
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilized lithium-ion battery anode performance by calcium-bridging of two dimensional siloxene layers†
Haruo
Imagawa
 * and
Hiroshi
Itahara
* and
Hiroshi
Itahara
Toyota Central R&D Labs., Inc., Nagakute, Aichi, 480-1192 Japan. E-mail: e1152@mosk.tytlabs.co.jp
First published on 2nd March 2017
Abstract
A Ca-bridged siloxene (Ca-siloxene) composed of two-dimensional siloxene planes with Ca bridging was synthesized via a solid state metathesis reaction using TaCl5 to extract Ca from CaSi2. Three different Ca-siloxenes synthesized at Cl2/Ca molar ratios of 0.25, 1.25 and 2.5 (CS0.25, CS1.25 and CS2.5, respectively) were fabricated and investigated as anode active materials for lithium-ion batteries. Both secondary and primary Ca-siloxene particles, which serve to increase the contact interfaces with conductive materials and to generate accessible sites for lithium ions, respectively, were found to become smaller and to have increased pore volumes as the Cl2/Ca molar ratio was increased. These Ca-siloxenes exhibited stable charge/discharge performance as anode materials, with 69–99% capacity retention after 50 charge/discharge cycles (compared with 36% retention for a conventional Kautsky-type siloxene). The charge capacity also increased with increases in the Cl2/Ca molar ratio, such that the CS2.5 showed the highest capacity after 50 charge/discharge cycles. This may reflect the formation of Si6Li6 rather than SiLi4.4 and suggests the maintenance of layered Si planes for large capacity retention after charge/discharge cycling. The increase of contact interfaces between acetylene black (as a conductive material) and Ca-siloxenes was found to effectively increase the lithium-ion capacity of Ca-siloxene during high rate charge/discharge cycling.
1. Introduction
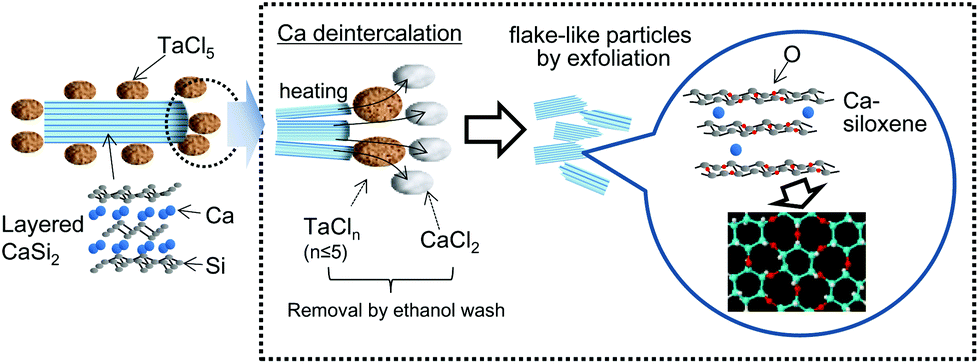
Certain anode materials have the potential to increase the capacity of lithium-ion batteries, so various graphite alternatives have been extensively studied for this purpose because of the limited theoretical capacity of graphite (372 mA h g−1).1,2 Silicon (Si)-based compounds, such as SiLi4.4, are among the possible alternatives and have shown high capacity values as anode materials (4200 mA h g−1).3 One drawback associated with Si-based materials is structural decomposition in conjunction with volume expansion during charge/discharge cycling, leading to a loss of conductivity between the Si-based materials and conductive cell components. As a result, effects of binder have been reported for the prevention of loss of conductivity. For example, polyacrylate binders have been applied for improvement in the electrochemical performance of Si-based materials.4 Furthermore, Si-based materials having different structures and compositions have also been investigated as a means of preventing this structural damage.5–14 For example, Si nanomaterials, including nanoparticles,5,6 nanowires7 and nanotubes,8 have been reported to exhibit effective capacity retention, especially in the presence of an active carbon matrix. Amorphous Si also shows promise in this regard,9,10 unlike crystalline Si, which is prone to high stresses and associated cracking. An interesting approach to fabricating porous Si materials has also been reported, with the aim of accommodating volume expansion during lithium insertion, using internal pores.11,12 Layered polysilane ((Si6H6)x) has also been studied because its two-dimensional (2D) layered structure can serve as an analogue for graphite and contains sites that are accessible for lithium ion insertion/extraction.13,14Layered siloxene is another promising material since it incorporates 2D Si-based planes15–18 that may be applicable to the lithium storage reaction. Si planes with Kautsky-type siloxene structures are composed of Si6O3H6 with Si6 rings connected through Si–O–Si bridges.17,18 Unfortunately, in this structure, there are no bridges connecting the layered Si planes other than van der Waals forces, which may lead to cleavage between planes during charge/discharge cycles.19 In a previous study, we reported a convenient method for the synthesis of calcium-bridged siloxene (Ca-siloxene) via a solid state reaction20 incorporating an ethanol wash19 (Scheme 1). This material shows potential for application as an anode active material in lithium-ion batteries.19 In this solid-state metathesis reaction, Ca is extracted from layered CaSi2 with the aid of TaCl5, acting as a Cl source to form CaCl2, thus maintaining a partial Ca-bridging between the Si layers, the extent of which is controlled by varying the TaCl5 concentration. This process forms Ca-siloxene owing to trace amounts of H2O in the wash ethanol and HCl derived from tantalum chloride species, similar to the siloxene synthesis mechanism.17,18 Because of the Ca-bridging, this Ca-siloxene has demonstrated improved cyclability as an anode active material compared with the Kautsky-type siloxene.19
 | ||
| Scheme 1 The solid state reaction employed for Ca-siloxene synthesis. | ||
Herein, we report the effects of the morphology and composition of Ca-siloxene on its performance as an anode for lithium-ion batteries. In this work, primary Ca-siloxene particles were reduced in size to generate flake-like particles, varying with the extent of Ca extraction in the synthesis step, using a technique based on our previous work.19 This may not only facilitate internal pore formation to accommodate volume expansion, but could also improve cell performance by producing accessible sites for lithium ion insertion/extraction into the Si planes during charge/discharge cycles. In order to focus on the intrinsic anode performance, no binders were added for the comparison of capacity. The effect of the amount of conducting materials on capacity increase is also discussed herein in order to assess the usefulness of Ca-siloxene as a high capacity anode active material.
2. Experimental
2.1 Synthesis of Ca-siloxenes19
Samples were synthesized based on Cl2/Ca molar ratios of 0.25, 0.5 and 2.5, and are denoted herein as CS0.25, CS1.25 and CS2.5, respectively. In these syntheses, CaSi2 (3.2, 1.6 and 0.32 mmol for CS0.25, CS1.25 and CS2.5, Rare Metallic) and TaCl5 (0.32 mmol, Wako Pure Chemical) were mixed together and packed in boron nitride crucibles. These mixtures were calcined at 215 °C for 5 h at a heating rate of 100 °C h−1, which was sufficient to prevent overshooting the desired temperature. The mixtures, still in the boron nitride crucibles, were subsequently stored within sealed stainless steel cells, each with an inner volume of 10 cm3. The products were washed with anhydrous ethanol in order to remove the by-product CaCl2 and excess TaCl5 or their derivatives, followed by drying under vacuum at 80 °C. All synthesis steps were performed under an Ar atmosphere. The Kautsky-type siloxene was synthesized for a reference sample as described in the literature.17,182.2 Characterization of Ca-siloxenes
Scanning transmission electron microscopy (STEM) performed with an energy dispersive X-ray spectrometer (EDX) (JEOL 2010) was used to analyze the microstructures of the Ca-siloxenes. The secondary particle sizes were evaluated using a Microtrac MT3300EX particle size distribution analyzer, while the pore volumes were calculated by applying the N2 adsorption method, employing a Quantachrome Autosorb-1. The crystalline structures of the samples were characterized by X-ray diffraction (XRD) using a Rigaku RINT-TTR (CuKα radiation, λ = 1.5418 Å, 50 kV, 300 mA). CaSi2 was used as the reference sample, corresponding to Cl2/Ca = nil.2.3 Electrochemical properties
The lithium ion capacities of the Ca-siloxenes over 50 charge/discharge cycles were assessed using a Hokuto Denko HJ1010 galvanostat at 25 °C. The capacity was calculated based on a per Ca-siloxene mass basis with the subtraction of that for acetylene black under the same experimental conditions from the measured capacity, in order to confirm the capacity for Ca-siloxene. In these trials, each Ca-siloxene was mixed with conductive acetylene black (HS100, 39 m2 g−1, Denka Co., Ltd) at a Ca-siloxene/acetylene black ratio of 70/30 by weight, and then pressed onto a Ni foam to obtain electrodes in order to assess the essential Ca-siloxene performance. Li foil was used as the counter electrode and a 1 M solution of LiPF6 in an ethylene carbonate/diethyl carbonate mixture (50/50 vol%) was employed as the electrolyte. A constant current of either 0.1 or 1 A g−1 Ca-siloxene was applied with a voltage window of 0.02 to 1.50 V vs. Li/Li+. Scanning electron microscopy (SEM) images of electrodes composed of CS2.5/acetylene black with 5 wt% PTFE were acquired to assess volume expansion after 50 charge/discharge cycles, using a Hitachi SU-3500.To determine the acetylene black proportion required to obtain optimal capacity and conductivity, CS2.5/acetylene black mixtures with mass proportions of 90/10, 80/20, 70/30, 50/50 and 30/70 were prepared, denoted as CS2.5-10, CS2.5-20 CS2.5-30, CS2.5-50 and CS2.5-70, respectively.
The electrical conductivity values of the CS2.5/acetylene black powder mixtures were measured using the two-probe method following uniaxial pressing at 10 MPa.
3. Results and discussion
3.1 Characterization of Ca-siloxene
The XRD spectra of the samples exhibit broad peaks derived from CaSi2 in the region from 25 to 40°, and these peaks clearly increase in intensity according to the increase in the Cl2/Ca ratio (Fig. S1†). This reflects the amorphous phase formation resulting from the fragmentation of layered CaSi2 by exfoliation, as has been reported in the literature.19 The presence of a Ca atom was confirmed in all Ca-siloxenes. The average Ca/Si molar ratios determined by EDX are 1.8/6, 0.8/6, and 0.3/6 for CS0.25, CS1.25, and CS2.5, respectively. The presence of Si–Ca bonding has been also confirmed by extended X-ray absorption fine structure spectra.19Such a phenomenon is often accompanied by small particle formation and larger pore volume. Therefore, STEM images were used to examine the detailed morphology of the Ca-siloxene particles, as shown in Fig. 1(A)–(D). The secondary particles of CS0.25 consist of agglomerated primary particles with sizes on the order of several hundred nm. These are similar in shape to the CaSi2 particles, although the secondary particles are evidently smaller than the CaSi2 particles. The morphology is seen to gradually change with greater Cl2/Ca molar ratios, due to the ongoing aggregation of smaller exfoliated primary particles, as seen in the case of the CS1.25 image in Fig. 1(B). Eventually, flake-like primary particles become dominant as in the CS2.5 (Fig. 1(C)), due to the exfoliation of the layered CaSi2. The magnified image of the CS2.5 around flake-like primary particles is shown in Fig. 1(E). The thickness of flake-like primary particles is almost 15–20 nm. This flake-like structure has been also confirmed by an N2 adsorption–desorption isotherm based on a typical H3-type hysteresis loop for a slit-like pore structure (Fig. S2†).19
 | ||
| Fig. 1 STEM images of (A) CS0.25, (B) CS1.25, (C) CS2.5 (D) CaSi2 and (E) CS2.5 (magnified image). | ||
The secondary particle sizes of the samples are shown in Fig. 2(A). The secondary particle size becomes smaller than that of the CaSi2 (for which the Cl2/Ca molar ratio is nil) as the Cl2/Ca molar ratio is increased. This effect is controlled by the amount of TaCl5 employed in the solid-state metathesis reaction, which in turn affects the exfoliation of the CaSi2.19Fig. 2(B) summarizes the pore volumes of the samples, which are seen to increase with increases in the Cl2/Ca molar ratio. This transition also results from the morphological changes due to the formation of flake-like particles in the solid state exfoliation reaction of the CaSi2. The peak of the pore size distribution is at approximately 4 nm (Fig. S2†), which may correspond to the size of the internal pores between the flake-like particles.19
 | ||
| Fig. 2 (A) Secondary particle sizes, and (B) pore volumes of samples as functions of the Cl2/Ca molar ratio. | ||
These data demonstrate that it is possible to control the formation of nano-sized structures with increased Si-planes and pore volumes as a synergetic effect by varying the synthesis conditions, particularly the Cl2/Ca molar ratio. This tailoring is capable of generating accessible sites for lithium ions within the Si planes by forming nano-sized flakes as primary particles. In addition, this technique produces increased contact interfaces between conductive particles through the formation of small secondary particles. Both these effects show promise in increasing the utility of this material as an anode in lithium-ion batteries, which will be discussed in the next section.
3.2 Charge/discharge cycling performance
The charge capacity values of the Ca-siloxene samples on a per Ca-siloxene mass basis over 50 charge/discharge cycles at a rate of 0.1 A g−1, providing an indication of the degree of delithiation of the specimens, are shown in Fig. 3. All capacity data were derived from the Ca-siloxene capacity obtained after subtraction of the acetylene black capacity. All Ca-siloxenes exhibit stable charge/discharge performance as an anode material, and do not show rapid capacity fading after the first few cycles. In contrast, c-Si and Kautsky-type siloxenes are known to undergo capacity fading within the initial ten charge/discharge cycles. | ||
| Fig. 3 Charge capacity per Ca-siloxene mass (i.e., the capacity of acetylene black was subtracted) over 50 charge/discharge cycles at a rate of 0.1 A g−1 (inset: capacity retention after 50 cycles). Legend: (a) CS0.25, (b) CS1.25, (c) CS2.5, (d) Kautsky-type siloxene, and (e) c-Si. | ||
The capacity of these Ca-siloxenes was evidently increased on increasing the degrees of Ca extraction owing to the exposed Si-planes and the larger pore volume that became available for lithiation/delithiation, such that the CS2.5 exhibited the highest capacity after 50 charge/discharge cycles. Interestingly, the capacity of the CS2.5 was higher than that of a Kautsky-type siloxene after 50 charge/discharge cycles, indicating the structural stability of the CS2.5 resulting from Ca-bridging between Si planes,19 even though the Kautsky-type siloxene and the Ca-siloxene have similar Si plane structures. The CS0.25 maintained almost all of its original capacity throughout 50 charge/discharge cycles, exhibiting 99% capacity retention (Fig. 3, inset), although it did not show the same capacity as the CS2.5, primarily due to the morphological differences in the primary particles of these two materials. For example, the CS0.25 retained a high concentration of Ca atoms, which prevented the formation of Si–Li bonds, and was chiefly composed of large particles with lower pore volumes, as shown in Fig. 2. These conditions do not favor the approach of lithium ions towards Si atoms. The capacity retention of the CS1.25 was not increased to the same extent as that of the CS2.5, likely because of the balance between the formation of flake-like particles and the extent of Ca extraction required for effective lithium ion storage in the Si planes in this material. Therefore, it can be said that the morphology of the Ca-siloxene affects the capacity largely because the available Si-plane formation with larger pore volumes provides more lithium ion accessible sites in the flake-like primary particles, as discussed in section 3.1.
Fig. 4(A) shows the charge/discharge capacities and coulombic efficiency of CS2.5 over 50 cycles at a rate of 0.1 A g−1. Some amount of irreversible capacity was observed in the first discharge step, probably due to solid electrolyte interface (SEI) formation on the CS2.5 surface, as often seen in the Si-based compounds.21,22 The voltage plateau was present at around 0.7 V, probably corresponding to the SEI formation (Fig. 4(B), charge/discharge curves). Therefore, such a formation also seems to contribute stable charge/discharge cycling in CS2.5. Although low coulombic efficiency at the first cycle was found in CS2.5 due to the SEI formation, a value more than 97% was maintained after the first several cycles.
 | ||
| Fig. 4 (A) Charge/discharge capacity per Ca-siloxene mass and coulombic efficiency, (B) charge discharge curves of CS2.5 over 50 cycles, (C) SEM images of the CS2.5 anode before cycling, and (D) after 50 cycles (inset: magnified CS2.5 particles). | ||
Fig. 4(C) and (D) present the SEM images of the CS2.5 anode before and after 50 charge/discharge cycles, respectively. No obvious cracks are seen between the Ca-siloxene particles and the acetylene black after cycling. Although it is possible that volume expansion of the Ca-siloxene samples could occur during charge/discharge cycles, since this phenomenon is often seen with Si-based materials, this volume change may be accommodated owing to the internal pores between the flake-like primary particles. Concerning the morphology change of the CS2.5 particles after charge/discharge cycling, pulverization or cracking of particles was not observed (Fig. 4(D) inset, magnified image of CS2.5 particles), and a Ca atom was also present. In the case of the Si particles, clear particle pulverization due to SiLi4.4 formation with huge volume expansion (>400%) during the charge/discharge cycling was reported to be one of the reasons for the Si anode deterioration.23 Therefore, the rapid decrease in the charge/discharge capacity is believed to have been prevented by successfully maintaining the contact interfaces between the Ca-siloxene and the conducting particles and also inside the particles, which may be one of the advantages over the Si anode.
Interestingly, the maximum capacity of the CS2.5 (524 mA h g−1 Ca-siloxene, corresponding to 692 mA h g−1 (Si mass basis)) was closer to the capacity of the Si6Li6 (approximately 957 mA h g−1) based on the Si planes of a polisilane,14 rather than the 4200 mA h g−1 value for SiLi4.4. The smaller CS2.5 capacity relative to that of the Si6Li6 may be due to Si–Ca bonding, which is not available for Li storage. This result is in agreement with a Ca-siloxene structure including Si planes, and also suggests that these Si planes are sustained during the charge/discharge cycling compared with c-Si and Kautsky-type siloxenes. Similar results were theoretically predicted in the previous report: single and double layer silicone (Si monolayer sheet) showed little irreversible structural change during the charge and discharge cycles.24
3.3 Effect of the conductive materials on capacity
The electrical conductivity of Ca-siloxene is known to be low: 6.4 × 10−4 S cm−1 for CS0.25 and 4.8 × 10−8 S cm−1 for CS2.5.19 Hence, the mixing of conductive materials with Ca-siloxene is an effective means of increasing the conductivity between the conductive material and the Ca-siloxene particles and also between the anode and the collector in a lithium-ion battery. This should lead to an increase in the charge/discharge capacity through optimization of the proportion of conductive material. For this reason, acetylene black, acting as a conductive material, was mixed with the CS2.5, which had demonstrated the best capacity among the Ca-siloxenes (see section 3.2). The charge capacity of CS2.5 with varying amounts of acetylene black is summarized in Fig. 5. The capacity was evidently increased with increases in the ratio of acetylene black to CS2.5, and the capacity was maintained in the CS2.5-30, -50 and -70 after 50 cycles. The CS2.5-70 also exhibited a constant charge capacity without any fading. In contrast, the CS2.5-20 and -30 showed a gradual decrease in capacity with prolonged cycling relative to the CS2.5-70, likely due to the slight ongoing decrease in the contact interface between the particles of CS2.5 and acetylene black during these charge/discharge cycles. The CS2.5-50 showed a slight decrease in capacity throughout 50 cycles, which is almost intermediate between CS2.5-30 and -70, just corresponding to the conductive material ratio. The CS2.5-10 had a rather small capacity, presumably because the contact interface between the CS2.5 and the acetylene black particles was insufficient to maintain electrical conductivity when employing the small amount of acetylene black in this material. | ||
| Fig. 5 Charge capacity per Ca-siloxene mass over 50 charge/discharge cycles at a rate of 0.1 A g−1 for samples with varying CS2.5/acetylene ratios. Legend: (a) CS2.5-10, (b) CS2.5-20, (c) CS2.5-30, (d) CS2.5-50 and (e) CS2.5-70. | ||
The conductivity of the CS2.5 pellets made with over 10 wt% acetylene black was almost 0.2 S cm−1, a value that is almost identical to that of pure acetylene black under the same experimental conditions. Therefore, a conductive pathway is evidently present in samples containing at least 10 wt% acetylene black, although intimate contact between the CS2.5 and the acetylene black particles may not occur until 20 wt% acetylene black is present, as seen in Fig. 5.
The formation of a contact interface between electrode active materials and conducting materials plays an important role, especially in a high charge/discharge rate, suggesting that the CS2.5-70 is expected to prevent capacity decrease under such a rate condition. Thus the effects of the contact interface between the Ca-siloxene and acetylene black particles on the current density of the CS2.5-30 and CS2.5-70 specimens are presented in Fig. 6. The capacity of the CS2.5-70 at a charge/discharge rate of 1 A g−1 remained at almost 90% of the value obtained at 0.1 A g−1. In contrast, the capacity of the CS2.5-30 at 1 A g−1 dropped to only 15% of the capacity measured at 0.1 A g−1. These data suggest that it is important to maintain the contact interface between the Ca-siloxene and the conducting material, especially during charge/discharge cycling at a high current density.
 | ||
| Fig. 6 Charge capacity per Ca-siloxene mass over 50 charge/discharge cycles at a rate of 0.1 A g−1 with sample/carbon mass ratios of (a) 70/30 and (b) 30/70, and at a rate of 1 A g−1 with ratios of (c) 70/30 and (d) 30/70. | ||
Ca-siloxene has the potential to generate a larger capacity than that of carbon, and exhibits a fast charge/discharge rate with structural stability during the charge and discharge process, as described above. Thus, an interface between the Ca-siloxene and the conducting material may be a key factor related to the future practical application of this material to lithium-ion batteries. Physical mixtures need a large amount of conducting material. Fabricating Ca-siloxene/carbon composite materials by adding organic precursors in the preparation step prior to calcination25,26 would be useful to reduce the amount of carbon. It is important to explore such Ca-siloxene coated with thin carbon layers in a further study.
4. Conclusions
Three different Ca-siloxenes, synthesized via a solid state metathesis reaction using TaCl5 and CaSi2, were assessed as anode active materials for lithium-ion batteries. Decreases in the primary and secondary particle sizes of the Ca-siloxene and increases in the pore volume were observed at higher Cl2/Ca molar ratios, leading to improved capacity after 50 charge/discharge cycles. In order to judge the maximum capacity and cyclability of Ca-siloxene, the mixing amount of acetylene black as a conductive material was changed. The formation of Si6Li6 and the structural stability of the Si planes during charge/discharge cycling were suggested. These findings reveal some of the fundamental properties of Ca-siloxenes with respect to their structure and composition, and should allow optimization of these compounds for future applications as anode materials in lithium-ion batteries.Acknowledgements
The authors thank Ms Akiko Ueki of Toyota central R&D Labs. for the assistance with TEM analysis.Notes and references
- H. Xia, W. Xiong, C. K. Lim, Q. Yao, Y. Wang and J. Xie, Nano Res., 2014, 7, 1797–1808 CrossRef CAS.
- H. Xia, Y. Wan, G. Yuan, Y. Fu and X. Wang, J. Power Sources, 2013, 241, 486–493 CrossRef CAS.
- H. Wu and Y. Cui, Nano Today, 2012, 7, 414–429 CrossRef CAS.
- S. Komaba, T. Ozeki, N. Yabuuchi and K. Shimomura, Electrochemistry, 2011, 79, 6–9 CrossRef CAS.
- S. Zeng, D. Liu, Y. Chen, J. Qian, Y. Cao, H. Yan and X. Ai, J. Mater. Chem. A, 2015, 3, 9938–9944 CAS.
- H. Kim, M. Seo, M.-H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS PubMed.
- M. R. Zamfir, H. T. Nguyen, E. Moyen, Y. H. Lee and D. Pribat, J. Mater. Chem. A, 2013, 1, 9566–9586 CAS.
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS PubMed.
- U. Kasavajjula, C. S. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS.
- M. T. McDowell, S. W. Lee, J. T. Harris, B. A. Korgel, C. Wang, W. D. Nix and Yi Cui, Nano Lett., 2013, 13, 758–764 CrossRef CAS PubMed.
- A. Magasinski, P. Dixon, B. Hertzberg, A. Kvit, J. Ayala and G. Yushin, Nat. Mater., 2010, 9, 353–358 CrossRef CAS PubMed.
- Z. Zhang, Y. Wang, W. Ren, Q. Tan, Y. Chen, H. Li, Z. Zhong and F. Su, Angew. Chem., Int. Ed., 2014, 53, 5165–5169 CAS.
- Y. Kumai, H. Kadoura, E. Sudo, M. Iwaki, H. Okamoto, Y. Sugiyama and H. Nakano, J. Mater. Chem., 2011, 21, 11941–11946 RSC.
- M. Ohashi, H. Nakano, T. Morishita, M. J. S. Spencer, Y. Ikemoto, C. Yogi and T. Ohta, Chem. Commun., 2014, 50, 9761–9764 RSC.
- A. Weiss, G. Beil and H. Meyer, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1979, 34, 25–30 Search PubMed.
- H. Nakano, M. Ishii and H. Nakamura, Chem. Commun., 2005, 2945–2947 RSC.
- S. Yamanaka, H. Matsu-ura and M. Ishikawa, Mater. Res. Bull., 1996, 31, 307–316 CrossRef CAS.
- H. Kautsky, W. Vogell and F. Oeters, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1955, 10, 597–598 Search PubMed.
- H. Imagawa, N. Takahashi, T. Nonaka, Y. Kato, K. Nishikawa and H. Itahara, J. Mater. Chem. A, 2015, 3, 9411–9414 CAS.
- S. Y. Oh, H. Imagawa and H. Itahara, J. Mater. Chem. A, 2014, 2, 12501–12506 CAS.
- S. Y. Oh, H. Imagawa and H. Itahara, Chem. – Asian J., 2014, 9, 3130–3135 CrossRef CAS PubMed.
- X. Wu, Z. Wang, L. Chen and X. Huang, Electrochem. Commun., 2003, 5, 935–939 CrossRef CAS.
- J. Wang, T. Xu, X. Huang, H. Li and T. Ma, RSC Adv., 2016, 6, 87778–87790 RSC.
- G. A. Tritsaris, E. Kaxiras, S. Meng and E. Wang, Nano Lett., 2013, 13, 2258–2263 CrossRef CAS PubMed.
- M. Thakur, S. L. Sinsabaugh, M. J. Isaacson, M. S. Wong and S. L. Biswal, Sci. Rep., 2012, 2, 795 Search PubMed.
- G. Liu, S. Xun, N. Vukmirovic, X. Song, P. Olalde-Velasco, H. Zheng, V. S. Battaglia, L. Wang and W. Yang, Adv. Mater., 2011, 23, 4679–4683 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction data and N2 adsorption–desorption isotherm of Ca-siloxenes. See DOI: 10.1039/c6dt03837a |
| This journal is © The Royal Society of Chemistry 2017 |