and the fac-[99mTcI(OH2)3(CO)3]+ complex was prepared using a commercially available Isolink® kit from Covidien. UV-Vis spectra were obtained using a Varian Cary 50 spectrophotometer (1 cm path-length). 1H and 13C NMR spectra were recorded on a Varian 300 or 400 MHz instrument at 25 °C in CD3OD or CDCl3. FT-IR spectra were obtained on a Thermo Nicolet 6700 FT-IR using NaCl plates and analyzed with OMNIC 7.1 software. Elemental analyses were performed on a Perkin Elmer 2400 Series II System. Mass spectrometry was performed on a Xevo TQ M using ESI.Separation and identification of compounds were conducted on a Perkin Elmer Series 200 High Pressure Liquid Chromatograph (HPLC) equipped with a UV/VIS Series 200 detector and a Radiomatic 610TR detector. Utilizing a Varian Pursuit XRs 5 μm particle and 250 × 4.6 mm C-18 column, the compounds were separated with a reverse phase gradient system using either 0.05% trifluoroacetic acid (TFA)/water (Method A) or 2 mM pH 7.4 phosphate buffer (Method B) switching to methanol (MeOH); 0–1.0 min (50% aqueous, 50% MeOH), 1.0–19.0 min (50% to 100% MeOH linear gradient), 19–22 min (100% MeOH), 22–30 min (50% aqueous, 50% MeOH) at a flow rate of 1.0 mL min−1. Detection of compounds was performed at 254 nm.
HPLC purification was performed on a Hitachi preparatory HPLC. Method 1 utilizing an Agilent Zorbax SB-C18 7 μm particle size 250 × 21.2 mm column, compounds were separated with a reverse phase gradient system beginning with 0.1% TFA aqueous eluent gradually shifting to methanol. The HPLC method used 0–6 min (50% TFA, 50% MeOH), 6–19 min (50% to 100% MeOH linear gradient), and 19–25 min (100% MeOH) at a flow rate of 10 mL min−1. Detection of compounds was using UV-Vis at a 220 nm wavelength. Method 2 utilized a Phenomenex Gemini C18 5 μm particle size 250 × 21.2 mm column. Separation was achieved using the same gradient as above with H2O shifting to methanol.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 383), 305 (12493), 319 (13310), 355 (4216) IR (NaCl, νmax/cm−1): 2030, 1914. MS (m/z): [M]+ 535.97.
688), 311 (18861)). IR (NaCl, νmax/cm−1): 2029, 1907. MS (m/z): [M]+ 509.00.
383), 305 (12493), 319 (13310), 355 (4216)). IR (NaCl, νmax/cm−1): 2030, 1914. MS (m/z): [M]+ 539.09.
503), 312 (11519)). IR (NaCl, νmax/cm−1): 2024, 1910. MS (m/z): [M]+ 552.03.
780); 242 (71990); 300 (sh, 25670); 320 (sh, 14170)). IR (NaCl, νmax/cm−1): 2034.3, 1920.6. MS (m/z): [M]+ 616.12.
363). IR (NaCl, νmax/cm−1): 2030, 1911. MS (m/z): [M]+ 629.12.
994). IR (NaCl, νmax/cm−1): 2031, 1913. MS (m/z): [M]+ 589.01.
527), 320 (5385) IR (NaCl, νmax/cm−1): 2019, 1882. MS (m/z): [M + H]+ 476.16.
404). IR (NaCl, νmax/cm−1): 2021, 1888. MS (m/z): [M + H]+ 476.06.
420); 254 (10900). IR (NaCl, νmax/cm−1): 2020.3, 1887.7. MS (m/z): [M + Na]+ 527.98.
660). IR (NaCl, νmax/cm−1): 2017.2, 1883.1. MS (m/z): [M + Na]+ 541.04.
113). IR (NaCl, νmax/cm−1): 2027, 1897. MS (m/z): [M + Na]+ 574.91.
463). IR (NaCl, νmax/cm−1): 2023, 1892. MS (m/z): [M + Na]+ 617.94.
527), 320 (5385). IR (NaCl, νmax/cm−1): 2019, 1882. MS (m/z): [M + Na]+ 578.03.
383), 305 (12493), 319 (13310), 355 (4216) IR (NaCl, νmax/cm−1): 2030, 1914. MS (m/z): [M]+ 535.97.
688), 311 (18861)). IR (NaCl, νmax/cm−1): 2029, 1907. MS (m/z): [M]+ 509.00.
383), 305 (12493), 319 (13310), 355 (4216)). IR (NaCl, νmax/cm−1): 2030, 1914. MS (m/z): [M]+ 539.09.
503), 312 (11519)). IR (NaCl, νmax/cm−1): 2024, 1910. MS (m/z): [M]+ 552.03.
780); 242 (71990); 300 (sh, 25670); 320 (sh, 14170)). IR (NaCl, νmax/cm−1): 2034.3, 1920.6. MS (m/z): [M]+ 616.12.
363). IR (NaCl, νmax/cm−1): 2030, 1911. MS (m/z): [M]+ 629.12.
994). IR (NaCl, νmax/cm−1): 2031, 1913. MS (m/z): [M]+ 589.01.
527), 320 (5385) IR (NaCl, νmax/cm−1): 2019, 1882. MS (m/z): [M + H]+ 476.16.
404). IR (NaCl, νmax/cm−1): 2021, 1888. MS (m/z): [M + H]+ 476.06.
420); 254 (10900). IR (NaCl, νmax/cm−1): 2020.3, 1887.7. MS (m/z): [M + Na]+ 527.98.
660). IR (NaCl, νmax/cm−1): 2017.2, 1883.1. MS (m/z): [M + Na]+ 541.04.
113). IR (NaCl, νmax/cm−1): 2027, 1897. MS (m/z): [M + Na]+ 574.91.
463). IR (NaCl, νmax/cm−1): 2023, 1892. MS (m/z): [M + Na]+ 617.94.
527), 320 (5385). IR (NaCl, νmax/cm−1): 2019, 1882. MS (m/z): [M + Na]+ 578.03.
X-ray crystal structure determination
For all structures intensity data were obtained on a Bruker APEX II CCD Area Detector system using the ω scan technique with Mo Kα radiation from a graphite monochromator. Data were collected at −173 °C. Intensities were corrected for Lorentz and polarization effects. Equivalent reflections were merged, and absorption corrections were made using the multi-scan method.21 Space group, lattice parameters and other relevant information are given in ESI.† The structures were solved by direct methods with full-matrix least-squares refinement, using the SHELX package22–24 with the aid of the program X-SEED.25 All non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms were placed at calculated positions and included in the refinement using a riding model, with fixed isotropic U.Computational methods
The unrestricted (U) B3LYP combination of density functionals was employed for the geometry optimization of: DMAP, [Re(CO)3(L1)]+, [Re(CO)3(L2)]+, [Re(CO)3(L3)]+ [Re(CO)3(L4)], [Re(CO)3(L5)], [Re(CO)3(L6)], 1c, 2c, 3c, 4c, 5c, and 6c using the Gaussian09 software package.26,27 The crystallographic data from each of the structures with DMAP was used as the starting point for geometry optimization in the gas phase. Comparison of the high and low spin (LS) states of all Re(I) containing compounds revealed substantial energetic preference for the LS states, and thus only those are discussed herein. The aug-cc-pVDZ-PP pseudopotential and associated basis set was used to describe the atomic orbitals of Re(I),28 while aug-cc-PVDZ was used to describe all other atoms.29 The combined method and basis set is denoted as (U)B3LYP/aug-cc-pVDZ-PP/aug-cc-pVDZ. Frequency calculations were performed on all optimized structures to obtain thermochemical corrections and ensure that they correspond to a local minima. The gas-phase 0 K ligand binding energies were obtained as the difference of product and reactant energies via the following reactions:| [ReI(CO)3(L1–L3)DMAP]+ → [ReI(CO)3(L1–L3)]+ + DMAP | (r1) |
| [ReI(CO)3(L4–L6)DMAP] → [ReI(CO)3(L4–L6)] + DMAP | (r2) |
99mTc labeling studies
Labeling with L6 was performed as previously described.18 Briefly, A solution of L6 in EtOH (100 μL, 50 mM) was added to a solution of NaHCO3 (800 μL, 10 mM) in a sealable vial. The vial was sparged with N2 for 5 min and a solution of fac-[99mTcI(OH2)3(CO)3]+ (100 μL) was added and the solution was heated to 50 °C for 1 h. 6′ was then purified using HPLC method B.
Results and discussion
A series of bidentate ligands (L1–L6) with the fac-[MI(CO)3]+ (M = Re, 99mTc) core was explored using the 2 + 1 methodology to evaluate the overall nature of complexes, synthetic yields, and stability. Within the series, the ligands were segregated into two classes (neutral and anionic) based on the overall charge of the fully deprotonated ligand (Fig. 1). In the neutral ligand series (L1–L3), each of the bidentate ligands consisted of a pyridine donor and a second aromatic donor (e.g., triazole, imidazole, pyridine). In the anionic ligand series (L4–L6), the bidentate ligands consisted of a carboxylate donor paired with an aromatic donor (e.g., triazole, imidazole, pyridine). Based on our previous studies illustrating the influence of monodentate ligands on the yields and stability of the complex, each ligand series was further evaluated as a 2 + 1 complex with a functionalized pyridine analog (e.g., pyridine (py), 4-methoxypyridine (4-MP), 4-dimethyaminopyridine (DMAP)). Methyl imidazole was also examined representing a separate class of aromatic amine donors due to promising stability results from recent literature studies.11,15 Neutral ligands (L1–L3) coordinated with the fac-[MI(CO)3]+ core and a monodentate ligand (Y) gave cationic complexes of the general form fac-[MI(CO)3(L)(Y)]+. Anionic ligands (L4–L6) coordinated with the fac-[MI(CO)3]+ core and a monodentate ligand gave neutral complexes of the general form fac-[MI(CO)3(L)(Y)]. Probing the differences within the bidentate ligand series and the monodentate functionalized pyridine allow the overall assessment of the systems with stable nonradioactive (Re) and radioactive (99mTc) complexes.Re 2 + 1 complexes with ligands (L1–L6) were synthesized for characterization and comparison with the analogous 99mTc analogs. With neutral ligands (L1–L3), a two-step, one-pot synthesis provided the general synthetic route to obtain cationic complexes (fac-[ReI(CO)3(L)(Y)]+). In the first step, the [Re(CO)5OTf] starting material was decarbonylated with a slight excess of ligand (L1–L3) in MeOH at 70 °C to generate the intermediate fac-[ReI(CO)3(sol)(L)]+ after 2 h. The second step involved the direct addition of the monodentate ligand (2 equivalents) with additional heating overnight to give the rhenium complexes 1a–d, 2a–d and 3a–d in moderate to good yields (35–89%) after isolation (Scheme 1). Preparation of the Re 2 + 1 complexes with the anionic ligand series (L4–L6) was also attempted using the previously mentioned two-step in one pot route. However, it was unsuccessful to obtain a single well defined product by HPLC analysis. The requirement of additional base to deprotonate the carboxylic acid group in the bidentate ligands (L4–L6) may have contributed to the incomplete intermediate fac-[ReI(CO)3(L)(OH2)] formation and the observation of several species in the reaction mixture chromatogram upon addition of the monodentate ligand. An alternative synthetic route with ligands (L4–L6) involved a two-step route including the isolation of the intermediate fac-[ReI(OH2)(CO)3(L)] prior to addition of the pyridine ligands. It was found that the aqueous starting material fac-[ReI(OH2)3(CO)3]OTf(aq) provided the best route to obtain the intermediates fac-[ReI(OH2)(CO)3(L4,L6)] in agreement with previous preparation for 4 and 6.6,18 However, the isolation of fac-[ReI(OH2)(CO)3(L5)], 5, via this route was observed in lower yields (24%). The second step involved the addition of a monodentate ligand (Y) to the intermediate complex in methanol at 70 °C. Isolation via precipitation or preparatory HPLC gave the desired fac-[ReI(CO)3(L)(Y)] complexes 4a–d, 5a–d and 6a–d in moderate to good yields (47–86%) (Scheme 1). Each of the new Re complexes were characterized by 1H and 13C NMR, UV-Vis, IR, MS and elemental analysis. While the analytical analysis details for each complex are reported within, the similar structural nature of the aromatic nitrogen rings and carboxylic acids in the bidentate ligand series (L1–L6) did not have significant variances in the data collected between the compound series. We had anticipated the basicity of the pyridine analogs (DMAP > 4-MP > Py) to potentially impact the data in the respective bidentate ligands in the 2 + 1 complexes, but no significant trend was observed within statistical error for each ligand series. IR spectra collected confirmed the presence and conformation of the carbonyls in each of the 2 + 1 rhenium complexes by exhibiting the characteristic two peaks of a pseudo C3v symmetry of the Re(CO)3 core. In each of the complexes, a sharp peak was observed at ∼2050 cm−1 along with a broad peak 1850–1900 cm−1 was consistent in each complex. 1H and 13C NMR analysis of the 2 + 1 rhenium complexes exhibited a downfield shift due to the electron withdrawing role of the metal of the complexed ligands (bidentate (L1–L6) and monodentate) from the respective free ligand chemical shifts. Within the bidentate ligand series in both the cationic and neutral complexes, the singlet 1H NMR shifts of the triazole (L3, L6) and imidazole (L2, L5) protons on the coordinating ring to the metal were specifically examined to ascertain the influence of basicity of the monodentate pyridine on their shifts. Unfortunately, no conclusive trend comparing pyridine basicity and NMR shifts was observed in the downfield shifts of the singlets (triazole 7.95–8.00 ppm, imidazole 7.60–7.65 ppm) within the rhenium 2 + 1 complexes. Mass spectra analysis in positive mode was conducted on the rhenium 2 + 1 complexes. In general, the cationic complexes with L1–3 gave the expected molecular ion peak [M+]. In the neutral 2 + 1 complexes with L4–6, m/z values of [M + H]+ or [M + Na]+ species were observed. Both series of complexes had relatively stable 2 + 1 complex ions in the gas phase with minimal loss of the monodentate ligand. One notable exception was fac-[ReI(CO)3(L5)(OH2)] (5) with a m/z resulting from the loss of coordinated water and the addition of a sodium ion.
 | ||
| Scheme 1 Synthesis of 2 + 1 complexes, fac-[ReI(CO)3(L)(Y)]0/+, with bidentate ligands (L1–L6) and monodentate aromatic amine ligands (Y). Top: a two-step one pot procedure for neutral ligands (L1–L3) and Y. Bottom: second step using the intermediate fac-[ReI(CO)3(L)(OH2)] of anionic ligands (L4–L6) with Y. | ||
To evaluate the structural differences observed in the bidentate series, the single crystals of the 2 + 1 Re complexes with DMAP (1c–6c) were obtained and analyzed by X-ray diffraction analysis (Fig. 2). Complete experimental parameters and tables of bond lengths and angles for each of the structures can be found in the CIF and ESI.† Commensurate with other 2 + 1 fac-[ReI(CO)3]+ complexes,10,15,17 the bidentate ligands (L1–L6) and DMAP saturate the coordination sphere to present distorted octahedral complexes (1c–6c) with similar bond angles and distances (Table 1). The geometries of the bidentate ligands within both ligand classes can vary slightly based on the type and orientation of the nitrogen donor in the ligands (5-member: triazole, imidazole vs. 6-member: pyridine). The bite angles of the bidentate ligands ranged from 74.16–75.48°. In general, the neutral ligands (L1–L3) exhibited more constrained values than the anionic ligands (L4–L6). Within the series, the aromatic amine donors within the bidentate ligands (L1–L6) had similar bond distances throughout (2.118–2.177 Å) the complexes. The coordinated carboxylate in anionic ligands had relatively consistent distances (2.138–2.161 Å) within the structures 4c–6c for a Re carboxylate bond. In general, Re(1)–N(DMAP) bond lengths are slightly longer in the neutral ligand complexes (1c–3c) than in the corresponding anionic ligand complexes (4c–6c) with the exception of 5c (Table 1), however; this does not represent a significant difference in bond lengths between the two classes of complexes.
 | ||
| Fig. 2 Crystal structures of 1c–6c with thermal ellipsoids at 30% probability. Hydrogen atoms, anions, and included solvent have been omitted for clarity. 4c was reproduced from literature.10 | ||
| Complex | Re–X bond length (Å) | Angle (°) | |||
|---|---|---|---|---|---|
| Re(1)–N(1) | Re(1)–N(2) | Re(1)–O(1) | Re(1)–N(DMAP) | Bite angle | |
| 1c | 2.170 | 2.165 | — | 2.205 | 74.96 |
| 2c | 2.118 | 2.211 | — | 2.222 | 74.16 |
| 3c | 2.140 | 2.194 | — | 2.206 | 74.49 |
| 4c | 2.177 | — | 2.138 | 2.191 | 75.48 |
| 5c | 2.151 | — | 2.161 | 2.226 | 75.32 |
| 6c | 2.152 | — | 2.161 | 2.190 | 75.23 |
The calculated gas-phase DFT geometries of 1c–6c are in excellent agreement with the X-ray crystallographic information. All Cartesian coordinates are provided in the ESI.† The average deviation in calculated versus experimental Re–N bond lengths is <0.05 Å, Re–C is 0.01 Å, and Re–O is 0.004 Å. The relative ligand binding energies were examined by DFT calculations using (U)B3LYP/aug-cc-pVDZ-PP/aug-cc-pVDZ gas-phase analysis to determine ligand binding energy of the DMAP with respect to the bidentate ligand (Table 2). Cationic complexes fac-[ReI(CO)3(L)(DMAP)]+ formed with neutral ligands (L1–L3) had the highest binding energies for DMAP for the series, where complex 1c had a binding energy at 52.5 kcal mol−1 with complexes 2c and 3c only slightly lower. Neutral complexes fac-[ReI(CO)3(L)(DMAP)] formed with anionic ligands (L4–L6) had lower binding energies for DMAP between 38.9 and 40.0 kcal mol−1 indicating significantly lower bond stability for the neutral complexes. The similarities within the groups show that the neutral ligand species (cationic complexes) allow the metal centers to attract more electron density from the DMAP ligand, creating stronger bonding. Conversely, the carboxylate in the anionic ligands donates significant electron density to the metal reducing the bonding interaction between DMAP and the metal. Interestingly, the type of aromatic nitrogen donor had very little effect overall on the binding energy of the complex, indicating that overall complex charge has a more significant effect than the nitrogen donor.
| Complex | Binding energy (kcal mol−1) | Q Re |
|---|---|---|
| 1c | 52.5 | −0.80 |
| 2c | 50.2 | −0.81 |
| 3c | 51.6 | −0.81 |
| 4c | 40.0 | −0.72 |
| 5c | 38.9 | −0.73 |
| 6c | 39.6 | −0.73 |
Synthesis and stability studies were conducted with the radioactive analog 99mTc for comparison to the rhenium data. Available from an Isolink® kit, the starting precursor fac-[99mTcI(OH2)3(CO)3]+ was used to prepare the 2 + 1 complexes in a two-step route. Initial complexation of fac-[99mTcI(OH2)3(CO)3]+ with the bidentate ligands (L1–L5) at 10−3 M was performed at pH 3 at 90 °C to yield the intermediate complexes fac-[99mTcI(CO)3(OH2)(L)]+/0 (1′–5′) in moderate to excellent (62–99%) yields. L6 required using our previously described method at pH 8 and 5 × 10−3 M concentration to obtain complex 6′ in good yields (62%).18 It is noteworthy to mention double addition of the bidentate ligands was observed in most of the systems. At neutral pH, the double addition products were more apparent in the HPLC chromatogram, but were significantly reduced (<10%) by conducting the reactions at a slightly acidic pH. In order to simplify identification, avoid competition reactions, and prevent peak overlap, the intermediates (1′–6′) were purified prior to addition of the monodentate ligands.
The 2 + 1 complexes were prepared from the intermediate complexes 1′–6′ by reacting with functionalized pyridine ligands (10−2 M) at 40 °C for 30 min (Scheme 2). The products (1a′–6d′) were analyzed by radio-HPLC and correlated to retention times of the respective Re analogs (1a–6d) for identification (ESI Table 1†) using HPLC method A for the neutral and method B for the anionic ligand systems (Fig. 3). Product yields ranged from 20–99% throughout the series based on both the bidentate and the monodentate ligands combination (Table 3). Comparison of the two bidentate ligand classes found significant differences in the complexation of monodentate ligands between the neutral and anionic ligands. Across the pyridine series, 2 + 1 complexes formed with neutral bidentate ligands (L1–L3) had lower radiochemical labelling yields than the complexes formed with anionic bidentate ligands (L4–L5), with the exception of L6 (Fig. 4). Among the neutral ligands, L2 had the highest labeling yields with the monodentate ligands (68%, DMAP) over similar yields observed for L1 and L3 (53, 47%, DMAP). For 1a′ only an estimate was able to be obtained for radiolabeling yields due to overlap of the peaks between the bidentate intermediate 1′ and 1a′ (ESI Fig. 1†). With the anionic ligands, radiochemical yields obtained for 2 + 1 complexes with L4 and L5 were typically very good (>70%) for each of the pyridine analogs, but L6 had significantly lower yields (29–53%). Examination of the monodentate pyridines within the 2 + 1 series showed similar increased yields as a function of pKa. The general trend (DMAP (pKa 9.71) > 4-MP (pKa 6.55) > py (pKa 5.33)) for radiochemical labeling was observed similarly for both neutral and anionic ligands.
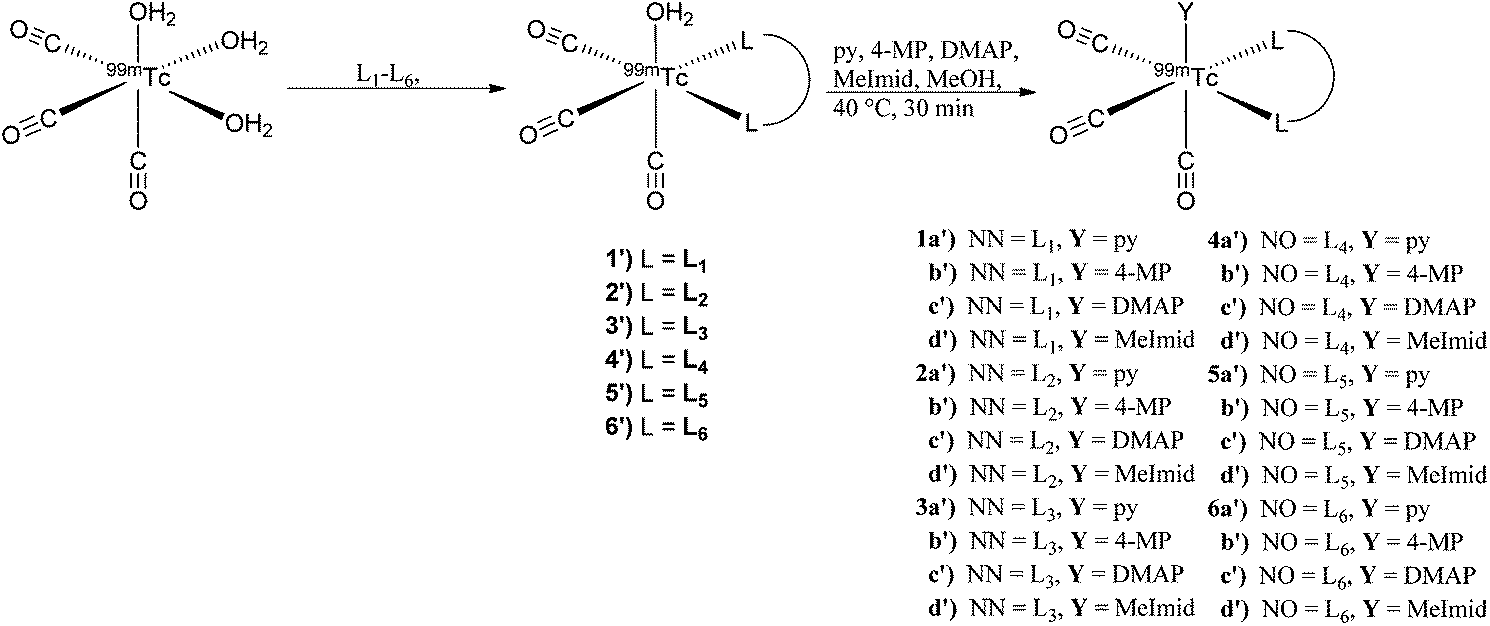 | ||
| Scheme 2 Synthesis of 2 + 1 complexes fac-[99mTcI(CO)3(L)(Y)]0/+ using a two step reaction from the starting material fac-[99mTcI(CO)3(OH2)3]+ through the intermediate fac-[99mTcI(CO)3(L)(OH2)]0/+ to the final product. | ||
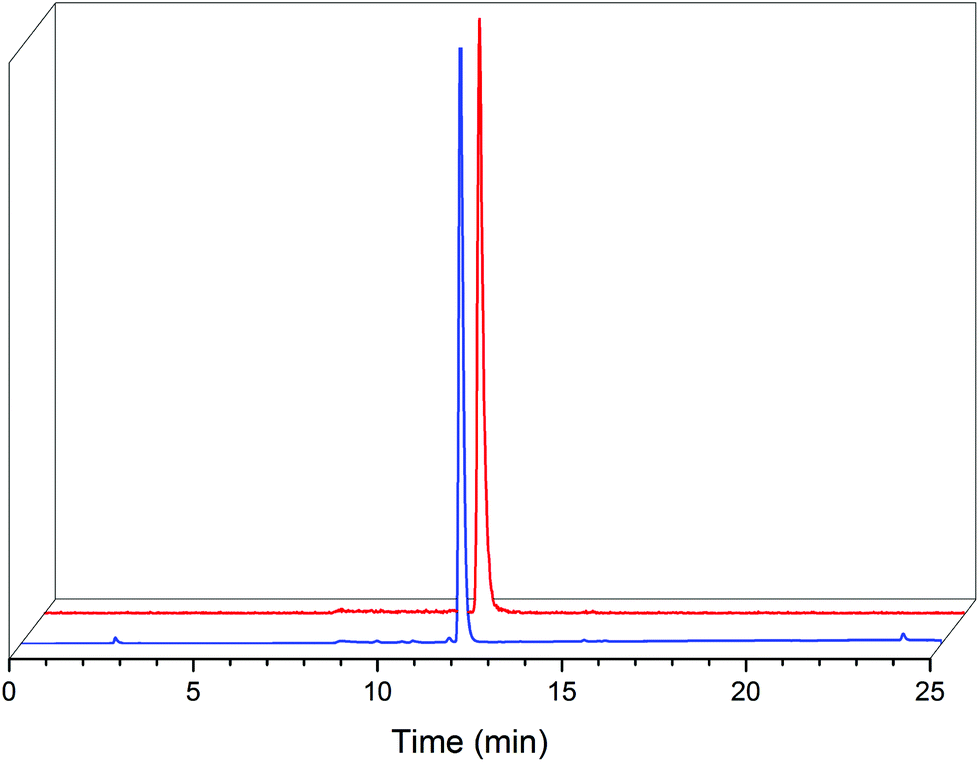 | ||
| Fig. 3 HPLC chromatogram of isolated fac-[ReI(CO)3(L2)(DMAP)] (2c, blue) with a UV detector (254 nm) and radiochromatogram (γ) of the fac-[99mTcI(CO)3(L2)(DMAP)] (2c′, red). | ||
 | ||
| Fig. 4 Plot of radiochemical yields of 2 + 1 formation with monodentate pyridine ligands (a–c) vs. pKa of monodentate pyridine ligands. Neutral bidentate ligands (L1–3) (top), anionic bidentate ligands (L4–6) (bottom) and pyridine ligands (py (pKa 5.33), 4-MP (pKa 6.55), DMAP (pKa 9.71)). *Yield for 1a′ estimated at 16% for this plot due to peak overlap. | ||
| Bidentate (L) | Monodentate (Y) | Complex | Yield | His stabilitya | Cys stabilitya | |
|---|---|---|---|---|---|---|
| a % of starting complex remaining in the presence of 1 mM amino acid after 1 hour at 37 °C. b Estimated value due to partial peak overlap. c Value not obtained due to complete peak overlap. d Reproduced from literature.10 | ||||||
| Neutral ligands | L1 | OH2 | 1′ | 91.3% | 71.6% | 59.0% |
| py | 1a′ | <20%b | NAc | NAc | ||
| 4-MP | 1b′ | 25.3% | 80.2% | 39.2% | ||
| DMAP | 1c′ | 53.6% | 93.3% | 84.7% | ||
| MeImid | 1d′ | 48.4% | 97.3% | 96.4% | ||
| L2 | OH2 | 2′ | 98.9% | 61.9% | 39.6% | |
| py | 2a′ | 31.1% | NAc | NAc | ||
| 4-MP | 2b′ | 46.2% | NAc | 38.9% | ||
| DMAP | 2c′ | 68.6% | 84.0% | 81.1% | ||
| MeImid | 2d′ | 85.0% | 89.4% | 87.5% | ||
| L3 | OH2 | 3′ | 80.0% | 55.0% | NAc | |
| py | 3a′ | 27.5% | 71.6% | 24.2% | ||
| 4-MP | 3b′ | 31.5% | 83.0% | 78.4% | ||
| DMAP | 3c′ | 47.0% | 93.5% | 88.1% | ||
| MeImid | 3d′ | 57.8% | 89.0% | 91.3% | ||
| Anionic ligands | L4 | OH2 | 4′ | >98% | — | — |
| py | 4a′ | 71.4% | 6.3%d | 2.9%c | ||
| 4-MP | 4b′ | 92.9% | 42.7%d | 15.7%d | ||
| DMAP | 4c′ | 96.4% | 82.3%d | 68.0%d | ||
| MeImid | 4d′ | >99% | 65.5% | 46.2% | ||
| L5 | OH2 | 5′ | 65.0% | — | — | |
| py | 5a′ | 80.1% | 17.0% | <1% | ||
| 4-MP | 5b′ | 91.6% | 31.5% | 12.3% | ||
| DMAP | 5c′ | 98.4% | 76.3% | 57.5% | ||
| MeImid | 5d′ | 98.0% | 87.4% | 70.7% | ||
| L6 | OH2 | 6′ | 62.0% | — | — | |
| py | 6a′ | 29.0% | 23.9% | 27.8% | ||
| 4-MP | 6b′ | 38.8% | 54.7% | 38.1% | ||
| DMAP | 6c′ | 53.3% | 86.9% | 41.0% | ||
| MeImid | 6d′ | 53.7% | 91.4% | 46.4% | ||
While complexation provided insight into the rate of coordination of monodentate ligands to the compounds, stability studies provide critical modeling of the overall behavior of the complexes for in vivo applications. Amino acid challenge assays using histidine and cysteine were performed with 99mTc complexes (1a′–6d′) to determine their stability to substitution and transchelation. HPLC purified complexes were incubated with 1 mM cysteine or histidine in pH 7.4 phosphate buffer for 1 h at 37 °C followed by HPLC analysis (Table 3). In general, the 2 + 1 complexes formed with neutral bidentate ligands (L1–L3) had a higher stability towards both histidine and cysteine than 2 + 1 complexes formed with anionic bidentate ligands (L4–L6). Analogous to the formation results, increasing pKa's in functionalized pyridine ligands (DMAP > 4-MP ≫ py) showed increased complex stability across all of the 99mTc complexes.10 At 1 h, DMAP complexes with neutral ligands (L1–L3) had excellent stability (81–94%) compared the anionic ligands (L4–L6) (41–87%) that showed greater transchelation, particularly with cysteine.
A distinct difference was observed in the transchelation HPLC chromatograms between the 2 + 1 complexes of neutral and anionic bidentate ligands. In a typical transchelation assay, the formation of the amino acid complex (e.g., fac-[99mTcI(CO)3(histidine)]) is monitored, indicating loss of both ligands of the 2 + 1 system. While 2 + 1 complexes with anionic ligands formed the respective cysteine and histidine products upon degradation, 2 + 1 complexes bearing neutral bidentate ligands (L1–L3) did not, indicating incomplete decomplexation of the ligands. Further evaluation of the complexes (1′–3′) under challenge assay conditions revealed that the neutral bidentate ligands did not transchelate, suggesting the formation of a monodentate bound amino acid 2 + 1 complexes (e.g., fac-[99mTcI(CO)3(L)(histidine)]). Due to this coordination, stability of the py coordinated complexes 1a′ and 2a′ were not obtained due to the difficulty approximating the close overlap of the 2 + 1 product and the 2 + 1 species formed with cysteine.
Methyl imidazole was examined due to increased interest in tridentate imidazole-based ligands for radiopharmaceuticals and high reported stability of methyl imidazole 2 + 1 complexes in comparison to pyridines.15,30–32 The 2 + 1 99mTc complexes were prepared from the intermediate complexes 1′–6′ by reacting MeImid under the same conditions as the pyridine analogs followed by HPLC analysis. MeImid 2 + 1 complexes (1d′–6d′) demonstrated good to excellent (48–98%) radiochemical yields across the bidentate series (Fig. 5). The combination of neutral ligands, L2 and L3, with MeImid exhibited higher labeling yields over DMAP analogs despite its lower basicity (pKa = 7.2). Amino acid challenge studies with 2 + 1 99mTc complexes were also conducted with MeImid complexes (1d′–6d′) exhibiting slightly higher stability than analogous DMAP complexes for cysteine and histidine in nearly all cases. Also in agreement with pyridine analogs, the MeImid containing 2 + 1 complexes exhibited higher stability with neutral bidentate ligands (L1–L3) over anionic ligands (L4–L6).
 | ||
| Fig. 5 Radiochemical labeling yields of the reaction of with monodentate ligands DMAP (gray) and MeImid (blue) with fac-[99mTcI(CO)3(L)(OH2)]0/+ containing bidentate ligands (L1–L6) to yield the respective product fac-[99mTcI(CO)3(L)(DMAP/MeImid)]0/+. | ||
Conclusion
Using a 2 + 1 strategy, a series of neutral and anionic bidentate ligands based on aromatic nitrogen donors were explored with monodentate pyridine analogs to determine the behavior on fac-[MI(CO)3]+ complexes with Re and 99mTc. Distinct differences were observed in the formation and the stability of the respective complexes based on the overall charge, whether cationic or neutral, under the conditions examined. In general, the 99mTc cationic complexes (fac-[MI(CO)3(L1–3)(Y)]+) formed with neutral ligands exhibited lower synthetic yields, but higher stability. Whereas, neutral complexes (fac-[MI(CO)3(L4–6)(Y)]) constructed with anionic bidentate ligands typically had higher yields, but significantly lower stability. Both computational and experimental studies confirm stronger binding of the complexes formed with the bidentate neutral ligands. Both classes of bidentate ligands were also examined with functionalized pyridine analogs as a function of pKa. Each of the 2 + 1 complexes bidentate ligand displayed a clear trend for formation and stability based on the monodentate pyridines (DMAP > 4-MP > py). A comparable aromatic amine, MeImid, was also evaluated under the same conditions to directly compare to the functionalized monodentate pyridine series. While the pKa (7.2) of MeImid is greater than the pKa for 4-MP, it had excellent formation yields and high stability across the bidentate series similar to DMAP suggesting the importance of donor type on the stability. The combination of pKa effect, ideal charge, and ligand types will help to more effectively design 2 + 1 fac-[99mTcI(CO)3]+ complexes with improved behavior.Despite the differences in the bidentate ligands reported, these results were in agreement with our previous studies illustrating electron donation influencing the preparation and stability of the 99mTc complexes. These results provide insight into designing tridentate ligands using this strategy. Use of neutral tridentate ligands may increase stability over ones containing carboxylate functionalities. Additionally, functionalized pyridines or imidazoles are likely to enhance the overall stability of newly developed tridentate ligands. These modifications may provide complexes with greater in vivo and in vitro stability, while providing an approach to modify pharmacokinetic properties.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
This worked was funded in part by the DOE, Radiochemistry and Radiochemistry Instrumentation Program (#DE-FG02-08-ER64672) and Washington State University.Abbreviations
| pic | Picolinic acid |
| py | Pyridine |
| 4-MP | 4-Methoxypyridine |
| DMAP | 4-Dimethylaminopyridine |
| MeImid | Methylimidazole |
| bipy | 2,2′-Birpyridine |
| DPA | 2,2′-Dipicolylamine. |
Acknowledgements
Isolink® kits were graciously provided by Dr Mary Dyszlewski at Covidien. MS analyses were done at the University of Idaho Mass Spectrometry Core facility which is supported in part by Idaho INBRE (NIH/NIGMS Grant # P20GM103408).References
- R. Alberto, Eur. J. Nucl. Med. Mol. Imaging, 2003, 30, 1299–1302 CrossRef PubMed.
- R. Alberto, R. Schibli, U. Abram, A. Egli, F. F. Knapp and P. A. Schubiger, Radiochim. Acta, 1997, 79, 99–103 CrossRef CAS.
- R. Alberto, R. Schibli, A. P. Schubiger, U. Abram, H. J. Pietzsch and B. Johannsen, J. Am. Chem. Soc., 1999, 121, 6076–6077 CrossRef CAS.
- R. Alberto, R. Schibli, R. Waibel, U. Abram and A. P. Schubiger, Coord. Chem. Rev., 1999, 190–192, 901–919 CrossRef CAS.
- S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205–2218 CrossRef CAS PubMed.
- R. Schibli, R. La Bella, R. Alberto, E. Garcia-Garayoa, K. Ortner, U. Abram and P. A. Schubiger, Bioconjugate Chem., 2000, 11, 345–351 CrossRef CAS PubMed.
- R. Alberto, J. K. Pak, D. van Staveren, S. Mundwiler and P. Benny, Biopolymers, 2004, 76, 324–333 CrossRef CAS PubMed.
- S. Mundwiler, M. Kuendig, K. Ortner and R. Alberto, Dalton Trans., 2004, 1320–1328, 10.1039/b400220b.
- B. B. Kasten, X. Ma, H. Liu, T. R. Hayes, C. L. Barnes, S. Qi, K. Cheng, S. C. Bottorff, W. S. Slocumb, J. Wang, Z. Cheng and P. D. Benny, Bioconjugate Chem., 2014, 25, 579–592 CrossRef CAS PubMed.
- T. R. Hayes, P. A. Lyon, C. L. Barnes, S. Trabue and P. D. Benny, Inorg. Chem., 2015, 54, 1528–1534 CrossRef CAS PubMed.
- T. S. Pitchumony, L. Banevicius, N. Janzen, J. Zubieta and J. F. Valliant, Inorg. Chem., 2013, 52, 13521–13528 CrossRef CAS PubMed.
- R. Wang, J.-C. Xiao, B. Twamley and J. N. M. Shreeve, Org. Biomol. Chem., 2007, 5, 671–678 CAS.
- T. U. Connell, D. J. Hayne, U. Ackermann, H. J. Tochon-Danguy, J. M. White and P. S. Donnelly, J. Labelled Compd. Radiopharm., 2014, 57, 262–269 CrossRef CAS PubMed.
- T. L. Mindt and R. Schibli, J. Org. Chem., 2007, 72, 10247–10250 CrossRef CAS PubMed.
- A. Yazdani, N. Janzen, L. Banevicius, S. Czorny and J. F. Valliant, Inorg. Chem., 2015, 54, 1728–1736 CrossRef CAS PubMed.
- M. E. Viguri, J. Pérez and L. Riera, Chem. – Eur. J., 2014, 20, 5732–5740 CrossRef CAS PubMed.
- S. V. Kumar, W. K. C. Lo, H. J. L. Brooks, L. R. Hanton and J. D. Crowley, Aust. J. Chem., 2016, 69, 489–498 CrossRef CAS.
- T. R. Hayes, B. B. Kasten, C. L. Barnes and P. D. Benny, Dalton Trans., 2014, 43, 6998–7001 RSC.
- H. He, M. Lipowska, X. Xu, A. T. Taylor, M. Carlone and L. G. Marzilli, Inorg. Chem., 2005, 44, 5437–5446 CrossRef CAS PubMed.
- W.-W. Qin, C.-Y. Sang, L.-L. Zhang, W. Wei, H.-Z. Tian, H.-X. Liu, S.-W. Chen and L. Hui, Eur. J. Med. Chem., 2015, 95, 174–184 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS. Version 2.10, University of Göttingen, Göttingen, Germany, 2003 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. G. Sheldrick, SHELXL-97, Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. G. Sheldrick, SHELXL-2013, Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 2013 Search PubMed.
- X-SEED: L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- G. Lu, K. P. Maresca, S. M. Hillier, C. N. Zimmerman, W. C. Eckelman, J. L. Joyal and J. W. Babich, Bioorg. Med. Chem. Lett., 2013, 23, 1557–1563 CrossRef CAS PubMed.
- G. Lu, S. M. Hillier, K. P. Maresca, C. N. Zimmerman, W. C. Eckelman, J. L. Joyal and J. W. Babich, J. Med. Chem., 2013, 56, 510–520 CrossRef CAS PubMed.
- R. W. Simms, D. H. Kim, D. M. Weaver, C. Sundararajan, M. Blacker, K. A. Stephenson and J. F. Valliant, Chem. – Eur. J., 2012, 18, 6746–6749 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete X-ray structural information for 2–6 are available as a CIF file, speciation diagrams and additional characterization data of selected complexes as a PDF. CCDC 1487875–1487879 for complexes 1c, 2c, 3c, 5c, and 6c. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt04282a |
| This journal is © The Royal Society of Chemistry 2017 |