
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of ring size on the selective carboxylation of cycloalkene oxides†
Raiedhah
Alsaiari
,
Luke T.
Perrott
,
Ewa
Nowicka
,
Rebecca V.
Engel
,
Peter J.
Miedziak
,
Simon A.
Kondrat
,
Jennifer K.
Edwards
,
David J.
Willock
 * and
Graham J.
Hutchings
*
* and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk; willockdj@cardiff.ac.uk; Fax: (+44) (0)2920 874 030; Tel: +44 (0)29 2087 4779 Tel: +44 (0)29 2087 4059
First published on 7th March 2017
Abstract
Carbon dioxide utilisation technology can contribute to the reduction of atmospheric CO2 levels both through its sequestration from flue gases and indirectly by relieving pressure on conventional feedstocks in chemical manufacturing. A promising approach is to employ CO2 to produce valuable cyclic carbonates (CCs) in reaction with suitable epoxides. This also has the advantage that carbon dioxide replaces toxic and hazardous reactants such as phosgene. In earlier work we have investigated the synthesis of epoxides from cycloalkenes using supported gold and gold–palladium nanoparticles as catalysts and oxygen from air as the oxidant under solvent free conditions. A strong dependence of epoxide selectivity on ring size was observed with C5 < C6 < C7 ≪ C8. In this study we extend this work to the investigation of cycloaddition of CO2 to different cycloalkene oxides with the ultimate aim of designing a process in which both epoxidation of an alkene and incorporation of CO2 could be achieved in a single process. However, we have found the opposite trend for the selectivity to carbonates: smaller ring cycloalkene oxides giving the highest carbonate selectivities while large rings do not yield CCs at all. The product distributions suggest that an alternative ring opening of the epoxides to yield alcohols and ketones is preferred under all the experimental conditions explored for larger ring systems. Additionally, the mechanism of the CC synthesis using a quaternary ammonium salt and ZnBr2 as the catalyst system was investigated using DFT methods. The results of the calculations support the experimental findings.
Introduction
The main sources of anthropogenic CO2 emissions are from the use of fossil fuels such as natural gas, oil and coal. These emissions lead to an increase in the concentration of carbon dioxide in the atmosphere. Carbon dioxide utilisation technology can contribute to reducing the CO2 level by using carbon dioxide as a starting material and transforming it into valuable chemicals such as cyclic carbonates.1 There is a wide range of applications of cyclic carbonates; they are extensively used as intermediates in the synthesis of pharmaceuticals, raw materials for engineering plastics, aprotic polar solvents and electrolytes for lithium-ion batteries.2–6 The use of cyclic carbonates (CCs) is one of the most effective routes for carbon dioxide fixation, and is usually prepared via the cycloaddition of carbon dioxide with epoxides to form a five-membered ring.3,7–10 Carbon dioxide is a C1 building block with 100% atom economy and thus, a better alternative than toxic and hazardous reagents such as phosgene.11 Many studies have reported quaternary ammonium salts to be active for the cycloaddition of CO2 to epoxide.12–15 These are thought to act through nucleophilic attack by the anion of the salt to ring open the epoxide which then allows CO2 to react with the resulting oxyanion.16 The nucleophile is eliminated on ring closure to form the cyclic carbonate. This requirement of the nucleophile to both facilitate ring opening and act as a leaving group on cyclic carbonate ring closure has been used to explain why Br− is a good choice of nucleophile.17 DFT calculations using ethene as a model reagent and NEt4 Br have shown that the rate limiting step (calculated barrier 30.7 kcal mol−1) is the initial nucleophilic attack for ring opening of the epoxide.18 Experimentally NBu4 Br is commonly used as the longer alkane chains shield the cation positive centre more effectively and so increase the nucleophilicity of the anion. The use of co-catalysts such as Mg porphyrins19 and ionic liquids20 have been shown to reduce the barrier to ring opening to 13.5 and 12.0 kcal mol−1 with respect to reagents, respectively. The calculations show that these act to stabilise the forming oxyanion in the epoxide ring opening transition state. A similar role also been found to be important for amino acid co-catalysts.21 Alternative nucleophiles such as (4-dimethylamino)pyridine (DMAP) with (salen)Cr(III)22 and hydroxide anions with NBu4 (ref. 23) have also been investigated and shown to follow similar mechanisms. For systems containing redox active metals such as Co in α-Keggin-type structures calculations have also shown that ring opening of the epoxide can occur without the intervention of a nucleophilic agent through electron transfer and oxidation of the metal cente.24To realise cyclic carbonate production on a large scale, epoxides themselves will have to be synthesised by the oxidation of alkenes. The direct synthesis of cyclic carbonates from olefins, avoiding additional work-up procedures, would be an interesting and economically feasible route. Cyclic carbonates can be produced directly from CO2 and activated alkenes such as styrene using a supported Au catalyst to form the epoxide and zinc bromide with tetrabutylammonium as catalyst for the CO2 insertion step, in one-pot.25 However, to date the extension of this technology to the more challenging cycloalkenes has not been successful. In spite of the fact that the oxidative-carboxylation of olefins has been known since 1962,26 little attention was paid to this method compared with the route that employs epoxide as a starting material. The combination of two reactions in a one-pot process requires compatibility between the reaction conditions such as temperature, pressure and suitable catalysts for each reaction. In previous studies, we have investigated the epoxidation of cycloalkenes, using supported gold and gold–palladium nanoparticles as catalysts under solvent free conditions with air as the oxidant and small amounts of a radical initiator.27 A range of substrates was tested with varying ring size (C5 to C12). It was found that the selectivity to epoxide increases with increasing ring size of the cycloalkene. This has lead us to re-investigate the concept of coupling an epoxidation and carbonylation catalyst for these substrates. As a next step in that direction we now report the ring size dependence of the carbonylation stage of the reaction for a series of cyclic alkene oxides. We will show that cycloaddition of CO2 to various sized cyclic alkene oxides can be completed using a quaternary ammonium salt and ZnBr2 under green conditions, without solvent. We show that the reactivity of the cyclic alkene oxides is related to the ring size, with the smaller rings more reactive than larger ones at the same temperature (90 °C). Additionally, the selectivity to the cyclic carbonate also depends on the size of the cyclic alkene oxide ring; selectivity is reduced as the ring size of cycloalkene oxide is increased.
We combined our experimental data with DFT calculations for the example of cyclopentene oxide in order to obtain a complete understanding of the reaction route. This allows us to highlight the role of ZnBr4 as a co-catalyst and to discuss the origin of the side products observed.
Experimental
Chemicals
Tetrabutylammonium bromide Bu4NBr (98+%, Alfa Aesar), zinc bromide ZnBr2 (99.9%, Alfa Aesar), cyclopentene oxide C5H8O (98%, Aldrich) cyclohexene oxide C6H10O (98%, Aldrich), cyclooctene oxide C8H14O (99%, Aldrich), cyclododecene oxide C12H22O (95%, Aldrich) were used as received from the suppliers without further purification.Cycloalkene oxide reactions with CO2
Catalytic experiments were performed in a stainless steel autoclave reactor (50 cm3, Parr Instrument), fitted with an inner Teflon liner. Typically, a specified amount of the Bu4NBr and ZnBr2 catalysts and the epoxide (5 ml) were added into the autoclave. The reactor was purged 3 times with CO2 before being pressurized with CO2 to the desired pressure. The autoclave was heated to required temperature, under continuous stirring. At the end of the reaction the reactor was cooled to 5 °C in an ice water bath. Products from the reaction were analysed using gas chromatograph (Varian star 3400 CX) fitted with a DB-5 column and a flame ionization detector (FID). For compounds identification GC-MS was used (Walters GCT Premier GC fitter with a HP 6890 N Mass spectrometer) which allowed further product quantification for some selected experiments. The experiments were repeated up to three times and the standard deviation was 1%.DFT calculations
All calculations presented were carried out with the Gaussian09 software package.28 We use the B3LYP hybrid functional29–32 with a 6-31G(d,p) basis33–38 throughout. Transition state geometries were identified through a combination of constrained co-ordinate scans and standard transition state search optimisation. A transition state was accepted only when a single negative mode corresponding to the expected reaction co-ordinate was found in a frequency calculation.Results and discussion
Mechanism of cycloaddition of CO2 to cyclopentene oxide
Darensbourg and Holtcamp have suggested that the role of the ZnBr2 based catalyst in this carbonylation reaction is to act as a Lewis acid stabilising the epoxide oxygen atom during the key epoxide ring opening step. CO2 is then activated forming a linear carbonate intermediate before cyclization to a cyclic carbonate.39Scheme 1 shows an outline of the steps envisaged for the incorporation of CO2 into cyclopentene oxide (as an example epoxide) catalysed by the quaternary ammonium salt and ZnBr2 catalyst system.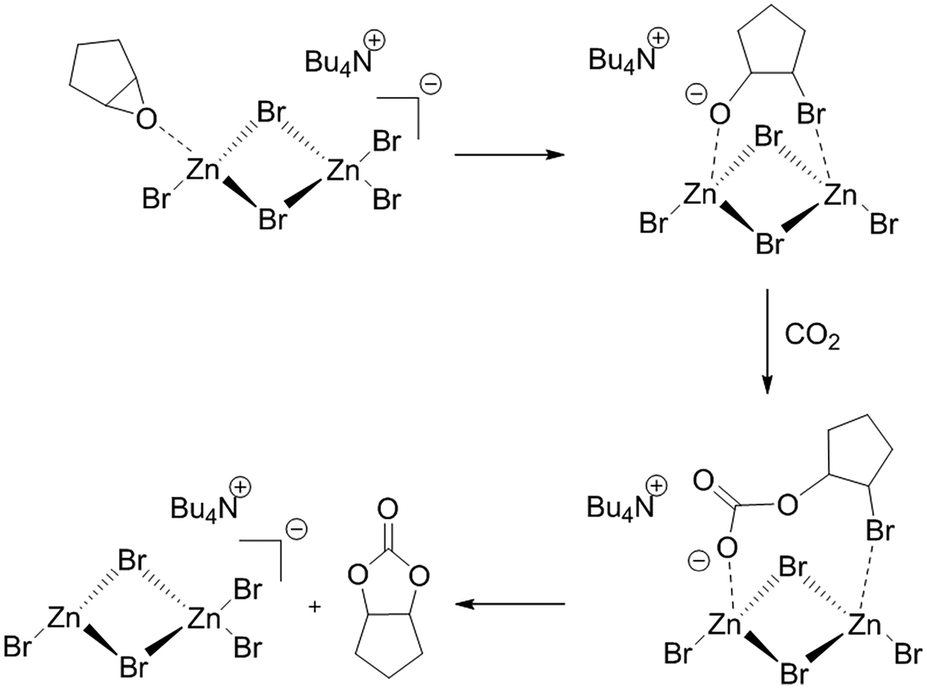 | ||
| Scheme 1 Reaction sequence for the incorporation of CO2 into cyclopentene oxide (as an example cyclic epoxide) to form a cyclic carbonate catalysed by Bu4NZn2Br5. | ||
This follows a similar mechanism to that proposed by North and Pasquale for the Al(Salen) catalysed incorporation of CO2 into alkene oxides.40 In the active form Br− from the quaternary ammonium salt is added to one of the Zn centres of a dimer to form a [Zn2Br5]− anion. The epoxide first co-ordinates to the [Zn2Br5]− species via a dative bond with a Zn Lewis acid centre. Transfer of Br− to one of the carbon atoms of the epoxide leads to epoxide ring opening with the resulting intermediate stabilised by interaction with both of the Zn Lewis acid sites. CO2 can now be incorporated to form a carbonate intermediate which ring closes to form the cyclic carbonate product. At the same time, Br− is returned to the Zn dimer to reform the catalyst.
We initially used DFT calculations to probe the detail of this reaction scheme. The catalyst is modelled by a [Zn2Br5]− cluster with a charge balancing [NEt4]+ cation, we have used tetraethylamnmonium rather than tetrabutylammonium as the counter ion for computational efficiency. We also considered cyclopentene oxide as a model reagent. The calculated potential energy for each step of the reaction is shown in Fig. 1. Chemical structure diagrams and atomic co-ordinates for each structure are available from the ESI.†
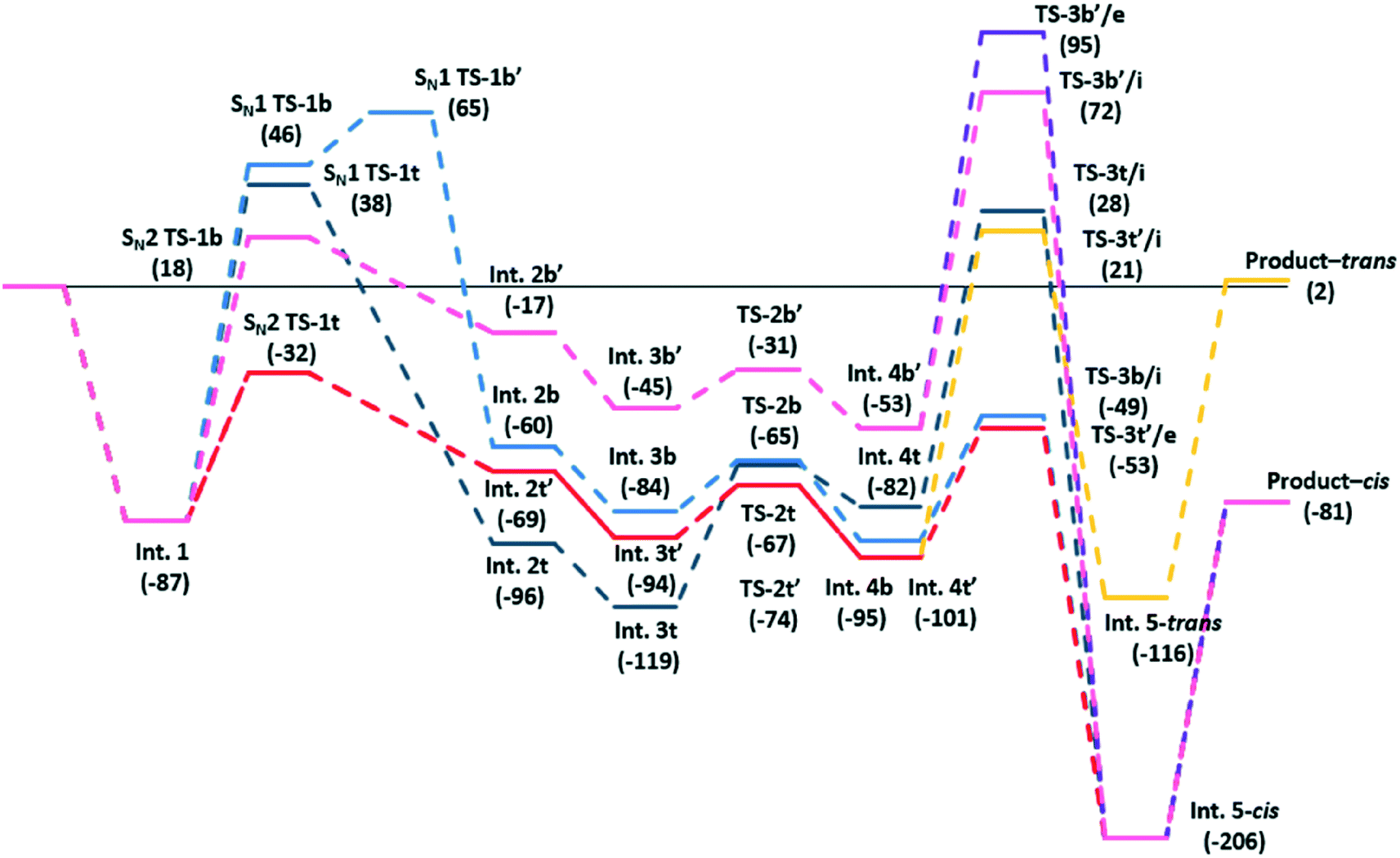 | ||
| Fig. 1 Calculated pathways for the addition of CO2 to pentene oxide to form a cyclic carbonate. Bracketed numbers are energies in kJ mol−1 relative to the starting reagents shown to the left. | ||
The initial co-ordination of the epoxide to a Zn centre is energetically favourable, resulting in a lowering of the system energy by some 87 kJ mol−1 (Int. 1). From this point, the ring opening of the epoxide group in isolation, following an SN1 pathway, was found to lead to high energy transition states. The exact energy depends on the orientation of the resulting carbocation with respect to the [Zn2Br5]− complex with energies ranging between 38 and 46 kJ mol−1 higher than the reference state of the isolated reagents (SN1, TS-1t and SN1, TS-1b) and 133 kJ mol−1 above the co-ordinated epoxide (Int. 1). Co-ordination of a Br− ion to the three co-ordinate C atom which is exposed on epoxide ring opening can then occur based on the addition of a terminal or a bridging Br− ion. The addition of “t” and “b” labels to structural labels in Fig. 1 has been used to distinguish these cases throughout. Addition of a bridging Br− along the SN1 route requires a second barrier at 65 kJ mol−1 (SN1, TS-1b′) to be surmounted, presumably due to breaking open of the Zn–Br–Zn bridge in this structure (Fig. 2b). The addition of a terminal Br− is found to be barrierless leading to Int. 2t. Alternatively, the epoxide ring opening from Int. 1 can follow pathways leading to transition states for a concerted SN2 type mechanism in which the new C–Br bond is formed as the epoxide ring opens and these were found to be considerably lower in energy. The bromination of the epoxide carbon atom can, again, takes place either using a bridging or terminal Br− ion.
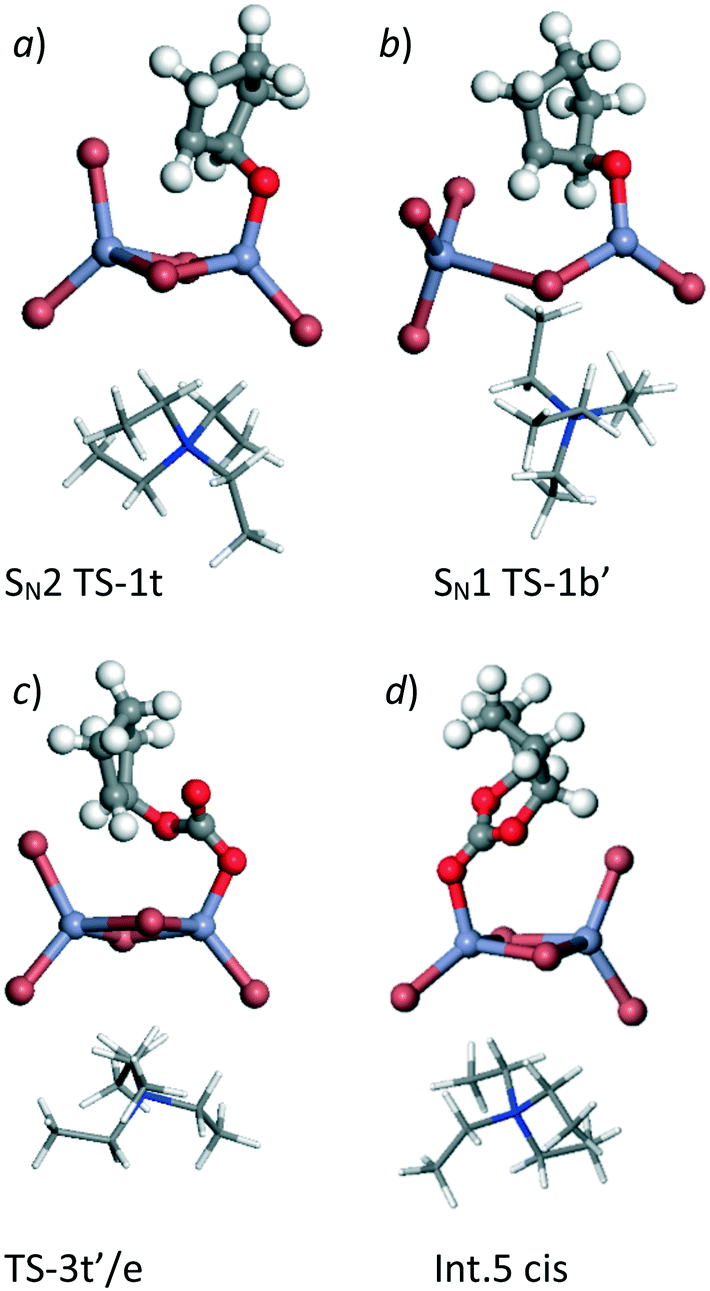 | ||
| Fig. 2 Calculated structures for example points on the PES shown in Fig. 1. a) Lowest energy ring opening TS, SN2 TS-1t′, b) second part of SN1 type epoxide ring opening mechanism SN1 TS-1b′, c) lowest calculated barrier for carbonate ring closure, TS-3t′/e and d) product carbonate co-ordinated to Lewis acid centre, Int.5 cis. The NEt4 cation is shown as wire all other atoms using ball and stick representations. Atom colours follow: C: grey, H: white, N: dark blue, O: red, Br: brown and Zn: light blue. | ||
Reaction using a terminal Br− ion was found to be preferred leading to a transition state in which the Br− nucleophile can align with the receiving C atom at the same time that the epoxide oxygen atom is stabilised on the Lewis acid Zn centre, as shown in Fig. 2a. This type of transition state geometry has previously been shown to be important using Mg porphyrin based catalysts for the ring opening of the epoxide,19 in this case the structure has an energy of −32 kJ mol−1 (SN2, TS-1t) relative to the initial reference state compared to 18 kJ mol−1 when a bridging Br− ion is used in the SN2 step (SN2, TS-1b). The intermediates formed from this point have both O and Br substituents on the cyclopentane ring stabilised by interaction with Zn centres. There are two possible chemical structures for the intermediate formed following the bromination of the epoxide, depending on the formation of the new C–Br bond from a bridging Br− ion (Int. 2b) or from a terminal Br− ion (Int. 2t). In Fig. 1 four distinct intermediates are shown as the detail of the configuration depends on the route taken to form the intermediate. Even so, both intermediates with a terminal Br− added are lower in energy than those forming a bridging Br–C bond. We have followed the CO2 insertion step for all four of the calculated structures. Initial co-ordination of CO2 leads to a lowering in energy for all four cases to give the Int. 3 structures and relatively low barriers are found for formation of a C–O bond between the C atom of the carbon dioxide and the oxygen atom originating from the epoxide (the TS-2 structure set). This addition of CO2 takes place via insertion of the molecule into the O–Zn bond with the simultaneous co-ordination of a CO2 oxygen atom to the Zn centre, so that the transition state is stabilised by interaction with the Lewis acid catalyst. The resulting structures contain a carbonate group which bridges between the cyclopentane and the Zn centre (Int. 4t and Int. 4b). Finally ring closure of the cyclic carbonate takes place and the Br− anion returns to the [ZrBr5]− cluster as a leaving group on the formation of the second C–O bond in the cyclic carbonate product. For this step, there is the additional choice of the oxygen atom that forms the second C–O bond in the cyclic carbonate product. From any of the Int. 4 structures we can envisage the O atom endocyclic in the metallocycle of the intermediate, i.e. co-ordinated to Zn, shifting to bond to the carbon atom. Alternatively a rotation of the C–O bond in the intermediate can bring the exo-cyclic oxygen atom into position for ring closure. This choice is recognised in Fig. 1 by the addition of /e or /i to the TS3 transition state structure labels for exo and endo-cyclic oxygen, respectively. The use of the exo-cyclic oxygen maintains co-ordination to the Zn Lewis acid centre in the transition state and, if this takes place from Int 4t the lowest energy transition state is found, at −53 kJ mol−1 (TS-3t′/e, Fig. 2c) relative to the starting point of the potential energy diagram in Fig. 1. The first endocyclic transition state is only marginally higher at −49 kJ mol−1 but this is arrived at following the SN1 route which has a high initial epoxide ring opening barrier (SN1 TS-1b′). Alternatives using the Zn co-ordinating oxygen for ring closure and/or the bridge site Br− ion lead to significantly higher barriers which would imply that these pathways are not kinetically relevant.
For this cyclopentene oxide example the low energy route through the potential energy diagram consists of an SN2 ring opening step with a terminal Br− ion and an exocyclic oxygen ring closure. This leads to a cis-arrangement of the cyclic carbonate and alkyl ring structures in the product. The trans-structure is only found for pathways containing high barriers to reaction.
Experimental results
Cyclic carbonate synthesis from epoxide and carbon dioxide has been an active field of research for a number of years.7,12–15 Here we investigate the effect of various reaction parameters such as reaction temperature and reaction time on the cycloaddition of carbon dioxide to cycloalkene oxides using a simple Zn based Lewis acid catalyst. These results will determine the most appropriate conditions for high yields of cyclic carbonate.Table 1 summarises the experimental results obtained in this study using cyclohexene oxide as substrate. In the absence of Bu4NBr and ZnBr2 no reaction takes place at 80 °C and only a 2% conversion with no observable cyclic carbonate product is seen at 125 °C (Table 1, entries 1 and 2), confirming that the catalyst is required to observe carbonate formation. When the catalyst is present a conversion of 51% with 85% selectivity is found at a temperature of 90 °C. Increasing the temperature to 125 °C leads to a conversion of 80%, while the selectivity decreases slightly to 83% (Table 1, entries 3 and 4). By allowing longer reaction times conversions up to 98% can be obtained after 16 h (Table 1, entries 5 and 6).
| Entry | Catalyst | Temp./°C | Time/h | Conv./% | CC sel./% |
|---|---|---|---|---|---|
| Reaction conditions: 5 ml (49.4 mmol) cyclohexene oxide, 0.4 g Bu4NBr, 0.16 g ZnBr2, 20 bar CO2. | |||||
| 1 | Blank | 80 | 4 | 0 | 0 |
| 2 | Blank | 125 | 4 | 2 | 0 |
| 3 | Bu4NBr + ZnBr2 | 90 | 4 | 51 | 85 |
| 4 | Bu4NBr + ZnBr2 | 125 | 4 | 80 | 83 |
| 5 | Bu4NBr + ZnBr2 | 125 | 8 | 89 | 81 |
| 6 | Bu4NBr + ZnBr2 | 125 | 16 | 98 | 80 |
However, at such long times, the selectivity to cyclic carbonate dropped to 80% and the formation of small amounts of other by-products were identified by GC-MS. These products were for example 1,2-cyclohexanediol and 2-cyclohexene-1-ol. The spectroscopic data can be found in the ESI.† These products were observed as impurities in the starting materials, however at significantly lower quantities than those observed in a reaction run.
The overall goal is the direct synthesis of cyclic carbonate from cycloalkenes. We envisage a two stage process in which the alkene is first oxidised to the epoxide and then the cyclic carbonate is formed by reaction with CO2. These initial reactions show how the cycloaddition step can be optimised for cyclohexene oxide. However, earlier work in our group has shown that epoxidation of cyclohexene is not very selective to cyclohexene oxide as the allylic ketone or alcohol is the preferred product using atmospheric oxygen with a radical initiator over a supported Au or Au–Pd catalyst.14 High epoxide selectivities were obtained when larger ring sizes, such as cyclooctene, were used as substrates in the oxidation step. Therefore, the cycloaddition of CO2 to cyclooctene oxide was investigated next.
Reaction of cyclooctene oxide
Following a similar approach to that employed for cyclohexene oxide we attempted to study the effects of reaction temperature and reaction time on the cycloaddition of CO2 to cyclooctene oxide (Table 2). In this case no cyclooctene carbonate was observed under any of the conditions used and no consumption of CO2 during the reactions was observed. The conversion of cyclooctene oxide requires higher temperatures when compared to cyclohexene oxide. After 4 h of reaction at 90 °C no conversion was observed (Table 2, entry 1). A conversion of 3% is seen at 130 °C after 4 h reaction time and at this temperature the reaction is practically completed after 48 h (Table 2, entries 2–4). Product identification by GC-MS indicated that cyclooctanone, cycloocatanol, and 1,2-cyclooctanediol are the major products (see ESI†). Furthermore, trace amounts of tributylamine, a decomposition product of the quaternary amine catalyst, were found, which may indicate reaction with the Bu4N+ cation has taken place. The availability of a reference standard for cyclooctanone allowed us to quantify this product routinely and under long reaction times selectivity to the cyclooctanone is high. The reaction proceeds at a greater rate at 150 °C (Table 2, entries 5–8). At both 130 °C and 150 °C the selectivity to the cyclooctanone product at the end of the reaction maximises at 61%. Lewis acids are known to catalyse the conversion of epoxides to ketones following the Meinwald rearrangement mechanism. DFT calculations suggest that this can take place through an H shift in intermediates such as Int 2 (Fig. 1).41 Our results suggest that, under the reaction conditions used here, this H shift is considerably faster than CO2 insertion for the cyclooctene oxide substrate.| Entry | Temp./°C | Time/h | Conv./% | Selectivity/% | ||
|---|---|---|---|---|---|---|
| Cyclooctene carbonate | Ketone | 1,2-Diol | ||||
| Reaction conditions: 5 g (39.6 mmol) cyclooctene oxide, 0.4 g Bu4NBr, 0.16 g ZnBr2, 20 bar CO2; (—) no GC-MS measurement made. Ketone = cyclooctanone, 1,2-diol = 1,2-cyclooctanediol. | ||||||
| 1 | 90 | 4 | 0 | 0 | 0 | — |
| 2 | 130 | 4 | 3 | 0 | 20 | 59 |
| 3 | 24 | 67 | 0 | 58 | 23 | |
| 4 | 48 | 99 | 0 | 61 | — | |
| 5 | 150 | 4 | 10 | 0 | 29 | — |
| 6 | 8 | 22 | 0 | 36 | — | |
| 7 | 16 | 79 | 0 | 62 | — | |
| 8 | 24 | 98 | 0 | 61 | — | |
Cycloaddition of CO2 to different cycloalkene oxides
So far, cycloaddition of CO2 to cyclohexene oxide and cyclooctene oxide have been studied and it was found that smaller ring size of the cycloalkene oxide convert easily to the corresponding cyclic carbonate. In order to confirm the observation of the effect of the ring size of cycloalkene oxide in this reaction, a range of different cycloalkene oxides have been studied (Table 3). As expected, smaller ring sizes such as cyclopentene oxide show high selectivity to cyclic carbonates (91%) at 90 °C after 4 h reaction time (Table 3, entry 1). Only small amounts of by-products were detected by GC-MS such as cyclopentanone, 1,2-cyclopentanediol and 2-bromocyclopentanol which may result from conversion of intermediates such as Int 2 in Fig. 1 to alcohols before the Br− anion is returned to the catalyst. For the spectroscopic data please see the ESI.† In the case of the larger ring sizes, cyclododecene oxide, even with increasing the reaction time from 8 to 16 h, showed no cyclic carbonate product (Table 3, entries 4 and 5). Cyclododecanone was found to be the main product for this reaction (selectivity greater than 95%). This is in line with the cyclooctene oxide results. In general, it can be said that with increasing ring size of the cycloalkene oxide the selectivity towards the corresponding cyclic carbonate is decreasing and with ring sizes larger than C8 there is no formation of the cyclic carbonate at all.| Entry | Substrate | Temp./°C | Time/h | Conv./% | CC sel./% |
|---|---|---|---|---|---|
| Reaction conditions: 5 ml (26.0–57.3 mmol) cycloalkene oxide, 0.4 g Bu4NBr, 0.16 g ZnBr2, 20 bar CO2. a 5 g (39.6 mmol). | |||||
| 1 | Cyclopentene oxide | 90 | 4 | 58 | 91 |
| 2 | Cyclohexene oxide | 125 | 4 | 80 | 83 |
| 3a | Cyclooctene oxide | 150 | 8 | 22 | 0 |
| 4 | Cyclododecene oxide | 150 | 8 | 13 | 0 |
| 5 | Cyclododecene oxide | 150 | 16 | 48 | 0 |
Conclusions
We have studied the mechanism of carboxylation of cyclopentene using DFT methods which indicate that the Zn Lewis acid centres in the Bu4NBr and ZnBr2 catalyst system play an important role in stabilising the intermediates and transition states. The rate-determining step appears to be the ring opening of the epoxide in this case with a concerted ring opening and bromination from a terminal Br− anion in the [ZnBr5]− catalyst. Insertion of CO2 into this intermediate is relatively straight forward with the second significant barrier for ring closure of the cyclic carbonate taking place via an exo-cyclic oxygen mechanism.Experimentally, the carboxylation process for a range of different cycloalkene oxides has been studied and it has been found that more forcing conditions are required for the larger ring sizes with the consequence that ketone and diol side products become dominant for the ring sizes of cyclooctene oxide and above. Reference to the DFT derived mechanism suggests that the ketone product can be formed by an H shift in the first intermediate formed after epoxide ring opening. The trend with ring size for selectivity to carbonate is the opposite to that observed in our earlier work on the oxidation of cycloalkenes in which epoxide selectivity increased with ring size.14 This suggests that the direct conversion of cycloalkenes to carbonates is probably only possible for cycloalkenes with a ring size below 8 using this Lewis acid catalyst. Although currently the temperatures required for the two steps (epoxidation: 30–70 °C,14 carboxylation: 90–125 °C, Table 3) make these stages of the reaction incompatible for a one-pot process. Progress may be made by identifying alternative Lewis acid catalysts for the carboxylation step or identifying reaction conditions which favour carboxylation over re-arrangement of the intermediate to form ketones.
Acknowledgements
RA thanks Najran University (Saudi Arabia) for financial support. The authors acknowledge funding from the European Research Council (“After the Goldrush” ERC-2011-AdG-291319). Additionally, we want to thank the EPSRC for funding the mass spectrometry facilities in Cardiff School of Chemistry (EP/L027240/1). Via our membership of the UK's HPC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202), this work made use of the facilities of ARCHER. Computing resource was also provided by Advanced Research Computing at Cardiff (ARCCA) and the HPC-Wales supercomputer facilities.Notes and references
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497–507 RSC.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312–1330 RSC.
- A. Khan, L. Yang, J. Xu, L. Y. Jin and Y. J. Zhang, Angew. Chem., Int. Ed., 2014, 53, 11257–11260 CrossRef CAS PubMed.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514–1539 RSC.
- J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- V. B. Saptal and B. M. Bhanage, Current Opinion in Green and Sustainable Chemistry, 2017, 3, 1–10 CrossRef.
- S.-I. Fujita, B. M. Bhanage, Y. Ikushima, M. Shirai, K. Torii and M. Arai, Catal. Lett., 2002, 79, 95–98 CrossRef CAS.
- J. Sun, S.-I. Fujita, F. Zhao and M. Arai, Appl. Catal., A, 2005, 287, 221–226 CrossRef CAS.
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143–148 CrossRef CAS.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- B. Song, L. Guo, R. Zhang, X. Zhao, H. Gan, C. Chen, J. Chen, W. Zhu and Z. Hou, J. CO2 Util., 2014, 6, 62–68 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- S. Foltran, R. Mereau and T. Tassaing, Catal. Sci. Technol., 2014, 4, 1585–1597 CAS.
- J.-Q. Wang, K. Dong, W.-G. Cheng, J. Sun and S.-J. Zhang, Catal. Sci. Technol., 2012, 2, 1480–1484 CAS.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J.-Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- A.-L. Girard, N. Simon, M. Zanatta, S. Marmitt, P. Goncalves and J. Dupont, Green Chem., 2014, 16, 2815–2825 RSC.
- K. R. Roshan, A. C. Kathalikkattil, J. Tharun, D. W. Kim, Y. S. Won and D. W. Park, Dalton Trans., 2014, 43, 2023–2031 RSC.
- D. Adhikari, S. T. Nguyen and M.-H. Baik, Chem. Commun., 2014, 50, 2676–2678 RSC.
- T. Ema, K. Fukuhara, T. Sakai, M. Ohbo, F.-Q. Bai and J.-Y. Hasegawa, Catal. Sci. Technol., 2015, 5, 2314–2321 CAS.
- F. Chen, X. Li, B. Wang, T. Xu, S.-L. Chen, P. Liu and C. Hu, Chem. – Eur. J., 2012, 18, 9870–9876 CrossRef CAS PubMed.
- J. Sun, S.-I. Fujita, F. Zhao, M. Hasegawa and M. Arai, J. Catal., 2005, 230, 398–405 CrossRef CAS.
- J. A. Verdol, U.S. Pat., 3025305, 1962 Search PubMed.
- H. Alshammari, P. J. Miedziak, D. W. Knight, D. J. Willock and G. J. Hutchings, Catal. Sci. Technol., 2013, 3, 1531–1539 CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Mol. Phys., 1974, 27, 209–214 CrossRef CAS.
- M. S. Gordon, Chem. Phys. Lett., 1980, 76, 163–168 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- D. J. Darensbourg and M. W. Holtcamp, Coord. Chem. Rev., 1996, 153, 155–174 CrossRef CAS.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed.
- J. M. A. Fraile, J. A. Mayoral and L. Salvatella, J. Org. Chem., 2014, 79, 5993–5999 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cy02448c |
| This journal is © The Royal Society of Chemistry 2017 |