
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure investigations of group 13 organometallic carboxylates†
Iwona
Justyniak
a,
Daniel
Prochowicz
a,
Adam
Tulewicz
b,
Wojciech
Bury
bc,
Piotr
Goś
d and
Janusz
Lewiński
*ab
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
cFaculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, Wrocław, Poland
dDepartment of Inorganic and Analytical Chemistry, Faculty of Pharmacy, Medical University of Warsaw, Banacha 1, 02-097 Warsaw, Poland
First published on 6th December 2016
Abstract
The octet-compliant group 13 organometallics with highly polarized bonds in the metal coordination sphere exhibit a significant tendency to maximize their coordination number through the formation of adducts with a wide range of neutral donor ligands or by self-association to give aggregates containing tetrahedral and higher coordinated aluminium centres, and even in some cases molecular complexes equilibrate with ionic species of different coordination numbers of the metal centre. This work provides a comprehensive overview of the structural chemistry landscape of the group 13 carboxylates. Aside from a more systematic approach to the general structural chemistry of the title compounds, the structure investigations of [R2M(μ-O2CPh)]2-type benzoate complexes (where M = B, Al and Ga) and their Lewis acid–base adducts [(R2M)(μ-O2CPh)(py-Me)] are reported. DFT calculations were also performed to obtain a more in-depth understanding of both the changes in the bonding of group 13 organometallic carboxylate adducts with a pyridine ligand.
Introduction
Many efforts have been made in developing convenient, efficient, and useful transformations taking advantage of the electron deficiency caused by the Lewis acidity on the coordination centre of group 13 organometallics.1,2 This concerns not only compounds with a low three-coordinate metal centre2 and the inherent Lewis acidity of electron-deficient metal centres but also a vast number of organometallics obeying the octet rule, for which the relative Lewis acidity of a metal centre is essentially not obvious.1 The latter compounds with a four-coordinate metal centre may rearrange to lower-coordinated species, and the ability of an electron precise molecule with intramolecular constraints to undergo cage opening via heteroleptic bond cleavage to generate a Lewis acidic site was termed “latent Lewis acidity”.3 On the other side, countless four-coordinate group 13 complexes obeying the octet rule are stable in solution and exhibit a tendency to maximize their coordination number under certain conditions by either self-association to give aggregates containing higher coordinate aluminium centres or by the formation of Lewis acid–base adducts with donor ligands.1a,c We term this feature as “dormant Lewis acidity”. The relative magnitude of the dormant Lewis acidity is related to the relative Lewis acidity of electron-deficient metal centres in the electron-precise group 13 organometallics.An intriguing case of the dormant Lewis acidity is associated with the chemistry of dichloroaluminium4 and dichlorogallium5 carboxylates. These compounds react with Lewis bases to form a large structural variety of adducts ranging from molecular complexes to ionic species exhibiting different coordination numbers of the metal centres and various coordination modes of carboxylate ligands (Scheme 1). For example, upon dissolution of a dimeric dichloroaluminium carboxylate [Cl2Al(μ-O2CR′)]2 followed by the addition of 4-methylpyridine (py-Me) a six-coordinate Lewis acid–base adduct [Cl2Al(μ-O2CR′)(py-Me)2] with a chelating carboxylate ligand was formed, whereas the dimer [Cl2Al(μ-O2CR′)]2 equilibrated with an ion-pair [(RCO2)2Al(THF)2+][AlCl4−] in the presence of THF (Scheme 1a).4 Thus, we were specifically curious about the reactivity of related four-coordinate [R2M(μ-O2CR′)]2-type complexes (M = B,6 Al7 or Ga8) towards Lewis bases, i.e., the group 13 organometallic carboxylate complexes with reduced Lewis acidity metal centres when compared to their inorganic homologues. It is also worth noting that despite unceasing investigations on [R2M(μ-O2CR′)]2 complexes, there are only a few structurally well-defined examples to date as well as essentially a lack of literature data on their reactivity with Lewis bases. Moreover, our previous study on the group 13 organometallic derivatives of dicarboxylic acids, i.e., tetranuclear molecular complexes [(Me2M)2(μ-O2C)2-1,2-C6H4]2 (where M = Al or In), demonstrated that the dormant Lewis acidity opened a route to the formation of coordination polymers in the presence of Lewis bases (Scheme 1b).9
 | ||
| Scheme 1 Effect of the character of a Lewis base on the molecular structure of (a) a dichloroaluminium carboxylate and (b) an organoaluminium phthalate. | ||
In the course of our systematic studies on organometallic carboxylates,1a we report on the structural investigations of a family of the group 13 [Me2M(μ-O2CPh)]2-type benzoates (where M = B, Al and Ga) to obtain a more in-depth understanding of both the changes in the bonding of group 13 organometallic carboxylate adducts with a pyridine ligand and the mode of dormant Lewis acidity realized for these complexes upon moving from the tetrahedral to higher coordinated moieties.
Results and discussion
Synthesis and structural characterization of [R2M(μ-O2CR′)]2 complexes
The group 13 organometallic benzoates were synthesized using a previously reported procedure.7,8 The reaction of Et3B and Me3M (M = Al, Ga) with an equimolar amount of benzoic acid resulted in a quantitative formation of [R2M(μ-O2CPh)]2-type complexes (Scheme 2). The resulting compounds [Et2B(μ-O2CPh)]2 (1), [Me2Al(μ-O2CPh)]2 (2) and [Me2Ga(μ-O2CPh)]2 (3) were characterized spectroscopically, and the molecular structure of 1 and 2 was determined by single crystal X-ray diffraction studies (due to the low quality of crystals of 3, we were unable to obtain a proper data set to perform a reliable X-ray analysis). | ||
| Scheme 2 The formation of [R2M(μ-O2CPh)]2-type complexes. | ||
The 1H NMR spectra are relatively simple and consistent with the formulation (see the Experimental section). The 11B NMR spectrum of 1 consists of a single resonance at 7.68 ppm corresponding to a four-coordinate boron centre.10 The 27Al NMR spectrum of 2 shows a single resonance at 151 ppm, typical of the four-coordinate aluminium centre.11 The IR spectrum of 1 shows bands at 1682 cm−1 [νasymm(CO2)] and 1453 cm−1 [νsymm(CO2)] indicating the presence of bridging carboxylate units. Similar characteristic patterns indicating the presence of bridging carboxylate groups can be observed in the IR spectra of 2 and 3 (see the Experimental section). The molecular structures of 1 and 2 consist of centrosymmetric dimers of two R2M units bridged by two carboxylate groups, as is shown in Fig. 1 (selected bond lengths and angles for 1 and 2 are given in Table 1). The central eight-membered M2O4C2 ring adopts a chair-like conformation (for an extended discussion on the dependence of the puckering of the chair-like conformation in this type group 13 carboxylates upon the steric bulk of the substituents on both the metal and the carboxylate, see ref. 6a). The observed M–O bond lengths are within the range expected for this type of boron6 and aluminium7 carboxylates.
 | ||
| Fig. 1 (a) Molecular structure of 1. (b) View of the chair-like conformation of 1. (c) Molecular structure of 2. (d) View of the chair-like conformation of 2. Hydrogen atoms have been omitted for clarity. | ||
| 1 | 2 | ||||||
|---|---|---|---|---|---|---|---|
| B(1)–O(1) | 1.557(2) | C(1)–B(1)–O(1) | 104.85(10) | Al(1)–O(1) | 1.807(1) | C(1)–Al(1)–O(1) | 105.84(7) |
| B(1)–O(2) | 1.558(2) | C(1)–B(1)–O(2) | 104.92(10) | Al(1)–O(2) | 1.811(1) | C(1)–Al(1)–O(2) | 105.96(7) |
| B(1)–C(1) | 1.597(2) | B(1)–O(1)–C(5) | 130.29(10) | Al(1)–C(1) | 1.946(2) | Al(1)–O(1)–C(3) | 151.93(11) |
| B(1)–C(3) | 1.600(2) | C(3)–B(1)–O(2) | 109.43(10) | Al(1)–C(2) | 1.949(2) | O(1)–Al(1)–O(2) | 108.70(6) |
Reactions of [R2M(μ-O2CR′)]2 complexes with Lewis bases
The dimeric compounds 1, 2 and 3 were used as basic model complexes to probe their dormant Lewis acidity in the presence of 4-methylpyridine (py-Me) or THF. The addition of 1 equiv. of py-Me to 1 dissolved in CH2Cl2 resulted in the quantitative isolation of a mononuclear Lewis-acid–base adduct [Et2B(μ-O2CPh)(py-Me)] (4). A similar synthetic procedure involving Me3M (M = Al, Ga) resulted in the formation of analogue adducts [Me2M(μ-O2CPh)(py-Me)] (M = Al (5), Ga (6)) (Scheme 3). | ||
| Scheme 3 The formation of Lewis acid–base adducts between the group 13 benzoates [R2M(μ-O2CPh)]2 and 4-methylpyridine. | ||
Novel complexes 4, 5 and 6 were characterized spectroscopically and their molecular structures were determined by single crystal X-ray diffraction studies. The IR spectrum of 4 shows bands at 1672 cm−1 [νasymm(CO2)] and 1448 cm−1 [νsymm(CO2)] characteristic for a monodentate carboxylate unit and are redshifted compared to that in the parent complex 1. In the IR spectrum of 5 and 6 similar characteristic patterns of the carboxylate bands can be observed (see the Experimental section). In turn, after the addition of THF to [R2M(μ-O2CPh)]2 complexes at ambient temperature, the IR spectra of the resulting mixtures exhibited a pattern characteristic for the parent dimeric structures. This data indicates that THF does not induce any transformations of the central eight-membered M2O4C2 ring.
The solid-state structures of 4, 5 and 6 are shown in Fig. 2 and the selected bond lengths and angles are given in Table 2. The molecular structures of these compounds consist of discrete mononuclear Lewis acid–base adducts with the pyridine ligand and the carboxylate group acting as an unsymmetrical chelate ligand with a relatively weak M⋯O interaction on the fifth coordination site. Compound 4 crystallizes in the monoclinic P21/c space group. The primary coordination sphere of a diethylboron unit is completed by one carboxylate oxygen atom and the pyridine ligand (Fig. 2a). The C(1)–B–C(3) angle is 114.7(2)° and the pyridine ring is coplanar with the B–O1 bond (with the O(1)–B–N angle of 99.89(13)°; the O(1)–C(5)–O(2) angle of the unsymmetrically coordinated carboxylate group is equal to 125.1(2)°). The B–C bond lengths (Table 1) are comparable to that observed for the parent four-coordinate dimer 1, and the B–N distance is 1.649(2) Å. The B–O(1) bond is slightly longer than the corresponding B–O bonds observed for 1, and the B–O(2) distance of 2.847(2) Å is shorter than the sum of the B and O van der Waals radii (3.44 Å).12 In addition, the carboxylate oxygen O(1) is involved in an intramolecular C–Hpyr⋯O1 hydrogen bonding interaction with the pyridine ligand (with the Hpyr⋯O distance of 2.310 Å), whereas the carboxylate oxygen O(2) and the pyridine hydrogen forms intermolecular C–Hpyr⋯O(2) interactions (vide infra). Thus, the addition of py-Me to the four-coordinate organoboron benzoate 1 leads to the disruption of the central B2O4C2 ring and the formation of a unique mononuclear Lewis-acid–base adduct featuring a relatively weak B⋯O interaction on the fifth coordination site.13
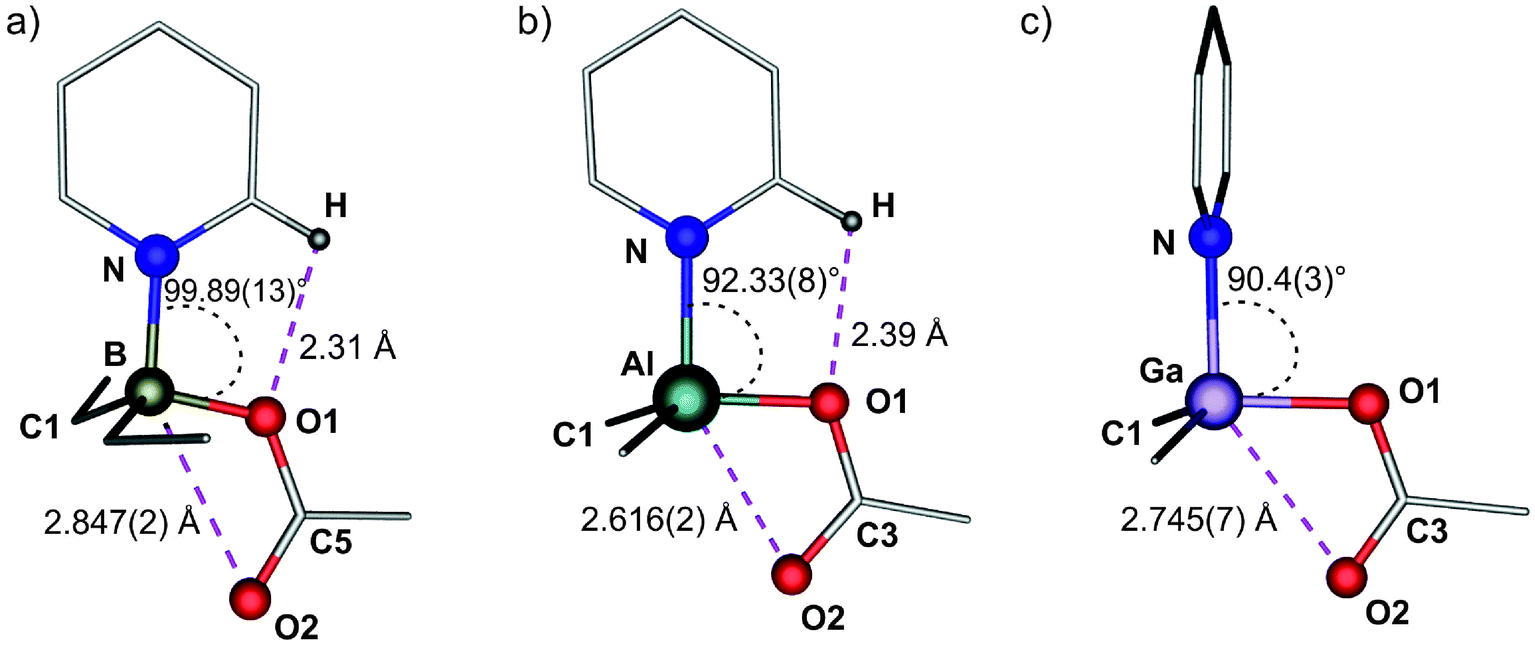 | ||
| Fig. 2 Molecular structures of 4–6 with the elected geometric parameters. | ||
| 4 | 5 | 6 | 4 | 5 | 6 | ||
|---|---|---|---|---|---|---|---|
| M–O(1) | 1.514(2) | 1.821(2) | 1.912(6) | C–M–C | 114.7(2) | 119.0(1) | 127.1(6) |
| M–O(2) | 2.847(2) | 2.616(2) | 2.748(6) | O(1)–M–C(1) | 113.76 | 116.15(10) | 110.5(3) |
| M–N(1) | 1.649(2) | 2.015(2) | 2.078(7) | O(1)–M–N(1) | 99.89(13) | 92.31(8) | 90.4(3) |
| M–C(1) | 1.613(3) | 1.956(3) | 1.959(7) | O(1)–C–O(2) | 125.1(2) | 120.8(2) | 123.1(2) |
| C(5)–O(1) | 1.313(2) | — | — | ||||
| C(3)–O(1) | — | 1.312(3) | 1.297(9) |
Compounds 5 and 6 crystallize in space groups P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and P21/c, respectively. In both structures, the central MMe2 moiety is chelated by the carboxylate group of the benzoate ligand, and its coordination sphere is completed by the pyridine ligand (Fig. 2b and c). The carboxylate group acts as an asymmetric chelate ligand (with the O(1)–C(3)–O(2) angle of 120.8(2)° and 123.1(2)°, respectively), and the coordinated pyridine ligand exhibits different spatial orientation in each complex; in 5 the pyridine ring is coplanar with the Al–O1 bond and involved in the intramolecular C–Hpyr⋯O (2.911(3) Å) with the Hpyr⋯O distance of 2.390 Å, and a perpendicular orientation of py-Me molecule in 6 is likely dictated by an intermolecular C–Hpyr⋯O interaction (vide infra). The C(1)–M–C(2) angle is 119.0(1)° and 127.1(6)° for 5 and 6, respectively. The observed M⋯O(2) distances 2.616(2) Å and 2.748(6) Å remain significantly below the sum of the van der Waals radii (3.36 Å and 3.39 Å for 5 and 6, respectively).12 Undoubtedly, the strength of the relatively weak M⋯O(2) interaction between the chelating carboxylate oxygen atom and the metal centre in [Me2M(μ-O2CPh)(py-Me)] adducts is controlled by the trans-influence of the axial substituents (this type of effect has previously been well documented for other group 13 hypercoordinate organometallic chelate complexes14) as well as intra- and intermolecular C–Hpyr⋯O hydrogen bond interactions.
and P21/c, respectively. In both structures, the central MMe2 moiety is chelated by the carboxylate group of the benzoate ligand, and its coordination sphere is completed by the pyridine ligand (Fig. 2b and c). The carboxylate group acts as an asymmetric chelate ligand (with the O(1)–C(3)–O(2) angle of 120.8(2)° and 123.1(2)°, respectively), and the coordinated pyridine ligand exhibits different spatial orientation in each complex; in 5 the pyridine ring is coplanar with the Al–O1 bond and involved in the intramolecular C–Hpyr⋯O (2.911(3) Å) with the Hpyr⋯O distance of 2.390 Å, and a perpendicular orientation of py-Me molecule in 6 is likely dictated by an intermolecular C–Hpyr⋯O interaction (vide infra). The C(1)–M–C(2) angle is 119.0(1)° and 127.1(6)° for 5 and 6, respectively. The observed M⋯O(2) distances 2.616(2) Å and 2.748(6) Å remain significantly below the sum of the van der Waals radii (3.36 Å and 3.39 Å for 5 and 6, respectively).12 Undoubtedly, the strength of the relatively weak M⋯O(2) interaction between the chelating carboxylate oxygen atom and the metal centre in [Me2M(μ-O2CPh)(py-Me)] adducts is controlled by the trans-influence of the axial substituents (this type of effect has previously been well documented for other group 13 hypercoordinate organometallic chelate complexes14) as well as intra- and intermolecular C–Hpyr⋯O hydrogen bond interactions.
A detailed inspection of the crystal structures of 4, 5 and 6 revealed that the intermolecular hydrogen bond systems play a substantial role in the molecular assembly of these complexes.15 As we noted earlier, for 4 and 5 the pyridine ligand, due to the appropriate orientation, is engaged in the intramolecular C–Hpyr⋯O interaction, and this type of interaction is not present for 6. Analysis of intermolecular contacts showed that adjacent monomeric moieties of 4 are held together in a 1D chain by C–Hpyr⋯O hydrogen bonds formed by the pyridine hydrogen and one carboxylate-oxygen (with the distances of Hpyr⋯O 2.50 Å and 2.31 Å), as illustrated in Fig. 3a. Further arrangement of molecules in the crystal structure is achieved by the complementary noncovalent C–Haliph⋯O interactions between the parallel H-bonded 1D chains leading to a 2D network (Fig. 3b). However, in the case of 5 the adjacent molecules are interconnected by a pair of intermolecular C–Har⋯O hydrogen bonds (C–H⋯O distance of 3.298(3) Å where the Har⋯O distance of 2.49 Å and the C–Har⋯O angle of 145.3°) leading to a H-bonded dimer, as depicted in Fig. 3c. In addition, the dimeric moieties are further linked by π⋯π interactions (with the shortest C⋯C distance of 3.519(3) Å) to form infinite layers parallel to the bc plane (Fig. 3d). The completely different supramolecular structure was found for the gallium analogue 6 where the monomeric moieties self-assemble via C–Har⋯O interactions to produce 1D zigzag chains (Fig. 3e). These H-bonded chains are further assembled by intermolecular C–Hpyr⋯O interactions into extended 2D layers featuring close packing of empty spaces in the crystal lattice (Fig. 3f). Thus, it seems likely that in the case of 6 the intermolecular C–Hpyr⋯O interaction is favored over the intramolecular C–Hpyr⋯O interaction and determines the spatial arrangement of the coordinated pyridine ligand.
 | ||
| Fig. 3 (a) View of the supramolecular arrangement in a 2D network of 4 along the b axis. (b) Crystal packing structure for 4 along the c axis. (c) View of the supramolecular arrangement of 5 in a H-bonded dimer. (d) Crystal packing structure for of 5 along the c axis. (e) View of the supramolecular arrangement in a 1D zig-zag chain of 6 along the b axis. (f) Crystal packing structure for 6 along the c axis. The purple lines represent cooperative noncovalent interactions. Hydrogen atoms are omitted, excluding those involved in the noncovalent interactions. | ||
Density functional theory calculations for [Me2M(μ-O2CPh)(py)] adducts
To provide a better basis for discussion concerning relative stability of the [R2M(μ-O2CPh)(py-Me)] adducts, density functional theory (DFT) calculations for [Me2M(μ-O2CPh)(py)] (where py = pyridine and M = B(4′), Al(5′) and Ga(6′)) as model systems were carried out. The optimized structures of 4′, 5′ and 6′ at the PBE0-D3/6-31G++(2d,2p) level of theory are presented in Fig. 4. An inspection of the geometry of 4′ reveals the semi-chelated Lewis acid–base adduct with a relatively weak B⋯O interaction on the fifth coordination site, which corresponds well to the molecular structure of 4. Substantial differences between the geometric parameters of 4′ and 4 are only observed for the value of the dihedral angle (27.5° for 4′ and 13.1° for 4), that is defined within the C–N–B–O atoms, and, as a result, for the value of the intramolecular C–Har⋯O(1) hydrogen bond distance.16 Conversely, a comparison between the structures 5 and 5′ or 6 and 6′ reveals more changes between the experimental and theoretically predicted geometries. For the aluminium and gallium complexes, the metal centres become five-coordinate due to the bidentate character of the carboxylate ligand in these complexes. The calculated theoretical values of Al–O(2) and Ga–O(2) bonds are 2.12 Å and 2.26 Å (Fig. 4), while the X-ray data point the distances around 2.62 Å and 2.75 Å, respectively. We attribute these discrepancies to the presence of different configurations of hydrogen bonds around O(2) atoms in both periodic networks of 5 and 6, which is depicted in Fig. 3. | ||
| Fig. 4 The optimized geometries of 4′, 5′ and 6′ obtained at the PBE0-D3/6-31G++(2d,2p) level of theory. | ||
In an effort to ascertain the magnitude of intramolecular C–Hpyr⋯O hydrogen bonding, we have estimated the energy barriers for the rotation of py around the M–N bonds for each of the three [(MMe2)(μ-O2CMe)(py)] carboxylates: B, Al and Ga (where 4′′ refers to [(BMe2)(μ-O2CMe)(py)]). Strikingly, the barrier of rotation obtained for 6′ represents the lowest value among all of the three studied systems (around +9.8 kJ mol−1 for 6′ compared with ca. +13.8 kJ mol−1 for 5′ and ca. +20.3 kJ mol−1 for 4′′ as shown in Fig. 6S†). The latter result may thus, at least partially, explain some discrepancies found between the experimental and theoretical values of the C–N–M–O dihedral angles. In addition, the calculated rotation barriers may serve as a quick assessment of the intramolecular C–Hpyr⋯O1 contact strength.
In the next step, the NBO analysis was provided for 4′′, 5′ and 6′ to get an insight into the nature of the M–N bonding. We have found that the B–N bond order has the highest value (0.55) among the studied M–N bonds and is more than twice higher compared with the values obtained for 5′ and 6′ (0.21 and 0.22, respectively). Note that the analysis of the M–N bond strengths shows a similar profile as the M–N bond energy was found to rise in the following order: 4′′ (−71.4 kJ mol−1) < 5′ (−67.8 kJ mol−1) < 6′ (−62.8 kJ mol−1). Finally, the NBO analysis was employed to estimate how the presence of the pyridine ligand affects the charge distribution in the Lewis acidity centre. We have found that for the boron derivative the charge decreases by 0.22 while for the remaining complexes this effect is almost negligible showing different character of the B–N bonding compared with the related Al–N and Ga–N connections.
Conclusions
In conclusion, aside from the discussion of the structural chemistry landscape of the group 13 carboxylates, we focused on the structure investigations of [R2M(μ-O2CPh)]2-type benzoate complexes (where M = B, Al and Ga) and their reactivity towards monodentate Lewis bases. We show for the first time that [R2M(μ-O2CPh)]2 complexes (where M = B, Al and Ga) readily form stable discrete mononuclear Lewis acid–base adducts with 4-methylpyridine, [R2M(μ-O2CPh)(py-Me)], in which the carboxylate group acts as an unsymmetrical chelate ligand with a relatively weak M⋯O interaction on the fifth coordination site. We also revealed that dichloroaluminium carboxylates [Cl2Al(μ-O2CR′)]2 and analogous [R2M(μ-O2CPh)]2-type complexes nicely serve as a model system to demonstrate their dormant Lewis acidity in the presence of a Lewis base. Finally, the provided qualitative and convenient rationalization for various chemical phenomena concerning the group 13 carboxylates incorporating highly polarized bonds can be extended to look at a vast number of organometallic compounds with a metal centre in octet-compliant structures and these investigations are in progress.Experimental section
Experimental details. General remarks
All manipulations were conducted under a nitrogen atmosphere by using standard Schlenk techniques. All reagents were purchased from commercial vendors. Solvents were dried and distilled prior to use. NMR spectra were recorded on a Varian Mercury 400 Spectrometer. IR spectra were recorded on an FTIR Bruker-Tensor II System. Elemental analyses were performed on Vario EL apparatus (Elementar Analysensysteme GmbH).Crystallographic data
The crystals of all complexes were selected under Paratone-N oil, mounted on the nylon loops and positioned in the cold stream on the diffractometer. The X-ray data for complexes 1 and 2 were collected at 100(2) K on a SuperNova Agilent diffractometer using MoKα radiation (λ = 0.71073 Å). The data were processed with CrysAlisPro.17 The X-ray data for complexes 4, 5, and 6 were collected at 100(2) K on a Nonius KappaCCD diffractometer18 using graphite monochromated MoKα radiation (λ = 0.71073 Å). The unit cell parameters were determined from ten frames, then refined on all data. The data were processed with DENZO and SCALEPACK (HKL2000 package).19 The structures were solved by direct methods and refined using the SHELXL 97.20 All non-hydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were introduced at geometrically idealized coordinates with a fixed isotropic displacement parameter equal to 1.5 (methyl groups) times the value of the equivalent isotropic displacement parameter of the parent carbon. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1477242 (1), 1477243 (2), 1477244 (4), 1477245 (5) and 1477246 (6). (no. 2), a = 7.4170(3) Å, b = 8.6964(8) Å, c = 9.1984(8) Å, α = 117.254(9)°, β = 109.439(6)°, γ = 94.798(6)°, U = 477.25(7) Å3, Z = 1, F(000) = 188, Dc = 1.240 g cm−3, μ(Mo-Kα) = 0.169 mm−1, θmax = 26.37°, 1948 unique reflections. Refinement converged at R1 = 0.0425, wR2 = 0.1097 for all data and 111 parameters (R2 = 0.0386, wR2 = 0.1060 for 1769 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.051. The residual electron density = +0.64/−0.18 e Å−3.
(no. 2), a = 7.2140(5) Å, b = 10.3400(6) Å, c = 10.9050(7) Å, α = 81.727(3)°, β = 77.086(2)°, γ = 71.565(3)°, U = 749.81(8) Å3, Z = 2, F(000) = 288, Dc = 1.202 g cm−3, μ(Mo-Kα) = 0.133 mm−1, θmax = 25.68°, 2741 unique reflections. Refinement converged at R1 = 0.0678, wR2 = 0.1620 for all data and 175 parameters (R1 = 0.0612, wR2 = 0.1573 for 2456 reflections with Io > 2σ(Io)). The goodness-of-fit on F2 was equal to 1.086. The residual electron density = +0.60/−0.57 e Å−3.
Computational details
The geometries of 4′, 5′ and 6′ were optimized (in a vacuum) with the empirical dispersion corrected PBE0-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21,22 functional in the 6-31G++(2p,2d) basis set. The nature of minima, obtained after the optimization process, was confirmed by the frequency analysis. A rigid PES scan of the [(MMe2)(μ-O2CMe)(py)] systems was run for the C–N–M–O dihedral angle. The NBO analysis was used to determine the origins of the bonding.23 All calculations were performed with the Gaussian24 suite of codes. Figures were prepared with the CYLview25 visualization software.
21,22 functional in the 6-31G++(2p,2d) basis set. The nature of minima, obtained after the optimization process, was confirmed by the frequency analysis. A rigid PES scan of the [(MMe2)(μ-O2CMe)(py)] systems was run for the C–N–M–O dihedral angle. The NBO analysis was used to determine the origins of the bonding.23 All calculations were performed with the Gaussian24 suite of codes. Figures were prepared with the CYLview25 visualization software.
Acknowledgements
The authors would like to acknowledge the National Science Centre within Grant Opus (UMO-2014/15/B/ST5/04492; I. J., D. P.) and Grant Maestro (DEC-2012/04/A/ST5/00595; J. L., A. T.) for financial support. The ICM University of Warsaw Computer Centre is acknowledged for facilities and computer time allocation within the grant G44-29.Notes and references
- (a) J. Lewiński and A. E. H. Wheatley, Top. Organomet. Chem., 2013, 41, 1–58 CrossRef; (b) D. G. Tuck, in Comprehensive Organometallic Chemistry, ed. G. Wilkinson, Pergamon Press, Oxford, 1982, vol. 1, pp. 683–723 Search PubMed; (c) S. Aldridge, A. J. Downs and D. L. Kays, in The group 13 metals aluminium, gallium, indium and thallium-chemical patterns and peculiarities, Wiley, Chichester, 2011, pp. 148–245 Search PubMed.
- (a) M. D. Healey, M. B. Power and A. R. Barron, Coord. Chem. Rev., 1994, 130, 63 CrossRef; (b) P. J. Brothers and P. P. Power, Adv. Organomet. Chem., 1996, 39, 1 CrossRef CAS; (c) S. Saito and H. Yamamoto, Chem. Commun., 1997, 1585 RSC.
- C. J. Harlan, S. G. Bott and A. R. Barron, J. Am. Chem. Soc., 1995, 117, 6465 CrossRef CAS.
- (a) Z. Florjańczyk, W. Bury, E. Zygadło-Monikowska, I. Justyniak, R. Balawender and J. Lewiński, Inorg. Chem., 2009, 48, 10892 CrossRef PubMed; (b) W. Bury, E. Chwojnowska, I. Justyniak, J. Lewiński, A. Affek, E. Zygadło-Monikowska, J. Bąk and Z. Florjańczyk, Inorg. Chem., 2012, 51, 737 CrossRef CAS PubMed; (c) Interestingly, the solution behaviour of dichloroaluminium carboxylates exhibits a number of common features with that observed for a dichloroaluminium acetylacetonate complex, see: J. Lewiński, S. Pasynkiewicz and J. Lipkowski, Inorg. Chim. Acta, 1990, 178, 113 CrossRef; J. Lewiński, S. Pasynkiewicz and J. Lipkowski, Inorg. Chim. Acta, 1988, 143, 39 Search PubMed; S. Pasynkiewicz and J. Lipkowski, Inorg. Chim. Acta, 1986, 122, 225 Search PubMed.
- S. A. Duraj, A. F. Hepp, R. Woloszynek, J. D. Protasiewicz, M. Dequeant and T. Ren, Inorg. Chim. Acta, 2011, 365, 54 CrossRef CAS.
- (a) M. Yalpani, R. Boese, K. Seevogel and R. Koster, J. Chem. Soc., Dalton Trans., 1993, 47 RSC; (b) A. Lang, H. Noth and M. Schmidt, Chem. Ber., 1995, 128, 751 CrossRef CAS.
- (a) A. Pietrzykowski, S. Pasynkiewicz and J. Popławska, Main Group Met. Chem., 1996, 18, 651 Search PubMed; (b) C. E. Bethley, C. L. Aitken, C. J. Harlan, Y. Koide, S. G. Bott and A. R. Barron, Organometallics, 1997, 16, 329 CrossRef CAS; (c) D. A. Dickie, M. C. Jennings, H. A. Jenkins and J. A. C. Clyburne, J. Organomet. Chem., 2004, 689, 2184 CrossRef; (d) L. Kalita, R. Pothiraja, V. Saraf, M. G. Walawalkar, R. J. Butcher and R. Murugavel, J. Organomet. Chem., 2011, 696, 3155 CrossRef CAS.
- (a) A. Keys, T. J. Barbarich, S. G. Bott and A. R. Barron, J. Chem. Soc., Dalton Trans., 2000, 577 RSC; (b) A. Keys, S. G. Bott and A. R. Barron, Polyhedron, 1998, 17, 3121 CrossRef CAS.
- I. Justyniak, W. Bury, D. Prochowicz, K. Wójcik, J. Zachara and J. Lewiński, Inorg. Chem., 2014, 53, 7270 CrossRef CAS PubMed.
- J. Lewiński and D. Kubicki, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, ed. J. Reedijk, Elsevier, 2015, pp. 691–703 Search PubMed.
- R. Benn, A. Rufińska, H. Lehmkuhl, E. Janssen and C. Kruger, Angew. Chem., Int. Ed. Engl., 1983, 22, 779 CrossRef.
- van der Waals radius for oxygen (1.52 Å) and boron (1.92 Å) atoms was taken from: M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806 CrossRef CAS PubMed.
- For examples of a hypervalent pentacoordinate organoboron compound with a constrained coordination environment, see: (a) M. Yamashita, Y. Yamamoto, K. Akiba and S. Nagase, Angew. Chem., Int. Ed., 2000, 39, 4055 CrossRef CAS; (b) M. Yamashita, Y. Yamamoto, K. Akiba, D. Hashizume, F. Iwasaki, N. Takagi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 4354 CrossRef CAS PubMed.
- (a) J. Lewiński, J. Zachara and I. Justyniak, Organometallics, 1997, 16, 4597 CrossRef; (b) J. Lewiński, J. Zachara and K. B. Starowieyski, J. Chem. Soc., Dalton Trans., 1997, 4217 RSC; (c) J. Lewiński, J. Zachara, K. B. Starowieyski, Z. Ochal, I. Justyniak, T. Kopeć, P. Stolarzewicz and M. Dranka, Organometallics, 2003, 22, 3773 CrossRef; (d) J. Lewiński, P. Goś, T. Kopeć, J. Lipkowski and R. Luboradzki, Inorg. Chem. Commun., 1999, 2, 374 CrossRef.
- For supramolecular architecture diversity arising from interplay between intermolecular secondary donor–acceptor interactions and hydrogen bonding in the group 13 organometallic chelate complexes, see: (a) J. Lewiński, J. Zachara, P. Stolarzewicz, M. Dranka, E. Kołodziejczyk, I. Justyniak and J. Lipkowski, New J. Chem., 2004, 28, 1320 RSC; (b) J. Lewiński, W. Bury and I. Justyniak, Eur. J. Inorg. Chem., 2005, 4490 CrossRef; (c) J. Lewiński, J. Zachara, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185 CrossRef.
- Note that comparison between both experimental and theoretical values of C–Har⋯O distances would be troublesome since precise determination of the hydrogen atom positions using X-Ray measurements is naturally affected by an experimental error.
- Agilent Technologies, CrysAlisPro, Version 1.171.35.21b Search PubMed.
- KappaCCD Software, Nonius B.V., Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, Methods Enzymol., 1997, 276, 307 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data for 1–6. CCDC 1477242–1477246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03958h |
| This journal is © The Royal Society of Chemistry 2017 |