
Molecular engineering of organic electroactive materials for redox flow batteries
Yu
Ding†
,
Changkun
Zhang†
,
Leyuan
Zhang†
,
Yangen
Zhou
and
Guihua
Yu
 *
*
Materials Science and Engineering Program and Department of Mechanical Engineering, The University of Texas at Austin, Austin, TX 78712, USA. E-mail: ghyu@austin.utexas.edu
First published on 18th October 2017
Abstract
With high scalability and independent control over energy and power, redox flow batteries (RFBs) stand out as an important large-scale energy storage system. However, the widespread application of conventional RFBs is limited by the uncompetitive performance, as well as the high cost and environmental concerns associated with the use of metal-based redox species. In consideration of advantageous features such as potentially low cost, vast molecular diversity, and highly tailorable properties, organic and organometallic molecules emerge as promising alternative electroactive species for building sustainable RFBs. This review presents a systematic molecular engineering scheme for designing these novel redox species. We provide detailed synthetic strategies for modifying the organic and organometallic redox species in terms of solubility, redox potential, and molecular size. Recent advances are then introduced covering the reaction mechanisms, specific functionalization methods, and electrochemical performances of redox species classified by their molecular structures. Finally, we conclude with an analysis of the current challenges and perspectives on future directions in this emerging research field.
 Yu Ding | Yu Ding received his BS degree in chemistry from Nankai University in 2012. He is currently pursuing a doctoral degree under the supervision of Prof. Guihua Yu in Materials Science and Engineering at the University of Texas at Austin. His research is mainly focused on molecular engineering and structural design of electroactive organic and organometallic materials for novel energy conversion and storage devices. |
 Changkun Zhang | Dr Changkun Zhang received his PhD degree from the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences in 2014. He currently works as a postdoc under the supervision of Prof. Guihua Yu in Mechanical Engineering at the University of Texas at Austin. His research focuses on next-generation redox flow batteries with high energy density. |
 Leyuan Zhang | Leyuan Zhang received his BS degree in materials science and engineering from Zhejiang University in 2012 and then Master's degree from the University of Chinese Academy of Sciences in 2015. He is currently pursuing a doctoral degree under the supervision of Prof. Guihua Yu in Mechanical Engineering at the University of Texas at Austin. His work is mainly focused on designing new organic electrolytes and membranes for redox flow batteries. |
 Guihua Yu | Guihua Yu is an Assistant Professor of Materials Science and Engineering and Mechanical Engineering at the University of Texas at Austin. He is an elected Fellow of the Royal Society of Chemistry (FRSC), Sloan Research Fellow and Camille Dreyfus Teacher-Scholar. He received his BS degree (2003) with the highest honor from the University of Science and Technology of China (USTC) and PhD (2009) in chemistry from Harvard University, followed by postdoctoral research (2012) at Stanford University. His research focused on rational design, synthesis, and fundamental investigation of nanostructured material systems for advanced energy and environmental technologies. |
1. Introduction
Securing a sustainable and environmentally friendly energy future is of utmost importance in the current development of human society. Among different types of energy resources, renewable sources, such as solar, wind, hydroelectric, wave, and tidal energy, provide a significant opportunity to alleviate the potential energy crisis stemming from the continuous increase in world energy consumption. According to World Bank data, fossil fuels, including natural gas, coal, and petroleum, still dominate the energy consumption and make up approximately 81% of the world's primary energy use.1 Although fossil fuels can be regenerated through natural processes, their formation takes millions of years. Moreover, the carbon dioxide emissions from burning the remaining fossil fuels will worsen environmental issues, global warming in particular. Efforts to decrease the dependence of human society on fossil fuels eagerly await improved renewable energy harvesting and storage technologies. Given the intermittent nature of renewable sources, electrochemical energy storage devices such as batteries may play a critical role in effective round-the-clock delivery of their generated electricity.2Redox flow batteries (RFBs), in which chemical energy is provided by electroactive materials dissolved in liquids and stored in outer tanks, show significant potential for applications in grid-scale energy storage.3 The working mechanism of an RFB is akin to both a fuel cell and a battery. A continuous circulation of electroactive materials is required to sustain the chemical reaction, like a fuel cell. Meanwhile, the reversible redox reaction enables charging and discharging within the same system, like a battery. Because of the special battery structure, RFBs feature a decoupled control on energy and power. The energy capacity is dictated by the amount of electrolyte stored, and the power is determined by the size of the current collectors and the number of cell stacks. More importantly, RFBs can be almost instantly recharged by replacing the discharged electrolytes, while simultaneously recovering the exhausted electroactive materials separately, which provides beneficial flexibility for multiple energy storage applications. The first RFB-like cell was conceptualized in 1884, when Charles Renard developed a zinc–chlorine battery to power his airship “La France”.4 Research on this technology was revived in the early 1970s, and the first practical RFB was invented by Lawrence Thaller at NASA for solar photovoltaic energy storage.3 While the investigation of RFBs has been sporadic for the past 40 years, the recent renaissance of renewable energy harvesting has prompted remarkable innovation in this field and given rise to numerous exciting advances, including tailored redox species, novel battery structures, and high-performance membranes.5 The majority of RFBs explored to date are still based on inorganic redox species, especially metal-based electroactive materials, and the redox reactions are primarily dependent on the change in the valence state of metal centers. In some designs, precious-metal electrocatalysts are required to enhance the electrochemical kinetics. Moreover, similar to solid state batteries, the development of metal-based RFBs has encountered a bottleneck.6,7 The electrochemical performance is restricted by the intrinsic properties of inorganic redox species, making it hard to further improve energy density. As a result, the widespread implementation of conventional RFBs is hindered by the high cost, material scarcity, environmental concerns, and uncompetitive performance metrics. To overcome the challenges of metal-based redox species, there is an urgent need to find inexpensive and green electroactive alternatives for sustainable high performance RFBs.
Recently, organic redox molecules have emerged as a promising class of electroactive materials for energy storage applications, including Li ion batteries, supercapacitors, and RFBs.8 In contrast to metal-based redox species, in which the redox reaction relies on the valence change of metal centers, the reaction of organic materials is commonly governed by the charge state of carbon, oxygen, sulfur and nitrogen atoms.9 In addition to the merits of potentially low cost, scalability, efficient biodegradability, and minimal environmental footprint, organic molecules possess highly tailorable chemical and physical properties. Molecular engineering provides excellent opportunities for constructing sustainable and green energy-storage devices. Over the last few decades, tremendous progress has been made to address the limiting factors of organic molecules in solid state batteries via functional group modification. Furthermore, the working mechanism and design principle have also been systematically summarized in a number of review papers.10–12 Meanwhile, the exploitation of organic molecules in RFBs has also gained increasing attention.13,14 In stark contrast to decreasing the solubility of organic electrode materials in solid state batteries, it is required to optimize the high solubility of organics in the electrolyte of RFBs. Up to now, achievements have been made in functionalizing organic molecules for RFB application, but a comprehensive summary about the rationale and underlying chemistry behind the molecular engineering method is absent. In this review, we provide background introduction, latest progress, challenges and future perspectives in this emerging research field. We are promoting the concept of using molecular engineering methods as a general problem solving approach in energy storage systems, especially in RFBs. Systematic, function-oriented synthetic strategies are presented by functional group category to show the highly tailorable properties of organics. Although the redox reactions of metal complexes are mostly based on the metal centers, they are included in this review due to their highly tunable properties via molecular functionalization.
In spite of increasing attention, the research on organic-based RFBs is still at its early stage, and a great number of challenges remain to be addressed. Therefore, we hope this review will promote more interest in this interdisciplinary research field. Finally, we aim to provide general design principles and guidelines to build high-performance sustainable RFBs (Fig. 1).
 | ||
| Fig. 1 Molecular engineering of redox species for sustainable RFBs. | ||
1.1 Aqueous RFBs
The first modern RFB, also the first aqueous RFB, was invented by Lawrence Thaller at the Lewis Research Center in Cleveland.3 After rational screening of redox species and systematic optimization of battery components, Thaller selected the iron–chromium system, in which Fe3+/Fe2+ and Cr3+/Cr2+ were dissolved in an acid medium as catholyte and anolyte, respectively. This device represents the most typical RFB type, in which both the cathode-active and anode-active materials are dissolved in water and separated by an ionic conductive membrane. Thaller also proposed several criteria regarding the system design, most of which are still valid today, including the following: (a) both the oxidized and reduced species should be soluble; (b) the solvent should remain inert in the working potential range; (c) the membrane should prevent crossover of redox species in both states of charge, and allow the diffusion of either positive or negative ions as charge carriers to maintain charge neutrality.15Up to now, aqueous RFBs have been the most appealing type of RFB because of their low cost, high safety, non-volatility, and facile processing. From Table 1, which compares typical aqueous and non-aqueous RFBs, it is obvious that the conductivity of an aqueous electrolyte is typically much higher than that of a non-aqueous electrolyte.3,16Table 2 provides a more comprehensive comparison.13,17,18 The characteristics of electrolytes are mainly governed by the intrinsic properties of both the salts and the solvents. Table 3 lists the basic physical properties of water and some conventional non-aqueous solvents.17,19–21 Because of its high dielectric constant, water can dissolve conventional acids, bases, and salts as either charge carriers or redox-active materials. Equally important is its low viscosity, which can lower pumping costs and increase ionic conductivity. The most widely used vanadium RFB commonly uses 3–4 M H2SO4 in water as the electrolyte, which offers conductivity as high as 700 mS cm−1.22 While the battery is operating, hydronium ions diffuse back and forth to balance the total charge. Since the charge carriers are positive, Nafion (sulfonated tetrafluoroethylene based fluoropolymer–copolymer) is adopted as a proton conducting separator. Due to the high ionic conductivity of Nafion (200 mS cm−1) together with the high conductivity of the aqueous electrolyte, aqueous RFBs can achieve power densities higher than 500 mW cm−2.3,23,24
| Aqueous RFB | Non-aqueous RFB | |
|---|---|---|
| Redox species | Metal ions | Metal–ligand complexes |
| Electrolyte | H2SO4 in H2O | TEABF4 in acetonitrile (ACN) |
| κ of electrolyte/(mS cm−1) | 500–700 | 40–55 |
| Potential window/(V vs. SHE) | 0–1.93 | −2.6–3.5 |
| Membrane | NAFION | AEM |
| Charge carrier of membrane | H+ | BF4− |
| σ of membrane/(mS cm−1) | 50–200 | 0.1–20 |
| Power density/(mW cm−2) | 10–600 | 1–10 |
| Electrolytes | Ionic conductivity/mS cm−1 | |
|---|---|---|
| 1 | 1 M H2SO4/H2O | 390 |
| 2 | 2 M H2SO4/H2O | 630 |
| 3 | 1 M NaOH/H2O | 170 |
| 4 | 1 M KOH/H2O | 210 |
| 5 | 2 M NaCl/H2O | 145 |
| 6 | 1 M TEABF4/ACN | 56 |
| 7 | 1.5 M TEABF4/ACN | 60 |
| 8 | 1 M TEATFSI/ACN | 46 |
| 9 | 1 M LiBF4/ACN | 16 |
| 10 | 1 M LiClO4/ACN | 34 |
| 11 | 1 M LiPF6/DMSO | 10.82 |
| 12 | 1 M LiPF6/PC | 7.33 |
| 13 | 1 M LiPF6/PC:EC | 6.07 |
| 14 | 1 M LiPF6/ACN | 50 |
| 15 | 1 M LiTFSI/ACN | 36 |
| 16 | 1 M LiTFSI/GDME | 8 |
| 17 | 1 M LiTFSI/DME | 14 |
| 18 | 1 M TEATFSI/DME | 17 |
| 19 | 1 M TEATFSI/DMSO | 9 |
| 20 | 1 M TEATFSI/THF | 11 |
| Solvent | Boiling point (°C) | Freezing point (°C) | Viscosity (mPa s) | Relative permittivity | Dipole moment (D) | E red (V vs. SHE) | E oxi (V vs. SHE) |
|---|---|---|---|---|---|---|---|
| Water (H2O) | 100 | 0 | 0.89 | 78.39 | 1.77 | 0 | 1.23 |
| Alcohols | |||||||
| Methanol | 64.5 | −97.7 | 0.551 | 32.7 | 1.71 | ||
| Ethanol | 78.3 | −114.5 | 1.083 | 24.6 | 1.74 | ||
| 1-Propanol | 97.2 | −126.2 | 1.943 | 20.5 | 1.71 | ||
| 2-Propanol | 82.2 | −88 | 2.044 | 19.9 | 1.66 | ||
| Ethers | |||||||
| Dimethoxymethane | 41 | −105 | 0.33 | 2.7 | 2.41 | ||
| Anisole | 153.8 | −37.5 | 0.895 | 4.33 | 1.245 | ||
| Tetrahydrofuran (THF) | 66 | −108.4 | 0.46 | 7.58 | 1.75 | −3.0 | 2.7 |
| 2-Methyltetrahydrofuran | 80 | −137 | 0.47 | 6.2 | 1.6 | ||
| 1,4-Dioxane | 101.3 | 11.8 | 1.087 | 2.21 | 0.45 | ||
| 1,3-Dioxolane (DOL) | 78 | −95 | 0.59 | 7.1 | 1.25 | ||
| 4-Methyl-1,3-dioxolane | 85 | −125 | 0.60 | 6.8 | 1.43 | ||
| 1,2-Dimethoxyethane (DME) | 84.5 | −69 | 0.455 | 7.2 | 1.71 | −3.5 | 2.1 |
| Diglyme | 159.8 | −64 | 0.989 | 1.97 | |||
| Ketone | |||||||
| Acetone | 56.1 | −94.7 | 0.303 | 20.6 | 2.7 | ||
| Acetylacetone | 138.3 | −23.2 | 0.694 | 25.7 | 2.78 | ||
| Nitriles | |||||||
| Acetonitrile (ACN) | 81.6 | −43.8 | 0.341 | 35.9 | 3.53 | −2.6 | 3.5 |
| Methoxyacetonitrile | 120 | −35 | 0.7 | 36 | −2.5 | 3.2 | |
| Propionitrile | 97.4 | −92.8 | 0.389 | 28.9 | 3.5 | ||
| Butyronitrile | 117.6 | −111.9 | 0.515 | 24.8 | 3.5 | ||
| Isobutyronitrile | 103.8 | −71.5 | 0.456 | 20.4 | 3.61 | ||
| Benzonitrile | 191.1 | −12.7 | 1.237 | 25.2 | 4.01 | ||
| 3-Methoxypropionitrile | 165 | −57 | 1.1 | 36 | −2.5 | 3.3 | |
| Amines | |||||||
| Ethylenediamine | 116.9 | 11.3 | 1.54 | 12.9 | 1.9 | ||
| Pyridine | 115.3 | −41.6 | 0.884 | 12.9 | 2.37 | ||
| Amides | |||||||
| Formamide | 210.5 | 2.5 | 3.3 | 111 | 3.37 | ||
| N-Methylacetamide | 206 | 30.5 | 3.65 | 191.3 | 4.27 | ||
| N,N-Dimethylformamide (DMF) | 153 | −60.4 | 0.802 | 36.7 | 3.24 | −2.8 | 1.8 |
| N,N-Dimethylacetamide (DMA) | 166.1 | −20 | 0.927 | 37.8 | 3.79 | ||
| N-Methyl-2-pyrrolidinone (NMP) | 202 | −24.4 | 1.67 | 32.2 | 4.09 | ||
| Carbonates | |||||||
| Propylene carbonate (PC) | 241.7 | −54.5 | 2.53 | 64.92 | 4.94 | −2.8 | 3.8 |
| Ethylene carbonate (EC) | 248.2 | 36.4 | 1.9 | 89.8 | 4.9 | ||
| γ-Butyrolactone (γ-BL) | 204 | −43.4 | 1.73 | 39.1 | 4.12 | −2.8 | 5.4 |
| γ-Valerolactone (γ-VL) | 208 | −31 | 2.0 | 34 | 4.29 | −2.8 | 5.4 |
| Dimethyl carbonate (DMC) | 91 | 4.6 | 0.59 | 3.107 | 0.76 | ||
| Diethyl carbonate (DEC) | 126 | −74.3 | 0.75 | 2.805 | 0.96 | ||
| Ethyl methyl carbonate (EMC) | 110 | −53 | 0.65 | 2.958 | 0.89 | ||
| 1,2-Butylene carbonate | 240 | −53 | 3.2 | 53 | −2.8 | 4.4 | |
| Others | |||||||
| Hexane | 68.7 | −95.3 | 0.294 | 1.88 | 0.085 | ||
| Benzene | 80.1 | 5.5 | 0.603 | 2.27 | 0 | ||
| Toluene | 110.6 | −95 | 0.553 | 2.38 | 0.31 | ||
| Nitromethane | 101.2 | −28.6 | 0.614 | 36.7 | 3.17 | −1.0 | 2.9 |
| Nitrobenzene | 210.8 | 5.76 | 1.62 | 34.8 | 4.00 | ||
| 1,2-Dichloroethane | 83.5 | −35.7 | 0.73 | 10.37 | 1.86 | −2.1 | 2 |
| Dimethyl sulfoxide (DMSO) | 189 | 18.5 | 1.99 | 46.5 | 4.06 | −2.7 | 1.7 |
| Ethyl acetate | 77.1 | −83.6 | 0.426 | 6.02 | 1.82 | ||
| Nitroethane | 115 | −90 | 0.68 | 28 | −1.1 | 3.2 | |
Thus far, several types of aqueous RFBs have been commercially developed, but technical and economic challenges have prevented their world-wide implementation.25 Moreover, the electrochemical performance of an aqueous flow system is restricted by the intrinsic properties of water, such as the narrow potential window and limited operating temperature range, as shown in Table 3. When considering organic-based redox species, incompatibility between hydrophobic organic molecules typically requires logical molecular engineering to meet performance metrics. In this regard, non-aqueous solvents provide much more freedom for rational screening and selection given the diverse solvent alternatives.16
1.2 Non-aqueous RFBs
Non-aqueous RFBs have the potential to achieve much higher energy density than aqueous ones because of their significantly wider potential windows, not to mention the extensive options for both cathode and anode-active materials. The first non-aqueous RFB can be traced back to 1988, when Matsuda and co-workers utilized a ruthenium complex [Ru(bpy)3](BF4)2 in ACN as an active material.26 The proof-of-concept design demonstrated an open circuit voltage of 2.6 V obtained at a discharge current of 5 mA cm−2. Since then, most reported non-aqueous RFBs have been anion-exchange systems with metal–ligand complexes as electroactive materials.16 Typical parameters of such a non-aqueous RFB are given in Table 1. Following the work by Matsuda, TEABF4 salt in ACN is still a commonly used electrolyte with conductivity between 40 and 55 mS cm−1, which is one order of magnitude smaller than those of aqueous electrolytes. Moreover, anions usually function as charge carriers during the working process of non-aqueous RFBs, necessitating the use of anion exchange membranes as separators. In contrast to cation exchange membranes that rely on sulfonate groups anchored on tetrafluoroethylene backbones to conduct protons, anion exchange membranes are commonly based on quaternary ammonium groups and hydrocarbon polymer backbones, and they exhibit much smaller ionic conductivity.27Table 2 shows the comparison of electrolyte conductivity between aqueous and non-aqueous RFBs in detail. It is noteworthy that the ionic conductivity of non-aqueous electrolytes is far less than those of aqueous electrolytes. Given the two factors mentioned above, the power densities of non-aqueous RFBs are eclipsed by those of aqueous RFBs, as indicated in Table 1. To address this concern, future attention can be geared towards developing advanced membranes and electrolytes to boost the ion transport. Meanwhile, a 3D porous current collector with a high surface area is beneficial to promote the electrochemical kinetics. Table 3 gives the physical properties of alternative solvents for RFBs. Rational screening of organic solvents should prioritize a wide operating temperature range and potential window to enable a high-voltage RFB applicable in harsh environments. Meanwhile, organic solvents with low viscosity are propitious to higher ionic conductivity of electrolytes as guided by Stokes’ law.20 ACN is widely utilized as the solvent for non-aqueous RFBs owing to its lower viscosity than water. Another important parameter in selecting organic solvents is relative permittivity, which determines both the solubility and dissociation of supporting salts. Generally, higher relative permittivity in organic solvents can endow high-concentration organic electrolytes with higher ionic conductivity. In light of the elevated output voltage of non-aqueous systems, such designs coupled with suitable separators are anticipated to reach power densities comparable to those of aqueous RFBs. In some novel RFBs with hybrid electrolytes, particularly those using a Li metal anode, solid state electrolyte membranes serve as separators, which effectively prevent the crossover of redox species and withstand the dendrites of alkaline metals.28 Though the ionic conductivity cannot compete with Nafion in aqueous electrolytes, further improvement of both the conductivity and stability of solid state electrolytes could advance this promising replacement for current polymer-based separators.29Non-aqueous RFBs have rarely been commercialized mainly due to the high cost of raw materials and complex packaging process. However, the 3-factor increase in potential window indicates great promise for such emerging energy systems. Fig. 2 shows the allowable chemical cost factor versus open circuit voltage of both aqueous and non-aqueous RFBs.30 This cost factor is based on the positive and negative electroactive materials, salt, solvent, and storage vessel costs on a mass of active material basis. The lines correspond to a constant capital cost of $120 kW h. Compared with the larger lighter shaded triangles, the dark shaded triangles are regarded to have a higher chance to realize. It is noted that the chemical cost in aqueous RFBs is far less than non-aqueous systems even if the output voltage is not comparable. According to cost estimates, 4–5 M electroactive materials in non-aqueous solvents can meet the cost requirements for energy storage and compete with 1–2 M aqueous electrolytes.30 The structural diversity and immense alterability of organic and organometallic redox species, together with an abundance of non-aqueous solvent alternatives with wide potential windows, provide a pathway for achieving such a research goal.
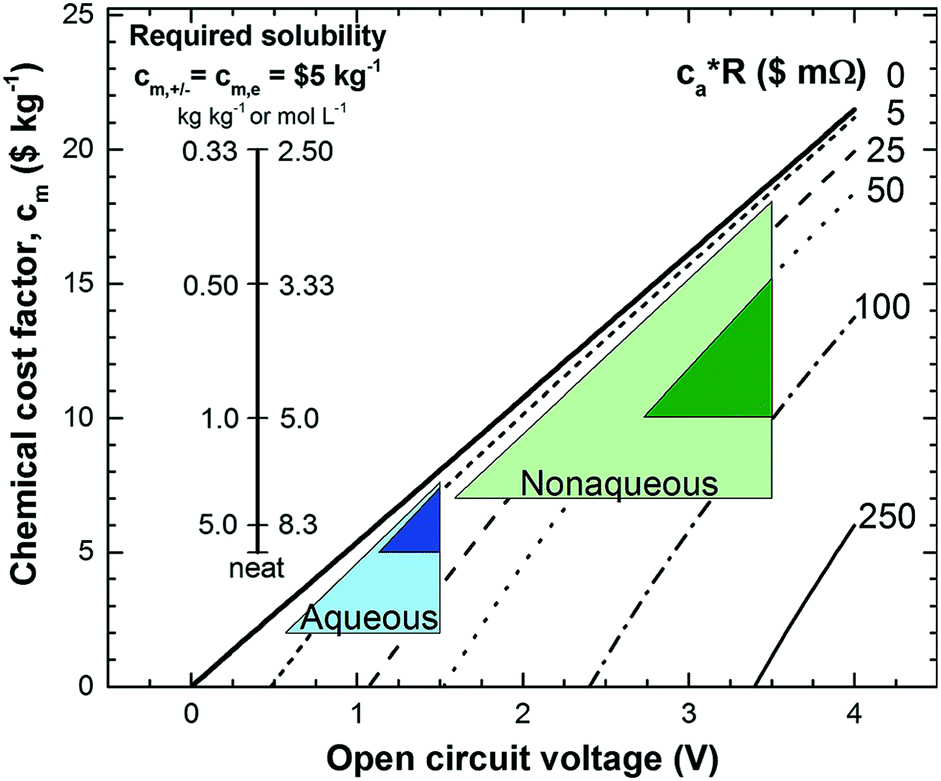 | ||
| Fig. 2 Allowable chemical cost factor versus open circuit voltage of aqueous and non-aqueous RFBs based on different reactor costs. cm,+, cm,− and cm,e stand for positive, negative redox species cost and electrolyte cost. ca is the reactor cost per unit area in $ m−2, and R is the area-specific resistance in Ω cm2. The leftmost inset vertical scale indicates the required solubility of a redox species in non-aqueous systems at a cost of $5 kg−1 for solute and solvent. The rightmost inset vertical scale on the right indicates the molar concentration. Reproduced with permission from ref. 30. Copyright 2014, Royal Society of Chemistry. | ||
2. Principle of molecular engineering
A typical RFB is composed of two storage tanks filled with electrolytes that are pumped across a membrane held between two electrodes serving as current collectors. An ideal membrane allows the fast transport of the supporting electrolyte, but prevents crossover of redox species. The electrochemical performance of an RFB depends on several parameters and can be evaluated by the following benchmarks. Energy density is the most important performance metric for energy storage devices, so as to store as much energy as possible in a given volume. In an RFB system, the energy density is dictated as below:where n is the number of electrons transferred in the reaction, C is the lower concentration of two electrolytes, F is Faraday's constant, V is the voltage of the battery, and μv is the volume factor (μv = 1 + lower electrolyte concentration/higher electrolyte concentration).31 Thus, the energy density is mainly dependent on the number of electrons involved, solubility of redox species, and the potential difference between the cathode and anode-active materials. Consequently, when selecting redox couples for RFBs, the solubility should be as high as possible. This is exactly the opposite in solid state batteries, in which the solubility of electrode materials should be minimized to avoid the performance degradation caused by electrode dissolution. The molecular weight of redox species should also be taken into consideration, since small molecules are more likely to achieve high gravimetric energy density. In addition to solubility, the redox potential of redox species is of equal importance. To achieve the highest possible output potential within the intrinsic range of the solvent, the anolyte and catholyte potentials should be near the extremes of the operating potential of the electrolyte.
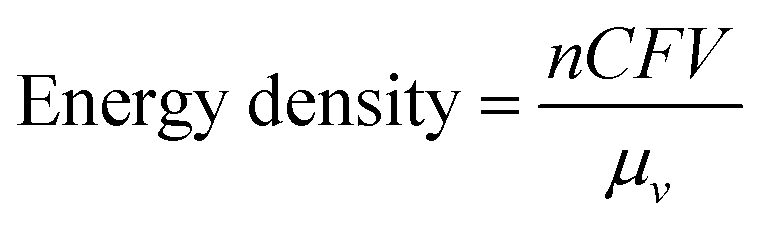
An equally important metric for consideration is power density, which is the amount of power per unit area and can be determined using the following equation:

The Coulombic efficiency (CE), also called current efficiency or faradaic efficiency, is another important parameter for evaluating battery performance. CE can be calculated using the same method as that for conventional solid state batteries, by determining the ratio of the charge output in the discharging process to the charge input in the charging process. Normally CE < 100% is attributed to either the crossover of redox species or the irreversibility of the redox reaction. Because high CE is indispensable in RFBs, cation or anion exchange membranes with high selectivity are commonly used to enable high efficiency despite their cost. In aqueous RFBs, Nafion is the most widely used separator, and it accounts for nearly 40% of the stack cost.33 In addition to the aforementioned performance metrics, it is also useful to analyze the voltage efficiency (VE), which is the ratio between the discharging and charging voltages, energy efficiency (EE), which equals CE times VE, and cycling stability.
In conventional RFBs based on metal ions, it is difficult to tune the intrinsic properties of redox species, leaving modification of other battery components as the only means for improving electrochemical performance. Inspired by the advances in synthetic chemistry, organic molecules emerge as promising alternatives with highly tailored properties. Solubility, redox potential, molecular size, and chemical reversibility can all be tuned by modification of the organic core component with particular functional groups to build a high-performance RFB (Fig. 1).
A general principle is provided here regarding functionalization of organic and organometallic molecules, including the relationship between functional groups and solubilization effects, redox potentials, and the size of redox species. In this review, electroactive materials are categorized according to active species types, including carbonyls, free radicals, polymers and metal complexes. As organic and organometallic-based redox species can be applied in both aqueous and non-aqueous systems, provided sufficient solubility and stability can be achieved, it is meaningful not to restrict the discussion to a certain type of solvent.
2.1 Solubility
As introduced above, the energy density of an RFB is proportional to the concentration of redox species. The solubility of the electroactive material is of vital interest in the scientific community of RFBs. Nevertheless, typical organic molecules, especially those with large molecular structures, maintain low solubility in water. Therefore, a rational molecular engineering method is usually required to tailor the structures of organic molecules for applications in RFBs. The solubility of a substance fundamentally depends on both the chemical and physical properties of the solute and solvents. Theoretically speaking, the solubility of a certain substance is governed by the balance of intermolecular interaction between the solute and solvent, and can be roughly estimated by the entropy change in the solvation process.34 Generally the solute molecules are held together by certain intermolecular forces, such as dipole–dipole, induced dipole–induced dipole, and ion–ion interactions. In the dissolution process, the interactive forces between solute–solvent molecules should be large enough to overcome the adhesive forces between solute–solute molecules. Thus strong solute–solvent attractions give rise to greater solubility than weak solute–solvent attractions.For example, in a typical dissolution process of ionic compounds, which contain periodic arrangements of charged ions bonded together via electrostatic force, the solvation energies of ions commonly need to be larger than the lattice energy.35 Therefore, the lattice energy of an ionic compound gives a rough indication of the solubility of the substance as it reflects the energy required to separate positive and negative ions. Meanwhile it is preferred for the solvent to have large relative permittivity, which can decrease the electrostatic force between positive and negative ions.36Table 3 includes the relative permittivity of various kinds of solvents, including water, alcohols, ethers, nitriles and so on. It shows that water possesses one of the highest relative permittivities, allowing salts to dissolve with ease. To achieve dissolution in organic solvents with low relative permittivity, the salts should possess large ion size and low lattice energies, for example tetraethylammonium tetrafluoroborate.37
Another general rule for predicting solubility is “like dissolves like”, which means that a solute tends to dissolve easily in solvents with a similar chemical structure.38 Specifically, polar solvents can dissolve ionic and other polar substances, whereas non-polar solvents tend to dissolve non-polar solutes. This principle can also predict the solubility of organic molecules in a certain solvent. Non-polar organic molecules are generally insoluble in water, but are highly soluble in non-polar solvents. The sequence of polarity of common organic functional groups is shown in Fig. 3.39,40 To increase the solubility of non-polar solutes in polar solvents, certain polar (and therefore hydrophilic) functional groups can be attached to the molecular chain, thus increasing the interaction between solutes and solvents. Likewise, if we want to improve the solubility of polar molecules in non-polar organic solvents, the solute molecules can be functionalized with non-polar groups. The relative permittivity of different solvents, as shown in Table 3, provides a rough measure of a solvent's polarity.41 In general, a solvent is regarded as nonpolar when it has a relative permittivity less than 15. Furthermore, the dipole moment of a solvent can be used to assess the solvent polarity as well.41 Many solvents with high relative permittivity also maintain high dipole moments. Nevertheless, the dipole moment sometimes underestimates the polarity of solvents with small molecules because it depends on the distance between the positive and negative charge centers in the molecule, and that's why the dipole moment of water is lower than expected.42
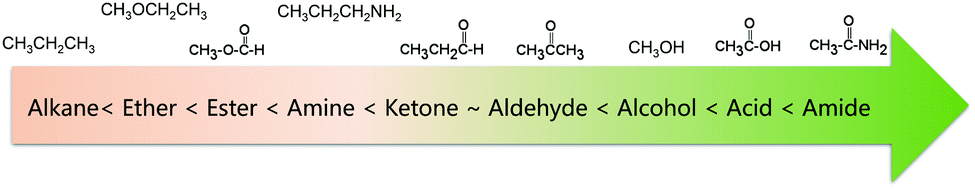 | ||
| Fig. 3 Polarity ranking of typical organic functional groups. | ||
In addition to the empirical principles introduced above, thermodynamic models provide a potentially efficient and cost-effective method for predicting a compound's solubility. Based on the Van’t Hoff equation and general solubility theory, a number of models have been constructed relating solubility to changes in Gibbs free energy, enthalpy, and entropy accompanying the solvation process.43,44 Compared with time-consuming and laborious experimental methods, the ability to accurately predict solubility represents a promising approach to rational design and screening of organic electroactive materials for RFBs. Notwithstanding recent progress, research is still needed to develop a reliable and fast model, capable of offering high levels of accuracy at a reasonable cost. Emerging scientific understanding with physical and chemical insight, combined with optimization of the parameters based on larger sets of experimental values may lead to breakthroughs in the computational modeling of organic electroactive materials.45
2.2 Redox potential
Energy density is dictated by the concentration of redox species and the cell voltage. Therefore one would seek cathode and anode-active materials with the greatest potential difference within the potential window of the electrolyte. In this regard, organic and organometallic-based molecules provide high flexibility to tune the redox potentials. Firstly, the redox potential is affected by both the chemical and physical conditions of the electrolyte. If protons are involved in the redox reaction of the organic electroactive materials, the redox potential will have a linear relationship with pH, as mapped out in the Pourbaix diagram.46 The slope of potential shift can be calculated from the Nernst equation, and the general trend is that increasing pH value will decrease the redox potential for a given proton-involved reaction.Additionally, the voltage of organics can be modified by means of molecular engineering, as has been done in Li ion batteries based on organic electroactive materials.47 The general principle behind the battery chemistry is that functionalization of electron-withdrawing groups, such as nitro and sulfonic acid, can increase the electron affinity of molecules, thus leading to increased redox potentials. In contrast, electron-donating groups, such as amino and hydroxyl, can release electrons to the redox centers and thus result in decreased redox potentials. Fig. 4 shows the substituent effects in sequence from deactivating to activating groups.48 In terms of redox potential, the electron-donating groups (activating groups) are applicable for the redox species in the anode side, while electron-withdrawing groups (deactivating groups) improve the potentials of molecules in the cathode side. The effect of functional groups has been verified by both experimental and computational results.49 In the calculated HOMO–LUMO energy levels, it is clear that frontier orbitals shift in the same direction according to electron-donating or electron-withdrawing groups, corresponding precisely to the potential tuning.50 Meanwhile, the redox potential derived from Gibbs free energy change also shows the same trend in accordance with the substituted groups.51
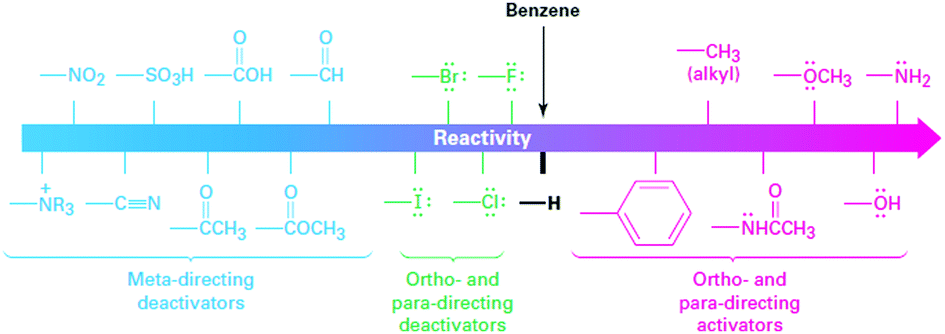 | ||
| Fig. 4 Electron donating and withdrawing groups. Reproduced with permission from ref. 48. Copyright 2012, Cengage Learning. | ||
Furthermore, the effect of functional groups on the redox potentials of some organic redox species can be quantified by the Hammett substituent constant, which indicates the dependence of substituent effects in aromatic molecules upon rates of reaction and equilibrium constants.52 A basic Hammett equation is shown below:
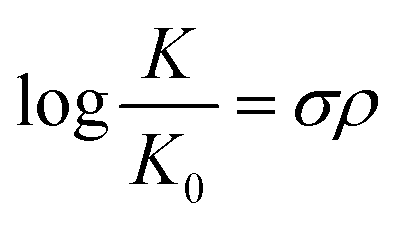
| E = 1.225 + 0.45σ |
| Substance | p-NH2 | p-OCH3 | p-CH3 | m-CH3 | m-OH |
| σ | −0.66 | −0.13 | −0.076 | −0.007 | −0.002 |
| Substance | H | m-OCH3 | m-OC2H5 | p-SO3− | m-CHO |
| σ | 0 | 0.115 | 0.15 | 0.38 | 0.381 |
| Substance | 3,4-(CH3)2 | 3,5-(CH3)2 | 3-OH,5-CH3 | p-Cl | 4-Cl,3-CH3 |
| σ | −0.083 | −0.014 | −0.009 | 0.227 | 0.22 |
2.3 Size
Another important criterion for quantifying the performance of an RFB is CE, which describes the efficiency of electron transfer within an electrochemical system. Unless using a ceramic-based separator to completely isolate the catholyte and anolyte, crossover of redox species is an inevitable issue.55 When electrochemically active species diffuse through separators, efficiency is not only lost in the charge–discharge cycle, but the cycling stability degrades due to the irreversible crossover process. The separator with high molecular selectivity is essential to alleviate the crossover issue of electrochemically active molecules. The selectivity of a membrane is commonly due to three effects, including physical blocking, electrostatic exclusion and Donnan exclusion, which permit many engineered processes to achieve better redox species isolation.56 The most widely used separator for RFBs is Nafion, which is a sulfonated tetrafluoroethylene based copolymer. According to the cluster-network model, Nafion consists of uniformly distributed sulfonate ion clusters with a diameter of 4 nm in the rigid fluorocarbon framework.23 Meanwhile, narrower channels of 1 nm length interconnect all the clusters continuously within the fluorocarbon matrix. Therefore, Nafion plays a critical role in regulating the operation of RFBs. However, the specialized structure of Nafion requires a costly manufacturing process, which hinders its wider industrial applications.The dimensions of common molecules range from a few angstroms to hundreds of angstroms. Thus, conventional polypropylene or polyethylene-based separators for Li ion batteries, with pores that are about 50 nanometers wide, are not suitable for RFB applications.57 Another promising type of separator is a dialysis membrane, which is only 5% to 10% of the cost of Nafion.58 Typically, the pore size ranges from ∼1 nm to 10 nm, and the separation effect is based on exclusion of molecules with large molecular masses. Therefore, the ion selectivity of separators can be enabled by size tuning of organic redox species, which should maintain molecular masses larger than the molecular-weight cutoffs of the membranes. Meanwhile, the separator must allow fast ion transport to ensure comparable power density.
To achieve an efficient size-exclusion effect, redox active monomers, oligomers, and polymers maintaining very wide size ranges can work as soluble nanoscopic energy storing units.59 As shown in Fig. 5, the tailorable size of organic-based redox species can be realized by anchoring redox active organic units onto polymer backbones with different sizes. In particular for macromolecule-based species, the main chains or backbones can undergo graft co-polymerization, and the pendant chains can be tailored to meet the required molecular size and electrochemical activity.60 Furthermore, the redox-active organic moieties can be functionalized on polymer colloids to make cross-linked polymer spheres with a uniform diameter range and reversible redox activity in the form of flowable dispersions.59 These methods demonstrate the diverse opportunities for tuning the sizes of organic species. It is noted that tethering redox active moieties to the polymer backbones may lead to decreased solubility of electroactive materials. Therefore, certain substituents with strong interactions with solvents are commonly functionalized to the polymer backbones as well to achieve reasonable concentration of the redox-active unit.61
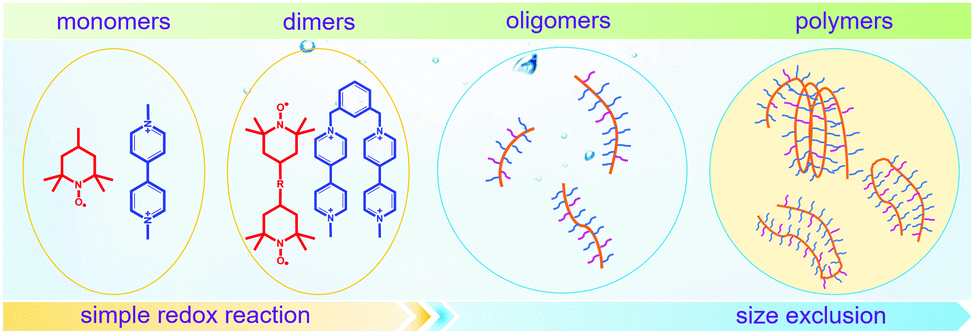 | ||
| Fig. 5 The tailored size of organic-based redox species by integrating redox active organic moieties into polymer backbones with different sizes. | ||
In rationally designing organic electroactive materials to match cost-effective size-exclusion membranes, size must be varied without reducing electrochemical activities of the redox moieties. Additionally, the organic molecule should remain highly soluble to meet energy density requirements. Lastly, the viscosity, rheological, and transport properties of the electrolytes should all be taken into consideration when implementing cost-effective flow systems based on inexpensive nanoporous commercial separators and size-tunable electroactive molecules.
So far we have summarized the general principle to design organic and organometallic electroactive molecules following fundamental laws of physics and chemistry of RFBs. The delicate synthetic strategies to prepare target molecules with different molecular structures and functional groups will be introduced in detail in the following sections.
3. Carbonyl-based redox species
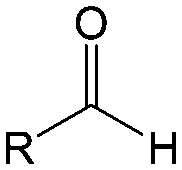
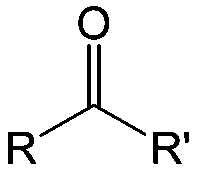
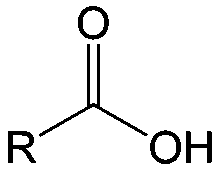
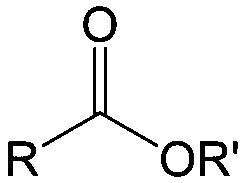
Carbonyl compounds are the most promising type of organic electroactive material. They feature fast reaction kinetics, great structural diversity, tailorability, and benefit from cost-effective synthesis processes.62 The first exploration of carbonyls for battery applications can be traced back to 1969, when Williams et al. built a primary battery with a dichloroisocyanuric acid cathode and a lithium anode in a non-aqueous electrolyte.63 Since then, continuous works have been done to broaden the applications of carbonyl compounds for either solid state batteries or RFBs with liquid form electrolytes.64A carbonyl group is an organic functional group composed of a carbon atom double-bonded to an oxygen atom.65 A compound containing such a carbon–oxygen double bond is called a carbonyl compound. Aldehydes and ketones are the simplest forms of carbonyl group, and these entities are commonly attached to larger carbon skeletons to make up a large group of organic molecules. Typical types of carbonyl compounds are presented in Table 5. Distinguished from the non-polar double bonds in alkenes, the difference between the electronegativity of carbon and oxygen induces the bond's polarity, in which the electronegative oxygen draws electron density away from carbon.66 As a result, the carbonyl compounds are much more reactive than alkenes. The partially negative oxygen can react with electrophiles, including positive ions as Lewis acids, and the positively charged carbon can be attacked by nucleophiles as Lewis bases. The properties of carbonyls are closely tied to their electronic structures and geometric positioning. In the atomic orbitals of a carbonyl group, one pair of the oxygen lone pairs occupies the 2s orbital, while the other is located in the 2p orbital, both of which contribute to the polarity of the carbon–oxygen double bond.67,68 Compared with alkenes, the double bond in carbonyls is shorter and the bond energy is higher.69 Therefore, the reactivity of carbonyls should be ascribed to the induced polarity, and shorter bond length usually correlates with even larger polarity. Among different kinds of carbonyls, amides are considered to be the most stable type of compound on account of the high-resonance stabilization effect between carbon–oxygen and carbon–nitrogen.70
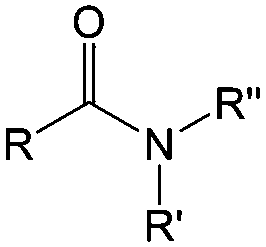
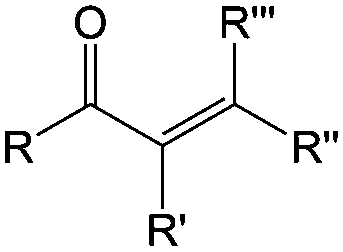
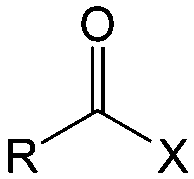
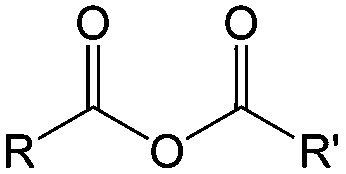
| Compound | Formaldehyde | Aldehyde | Ketone | Carboxylic acid | Ester | Amide | Enone | Acyl halide | Acid anhydride |
|---|---|---|---|---|---|---|---|---|---|
| Structure |
|
|
|
|
|
|
|
|
|
| General formula | CH2O | RCHO | RCOR′ | RCOOH | RCOOR′ | RCONR′R′′ | RCOCR′CR′′R′′′ | RCOX | (RCO)2O |
3.1 Reaction mechanism
In contrast to organic reactions in chemical synthesis, reversibility is essential in battery reactions. The redox reactivity of carbonyls is based on the enolization process, in which positive charge carriers, such as H+ or Li+, bind to negatively charged oxygen (Fig. 6).71 Therefore, nearly all carbonyl compounds can be categorized into n-type electroactive organic molecules, accepting one or more electrons to form anions during the reduction process. Carbonyl entities are commonly connected to conjugated structures to facilitate electron transport and delocalize the charge on redox products. In some molecules with multiple carbonyl groups, further reduction can take place to form multivalent anions under the stabilization of conjugated substituents. As shown in Fig. 7, depending on different kinds of stabilization structures, the carbonyl compounds can be classified into three groups.62 In Group I, adjacent carbonyl groups are present to form stable enolates upon reduction. In Group II, carbonyl groups are directly connected to the aromatic core, which can help disperse the negative charge over the ring structure. The molecules in Group III commonly contain quinone moieties with a fully conjugated cyclic dione structure. Group III compounds also share some similar structure characteristics with Groups I and II, but the main difference is that addition aromatic ring will be generated during reduction, which contributes to the primary stabilizing force.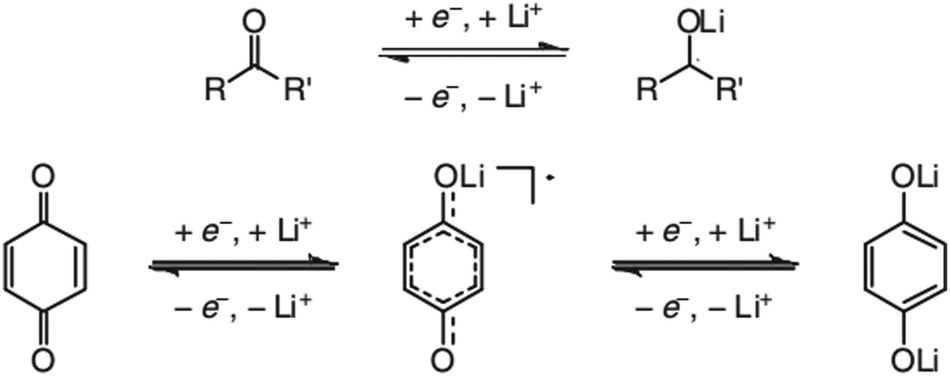 | ||
| Fig. 6 Reaction mechanism of general carbonyl compounds (up) and quinone (down). Reproduced with permission from ref. 71. Copyright 2012, Wiley. | ||
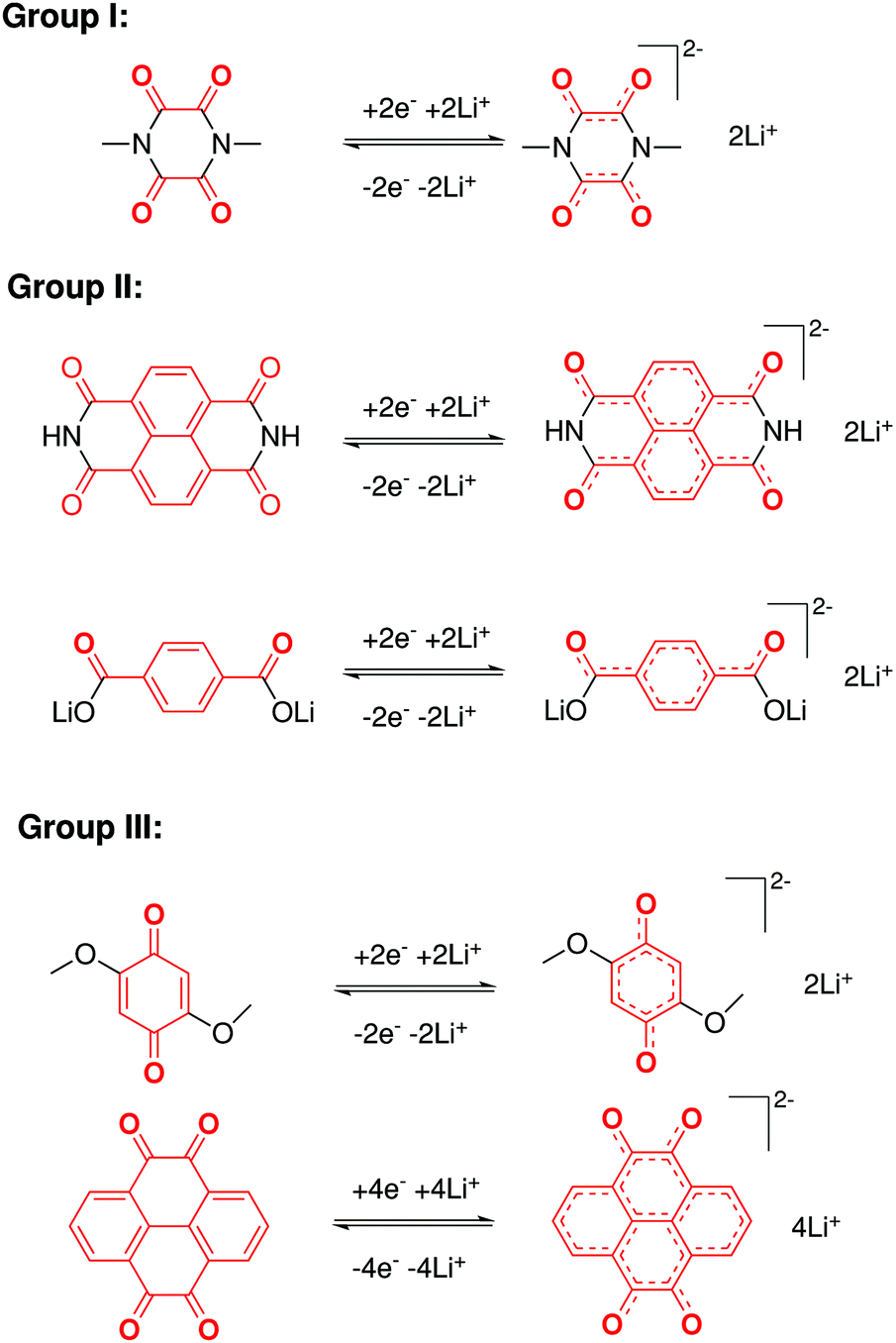 | ||
| Fig. 7 Stabilization effects of three types of carbonyl compounds. Reproduced with permission from ref. 62. Copyright 2015, Wiley. | ||
3.2 Molecular engineering method
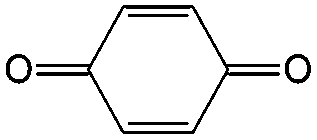
Though great advances have been made in using carbonyl compounds for solid state batteries, their application in RFB has not received comparable attention.64,72,73 Almost all types of carbonyl compounds in Table 5, such as imides, anhydrides, carboxylic acids, ketones and quinones, have been explored in Li ion batteries, but they cannot all be applied in RFBs due to limited solubility in aqueous and non-aqueous electrolytes.64 In order to compete with the performance of conventional metal-based aqueous RFBs, rational functionalization is normally required to modify these electrochemically active molecules. The design route and experimental approach vary based on requirements for solubility, redox potential, chemical stability, etc. This section will review the design principle and synthetic methods for tuning properties of carbonyl compounds for RFBs. Table 6 lists the solubility and redox potentials of certain carbonyl compounds published to date.| # | Molecule | Electrolyte | Solubility (M) | Redox potential V (vs. SHE) | Ref. |
|---|---|---|---|---|---|
| Aqueous electrolyte | |||||
| 1 |
|
1 M H2SO4 | 0.1 | 0.68 | 82 |
| 2 |
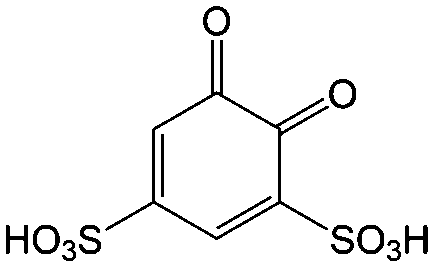
|
1 M H2SO4 | 1 M | 0.87 | 82 |
| 3 |
|
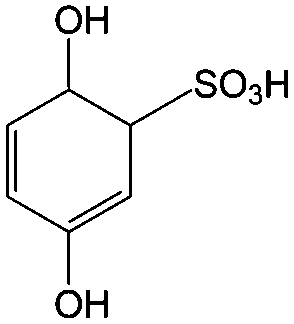
1 M H2SO4 | 0.8 | 0.7 | 82 |
| 4 |
|
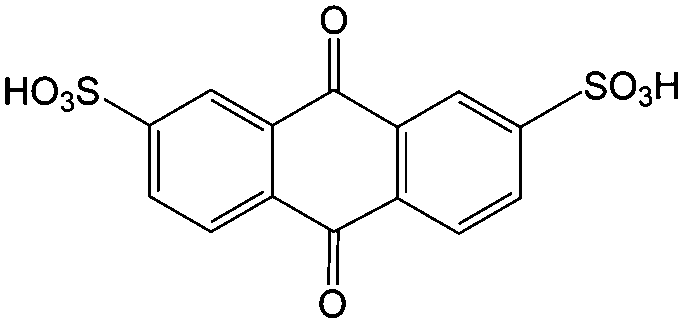
1 M H2SO4 | 1 | 0.22 | 79 |
| 5 |
|
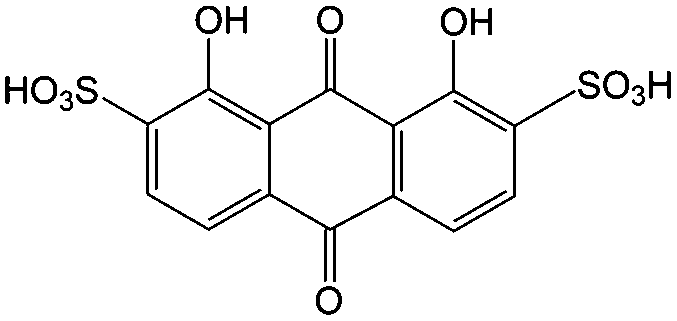
1 M H2SO4 | 1 | 0.12 | 80 |
| 6 |
|
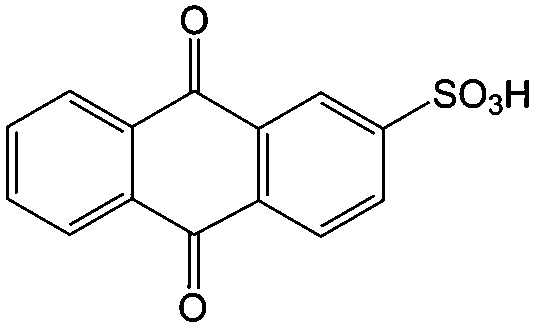
1 M H2SO4 | 0.2 | 0.15 | 82 |
| 7 |
|
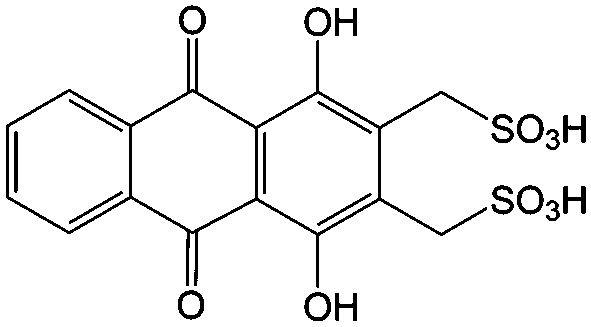
1 M H2SO4 | 1 | 0.02 | 80 |
| 8 |
|
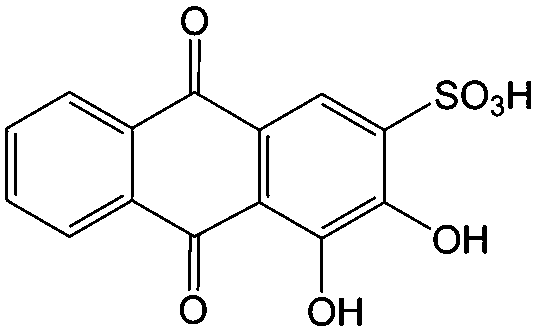
1 M H2SO4 | 0.05 | 0.082 | 85 |
| 9 |
|
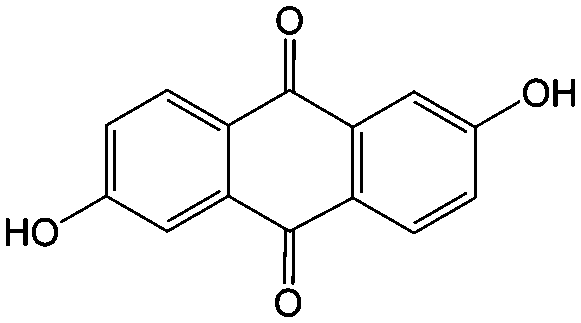
1 M KOH | 0.6 | −0.7 | 81 |
| 10 |
|
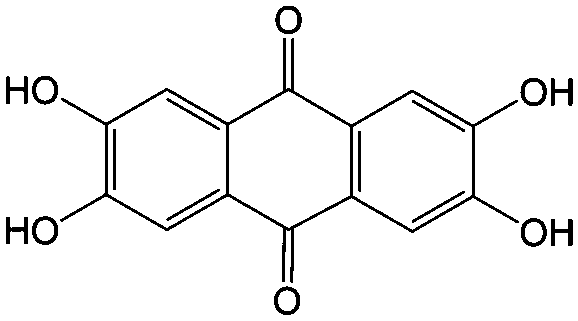
1 M KOH | N/A | ∼−0.8 | 81 |
| 11 |
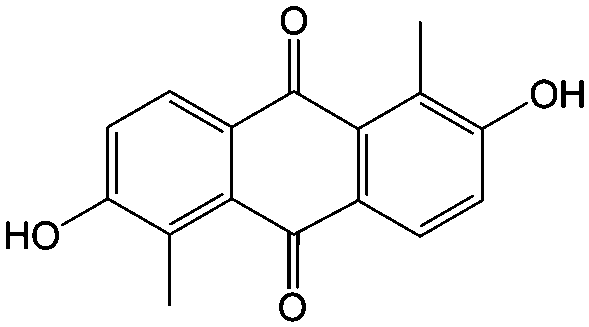
|
1 M KOH | N/A | ∼−0.8 | 81 |
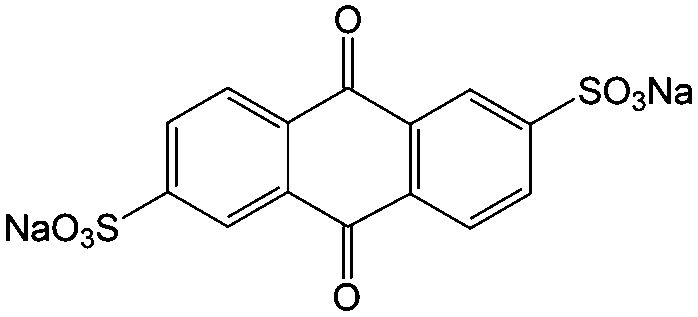
| 12 |
|
1 M H2SO4 | 0.5 | 0.12 | 82 |
| 13 |
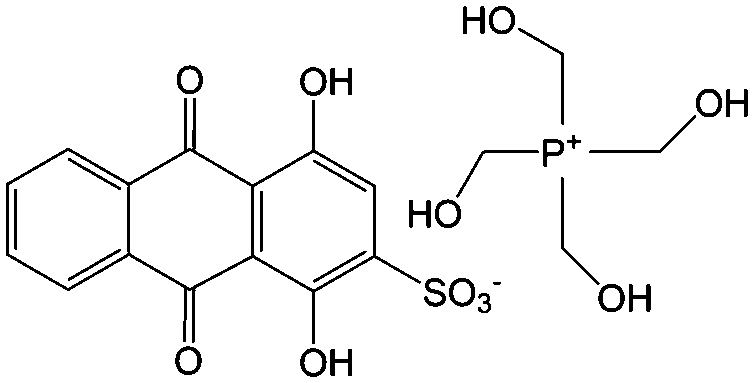
|
0.1 M HClO4 | 0.2 | 0.07, 1.07 | 86 |
| 14 |
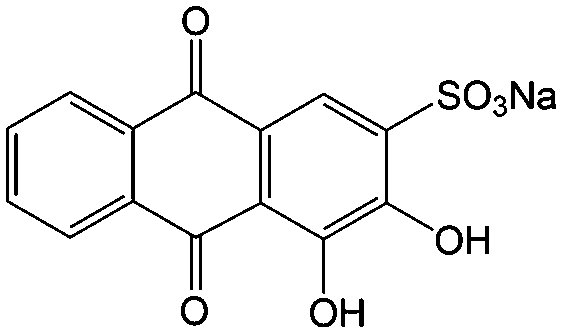
|
0.1 M HClO4 | 1.6 | 0.02 | 86 |
| 15 |
|
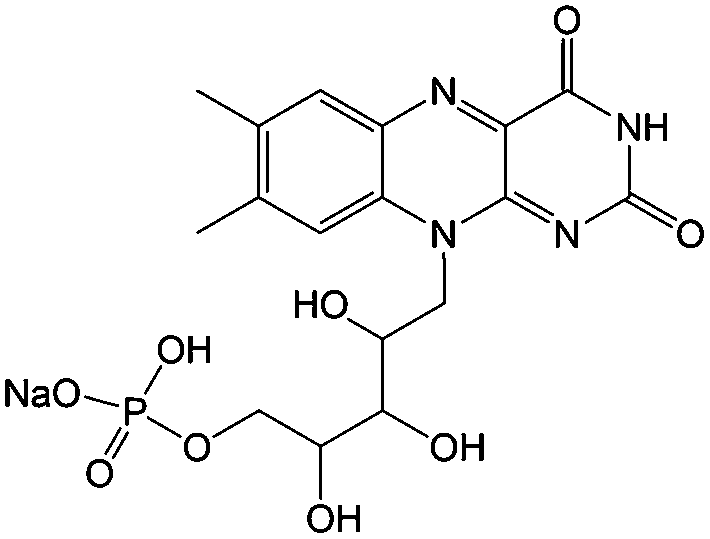
1 M KOH, 3 M nicotinamide | 1.5 | −0.52 | 94 |
| 16 |
|
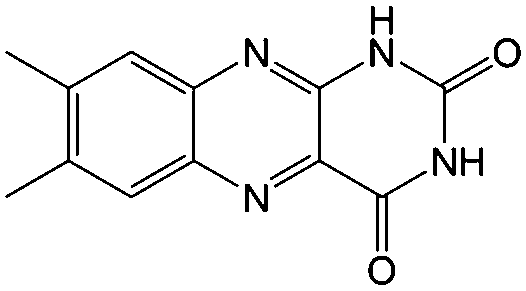
KOH | N/A | −0.7 | 95 |
| 17 |
|
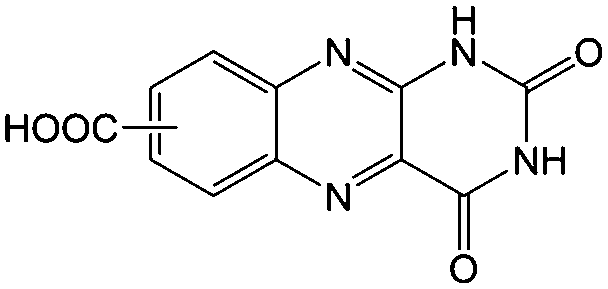
KOH at pH 14 | 2 | −0.62 | 95 |
| 18 |
|
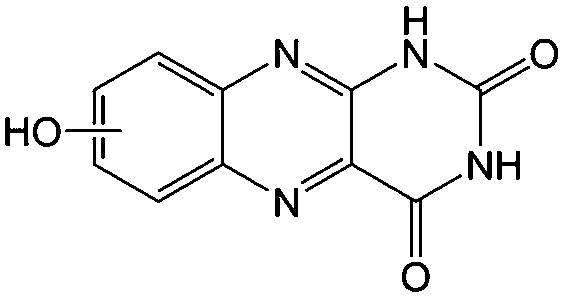
KOH | N/A | −0.73 | 95 |
| 19 |
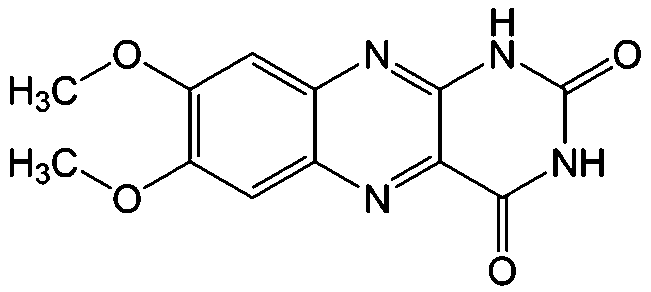
|
KOH | N/A | −0.75 | 95 |
| Non-aqueous electrolyte | |||||
| 20 |
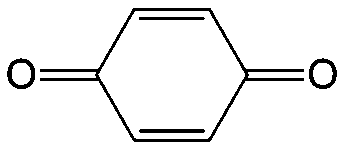
|
1 M LiClO4/butyrolactone | 2.6 | −0.1 | 83 |
| 21 |
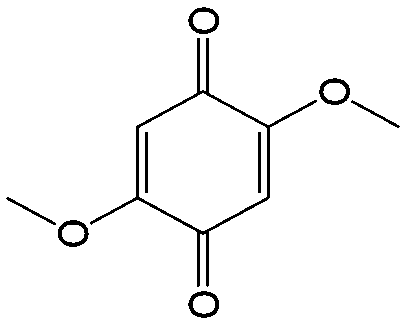
|
1 M LiClO4/butyrolactone | 0.006 | −0.3 | 83 |
| 22 |
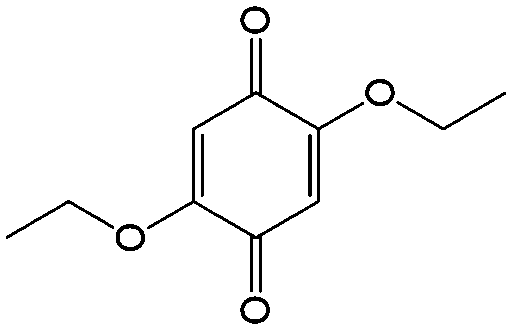
|
1 M LiClO4/butyrolactone | 0.3 | −0.3 | 83 |
| 23 |
|
1 M LiClO4/butyrolactone | 0.06 | −0.4 | 83 |
| 24 |
|
1.0 M LiPF6/PC | 0.25 | −0.7 | 84 |
| 25 |
|
0.1 M TBAP/ACN | 0.02 | −1.7 and 0.5 vs. Ag/Ag+ | 87 |
| 26 |
|
0.2 M LiTFSI/DMA | 1.2 | −0.29 | 88 |
| 27 |
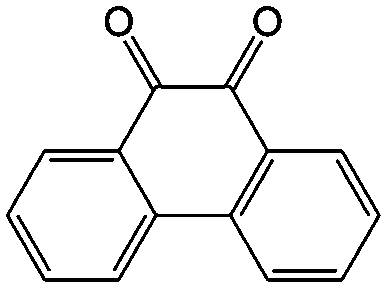
|
0.2 M LiTFSI/DMA | 0.3 | −0.26 | 88 |
| 28 |
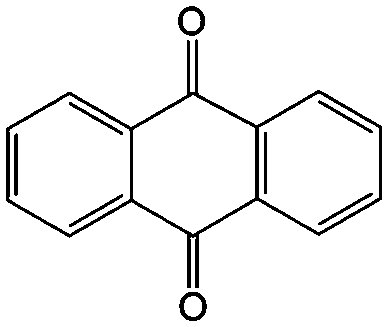
|
0.2 M LiTFSI/DMA | 0.05 | −0.54 | 88 |
| 29 |
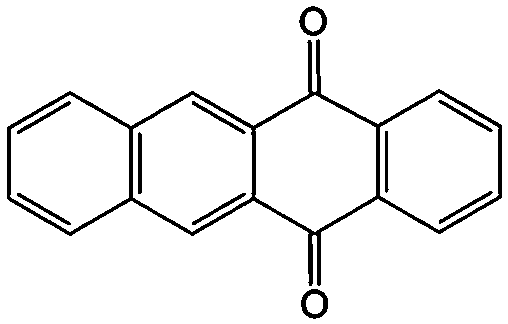
|
0.2 M LiTFSI/DMA | 0.05 | −0.77 | 88 |
| 30 |
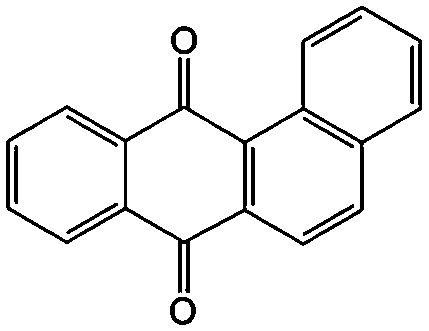
|
TEGDME | 0.083 | −0.66 | 89 |
| 31 |
|
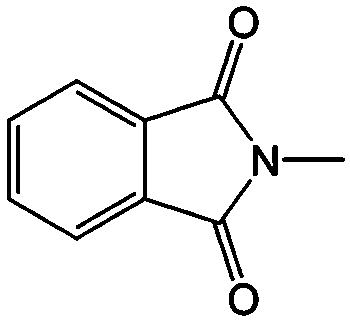
DME | 0.7 | −1.3 vs. Ag | 92 |
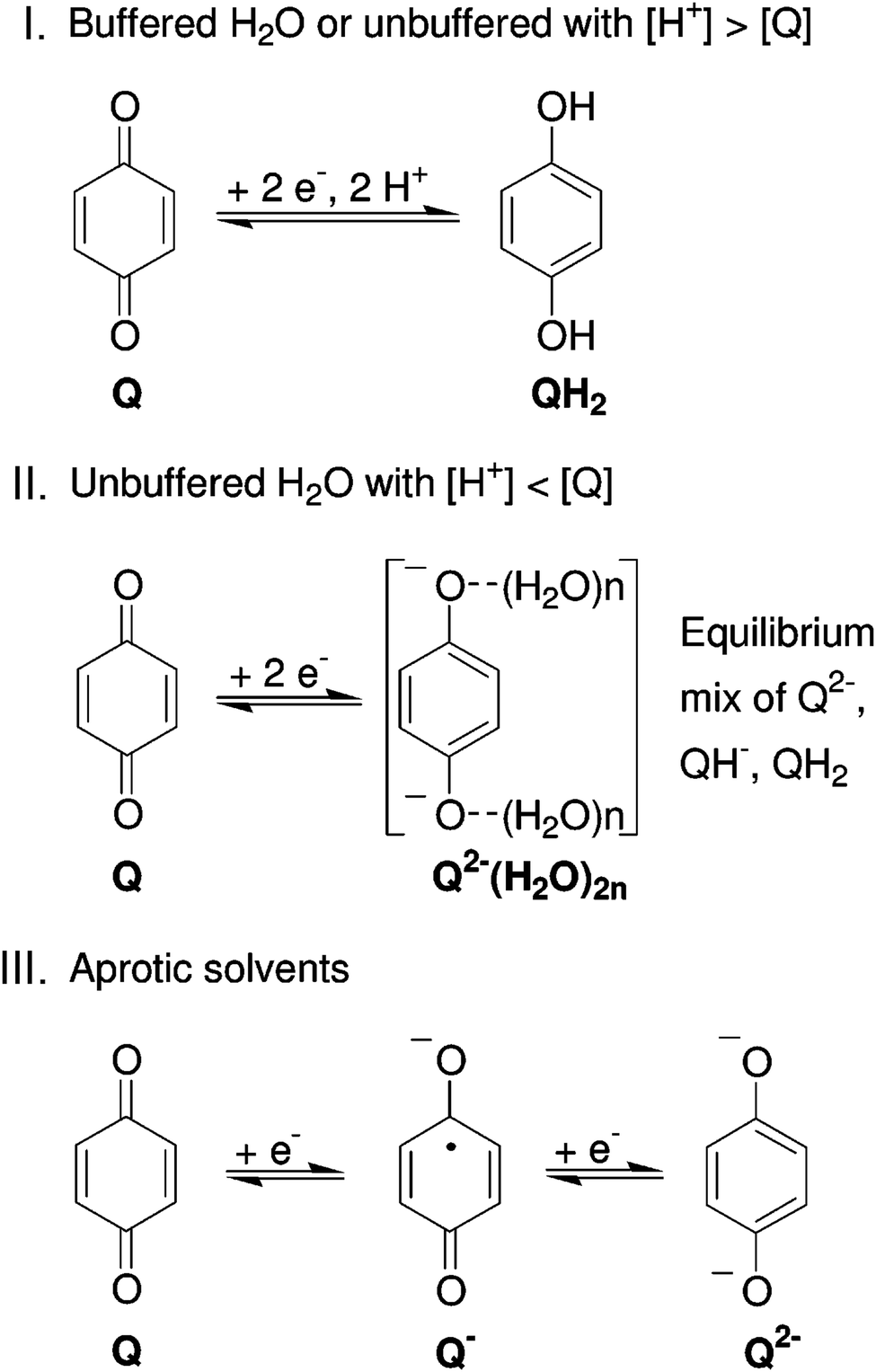 | ||
| Fig. 8 Reaction mechanism of BQ in different electrolytes. Reproduced with permission from ref. 76. Copyright 2007, American Chemical Society. | ||
Quinones in RFBs have received significant attention in the past decade. Fig. 9 shows a quinone library for both aqueous and non-aqueous RFBs. As one of the simplest organic redox species with small molecular weight, BQ stands out as the primary choice for organic RFBs.77 However, the solubility of BQ in water is only around 0.1 M, and a number of molecular engineering approaches have been conducted to overcome this limitation. The first quinone-based RFB was demonstrated in 2009 using 1,2-benzoquinone-3,5-disulfonic acid (BQDS, #2, Table 6) or sulfonic quinol (#3, Table 6) as catholytes and PbSO4/Pb as the anode.78 The sulfonic groups were grafted to increase both the redox potentials and solubility of the quinone bases. Due to the nucleophilic attack of water on BQDS, an irreversible oxidation takes place in the first cycle. As for sulfonic quinol, the cycling performance is more stable with average coulombic and energy efficiencies of up to 98% and 70%, respectively, over 100 cycles. Despite capacity loss, low energy density, and high IR drop at high current density, the exploitation of quinone-based redox species can be a new path towards sustainable energy storage.
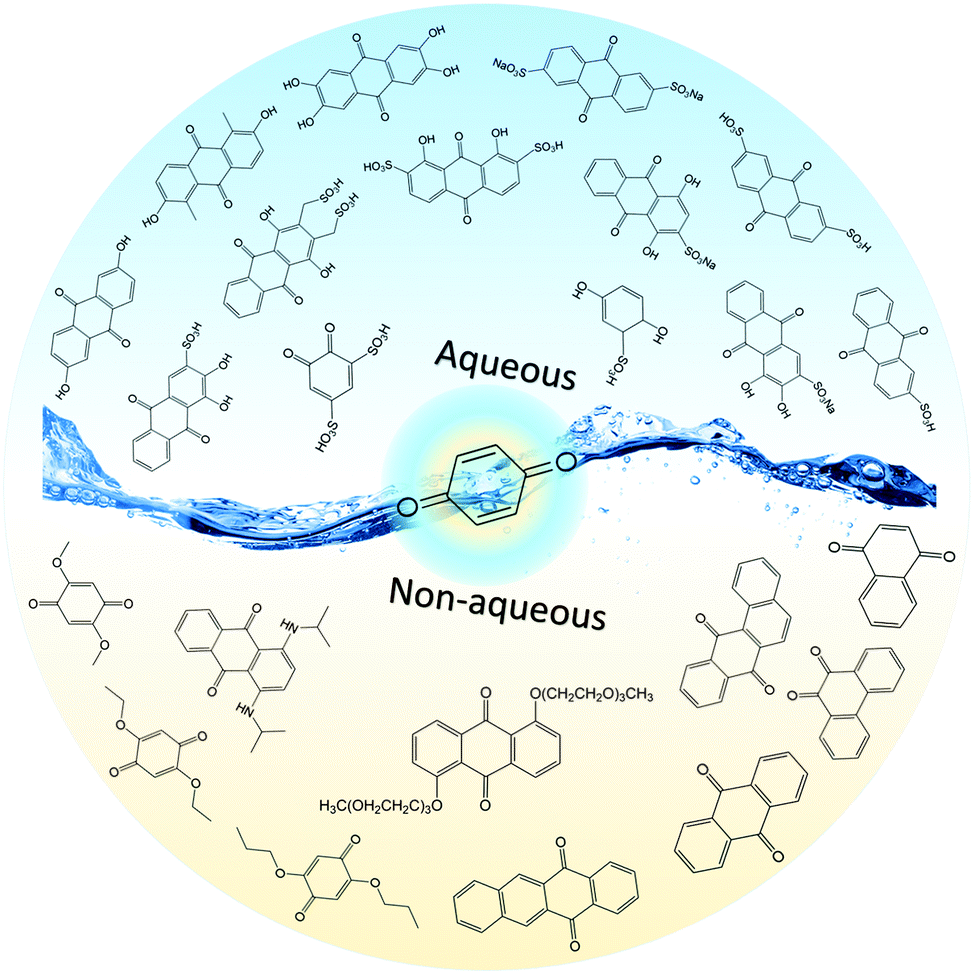 | ||
| Fig. 9 A quinone library for aqueous and non-aqueous RFBs. | ||
Anthraquinone (AQ, #28, Table 6), with two more aromatic rings that delocalize the negative charge, maintains higher chemical stability than BQ, but its intrinsic insolubility in water impedes application in aqueous RFBs. To address this issue, the Aziz group utilized 9,10-anthraquinone-2,7-disulphonic acid (AQDS, #4, Table 6) as the anolyte, which possesses an aqueous solubility of up to 1 M at pH 0 due to the hydrophilic effect of sulfonic groups.79 When paired with a bromine-based catholyte, such a metal-free RFB can deliver an energy density as high as 50 W h L−1. The cell voltage can be further improved by lowering the potential of the anolyte when the hydroxyl groups are anchored on the AQDS molecule as 1,8-dihydroxy-9,10-anthraquinone-2,7-disulphonic acid (DHAQDS, #5, Table 6), confirming the effect of –OH as an electron-donating group. A series of AQ derivatives were developed to tailor both the chemical and physical properties, including anthraquinone-2-sulfonic acid (AQS, #6, Table 6), 1,4-dihydroxyanthraquinone-2,3-dimethylsulfonic acid (DHAQDMS, #7, Table 6), alizarin red S (ARS, #8, Table 6) in addition to AQDS and DHAQDS.80 Among these five quinones, an AQDS-based RFB delivers the lowest voltage given the highest redox potential due to the strong electron-withdrawing effect of two sulfonic groups. Though DHAQDMS maintains the lowest reduction potential, its irreversible redox reaction precludes its use in RFBs. In view of the instability of DHAQDS and ARS in the presence of bromine, AQS was selected as the anolyte. When paired with a bromine-based catholyte, an optimized RFB can provide higher voltage and power density than the AQDS-bromide battery.
Another step forward is utilizing an alkaline electrolyte instead of the conventional sulfuric acid electrolyte. In this way, more quinone derivatives can be considered while rationally screening redox species. In the alkaline RFB by Lin et al., two hydroxyl groups with electron-donating ability and hydrophilicity were grafted on AQ molecular cores without introducing electron-withdrawing sulfonic acid groups.81 In an alkaline electrolyte, these hydroxyl groups are deprotonated to provide high solubility. Moreover, no protons are involved in the redox reaction, and the redox potential is further decreased in alkaline electrolytes compared with acidic electrolytes, in accordance with the Pourbaix diagram. The AQ derivative 2,6-dihydroxyanthraquinone (2,6-DHAQ, #9, Table 6) shows a room-temperature solubility of about 0.6 M in 1 M KOH electrolyte, and the cell voltage can reach 1.2 V using a ferro/ferricyanide catholyte.
Most of these reported RFBs still partially rely on metal redox species. To take it a step further, Yang and co-workers investigated a number of quinone alternatives in order to build an all-organic RFB. After studying the chemical and physical properties, BQDS and AQDS were employed as positive and negative redox species, respectively, delivering a working voltage of 0.4 V.82
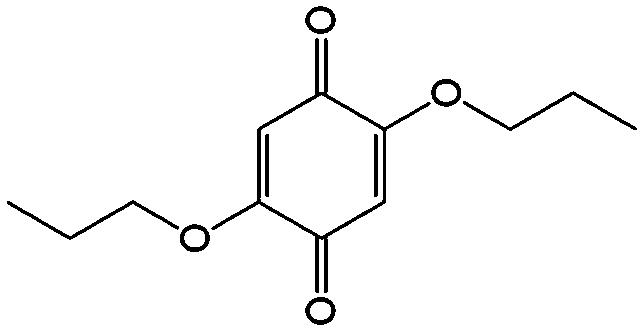
In comparison with aqueous electrolytes, non-aqueous solvents offer wider potential windows, broader operating temperature ranges, and more flexibility to tune the properties of electroactive molecules either by functionalization or solvent screening. In 2011, the Siroma group studied several BQ derivatives in non-aqueous hybrid RFBs, in which a solid state electrolyte functions as a separator to prevent the self-discharge reaction between quinones and a Li anode.83 Among all the species, BQ maintains solubility of up to 2.6 M with an average potential of 2.9 V. Given the two-electron transfer reaction, the theoretical energy density can reach approximately 400 W h L−1. Nevertheless, the discharge capacity decreased sharply with an increased concentration of BQ. Possible reasons include low electroactivity of reduction products, evaporation of quinone molecules, and instability of the reaction intermediates. To achieve better cycling stability, different peripheral substituents were functionalized on the BQ moiety, so as to restrain evaporation and stabilize the intermediate radicals. The following electrochemical test shows that 2,5-dipropoxy-1,4-benzoquinone (DPBQ, #23, Table 6) can deliver improved cycling stability even at a concentration exceeding the solubility.
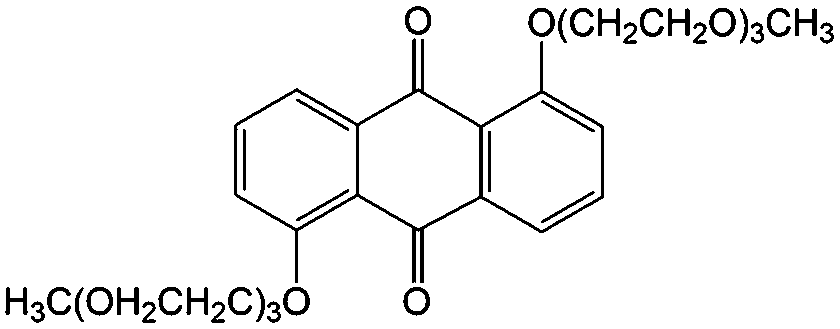
Subsequently, Wang and colleagues introduced two triethylene glycol monomethyl ether groups into the AQ molecular structure via nucleophilic aromatic substitution of 1,5-dichloroanthraquinone, leading to improved solubility in non-aqueous electrolytes from less than 0.1 M to over 0.25 M (#24, Table 6).84 In the proof-of-concept hybrid RFB test, the battery based on 0.25 M electroactive material in PC could provide an energy density of 25 W h L−1 at an energy efficiency of 82% over 9 cycles. The capacity decay was mainly due to the side reactions between the quinone molecules and the carbonate solvents.85,86
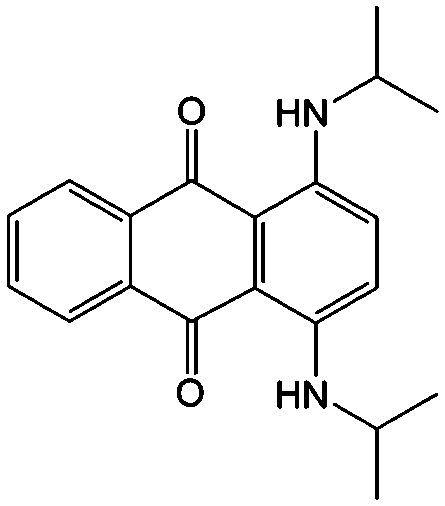
To further modify the redox activities of AQ-based molecules, the Abruna group introduced two amino groups into the quinone structure (#25, Table 6), which can accommodate up to five discrete redox states over a wide range of potentials.87 The bipolar property of such an AQ derivative makes it possible to construct a symmetric RFB employing functionalized AQ as both the positive and negative electroactive materials. The viability of such a battery was validated using an H-cell with a discharge voltage of 1.22 V, but the utility in a real battery device is restricted by low solubility. To achieve practical implementation of such a device, the bipolar molecule should have high solubility at all redox states.
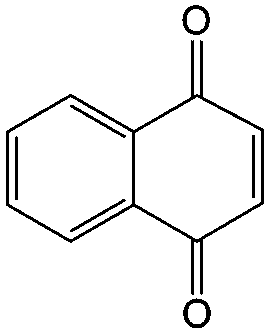
In addition to the functional groups, the properties of quinones are also strongly related to the aromatic cores. In this regard, the Yu group systematically studied a series of quinones, including BQ, 1,4-naphthoquinone (NQ, #26, Table 6), 9,10-phenanthrenequinone (PQ, #27, Table 6), AQ (#28, Table 6) and 5,12-naphthacenequinone (NAQ, #29, Table 6) via a combined experimental and computational method, as shown in Fig. 10.88 The redox potentials of different quinones, which can be calculated from the binding energy, are strongly related to the degree of aromaticity of reduced quinones following Clar's rule. In the hybrid RFBs test, it was found that the utilization of carbonyl groups in quinones could be affected by the HOMO–LUMO energy gap, with small gaps enhancing the electron transfer process and thus increasing reactivity. The correlation between HOMO–LUMO energy gap and the electrochemical performance was also confirmed in a study by the Cho group.89 They investigated two quinone-based constitutional isomers for RFBs, and found that 1,2-benzanthraquinone (BAQ, #30, Table 6) with a smaller energy gap presents better rate performance than NAQ.
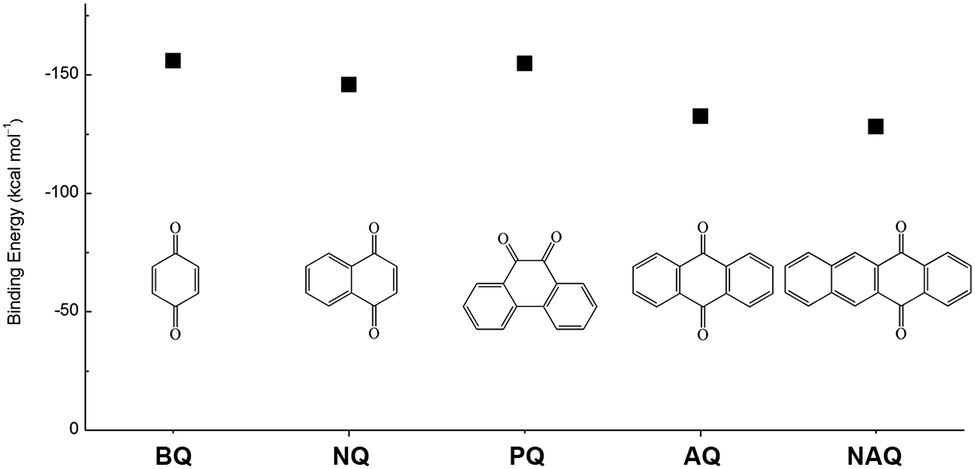 | ||
| Fig. 10 Chemical structures and calculated Li-binding energies of five quinone molecules. | ||
Advanced computational modeling is also developed to accelerate the prediction and screening of quinone derivatives for RFB applications.90 A virtual library of molecules can be generated by either altering the core structures, or decorating quinones with functional groups at specific positions. The Aspuru-Guzik group studied 1710 quinone derivatives from single to full substitutions on the quinone cores (Fig. 11).91 The potential can be predicted from the energy change due to hydroquinone formation from the quinone and hydrogen gas. The solvation energy, which is the total energy difference between the solvated form and the gas phase form, functions as an indicator of quinone solubility in aqueous solution. The high-throughput computational approach provides an efficient screening tool for the design of high-performance organic RFBs. Theoretical results can serve as pivotal reference data for refining feasible organic alternatives, but experimental assessment are also required. Moreover, the chemical stability and reaction reversibility, both of which govern the cycling stability of an organic-based RFB, should also be evaluated from the computational point of view.
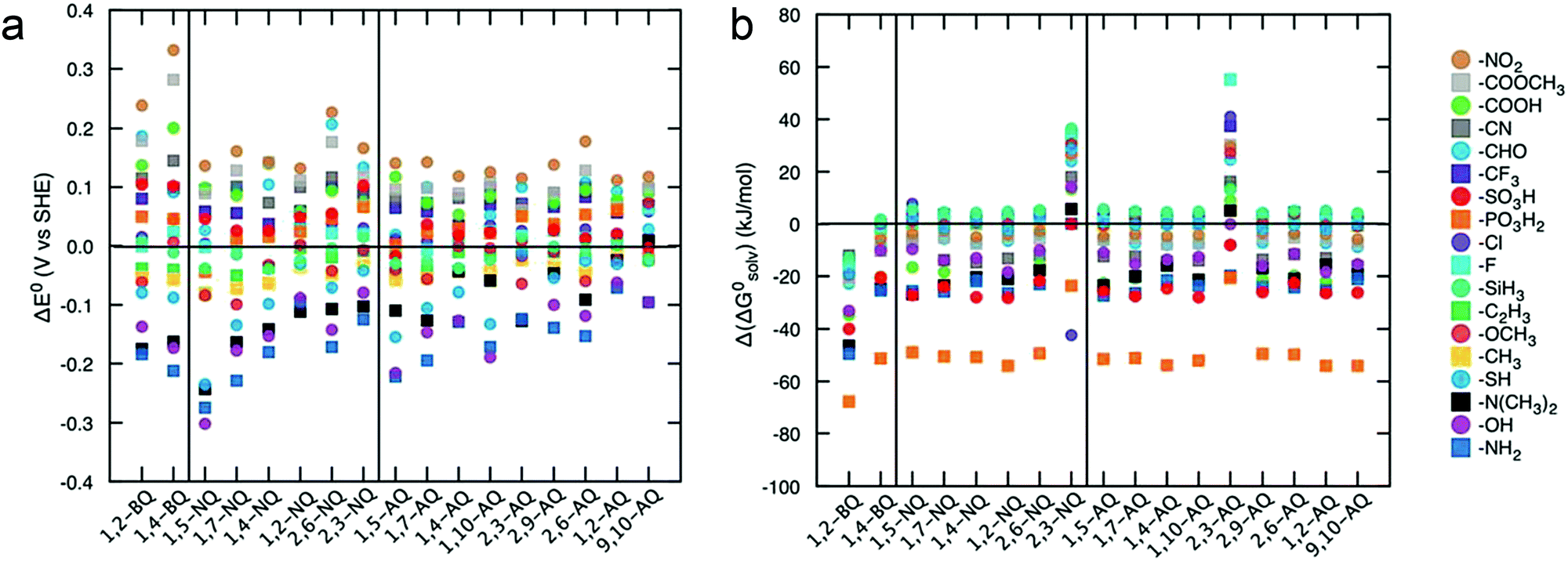 | ||
| Fig. 11 (a) Effects of functional groups on the redox potential of different quinone classes. (b) Effects of functional groups on the solubility of different quinone classes. Reproduced with permission from ref. 91. Copyright 2015, Royal Society of Chemistry. | ||
Flavin, a highly versatile electroactive cofactor in biological systems, represents another promising class of organic redox species. The flavin group contains a series of organic molecules with the basic planar alloxazine ring structure, but few of these bio-inspired species can meet the requirement of solubility for RFBs. The Meng group selected flavin mononucleotide (FMN-Na, #15, Table 6) as a negative-active material due to its relatively high solubility (0.1 M in 1 M KOH), that can be further enhanced by employing nicotinamide as a hydrotropic agent.94 In an aqueous solution containing 1 M KOH and 3 M nicotinamide, FMN-Na shows a maximum solubility of 1.5 M, which can lead to commendable volumetric capacity given the two-electron transfer reaction. It is worth noting that the electrochemistry of FMN-Na is dependent on the electrolyte. The deprotonated state of FMN-Na varies at different pH values, and the electron-transfer process in an alkaline electrolyte is illustrated in Fig. 12b. With K4[Fe(CN)6] as positive species, the FMN-Na-based biomimetic RFB shows a cycling performance with 99% capacity retention over 100 cycles. Lin et al. also investigated a series of alloxazine derivatives with functional substituents of carboxylic acid (#17 Table 6), hydroxyl (#18. Table 6), methoxy groups (#19, Table 6), and more (Fig. 12a).95 It was found that the carboxylic acid group renders the alloxazine molecule with a solubility of up to 2 M in alkaline electrolytes. Paired with K4[Fe(CN)6] solution as the catholyte, such an RFB can provide an open-circuit voltage approaching 1.2 V, while the capacity retention can reach 95% over 400 cycles. Further computational study revealed the effects of functional groups on the redox potential and chemical stability of alloxazine cores (Fig. 13).95
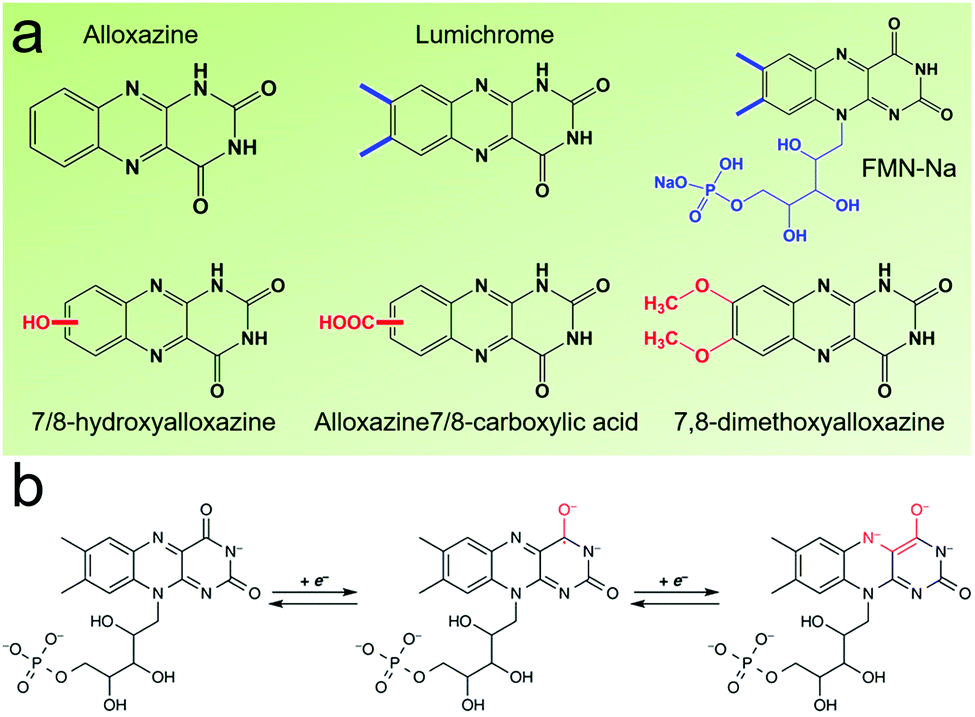 | ||
| Fig. 12 (a) Several typical alloxazine-based redox species. (b) The electron-transfer process of FMN-Na in an alkaline electrolyte. Reproduced with permission from ref. 94. Copyright 2016, Nature Publishing Group. | ||
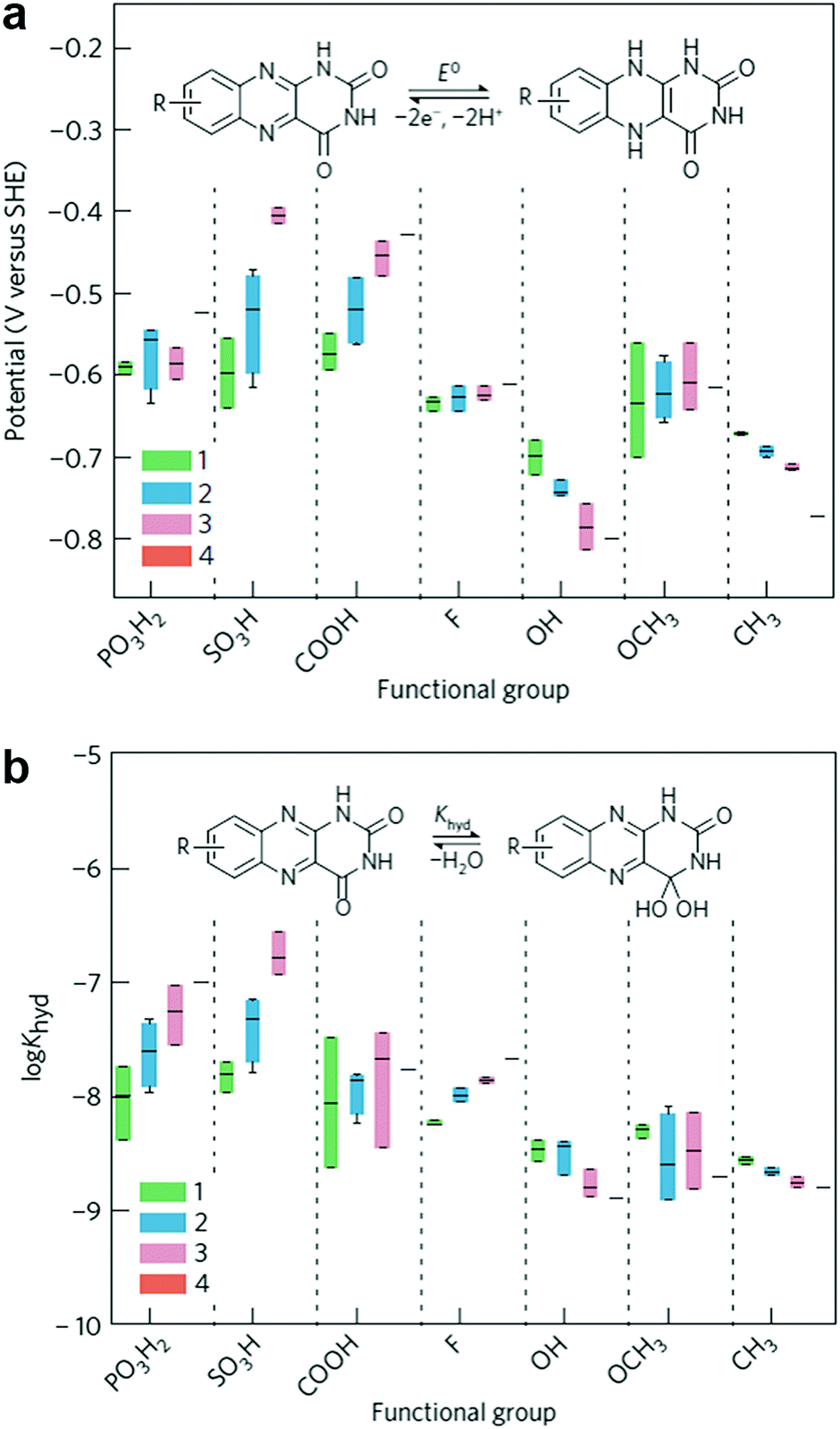 | ||
| Fig. 13 Computational modeling of alloxazine derivatives. (a) Effects of functional groups on the redox potential of alloxazine classes. (b) Effects of functional groups on the hydration equilibrium constant (Khyd) of alloxazine classes. Reproduced with permission from ref. 95. Copyright 2016, Nature Publishing Group. | ||
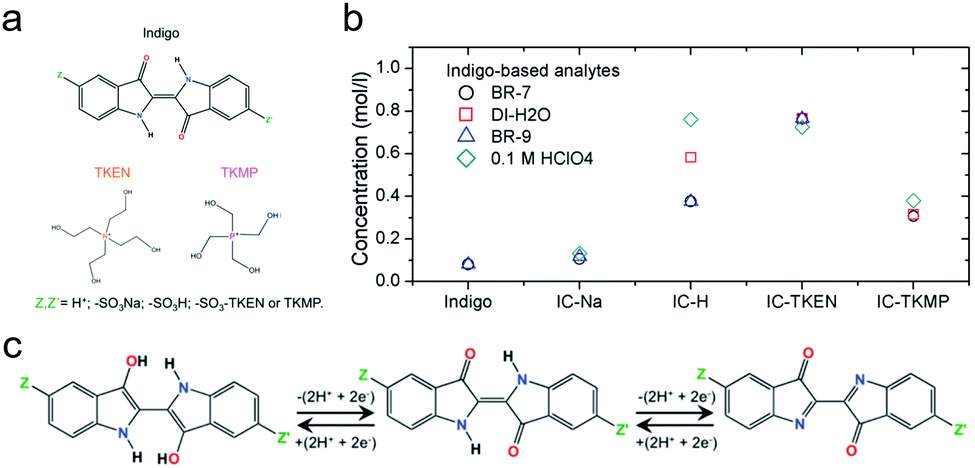 | ||
| Fig. 14 (a) Indigo derivatives with different functional groups and counter ions. (b) Solubility of indigo derivatives in different electrolytes. BR-7: Britton–Robinson buffer at pH 7. BR-9: Britton–Robinson buffer at pH 9. (c) Two-electron transfer reaction mechanism of indigo derivatives. Reproduced with permission from ref. 86. Copyright 2016, Royal Society of Chemistry. | ||
4. Radical-based redox species
In chemistry, radicals are chemical species bearing one or more unpaired or open-shell electrons. They typically possess high reactivity and tend to dimerize, polymerize, or even react with other molecules.96 They are often generated as intermediates in photochemical and thermal reactions, and can help initiate and propagate polymerization and combustion reactions.97–100 Though most radicals are regarded as unstable and intractable materials, they can be stabilized by steric and/or resonance effects.101 Typical examples are shown in Fig. 15.102 Tri(pentachlorophenyl)methyl and bis(diphenylenepropenyl)phenylmethyl are examples of carbon-centered radicals, while diphenylpicryl hydrazyl (DPPH) and triphenylaminium cationic radicals are nitrogen-centered. 2,4,6-Tri-t-butylphenoxyl, galvinoxyl, nitronyl nitroxide, and TEMPO are oxygen-centered radicals.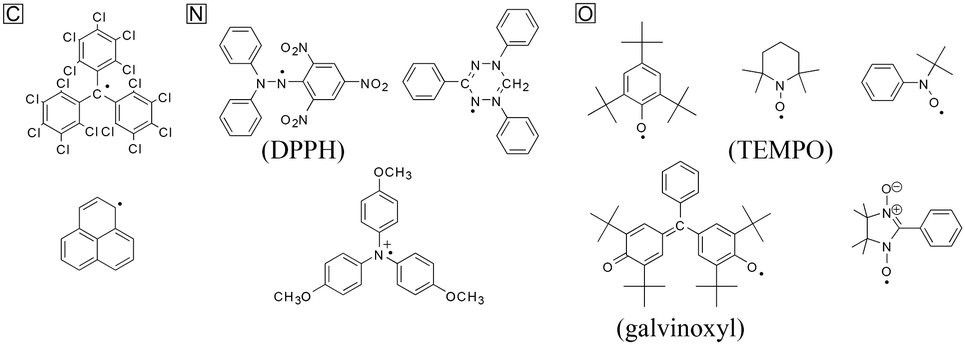 | ||
| Fig. 15 Some examples of stable radical molecules. Reproduced with permission from ref. 102. Copyright 2005, The Electrochemical Society. | ||
In this section, we will introduce four types of radical-based redox species. Although some types of molecules are not radicals themselves, they are still included considering that stable radicals may be generated as the redox reaction products. More broadly, it is common for organic molecules to generate radicals as redox reaction intermediates, similarly to the case of quinones in aprotic electrolytes.
4.1 Reaction mechanism
Free radicals tend to react with substrates via various modes, such as addition reactions, substitution reactions, and oxidation and reduction reactions.103–106 However, in organic chemistry, such oxidation and reduction processes differ from those reactions in inorganic chemistry. They commonly involve the gain or loss of oxygen or hydrogen instead of the electron transfer process in the electrochemical mode. In order to be applicable in energy storage systems, stable radical redox species must maintain reversible redox reactions accompanied by the formation of anions or cations after accepting or donating electrons. Because of the high reactivity of free radicals, they offer very fast kinetics, which is beneficial for improving power density.107 Some radicals can even undergo both oxidation and reduction reactions, opening the door for a bipolar battery.102 The specific reaction mechanisms of different radical-involved redox reactions will be introduced in detail in the next subsection.4.2 Molecular engineering method
The radical-based redox species have been utilized in solid state organic batteries.96 To extend their application to RFBs, the stability and solubility of radicals must be studied and enhanced. Moreover, the size of radical-based species can be tailored by appending radical moieties on non-conjugated polymer backbones. Essentially the general design principle and synthetic strategies on carbonyl compounds are also applicable to radical-based redox species. In the following subsections, specific functionalization methods for different types of radical-based redox species are discussed. | ||
| Fig. 16 The redox reaction mechanism of TEMPO. | ||
| Abbreviation | Structure | E/V | Concentration/M |
|---|---|---|---|
| TEMPO |
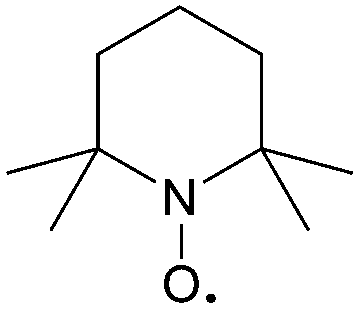
|
3.5 V vs. Li/Li+ | 2.0 M in EC/PC/EMC/LiPF6 |
| AcNH-TEMPO |
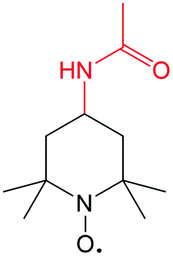
|
3.63 V vs. Li/Li+ | 0.5 M in 1 M LiBF4/PC |
| MeO-TEMPO |
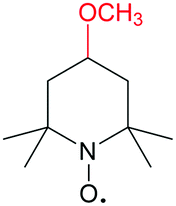
|
3.6 V vs. Li/Li+ | >2.5 M in LiTFSI/ACN |
| 4-HO-TEMPO |
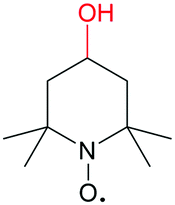
|
0.8 V vs. NHE | 2.1 M in water |
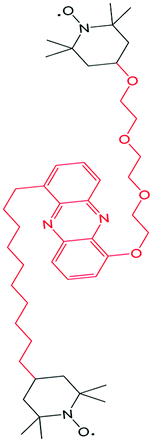
| TEMPO/phenazine combi-molecule |
|
0.6 V vs. AgCl/Ag | — |
The stability of the TEMPO radical is related to the delocalization of electrons from nitrogen onto oxygen and the steric protection by the four methyl groups. Due to the similarity in polarity with carbonate-based electrolytes, TEMPO has a high solubility of 5.2 M in the EC/PC/EMC solvent as reported by Wang et al.110 Even in the presence of LiPF6 as a supporting salt, TEMPO concentration can reach 2.0 M. When paired with Li metal as the anode, the battery voltage can reach up to 3.5 V, leading to a high theoretical volumetric energy density of 188 W h L−1. In a practical battery test, such a Li/TEMPO hybrid RFB delivers an energy density of 126 W h L−1 with an energy efficiency of about 70%. This high energy density is nearly five times that of conventional aqueous RFBs and exceeds most non-aqueous RFBs.5 Furthermore, the redox potential can be enhanced from 3.5 V to 3.63 V vs. Li/Li+ when using 4-acetamido substituted TEMPO (AcNH-TEMPO) in LiBF4/PC as the catholyte contributed to the electron-withdrawing effect of the functional group (Table 7). However, the solubility of both AcNH-TEMPO and AcNH-TEMPO+ species (0.5 M) is inadequate compared to TEMPO.111
Takechi et al. proposed another strategy to maximize the solubility of TEMPO derivative by forming a redox-active supercooled liquid.109 This “solvate ionic liquid” is a mixture of 4-methoxy-2,2,6,6-tetramethylpiperidine 1-oxyl (MT or MeO-TEMPO) as an organic radical and LiTFSI (LT) salt. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar coupling between MeO-TEMPO (at the N–O˙ site) and Li+ leads to the formation of a new ionic couple between (Li–MT)+ and TFSI−. The TFSI anion exhibits an uncommon plasticizing effect and induces the formation of this supercooled ionic liquid (MTLT). At a mole ratio of 1:1 with 17 wt% of water (MTLT (1/1) + H2O(17)), the concentration can be over 2 M, which is much higher than the solubility of MeO-TEMPO in water (≈0.3 M). In the catholyte, the addition of an appropriate amount of water helps decrease the viscosity, thus enhancing the electrochemical performance. In the proof-of-concept hybrid RFB test with Li metal as the anode and a solid electrolyte as the separator, a high energy density of 200 W h L−1 was delivered.
:1 molar coupling between MeO-TEMPO (at the N–O˙ site) and Li+ leads to the formation of a new ionic couple between (Li–MT)+ and TFSI−. The TFSI anion exhibits an uncommon plasticizing effect and induces the formation of this supercooled ionic liquid (MTLT). At a mole ratio of 1:1 with 17 wt% of water (MTLT (1/1) + H2O(17)), the concentration can be over 2 M, which is much higher than the solubility of MeO-TEMPO in water (≈0.3 M). In the catholyte, the addition of an appropriate amount of water helps decrease the viscosity, thus enhancing the electrochemical performance. In the proof-of-concept hybrid RFB test with Li metal as the anode and a solid electrolyte as the separator, a high energy density of 200 W h L−1 was delivered.
In addition to its application in organic RFB, TEMPO can be used in aqueous systems when modified with hydrophilic groups. For example, in 4-hydroxy-tetramethylpiperidin-1-oxyl(4-HO-TEMPO) the hydrophilic −OH group enhances its solubility in water up to 2.1 M.112 When coupled with methyl viologen (MV) as an anolyte, an all-organic-based RFB was built using cost-effective and environmentally benign NaCl as the supporting salt. The high ionic conductivity of the neutral electrolyte enabled the operation of the aqueous RFB at high current densities ranging from 20 to 100 mA cm−2. Furthermore, the battery delivered stable capacity for 100 cycles with nearly 100% Coulombic efficiency.112
Additionally, a novel combi-molecule with TEMPO with phenazine subunits was designed in a symmetric aqueous RFB.108 The redox-active subunits, TEMPO and phenazine, were covalently bound via TEG linkers. This combi-molecule features a redox voltage of 1.2 V in aqueous electrolytes. No capacity degradation was observed for over 1800 cycles with CE of up to 98.3%. However, the discharge voltage peaked at ∼500 mV with a voltage drop of ∼570 mV, which was attributed to the sluggish redox reaction of the phenazine subunit.108
The TEMPO redox moieties can also be affixed to polymer chains to prepare non-conjugated polymer-based redox species.61,113,114 This allows fine tuning of the molecular weight and exploitation of low-cost size-exclusion membranes instead of ion-exchange membranes (e.g. Nafion). The design principle and synthetic strategies are introduced in detail in Section 5.
For example, 2,5-di-tert-butyl-1,4-bis(2-methoxyethoxy)benzene (DBBB) has the solubility of only 0.4 M in carbonate-based electrolytes. It was found that redox center symmetry permits electrochemical activity and stability, while the incorporated poly ethylene oxide (PEO) chains help improve the solubility in carbonate-based electrolytes. Interestingly, the PEO substituents also change the physical forms of the functionalized molecules, from solid state (DBBB) to semi-liquid (ANL-10) to liquid (ANL-8, ANL-9), as shown in Fig. 17. The liquid form redox molecules open a new path towards RFBs with significantly increased energy density.117
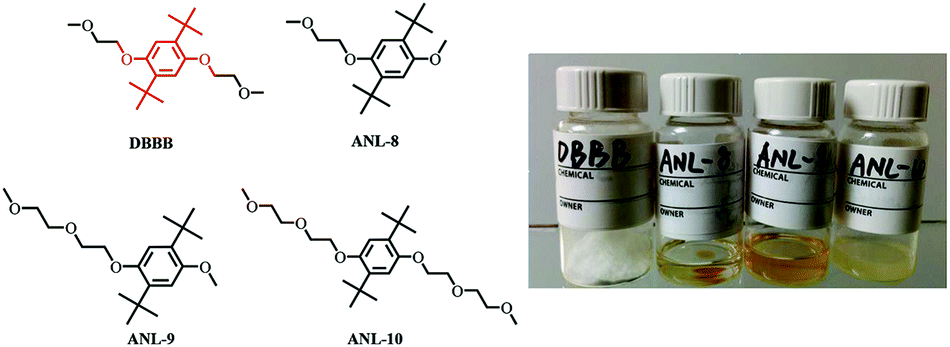 | ||
| Fig. 17 Chemical structures and images of a series of DBBB derivatives. Reproduced with permission from ref. 117. Copyright 2015, Wiley. | ||
Still using ANL-8 as the catholyte, Wang et al. investigated different types of all-organic nonaqueous RFBs.93,118 They found that different supporting electrolytes and salts have significant effects on the chemical stability of anions and cations. When 9-fluorenone (FL) was used as an anolyte, the TEA-TFSI/DME electrolyte offered the highest chemical stability to both radicals, and the flow cell maintained 90% of its original capacity over 50 cycles.118 Similar results were reported when using N-methylphthalimide as the anolyte.93
Furthermore, a minimalist structure with exceptional long-term stability in its oxidized form and high intrinsic capacity was generated from a combinatorial set of 1,4-dimethoxybenzene derivatives with different arrangements of substituents.119,120 Using a subtractive approach, it was found that the absence of the bulky non-polar tert-butyl groups in 2,3-dimethyl-1,4-dimethoxybenzene (23DDB) and 2,5-dimethyl-1,4-dimethoxybenzene (25DDB) increased the solubility in 0.5 M LiTFSI/PC electrolyte (2.0 M and 0.6 M, respectively). Moreover, methyl substitution was found to be sufficient to provide the steric protection against the radical addition reactions.120 CV measurements showed that 23DDB and 25DDB exhibit quasi-reversible redox reactions with a one-electron transfer. However, the electrochemical stability was substandard. The electron-withdrawing groups at the carbon-2 position were also synthesized (as compounds 9, 10, and 11 in Fig. 18). These electron-withdrawing groups were selected to uplift the redox potential and energy density. For –COOCH3 and –NO2 substituted compounds 9 and 10, the redox potential was enhanced to about 4.3 V and 4.45 V vs. Li+/Li, respectively. In the fluorine-substituted compound 11, the electrochemical oxidation was irreversible.120
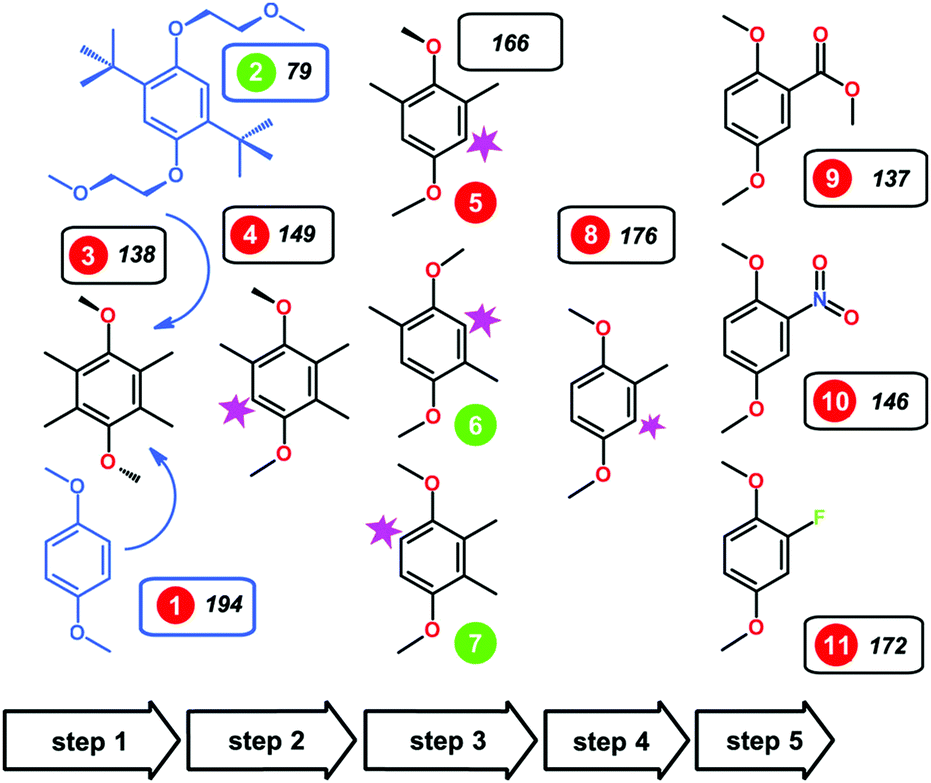 | ||
| Fig. 18 Alkoxybenzene-based redox compounds. The green/red circles indicate compounds that were reversibly/irreversibly oxidized in the CV trials. Reproduced with permission from ref. 120. Copyright 2016, Nature Publishing Group. | ||
Viologens have a long history as redox indicators in biological studies.123 The prototype viologen, 1,1′-di-methyl-4,4′-bipyridilium, is often known as MV, while other simple symmetrical bipyridilium species are commonly called substituent viologens.124
As shown in Fig. 19a, the viologen molecule exists in three main oxidation states.124 The colourless viologen is the most stable. It can accept one electron to form radical cations or two electrons to form the neutral state. The first reduction step is highly reversible, and the stability of the radical cation is attributed to the delocalization of the radical electron throughout the p-framework of the bipyridyl nucleus.124 Further reduction of the radical cations to the fully reduced state is less reversible.121 The 1,1′-substituents affect both the solubility and redox potential of viologens. Table 8 lists the effect of substituents and anions on the redox potential and solubility.31,121 In accordance with the increasing carbon number in the alky groups, the redox potential shifted negatively, correlating well with the design principle proposed above. 4,4′-Bipyridine has low solubility in water, but when it is methylated to MV, the aqueous solubility can reach up to 3.0 M.31 However, the functionalization of alkyl groups on the N position would lower the hydrophilicity of the corresponding viologen compounds. The maximum concentration of 4,4-diethyl bipyridinium diiodide (ethyl viologen, EV) and 4,4-dibenzyl bipyridinium dichloride (benzyl viologen, BV) decreases to 1.5 M and 0.04 M. As shown in Fig. 19c, both MV and EV display close redox potentials at −0.45 V vs. NHE. But BV exhibits an irreversible reaction with another anodic peak observed. Given its high solubility and low reduction potential, MV2+Cl2 can be used as an anolyte in RFBs. In the proof-of-concept demonstration by Liu et al., the MV-base anolyte can be paired with two kinds of redox species, 4-HO-TEMPO and ferrocene derivatives, as catholytes. The RFBs exhibit high theoretical energy density and excellent cycling performance.31,112 However, it is noteworthy that MV is toxic to human beings and animals, which may affect its practical applications in RFBs.
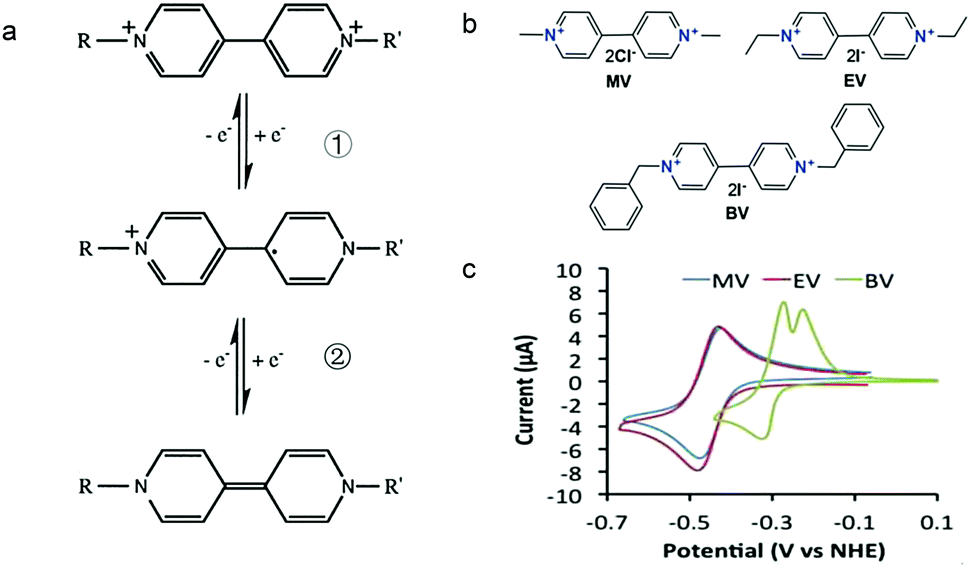 | ||
| Fig. 19 (a) Different oxidation states of a viologen molecule. (b) Three types of viologen derivatives. (c) CV of these three viologen compounds in (b). Reproduced with permission from ref. 112. Copyright 2016, Wiley. | ||
In addition to these chemical characteristics, the sizes of viologen-based redox species can also be tuned via synthetic modifications. Researchers have synthesized redox-active colloidal particles or redox-active polymers by copolymerization of 4-vinylbenzyl chloride and poly(vinylbenzyl chloride), which is reviewed in the next section.61,125
The Sanford group reported an evolutionary design of a series of pyridine-based anolyte materials for non-aqueous RFBs.126 The effect of functionalizing with an electron-withdrawing ester group in different positions is first examined using molecules 1a, 2a, and 3a in Fig. 20, in which 3a shows the highest reversibility. The electrochemical behavior was then optimized by varying the alkyl moieties in the ester substituents from molecules 3a–h. 3d maintained the highest stability because electron-donating groups can decelerate decomposition due to heterolytic cleavage of the (O)C–O bond. To further stabilize the redox reaction, a variety of N-functionalized pyridine molecules were prepared from 4a to 5d, and it is even possible to achieve a two-electron reduction process in some case. And a series of N-alkylpyridinium salts with various substituents and counter ions were synthesized to take advantage of the stronger N-alkyl bond, compared to that of N-BF3 or N-oxides. Among these species, 9a possesses the lowest equivalent weight and undergoes a highly reversible two-electron transfer reaction. Finally, a 9a analogue was prepared as 10a by replacing the esters with a ketone moiety. Because the fully reduced form of 10a is less basic than reduced 9a, possible parasitic reactions with water are restrained. The reaction mechanism of 10a is presented in Fig. 20, and this highly reversible two-electron transfer process offers accessibility to higher volumetric capacity anolytes for non-aqueous RFBs.
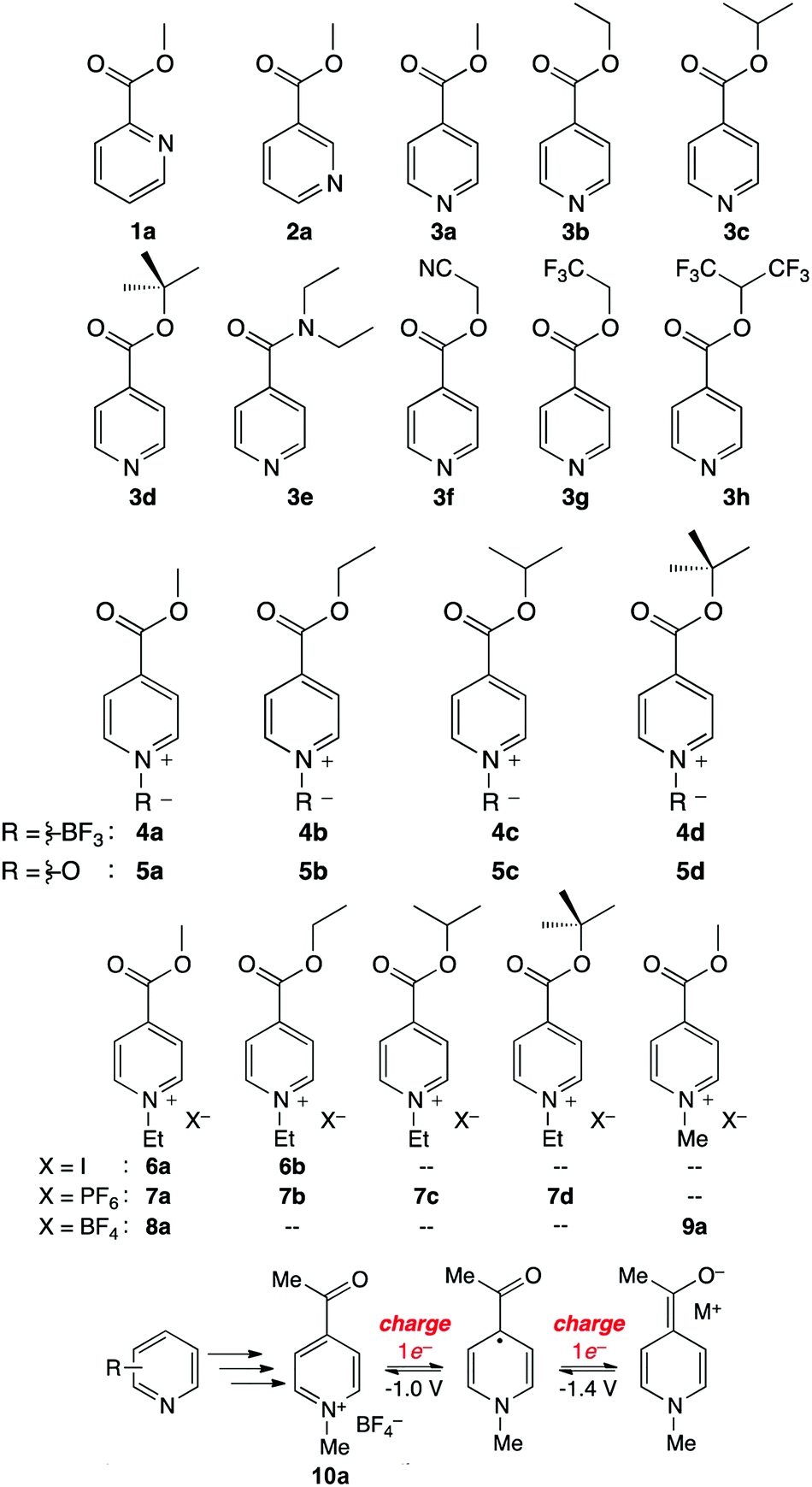 | ||
| Fig. 20 A series of pyridine-based redox species as anode-active materials for non-aqueous RFBs. The two-electron transfer mechanism of 10a is shown below. Reproduced with permission from ref. 126. Copyright 2015, American Chemical Society. | ||
Quinoxaline is another typical heterocyclic compound, and the pyrazine rings in quinoxaline act as the redox centers. When the pyrazine ring in the molecule is reduced, the non-π-conjugated pyrazine moiety is formed.127 It is considered as a promising redox species due to its high solubility (∼7 M in PC), low molecular weight, and multi-electron transfer reaction. Despite these features, the quinoxaline-based RFB is limited by the cell voltage given the high potential of quinoxaline as the negative active material. To address this issue, a series of substituents were functionalized on the quinoxaline molecule at different positions to tune its redox potential.128 As summarized in Fig. 21, substituting electron-donating methyl groups onto the 2 and 3 positions of the quinoxaline not only lowers the redox potential, but also enhances the redox activity. In comparison, when methyl groups were in the 5 and 6 positions, 6-methyquinoxaline showed slightly better performance than the quinoxaline, whereas the 5-methylquinoxaline demonstrated lower activity. The methyl group substitutions on the benzene ring had less impact on performance compared to substitutions on the nitrogen-bearing ring. While phenyl group substitutions on the nitrogen-bearing ring also enhanced the redox activity, its effects on redox potential were mixed. The upper redox event occurred at a lower potential while the lower redox event occurred at a higher potential compared to the pristine quinoxaline. After accounting for all the effects, 2,3,6-trimethylquinoxaline with methyl substitutions on the 2, 3, and 6 positions was selected as the anode-active material, which exhibits the lowest redox potential without sacrificing its electrochemical activity. When taking DBBB as the catholyte, the current efficiency and the energy efficiency of such an all-organic non-aqueous RFB were stabilized at 70% and 37% over 30 cycles, respectively.
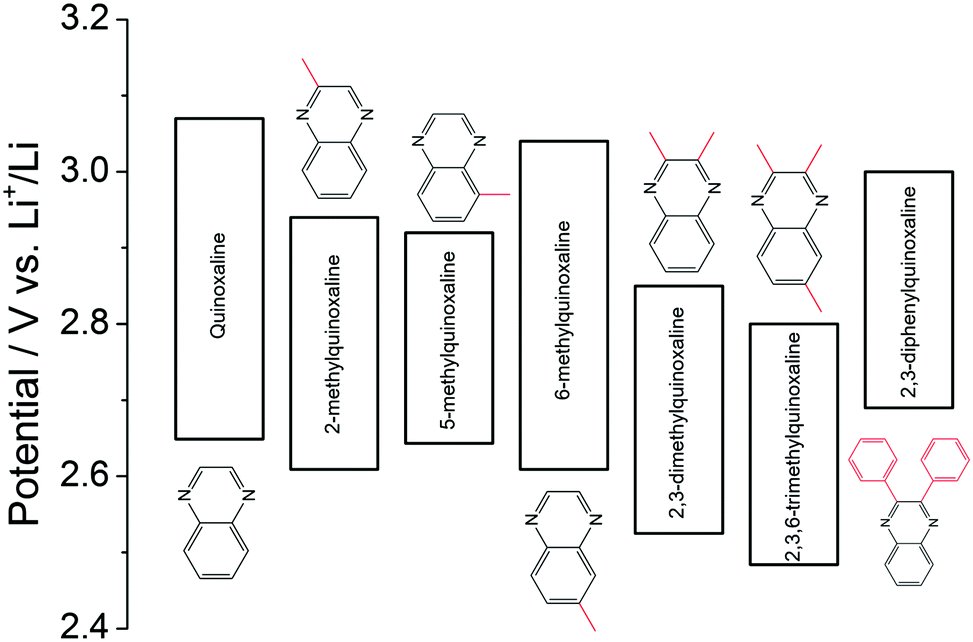 | ||
| Fig. 21 Effects of functional groups on the redox potential of quinoxaline classes. | ||
In addition to non-aqueous electrolytes, the chemistry of quinoxaline in aqueous electrolytes was also studied.129 It was found that the cations (Li+, Na+, K+), anions (Cl−, NO3−, OH−, SO42−, HCO3−, C2H3O2−), and pH values of electrolytes have a crucial impact on the electrochemical behaviour of quinoxaline. Among these factors, pH has the greatest impact. In chloride-containing supporting electrolytes (pH > 11), quinoxaline has a redox potential of −0.02 V vs. RHE, and it is soluble up to ∼4.5 M in water, exhibiting a two-electron transfer capability.129
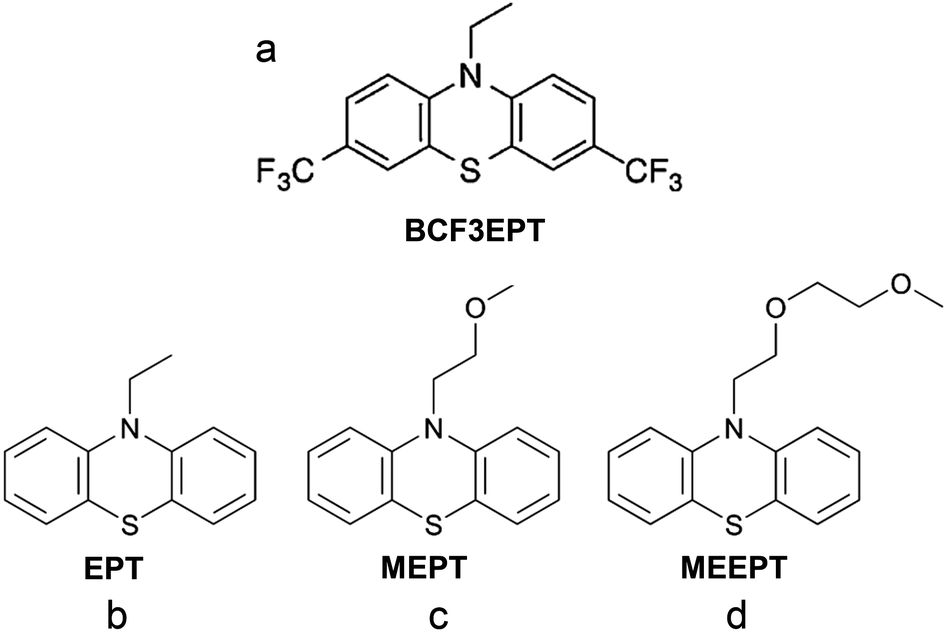 | ||
| Fig. 22 Chemical structures of (a) 3,7-bis(trifluoromethyl)-N-ethylphenothiazine (BCF3EPT), (b) N-ethylphenothiazine (EPT), (c) N-(2-methoxyethyl)phenothiazine (MEPT), and (d) N-(2-(2-methoxyethoxy)-ethyl)phenothiazine (MEEPT). Reproduced with permission from ref. 130. Copyright 2014, ref. 132. Copyright 2016, Royal Society of Chemistry. | ||
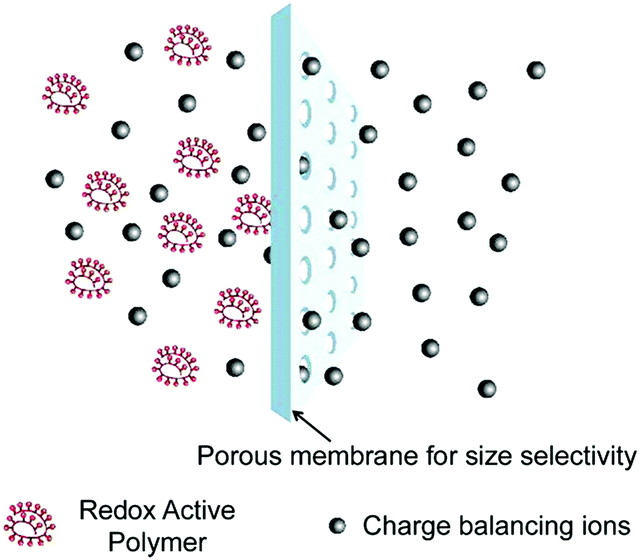 | ||
| Fig. 23 Schematic illustration of size exclusion in polymer-based RFBs. Reproduced with permission from ref. 125. Copyright 2014, American Chemical Society. | ||
5. Polymer-based redox species
Crossover of redox species is a critical issue to be taken into account for RFBs in large-scale use. A pervasive and irreversible crossover process always leads to capacity fade, low CE and underutilization of materials.13,59 A highly selective separator is needed to alleviate this issue. Meanwhile, the separator should also possess high ionic conductivity to meet the power density requirement.133,134 Ion-exchange membranes, consisting of polymeric backbones decorated with charged ion groups, represent the most widely used separators for RFBs.135 In spite of the high selectivity of an ion-exchange membrane contributed mainly by Donnan exclusion, the special structure often requires costly manufacturing processes, which preclude its wider applications.136 In this regard, cost-effective membranes based on physically blocking mechanisms, such as dialysis membranes, could potentially lower the capital cost of RFBs (Fig. 23).124,137 To achieve an efficient size-exclusion effect, redox active polymers possessing a wide size range and high tailorability can work as soluble or semi-solid charge storage materials for RFBs (Table 9).61,113,125,138–143| # | Molecule | Electrolyte | Solubility (M) | Redox potential | Ref. |
|---|---|---|---|---|---|
| 1 |

|
2 M NH4Cl/H2O | 150 g L−1 | 0.27 V vs. SCE | 138 |
| 2 |
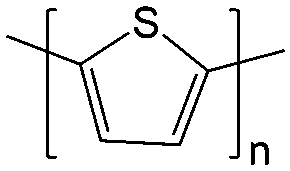
|
1.0 M TEABF4/PC | 0.1 eq. L−1 of thiophene |
Negative: −2 V vs. Ag/Ag+
Positive: +0.5 V vs. Ag/Ag+ |
139 |
| 3 |
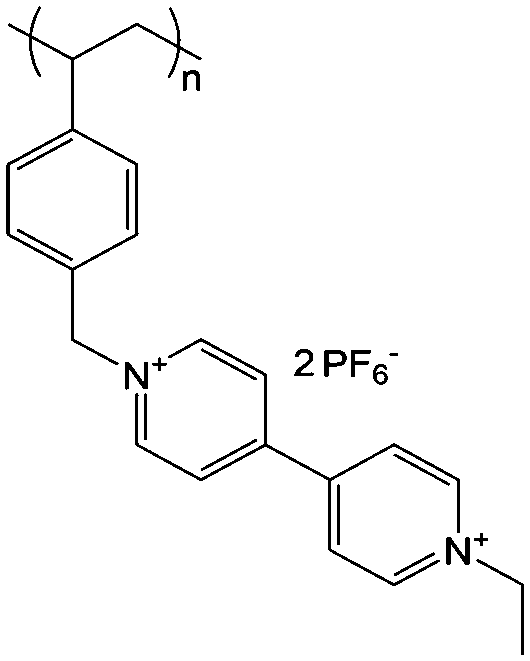
|
0.1 M LiBF4/ACN | 2 M in ACN or PC | −0.7 V vs. Ag/Ag+ | 125 |
| 4 |
|
0.1 M (n-C4H9)4NClO4/EC/DEC | 0.1 M | 1.0 V vs. Ag/AgCl | 140 |
| 5 |
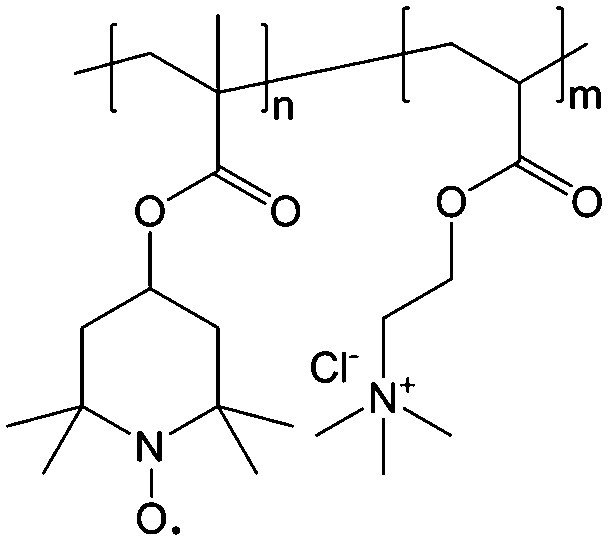
|
2 M NaCl/H2O | Equal to a charge storage capacity of about 10 A h L−1 | 0.7 V vs. Ag/AgCl | 61 |
| 6 |
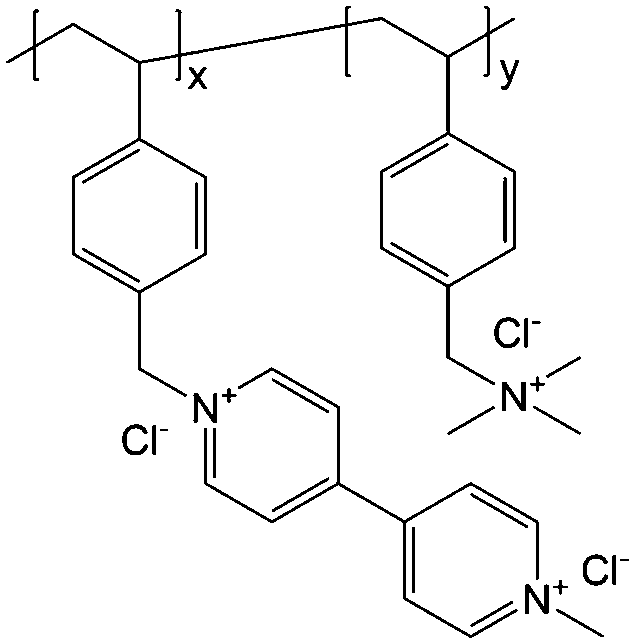
|
2 M NaCl/H2O | Equal to a charge storage capacity of about 10 A h L−1 | −0.4 V vs. Ag/AgCl | 61 |
| 7 |
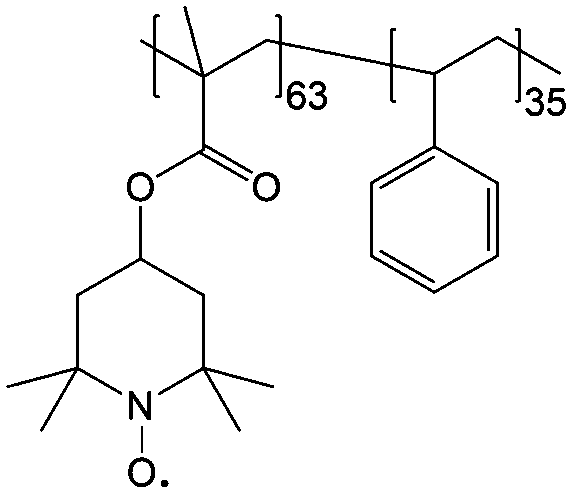
|
0.1 M Zn(ClO4)2 × 6H2O/EC/DMC/DEC | 0.1 M | 0.4 V vs. Ag/AgNO3 | 141 |
| 8 |
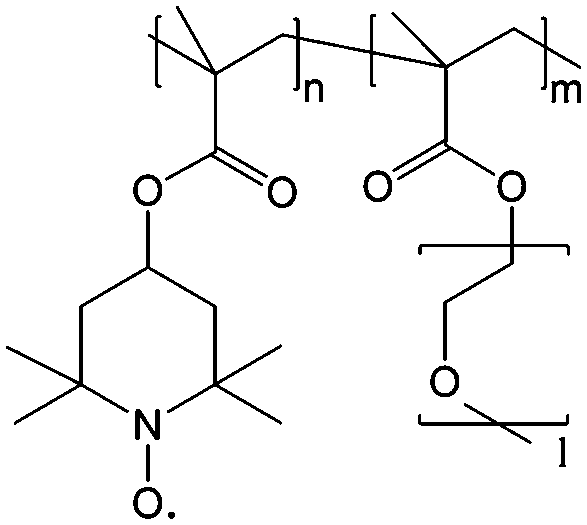
|
0.75 M Zn(ClO4)2 × 6H2O EC/DMC/DEC | 0.01 M | 0.4 V vs. Ag/AgNO3 | 113 |
| 9 |
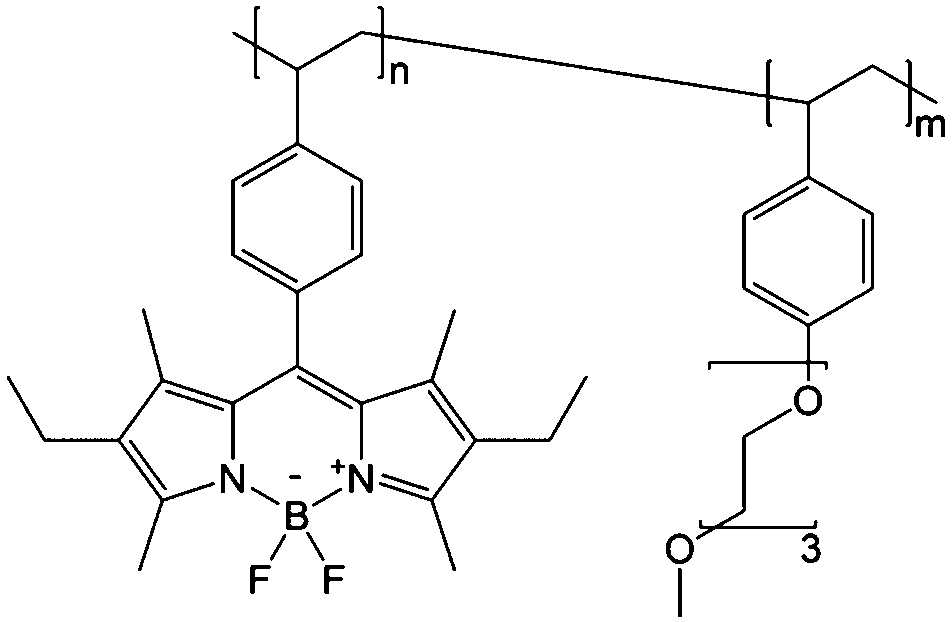
|
0.5 M Bu4NClO4/PC | 13 mg mL−1 | 0.7 V and −1.5 V vs. Ag/AgNO3 | 142 |
| 10 |
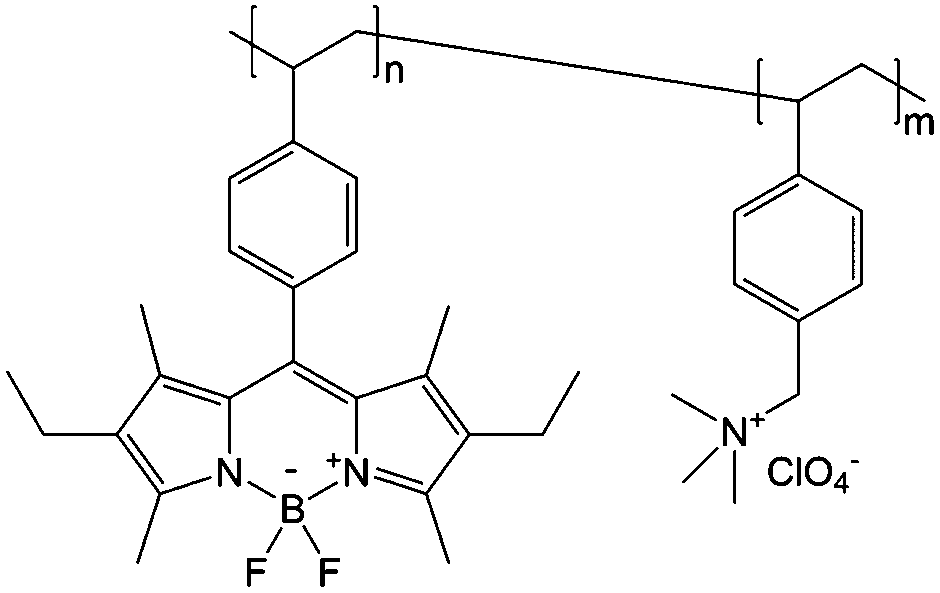
|
0.5 M Bu4NClO4/PC | 13 mg mL−1 | 0.7 V and −1.5 V vs. Ag/AgNO3 | 142 |
| 11 |
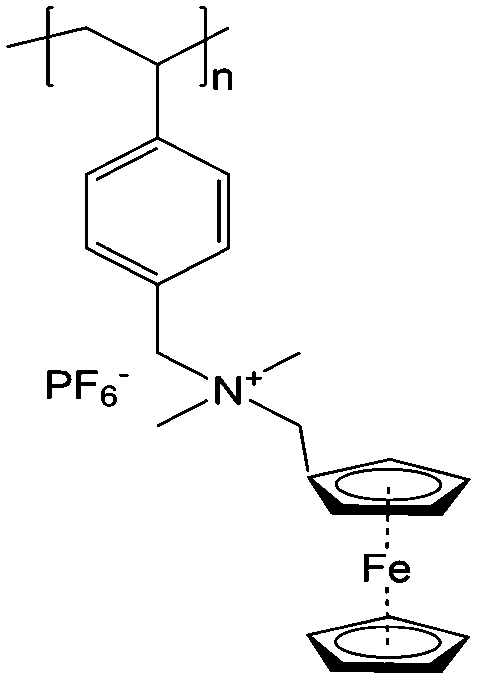
|
0.1 M LiBF4/ACN | 10 mM (by effective repeating unit) | 0.26 V vs. Ag/Ag+ | 143 |
5.1 Reaction mechanism
Polymer redox species can be classified into two groups, conjugated polymers and non-conjugated polymers.144,145 Conjugated polymer redox species have highly π-conjugated polymeric chains. Therefore, they can be reversibly doped to have negative or positive charges, while counter ions with opposite charges being trapped or released from the polymer matrix to maintain neutrality. Although intrinsic conjugated polymers have a low electrical conductivity, they may become highly conductive after doping.146 Since the discovery of conductive polymers in 1977, conjugated polymer redox species have been widely used as redox active organic molecules in supercapacitors and batteries.147Non-conjugated polymer redox species have redox active moieties covalently bonded to the polymer backbone as side chains rather than as part of the main chain. In a typical conjugated polymer, the fully planar structure leads to high conductivity at the doped state, but the doping level is commonly limited to 0.5 or less.71 In contrast, a redox-active non-conjugated polymer could have a consistently high doping level (0.8–0.9).148 The reaction mechanism of a non-conjugated polymer mainly relies on redox centers in the side chains.
5.2 Molecular engineering method
Molecular engineering to tune the size of organic molecules enables access to utilizing cost-effective membranes with alleviated crossover. Physically blocking mechanisms can be used to decrease the capital cost of RFBs, but the high molecular masses of redox active polymers also raise issues, such as low solubility, high viscosity, low energy density, and poor charge transport characteristics. Molecular engineering methods allow tailoring of redox active moieties, other functional pendant groups, macromolecular size or even backbone structure to achieve desirable properties. These diverse synthetic strategies make polymer-based redox species potentially attractive in RFBs.Oh and co-workers139 investigated polythiophene as a redox couple in RFBs. The polymer becomes conductive when electrons are added or removed from the thiophane unit in the conjugated p-orbitals via n- and p-doping, which corresponds to redox potentials of about −2.0 V vs. Ag/Ag+ and +0.5 V vs. Ag/Ag+, respectively. As a result, this material could be used as a bipolar redox active species and provide the high cell potential of 2.5 V in all-organic RFBs.139 The polythiophene microparticles dispersed in a 1 M TEABF4/PC solution displayed stable charge/discharge performance with an energy efficiency of 60.9% at 0.5 mA cm−2. Limited by the poor conductivity of the polythiophene suspension, the utilization of active materials could only reach 34.5%. Future improvement can be expected by molecular size tuning or battery structure optimization.
Furthermore, the redox active moieties can also be extended to other redox species in addition to viologen-based polymers. In the work by Nishide and co-workers, TEMPO-crowded bottlebrush polymers were synthesized and applied in RFBs.140 Polymer structures have notable effects on the charge transport properties. For example, it has been revealed that compared with linear polymers, network-type polymers increase the effective distance of the charge transport.152 Because of the defined molecular dimension and relatively low solution viscosity in comparison with corresponding linear polymers, this bottlebrush polymer in a RFB exhibited 95% efficiency of the theoretical charge capacity.
High molar masses induce redox active polymers dissolved in electrolytes to maintain high viscosity, which becomes a major challenge in RFBs. Special shaped polymers such as a micellar structure can be designed to counteract this phenomenon. A redox active polymer with micellar structures can be achieved by utilizing a diblock copolymer composed of a polar poly(TEMPO methacrylate) (PTMA) and a non-polar poly(styrene) (PS) block. This enables the formation of core-corona micelles in organic carbonates.141 The synthesized PTMA-b-PS can act as a catholyte in a polymer/Zn RFB and the constructed device features an excellent capacity retention of 95% after 1000 cycles.
In addition to the non-aqueous systems, molecular engineering on redox active polymers can also lead to soluble polymers in aqueous electrolytes, which enables higher conductivity and lower cost compared to non-aqueous electrolytes. When designing the target molecules, redox active polymers should behave as stable redox species in water, which requires hydrophilic units to enhance water solubility. The Schubert group studied various water-solubility-enhancing comonomers for polymers containing either TEMPO or viologen as redox active units.114 The optimized redox active polymers use a quaternary ammonium cation pendant group to enhance water-solubility. This aqueous all-polymer-based RFB has an energy density of 10 W h L−1, current densities of up to 100 mA cm−2, and stable long-term cycling capability.61 Moreover, it uses an environmentally benign sodium chloride solution and cheap, commercially available filter membranes rather than organic solvents or highly corrosive acid solutions as electrolytes and expensive membrane materials.
To further enhance the size-exclusion effect, another promising class of redox active materials based on cross-linked polymer spheres was developed with a wider range of sizes (from tens to thousands of nanometres).153 The microscale dimensions of these redox active colloids dramatically decrease crossover to near-zero. Ferrocene-based redox active colloids paired with viologen-based redox active colloids could cycle efficiently and reversibly in a non-aqueous RFB with a size-selective separator. Due to the modular nature of redox active colloid synthesis, this paradigm can be used for other redox active pendant moieties.
To gain insight into the chemistry of polymer-based RFBs, a systematic molecular design approach was used to study the impact of linker and redox-pendant electronic interactions on the performance of viologen redox active polymers.154 It was found that the tether length of redox active dimers controlled electron self-exchange kinetics and the rigidity influenced their charge transfer properties. The number of redox radical pendants per unit can be doubled for faster kinetics for enhanced performance, because of the stronger electronic interaction between radicals. However, for the polymer bound radical pendants, extreme proximity would induce a decrease in chemical reversibility. Therefore, in designing new redox active polymers, electronic interactions and backbone rigidity need to be balanced for optimized electrochemical performance.
Another notable work is a novel RFB designed by the Schubert group employing polymers relying on the same redox active moieties as both positive and negative species.142,155 Boron dipyrromethenes (BODIPYs), which are frequently used as fluorescent dyes,156 were selected as the redox active units. A styrene-containing monomer was copolymerized with two kinds of solubility-enhancing comonomers to prepare two different BODIPY copolymers. Characterization of the electrochemical properties of the styrene-containing monomer showed two quasi-reversible redox reactions at −1.51 and 0.69 V vs. Ag/AgNO3. The bipolar redox unit makes it possible to build the polymer RFB solely based on the same redox-active groups. In the battery test, a stable cycling over 90 consecutive cycles was achieved after the first 10 cycles, and the average discharge voltage reached 1.28 V. This work also demonstrated that molecular engineering methods enable preparation of novel polymer redox species for RFB applications.
In contrast to the large volume of literature on electrode-confined polymer redox species, studies on polymer redox species in RFBs are incipient because of some technical concerns caused by the increased viscosity of electrolytes. Rheological properties of polymer-based electrolytes should be taken into consideration in future research.
6. Metal complex-based redox species
Metal complexes, also known as coordination compounds, typically consist of a metal center and a surrounding array of bound ligands. Different from conventional metal ions as redox species, metal complexes maintain more flexibility to tailor both physical and chemical properties by organic ligand selection or functionalization. Sometimes the complexing agents can also undergo reversible redox reactions, which further promote the electroactivity of metal complex-based redox species.157 Moreover, metal complexes generally possess a larger molecular size compared with conventional metal ions in aqueous RFBs, leading to alleviated crossover issue in the battery test.158 Therefore, metal complexes are regarded as good examples of the evolution from metal-based to organic-based RFBs. In this section, we review a series of molecular engineering methods to tailor the properties of metal complexes, including redox potential, solubility, charge transfer and diffusion.6.1 Reaction mechanism
Metal complexes can be classified according to the nature of ligands. In this section, mainly two types of metal complexes will be introduced, namely conventional metal–ligand complexes and metallocene-based redox species. Generally speaking, the redox reaction of both of the two types of metal complexes relies on the metallic coordination center and the effect of ligands.159 In conventional metal–ligand complexes, the complexing agents bind to the metal centers via lone pairs of electrons residing on the main group atoms of the agents. The electron-transfer processes can be either inner sphere electron transfer or outer sphere electron transfer depending on the effects between metal centers and ligands.160 During an inner sphere electron transfer reaction, two coordination centers are bridged by a ligand with two lone electron pairs, and electrons are transferred via the bridging ligand. Any other electron transfer processes aside from inner sphere electrons can be assumed to be outer sphere. Because of the strong interaction between metal centers bridged by ligands, inner sphere electron transfer is usually enthalpically more favorable. Nevertheless, it is entropically less favorable given the more ordered structure in inner sphere electron transfer than in outer sphere electron transfer.161As a typical type of organometallic-based redox species, metallocenes represent another important class of electroactive materials for both aqueous and non-aqueous RFBs. A metallocene is a compound consisting of two cyclopentadienyl (Cp) ligands bound to a metal center in the oxidation state of II,162 and the Cp rings in metallocenes provide more flexibility to tune their physical and chemical properties via rational synthetic procedures.157 The intrinsic coordination and chemical reactivity of metallocenes are mainly regulated by the d-orbitals in the metal center and p-orbitals of the Cp rings.162 Different electronic structures and valence electrons along with the variation of metal–carbon bonds contribute to distinct properties. The sandwich structure of metallocene is very robust with a variety of metal centers with the number of d electrons ranging from 14 to 20. The redox reactions of metallocenes are accompanied by a change in the valence state of the metal centers.
6.2 Molecular engineering method
The tailorable properties of metal complexes are discussed to achieve better electrochemical performance of RFBs. For conventional metal-coordinated complexes, the transition metal usually acts as the charge transfer center, while the potential, solubility, kinetic rates, and stability can be tuned with different ligands. In complexes with redox-active ligands, charges can also be stored in complexing agents in addition to the central metals as redox moities.163As for metallocene-based redox species, they share many characteristics of organic materials and undergo reactions similar to aromatic compounds, enabling the preparation of a variety of substituted derivatives.158 Therefore the design principle for tailoring the properties of organic redox species is also applicable to metallocene-based electroactive molecules. On the other hand, the intrinsic properties of metallocenes mainly rely on the metal centers, and tailored properties of metallocenes can also be achieved by rational screening of the redox-active metal centers.
Since then, other metal complexes of Ni, Fe, Co, V, Cr, and Ce with various ligands, such as bpy,164 phenanthroline(phen),165 triethanolamine (TEOA),166 acetylacetonate (acac),167–171 and ethylenediaminetetraacetate (EDTA),165 have been applied in RFBs as well. A summary of metal–ligand complexes as electroactive materials for RFBs is shown in Table 10.26,163,164,167–181 Although the concentration of metal complexes has yet to be improved for practical applications, the delivered cell voltage in non-aqueous electrolytes is higher than those of conventional aqueous RFBs. Besides, with proper screening of the ligands, the redox potential of the metal complex with a certain coordination center can be tailored in a broad range. By doing this, the delivered cell voltage can be rationally optimized via increasing or decreasing the redox potentials of cathode-active or anode-active complexes.
| Anode redox reaction | Cathode redox reaction | Concentration (M) | Cell open circuit voltage (V) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| acac: acetylacetonate complexes. acac*: ester functionalized acetylacetonate complexes. acacen: bis(acetylacetone)ethylenediamine. mTEA: 1-[bis(2-hydroxyethyl)amino]-2-propanol. | |||||
| [Ru(bpy)3]2+ + e− ↔ [Ru(bpy)3]+ | [Ru(bpy)3]2+ ↔ [Ru(bpy)3]3+ + e− | 0.02 | 2.6 | 0.1 M Et4NBF4/ACN | 26 |
| [V(mnt)3]2− + e− ↔ [V(mnt)3]3− | [V(mnt)3]2− ↔ [V(mnt)3]−+ e− | 0.02 | 1.1 | 0.1 M TBAPF6/ACN | 163 |
| [Fe(bpy)3]2+ + e− ↔ [Fe(bpy)3]+ | [Fe(bpy)3]2+ ↔ [Fe(bpy)3]3+ + e− | 0.4 | 2.41 | 0.5 M TEABF4/PC | 172 and 173 |
| [Ni(bpy)3]2+ + e− ↔ [Ni(bpy)3]+ | 0.2/0.4 | 2.2 | 0.5 M TEABF4/PC | 164 | |
| [Co(bpy)3]2+ + e− ↔ [Co(bpy)3]+ | 0.2/0.2 | 2.16 | 0.5 M TEABF4/PC | 174 | |
| [Ru(acac)3] + e− ↔ [Ru(acac)3]− | [Ru(acac)3] ↔ [Ru(acac)3]+ + e− | 0.002 | 1.17 | 0.05 M TEABF4/ACN | 173 |
| [Mn(acac)3] + e− ↔ [Mn(acac)3]− | [Mn(acac)3] ↔ [Mn(acac)3]+ + e− | 0.05 | 1.1 | 0.5 M TEABF4/ACN | 175 |
| [Cr(acac)3] + e− ↔ [Cr(acac)3]− | [Cr(acac)3] ↔ [Cr(acac)3]+ + e− | 0.05 | 3.4 | 0.5 M TEABF4/ACN | 176 |
| [V(acac)3] + e− ↔ [V(acac)3]− | [V(acac)3] ↔ [V(acac)3]+ + e− | 0.05 | 2.2 | 0.5 M TEABF4/ACN | 167–171 |
| [Co(phen)3]+ + e− ↔ [Co(phen)3]2+ | [Fe(phen)3]2+ ↔ [Fe(phen)3]3+ + e− | 0.01/0.01 | 2.1 | 0.3 M TEAPF6/ACN | 178 |
| [V(acac*)3] + e− ↔ [V(acac*)3]− | [V(acac*)3] ↔ [V(acac*)3]+ + e− | 0.1 | 2.1 | 1 M TEABF4/ACN | 179 |
| [Co(acacen)] + e− ↔ [Co(acacen)]− | [Co(acacen)] ↔ [Co(acacen)]+ + e− | 0.01 | 2.0 | 0.1 M TEABF4/ACN | 180 |
| [Fe(TEOA)(OH)]−+ e− ↔ [Fe(TEOA)(OH)]2− | [Co(mTEA)(H2O)]− ↔ [Co(mTEA)(H2O)] + e− | 0.5/0.5 | 0.93 | 5 M NaOH/H2O | 181 |
The Yan group systematically compared a series of soluble iron complex-based redox species, as summarized in Fig. 24.166 It is shown that the redox potential of iron as the metal center ranges from ∼−0.8 V (vs. SHE) to ∼1.5 V (vs. SHE) when complexed with different ligands at different pH values. Considering the huge potential gap, it is possible to build an all-soluble and all-iron aqueous RFB. Among all the iron complexes listed, iron–triethanolamine (Fe–TEOA) and iron–cyanide (Fe–CN) molecules were taken as negative and positive redox species, respectively. Such a RFB, which integrates electroactive species with the same redox-active element and different coordination chemistries, has a cell voltage of 1.34 V. At a current density of 40 mA cm−2, the stabilized Coulombic efficiency is between 80% and 90%. The polarization curve test shows that the power density of such an all-iron RFB can reach 160 mW cm−2. Compared with conventional all-Fe-based RFB, the enhanced power density is mainly ascribed to the facile reaction kinetics corroborated by the following electrochemical analysis.
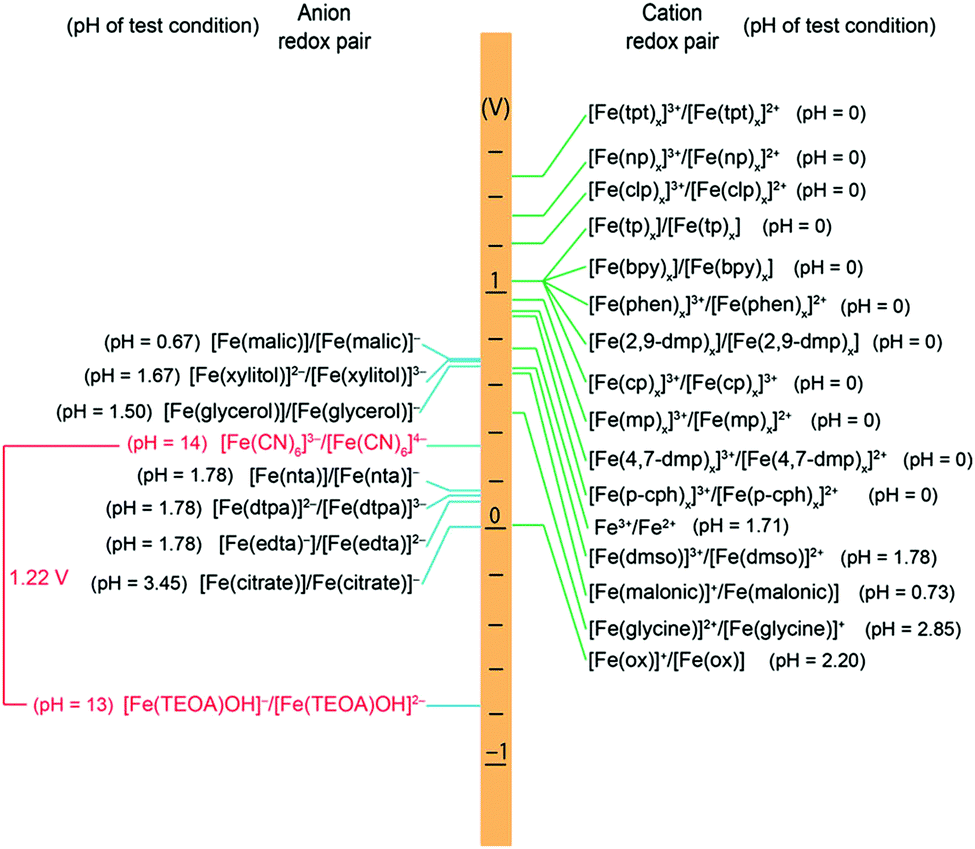 | ||
| Fig. 24 Redox pairs of iron complexes that have been tested for RFB applications. Each redox pair is represented by its formal potential vs. SHE at the corresponding pH of its test conditions. Reproduced with permission from ref. 166. Copyright 2016, American Chemical Society. Abbreviations: tpt: tripyridinetriazine. np: 5-nitro-o-phenanthroline. clp: 5-chloro-o-phenanthroline. 2,9-dmp: 2,9-dimethyl-o-phenanthroline. tp: terpyridine. cp: 4-cyanopyridine. mp: 4-methy-o-phenanthroline. 4,7-dmp: 4,7-dimethyl-o-phenanthroline. p-cph: 2-pyridinecarboxaldehyde. CN: cyanide. nta: nitrilotriacetic acid. dtpa: diethylenetriaminepentaacetic acid. ox: oxalate. | ||
In addition to redox potentials, the solubility of metal complexes can also be rationally tailored via molecular engineering. The Sanford group synthesized metal complexes of tridentate bipyridylimino isoindoline (BPI) ligands which can bind more intensely to the metal center so as to inhibit ligand dissociation and render complexes with enhanced stability and electrochemical reversibility.182Fig. 25 shows the complexes of BPI derivatives with various metals and ligands, and M(L1)2 indicates a metal center bearing an unsubstituted BPI ligand. The solubility of metal complexes can be improved by decreasing the molecular symmetry to suppress ordered crystal packing via functionalizing ether substituents. In addition to enhanced solubility, the BPI derivatives decorated with electron-donating alkoxy functional groups possess more negative redox potentials compared with the pristine metal complexes. As presented in Fig. 25b, more than 3 orders of magnitude larger solubility can be achieved via functionalizing a diethyleneglycoxy (ODG) substituent [complexes Ni(L4)2–Ni(L6)2] on the pristine complex Ni(L1)2. It is noted that Ni(L6)2 can be isolated as a viscous liquid that is partially miscible with ACN to prepare a flowable one-phase electrolyte with more than 700 mM metal complex as electroactive species. Moreover, the effect of charge shielding of ligands on various metallic coordination centers is also explored, which further dictates the solubility of metal complexes, as shown in Fig. 25c. It is found that the shielding effect on early transition metals is generally less strong than late transition metals. Consequently complexes based on early metals are generally more polar and exhibit higher solubility in polar solvents versus late metals.
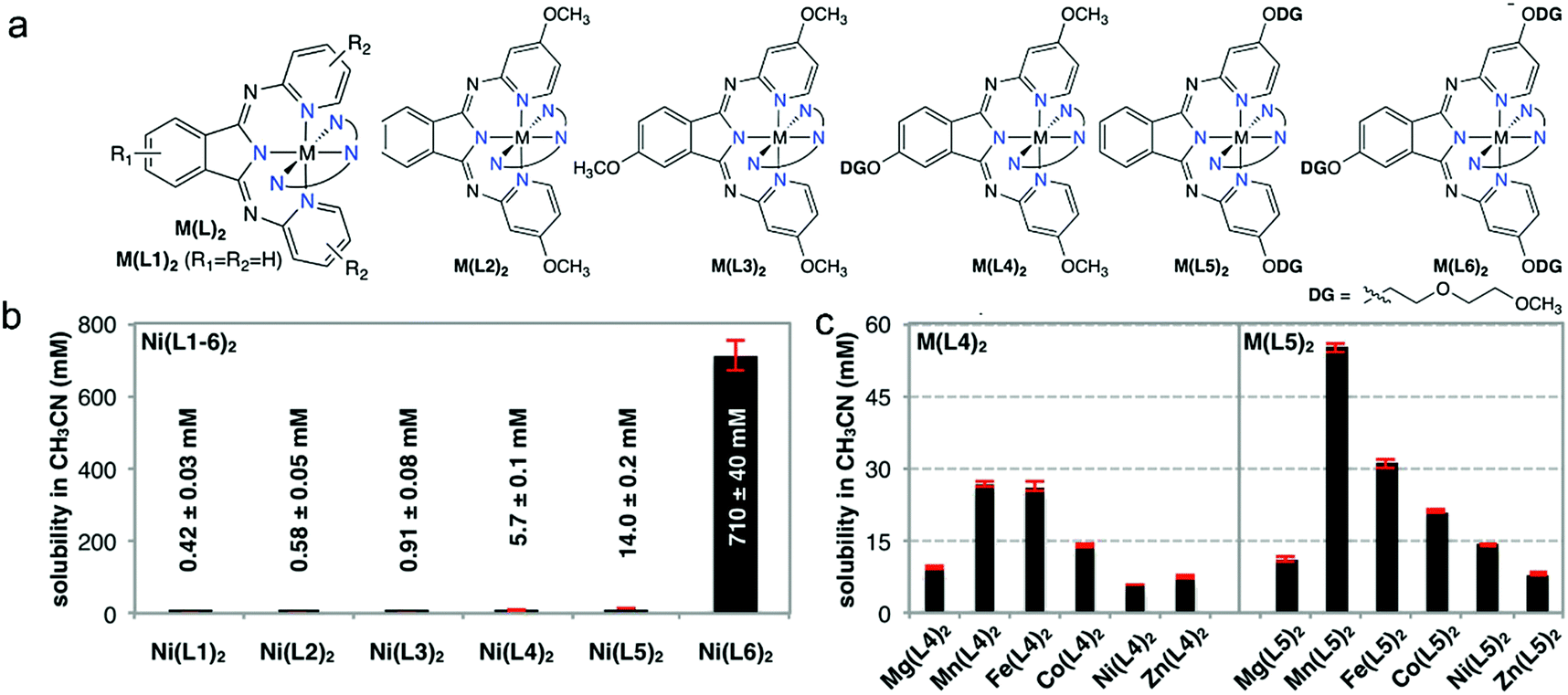 | ||
| Fig. 25 (a) Metal complexes of BPI derivatives with varying metals. (b) Effect of the ligand on the solubility of Ni complexes Ni(L1)2–Ni(L6)2. (c) Effect of the metal center on the solubility of complexes with ligand M(L4)2 (left) or M(L5)2 (right). Reproduced with permission from ref. 182. Copyright 2016, American Chemical Society. | ||
 | ||
| Fig. 26 A ferrocene library for aqueous and non-aqueous RFBs. | ||
| # | Molecule | Electrolyte | Solubility (M) | Redox potential V (vs. SHE) | Ref. |
|---|---|---|---|---|---|
| 1 |
|
1.2 M LiTFSI/EC/PC/EMC (4:1:5) |
0.04 | 0.3 | 188 |
| 2 |
|
1.2 M LiTFSI/EC/PC/EMC (4:1:5) |
0.85 | 0.5 | 188 |
| 3 |
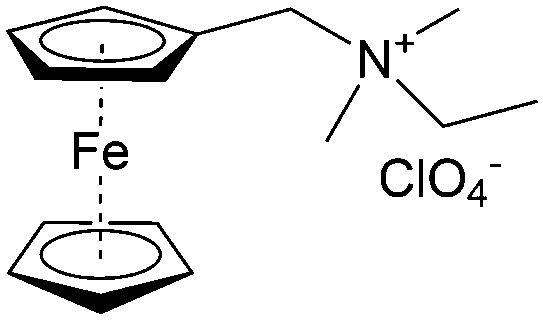
|
EC/PC/EMC (4:1:5) |
0.63 | 0.45 | 189 |
| 4 |
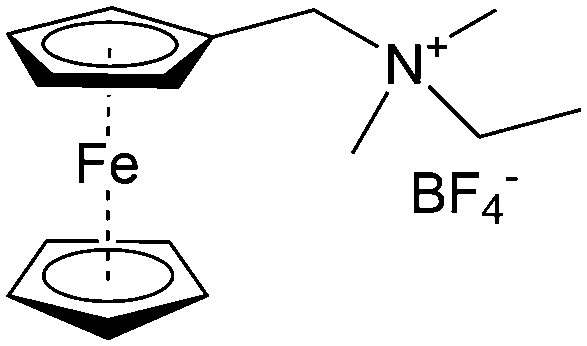
|
EC/PC/EMC (4:1:5) |
0.4 | 0.49 | 189 |
| 5 |
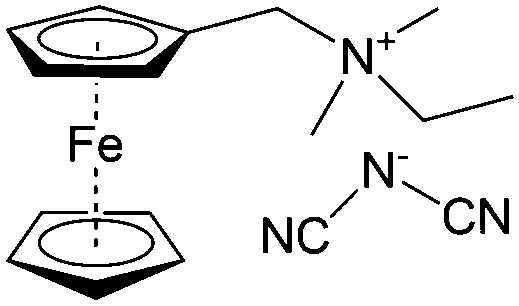
|
EC/PC/EMC (4:1:5) |
2.08 | N/A | 189 |
| 6 |
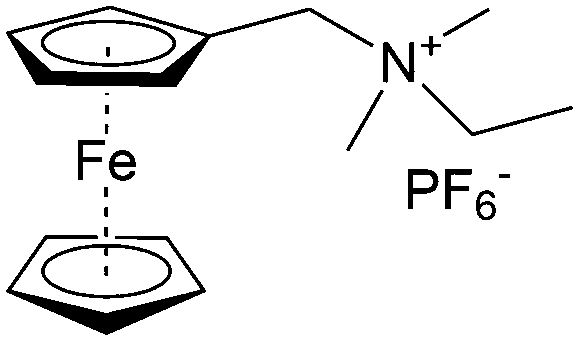
|
EC/PC/EMC (4:1:5) |
1.71 | 0.44 | 189 |
| 7 |
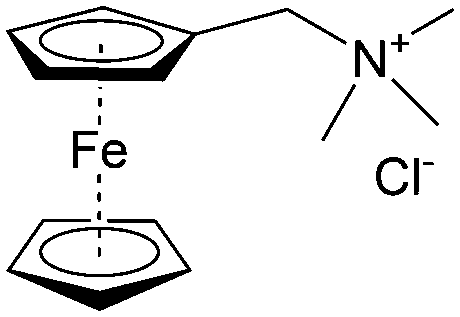
|
H2O | 4 | 0.61 | 31 |
| 8 |
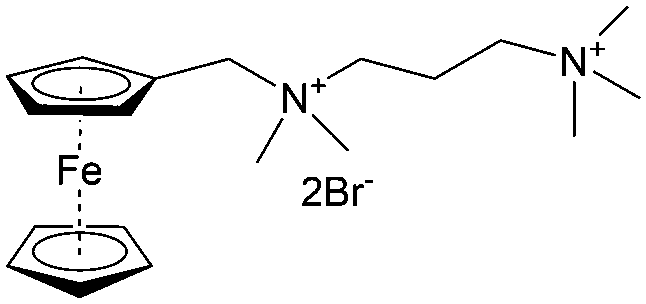
|
H2O | 3.1 | 0.61 | 31 |
| 9 |
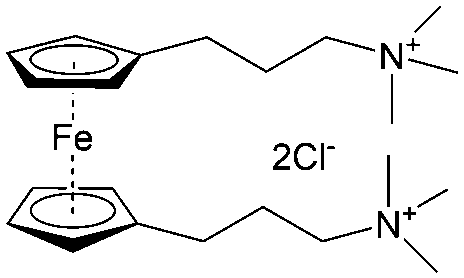
|
H2O | 1.9 | 0.39 | 191 |
| 10 |
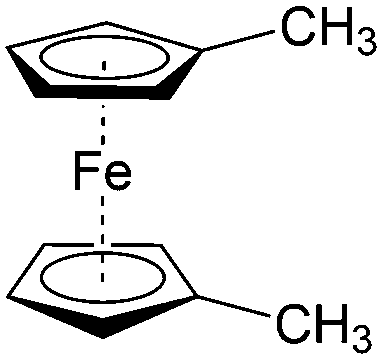
|
EC/DEC at 50 °C | 3 | 0.25 | 192 |
| 11 |
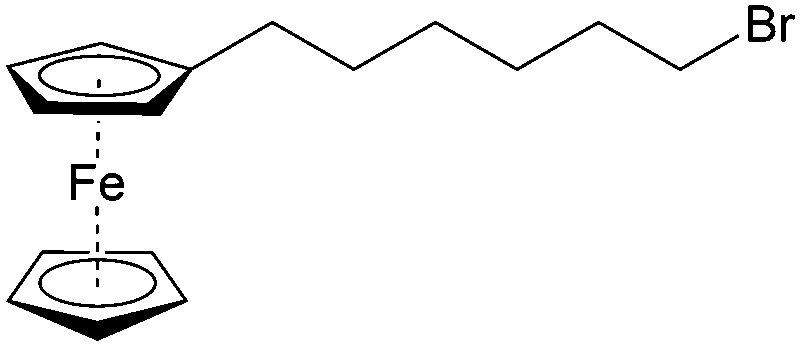
|
1 M LiTFSI/EC/DEC | N/A | 0.2 | 193 |
| 12 |
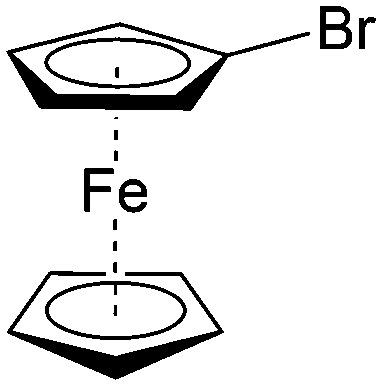
|
1.0 M TEAPF6/ACN | N/A | 0.25 V vs. Ag/Ag+ | 177 |
| 13 |
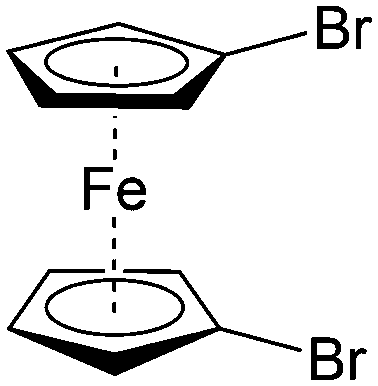
|
1 M LiPF6/DMC:EC (1:1) |
N/A | 0.55 | 194 |
| 14 |
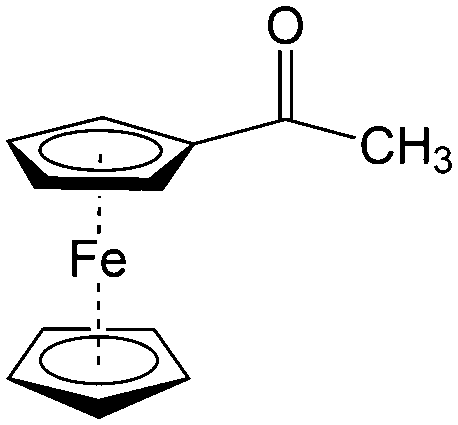
|
1.0 M LiPF6/PC | 0.81 | 0.65 | 195 |
| 15 |
|
0.5 M LiTFSI/DOL | 1.5 | −1.1 | 196 |
| 16 |
|
0.5 M LiTFSI/DOL | 0.1 | −1.65 | 196 |
Among various sandwich compounds, ferrocene attracts the most research attention in the field of energy storage.186 Golovin and co-workers initially employed ferrocene and its derivatives as effective electrolyte additives for overcharge protection given the appealing characteristics of ferrocene-based species, such as high reversibility, fast reaction kinetics, and compatible redox potential with Li ion batteries.187 However, the direct adoption of ferrocene as a redox active material for RFBs is limited mainly by its low solubility. In some typical carbonate-based organic electrolytes, the solubility of ferrocene is only about 0.04 M in the presence of Li salt, which is far from the practical requirement for RFBs.188 To address this issue, Wang and co-workers decorated an ionic charged tetraalkylammonium pendant on the Cp ring accompanied by a TFSI− counter anion (#2, Table 11), giving rise to a 20-fold increase in its solubility.188 The subsequent NMR and DFT test revealed a fundamental understanding of the solvation chemistry of the functionalized ferrocene, which is attributed to intensified solvent–solute interactions through the ionic charged pendant. The same groups then studied the effect of various counter anions on tetraalkylammonium ionized species appended to ferrocene, including TFSI−, perchlorate (#3, Table 11), tetrafluoroborate (#4, Table 11), dicyanamide (#5, Table 11), and hexafluorophosphate (#6, Table 11). The synthetic conditions are presented in Fig. 27.189 Though PF6− and BF4− anions can also improve solubility, the poor chemical stability precludes their use in RFBs. Therefore, when designing novel organic electroactive molecules, the reactivity should be taken into account in addition to solubility.
 | ||
| Fig. 27 Effects of counter ions on the properties of ferrocene-based redox species. | ||
Ferrocene is almost insoluble in water. Based on the same design principle and synthetic strategy, the aqueous solubility of ferrocene can also be boosted for application in aqueous RFBs.31,190 In the work by the Liu group, they synthesized (ferrocenylmethyl)trimethylammonium chloride (FcNCl, #7, Table 11) and N1-ferrocenylmethyl-N1,N1,N2,N2,N2-pentamethylpropane-1,2-diaminium dibromide (FcN2Br2, #8, Table 11) through a one-step N-alkylation reaction.31 Due to the strongly hydrophilic effect of the tetraammonium pendant and halide counter ion, the solubility of ferrocene was increased to 4 M and 3.1 M in water, respectively. When paired with MV as negative species, the FcNCl-based aqueous RFB has a theoretical energy density of 45.5 W h L−1. Such a RFB goes through 700 cycles with 99.99% capacity retention per cycle, and the power density can reach 125 mW cm−2. A similar strategy was also adopted by the Aziz group, who prepared bis((3-trimethylammonio)propyl)-ferrocene dichloride (#9, Table 11), for which the solubility was improved to nearly 2 M.191
Another approach to achieve high-concentration ferrocene catholyte is to prepare a low-melting-point ferrocene derivative. The Lu group has proposed that the liquid form ferrocene derivative not only contributes to a high concentration of redox active materials, but also acts as a solvating medium for supporting salts.192 By attaching two methyl groups to the two Cp rings of ferrocene, the molecular symmetry was broken, and the melting point decreased dramatically from ∼173 °C to ∼38 °C. Using the catholyte containing 3 M 1,1-dimethylferrocene (DMFc, #10, Table 11) in EC/DEC at 50 °C, the demonstrated volumetric capacity was as high as 68 A h L−1.
In addition to solubility, the molecular size and redox potential of metallocenes can be fine-tuned. When attaching a long side chain of 6-bromohexyl groups to ferrocene (#11, Table 11), crossover of ferrocene through the PEO-based separator membrane can be significantly reduced.193 Paired with a Li anode, such a non-aqueous RFB can be cycled over 50 cycles with little degradation. To enhance the cell voltage, electron-withdrawing substituents, such as bromo groups, can be connected to the Cp rings to improve the redox potential of ferrocene as positive species (#12, Table 11).177 A similar method was adopted by the Wang group, who designed a LiFePO4-based semi-liquid RFB employing ferrocene and 1,1′-dibromoferrocene (#13, Table 11) as redox mediators.194 Ferrocene possesses a redox potential at 3.25 V vs. Li+/Li, and the potential is lifted to 3.55 V vs. Li+/Li when substituted with two bromo groups. These two ferrocene derivatives have redox potentials that straddle that of LiFePO4 (3.45 V), allowing reversible chemical delithiation/lithiation of LiFePO4.
Moreover, simultaneous tuning of solubility and redox potential was realized by Kim et al., who utilized acetylferrocene (#14, Table 11) as positive species in an RFB.195 Compared to pristine ferrocene, whose solubility in PC is 0.2 M, the solubility of acetyl group substituted ferrocene can reach 4 times that value (0.81 M). Because of the electron-withdrawing effect of the acetyl substituent, the redox potential of acetylferrocene (3.65 V vs. Li+/Li) was also improved compared to ferrocene (3.44 V vs. Li+/Li). This method demonstrates an economic approach to meeting various requirements for RFBs by rationally employing multifunctional substituents.
Finally, the tuning of metallocenes can be achieved by metal center exchange. In a systematic research by the Yu group, a class of metallocenes were studied as alternatives for RFBs.196 It was found that metallocenes typically possess fast reaction kinetics ascribed to the simple electron-transfer process without significantly alternating the molecular structures. Cobaltocene was selected as the anode species given its reversible reaction at 1.9 V vs. Li+/Li. Furthermore, methyl substituents were utilized to further lower the potential to 1.35 V. Paired with a ferrocene-based catholyte, an all-metallocene-based RFB was constructed maintaining the working voltage up to 2.1 V (Fig. 28).196 In future work, more rational molecular engineering strategies can be adopted to achieve the required solubility, redox potential, and molecular size.
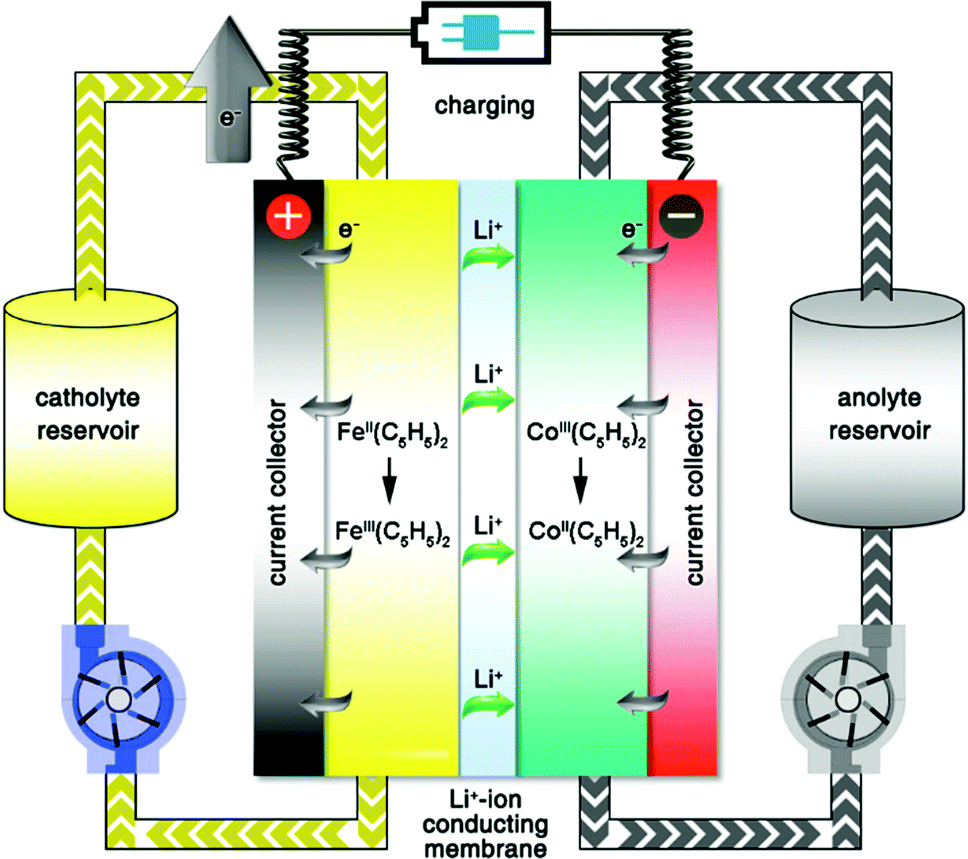 | ||
| Fig. 28 Working principle of the all-metallocene-based RFB. Reproduced with permission from ref. 196. Copyright 2017, Royal Society of Chemistry. | ||
7. Conclusion and perspectives
Large-scale exploitation of renewable energy sources calls for high-performance and cost-effective energy storage devices. The use of organic and organometallic electroactive redox species for RFBs represents a promising direction for environmentally sustainable energy storage.11,197 In this review, we summarize the design principles of organic redox species for such novel green energy systems and cover comprehensive synthetic strategies toward high electrochemical performance via function-oriented organic synthesis and molecular engineering.In spite of influential advances made in the past decade, tremendous effort is needed to realize the practical applications of organic RFBs. As a core element of an electrochemical energy storage system, electroactive materials play a critical role in the electrochemical performance. The key performance metrics of a RFB, including energy density, power density, cycle life, efficiency, capital cost and ecological footprint, should be taken into consideration when designing redox species.32Fig. 29 compares the voltage, effective molarity and energy density of typical organic RFBs and commercial aqueous RFBs.31,61,79,81,95,109,110,112,118,192,198–200 Except for some hybrid RFBs applying Li metal as the anode, the energy density of organic RFBs is still not competitive with traditional metal-based aqueous RFBs. Moreover, the power density of non-aqueous RFBs is far less than the aqueous systems limited by the membrane and electrolyte conductivity. The target concentrations of organic species should reach 2 M in aqueous systems and 5 M in non-aqueous systems to be practically useful. Furthermore, longer cycling stability is needed in order to rival current stationary energy storage systems. Deep eutectic solvents emerge as promising electroactive materials to meet the requirements in performance metrics owing to the appealing features such as low cost, high concentration, potential biodegradability, non-flammability and multi-electron transfer reactions.201–204
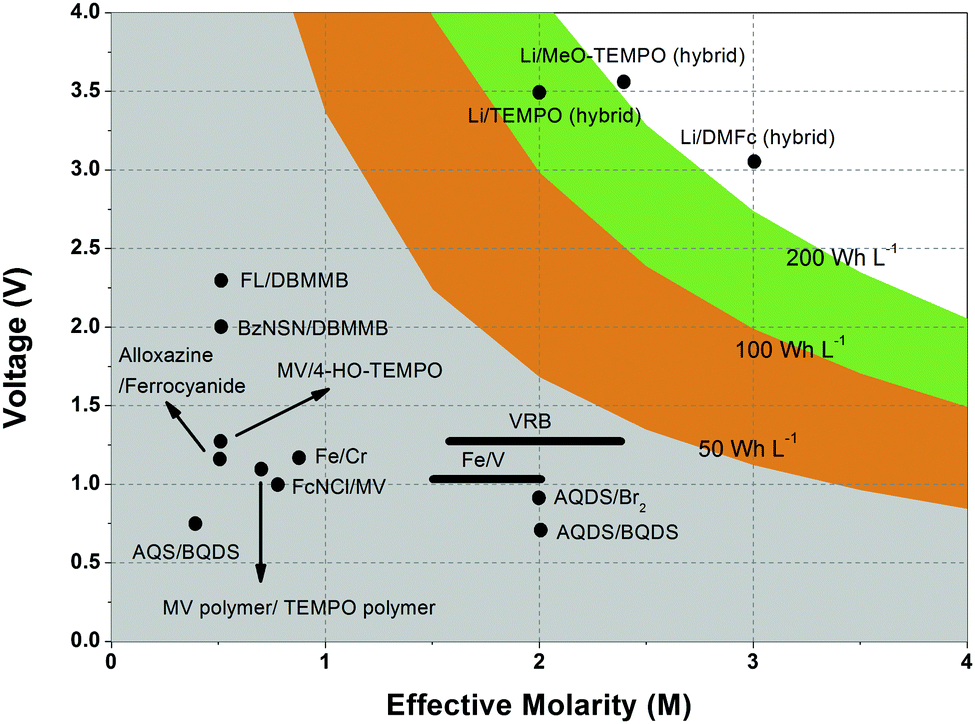 | ||
| Fig. 29 Comparison of performance metrics between typical organic RBFs and commercial RFBs. | ||
The well-developed organic synthesis techniques promote the development of novel organic electroactive species with high solubility, suitable redox potential, multi-electron transfer reaction, chemical stability, and fast reaction kinetics to meet the requirements of high-performance batteries. Moreover, the effective synthetic methodology spurs the discovery and optimization of the most efficient and economic synthesis route to obtain the target molecule. In this research direction, which combines the academic disciplines of organic chemistry and energy science, the advanced synthesis methods will accelerate the selection and screening of organic redox alternatives. Moreover, the characterization techniques for organic compounds will facilitate the comprehensive study of the chemistry of organic molecules, such as reaction reversibility, electron transfer, intermediate stability, and degradation mechanisms.
The rapid development of computational organic chemistry promotes its integration into designing organic and organometallic-based RFBs. On one hand, the computational models can offer a high-throughput screening approach to facilitate the selection of redox active molecules. On the other hand, it can provide insight into the reaction pathways, as well as geometric and electronic properties of reactants, products, intermediates, and transition-state structures, all of which are essential to understanding the working principles of organic and organometallic redox species.205 Experimental methods are still needed to align with computational modeling to elucidate the unknowns in organic systems.
The structural diversity of organic materials allows the tailoring of solubility, electrochemical activity, and redox potential by substitutional functionalization. Likewise, the synthetic strategies can also be extended to modify the characteristics of membranes, which play a critical role in regulating the power density, cycling performance and efficiency of RFBs. Therefore, the development of low-cost membranes with high ion selectivity, conductivity, and chemical stability is eagerly anticipated in future research.135,206
In addition to molecule and membrane modification, the improvement on battery performance also relies on advanced battery design. By using positive and negative electrodes composed of particles suspended in a carrier liquid and stored in outer tanks, such a semi-solid RFB combines the merits of both Li ion batteries and RFBs, including the high energy density and independent control of energy and power.207 Moreover, the photo-chargeable RFB provides an attractive means for in situ capture and storage of intermittent solar energy.208 When integrating an organic solar cell with an organic RFB, the flexibility of organic molecules enables their use in both energy conversion and storage, representing an encouraging direction for sustainable energy storage. The above two energy systems stretch the research boundaries of battery types, broaden the scope of novel energy devices with new battery chemistries, and exhibit the wider applicability of organic-based redox species via molecular engineering.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. Y. acknowledges the financial support from the National Science Foundation award NSF-CMMI-1537894, the Welch Foundation grant (F-1861), the Sloan Research Fellowship, and the Camille Dreyfus Teacher-Scholar Award.Notes and references
- Fossil fuel energy consumption, DOI: http://data.worldbank.org/indicator/EG.USE.COMM.FO.ZS.
- Z. Yang, J. Zhang, M. C. W. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- G. L. Soloveichik, Chem. Rev., 2015, 115, 11533–11558 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Adv. Energy Mater., 2012, 2, 770–779 CrossRef CAS.
- Y. Zhao, Y. Ding, Y. Li, L. Peng, H. R. Byon, J. B. Goodenough and G. Yu, Chem. Soc. Rev., 2015, 44, 7968–7996 RSC.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2016, 2, 16080 CrossRef.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- Y. Wang, Y. Ding, L. Pan, Y. Shi, Z. Yue, Y. Shi and G. Yu, Nano Lett., 2016, 16, 3329–3334 CrossRef CAS PubMed.
- Y. Lu, Q. Zhao, L. Miao, Z. Tao, Z. Niu and J. Chen, J. Phys. Chem. C, 2017, 121, 14498–14506 CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091–7106 CAS.
- P. Leung, A. A. Shah, L. Sanz, C. Flox, J. R. Morante, Q. Xu, M. R. Mohamed, C. Ponce de León and F. C. Walsh, J. Power Sources, 2017, 360, 243–283 CrossRef CAS.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2016, 56, 686–711 CrossRef PubMed.
- L. H. Thaller, 1974, http://ntrs.nasa.gov/search.jsp?R=19740013575.
- S.-H. Shin, S.-H. Yun and S.-H. Moon, RSC Adv., 2013, 3, 9095–9116 RSC.
- L. Su, R. M. Darling, K. G. Gallagher, W. Xie, J. L. Thelen, A. F. Badel, J. L. Barton, K. J. Cheng, N. P. Balsara, J. S. Moore and F. R. Brushett, J. Electrochem. Soc., 2016, 163, A5253–A5262 CrossRef CAS.
- Conductance Data For Commonly Used Chemicals, DOI: http://www2.emersonprocess.com/siteadmincenter/pm%20Rosemount%20Analytical%20Documents/LIQ_MAN_6039_Conductance_Data_Commonly_Used_Chemicals.pdf.
- K. Izutsu, Electrochemistry in nonaqueous solutions, John Wiley & Sons, 2009 Search PubMed.
- K. Gong, Q. Fang, S. Gu, S. F. Y. Li and Y. Yan, Energy Environ. Sci., 2015, 8, 3515–3530 Search PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- L. Hnedkovsky, R. H. Wood and V. N. Balashov, J. Phys. Chem. B, 2005, 109, 9034–9046 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS PubMed.
- D. S. Aaron, Q. Liu, Z. Tang, G. M. Grim, A. B. Papandrew, A. Turhan, T. A. Zawodzinski and M. M. Mench, J. Power Sources, 2012, 206, 450–453 CrossRef CAS.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS.
- Y. Matsuda, K. Tanaka, M. Okada, Y. Takasu, M. Morita and T. Matsumura-Inoue, J. Appl. Electrochem., 1988, 18, 909–914 CrossRef CAS.
- J. Wang, Z. Zhao, F. Gong, S. Li and S. Zhang, Macromolecules, 2009, 42, 8711–8717 CrossRef CAS.
- Y. Zhao, Y. Ding, J. Song, L. Peng, J. B. Goodenough and G. Yu, Energy Environ. Sci., 2014, 7, 1990–1995 CAS.
- Y. Li, B. Xu, H. Xu, H. Duan, X. Lü, S. Xin, W. Zhou, L. Xue, G. Fu, A. Manthiram and J. B. Goodenough, Angew. Chem., 2017, 129, 771–774 CrossRef.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 CAS.
- B. Hu, C. Debruler, Z. Rhodes and T. Liu, J. Am. Chem. Soc., 2016, 139, 1207–1214 CrossRef PubMed.
- J. Noack, N. Roznyatovskaya, T. Herr and P. Fischer, Angew. Chem., Int. Ed., 2015, 54, 9776–9809 CrossRef CAS PubMed.
- B. Schwenzer, J. Zhang, S. Kim, L. Li, J. Liu and Z. Yang, ChemSusChem, 2011, 4, 1388–1406 CrossRef CAS PubMed.
- A. F. M. Barton, Chem. Rev., 1975, 75, 731–753 CrossRef CAS.
- L. N. Kuleshova, D. W. M. Hofmann and R. Boese, Chem. Phys. Lett., 2013, 564, 26–32 CrossRef CAS.
- A. N. Paruta, B. J. Sciarrone and N. G. Lordi, J. Pharm. Sci., 1964, 53, 1349–1353 CrossRef CAS PubMed.
- E. G. Yanes, S. R. Gratz and A. M. Stalcup, Analyst, 2000, 125, 1919–1923 RSC.
- R. Schmid, Monatsh. Chem., 2001, 132, 1295–1326 CrossRef CAS.
- T. Shikata and M. Okuzono, J. Phys. Chem. B, 2013, 117, 7718–7723 CrossRef CAS PubMed.
- Polarity of Organic Compounds, DOI: http://chemistry.elmhurst.edu/vchembook/213organicfcgp.html.
- T. R. Griffiths and D. C. Pugh, Coord. Chem. Rev., 1979, 29, 129–211 CrossRef CAS.
- J. K. Gregory, D. C. Clary, K. Liu, M. G. Brown and R. J. Saykally, Science, 1997, 275, 814–817 CrossRef CAS PubMed.
- M. Tao, Z. Wang, J. Gong, H. Hao and J. Wang, Ind. Eng. Chem. Res., 2013, 52, 3036–3041 CrossRef CAS.
- H. Modarresi, E. Conte, J. Abildskov, R. Gani and P. Crafts, Ind. Eng. Chem. Res., 2008, 47, 5234–5242 CrossRef CAS.
- R. E. Skyner, J. L. McDonagh, C. R. Groom, T. van Mourik and J. B. O. Mitchell, Phys. Chem. Chem. Phys., 2015, 17, 6174–6191 RSC.
- S. I. Bailey, I. M. Ritchie and F. R. Hewgill, J. Chem. Soc., Perkin Trans. 2, 1983, 645–652, 10.1039/P29830000645.
- M. Lee, J. Hong, D.-H. Seo, D. H. Nam, K. T. Nam, K. Kang and C. B. Park, Angew. Chem., Int. Ed., 2013, 52, 8322–8328 CrossRef CAS PubMed.
- J. E. McMurry, Organic Chemistry, Cengage Learning, 2012 Search PubMed.
- J. Lee and M. J. Park, Adv. Energy Mater., 2017, 7, 1602279 CrossRef.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef CAS PubMed.
- K. C. Kim, T. Liu, S. W. Lee and S. S. Jang, J. Am. Chem. Soc., 2016, 138, 2374–2382 CrossRef CAS PubMed.
- J. C. Bachman, R. Kavian, D. J. Graham, D. Y. Kim, S. Noda, D. G. Nocera, Y. Shao-Horn and S. W. Lee, Nat. Commun., 2015, 6, 7040 CrossRef PubMed.
- L. R. Domingo, P. Pérez and R. Contreras, J. Org. Chem., 2003, 68, 6060–6062 CrossRef CAS PubMed.
- D. Job and H. B. Dunford, Eur. J. Biochem., 1976, 66, 607–614 CrossRef CAS PubMed.
- Y. Zhao, L. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- S. Sarkar, A. K. SenGupta and P. Prakash, Environ. Sci. Technol., 2010, 44, 1161–1166 CrossRef CAS PubMed.
- S. Yan, X. Xiao, X. Huang, X. Li and Y. Qi, Polymer, 2014, 55, 6282–6292 CrossRef CAS.
- M. Mavukkandy, M. Bilad, J. Kujawa, S. Al-Gharabli and H. Arafat, Sep. Purif. Technol., 2017, 187, 365–373 CrossRef CAS.
- M. Burgess, J. S. Moore and J. Rodríguez-López, Acc. Chem. Res., 2016, 49, 2649–2657 CrossRef CAS PubMed.
- M. Burgess, K. Hernández-Burgos, B. H. Simpson, T. Lichtenstein, S. Avetian, G. Nagarjuna, K. J. Cheng, J. S. Moore and J. Rodríguez-López, J. Electrochem. Soc., 2016, 163, H3006–H3013 CrossRef CAS.
- T. Janoschka, N. Martin, U. Martin, C. Friebe, S. Morgenstern, H. Hiller, M. D. Hager and U. S. Schubert, Nature, 2015, 527, 78–81 CrossRef CAS PubMed.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116, 2–4 CrossRef CAS.
- Q. Zhao, Z. Zhu and J. Chen, Adv. Mater., 2017, 1607007, DOI:10.1002/adma.201607007.
- L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757–824 CrossRef CAS PubMed.
- H. Dahn, P. Péchy and V. Van Toan, Magn. Reson. Chem., 1997, 35, 589–592 CrossRef CAS.
- O. A. Gansow, D. A. Schexnayder and B. Y. Kimura, J. Am. Chem. Soc., 1972, 94, 3406–3408 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- E. R. Davidson, K. L. Kunze, F. B. C. Machado and S. J. Chakravorty, Acc. Chem. Res., 1993, 26, 628–635 CrossRef CAS.
- H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 CAS.
- T. Sun, Z.-j. Li, H.-g. Wang, D. Bao, F.-l. Meng and X.-b. Zhang, Angew. Chem., 2016, 128, 10820–10824 CrossRef.
- H. Kurreck and M. Huber, Angew. Chem., Int. Ed., 1995, 34, 849–866 CrossRef CAS.
- D. T. Scott, D. M. McKnight, E. L. Blunt-Harris, S. E. Kolesar and D. R. Lovley, Environ. Sci. Technol., 1998, 32, 2984–2989 CrossRef CAS.
- M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed.
- Y. Ding and G. Yu, Angew. Chem., Int. Ed., 2016, 55, 4772–4776 CrossRef CAS PubMed.
- Y. Xu, Y. Wen, J. Cheng, Y. Yanga, Z. Xie and G. Cao, World Non-Grid-Connected Wind Power and Energy Conference, IEEE, 2009, DOI:10.1109/WNWEC.2009.5335870.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- M. R. Gerhardt, L. Tong, R. Gómez-Bombarelli, Q. Chen, M. P. Marshak, C. J. Galvin, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Adv. Energy Mater., 2017, 7, 1601488 CrossRef.
- K. Lin, Q. Chen, M. R. Gerhardt, L. Tong, S. B. Kim, L. Eisenach, A. W. Valle, D. Hardee, R. G. Gordon, M. J. Aziz and M. P. Marshak, Science, 2015, 349, 1529–1532 CrossRef CAS PubMed.
- B. Yang, L. Hoober-Burkhardt, F. Wang, G. K. Surya Prakash and S. R. Narayanan, J. Electrochem. Soc., 2014, 161, A1371–A1380 CrossRef CAS.
- H. Senoh, M. Yao, H. Sakaebe, K. Yasuda and Z. Siroma, Electrochim. Acta, 2011, 56, 10145–10150 CrossRef CAS.
- W. Wang, W. Xu, L. Cosimbescu, D. Choi, L. Li and Z. Yang, Chem. Commun., 2012, 48, 6669–6671 RSC.
- S. Zhang, X. Li and D. Chu, Electrochim. Acta, 2016, 190, 737–743 CrossRef CAS.
- J. Carretero-Gonzalez, E. Castillo-Martinez and M. Armand, Energy Environ. Sci., 2016, 9, 3521–3530 CAS.
- R. A. Potash, J. R. McKone, S. Conte and H. D. Abruña, J. Electrochem. Soc., 2016, 163, A338–A344 CrossRef CAS.
- Y. Ding, Y. Li and G. Yu, Chem, 2016, 1, 790–801 CAS.
- M. Park, D.-S. Shin, J. Ryu, M. Choi, N. Park, S. Y. Hong and J. Cho, Adv. Mater., 2015, 27, 5141–5146 CrossRef CAS PubMed.
- J. E. Bachman, L. A. Curtiss and R. S. Assary, J. Phys. Chem. A, 2014, 118, 8852–8860 CrossRef CAS PubMed.
- S. Er, C. Suh, M. P. Marshak and A. Aspuru-Guzik, Chem. Sci., 2015, 6, 885–893 RSC.
- Z. Li, S. Li, S. Liu, K. Huang, D. Fang, F. Wang and S. Peng, Electrochem. Solid-State Lett., 2011, 14, A171 CrossRef CAS.
- X. Wei, W. Duan, J. Huang, L. Zhang, B. Li, D. Reed, W. Xu, V. Sprenkle and W. Wang, ACS Energy Lett., 2016, 1, 705–711 CrossRef CAS.
- A. Orita, M. G. Verde, M. Sakai and Y. S. Meng, Nat. Commun., 2016, 7, 13230 CrossRef CAS PubMed.
- K. Lin, R. Gómez-Bombarelli, E. S. Beh, L. Tong, Q. Chen, A. Valle, A. Aspuru-Guzik, M. J. Aziz and R. G. Gordon, Nat. Energy, 2016, 1, 16102 CrossRef CAS.
- H. Nishide, S. Iwasa, Y.-J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827–831 CrossRef CAS.
- L. S. Kaanumalle, J. Nithyanandhan, M. Pattabiraman, N. Jayaraman and V. Ramamurthy, J. Am. Chem. Soc., 2004, 126, 8999–9006 CrossRef CAS PubMed.
- R. R. Jones and R. G. Bergman, J. Am. Chem. Soc., 1972, 94, 660–661 CrossRef CAS.
- J. A. Pojman, V. M. Ilyashenko and A. M. Khan, J. Chem. Soc., Faraday Trans., 1996, 92, 2825–2837 RSC.
- S. W. Benson, J. Am. Chem. Soc., 1965, 87, 972–979 CrossRef CAS.
- F. G. Bordwell and T. Y. Lynch, J. Am. Chem. Soc., 1989, 111, 7558–7562 CrossRef CAS.
- H. Nishide and T. Suga, Electrochem. Soc. Interface, 2005, 14, 32–36 CAS.
- K. Nozaki, K. Oshima and K. Uchimoto, J. Am. Chem. Soc., 1987, 109, 2547–2549 CrossRef CAS.
- J. M. Tedder, Angew. Chem., Int. Ed., 1982, 21, 401–410 CrossRef.
- J. M. Braganza, D. G. Wickens, P. Cawood and T. L. Dormandy, Lancet, 1983, 322, 375–379 CrossRef.
- A. Gansäuer, C.-A. Fan and F. Piestert, J. Am. Chem. Soc., 2008, 130, 6916–6917 CrossRef PubMed.
- J. H. Fuhrhop, P. Wasser, D. Riesner and D. Mauzerall, J. Am. Chem. Soc., 1972, 94, 7996–8001 CrossRef CAS PubMed.
- J. Winsberg, C. Stolze, S. Muench, F. Liedl, M. D. Hager and U. S. Schubert, ACS Energy Lett., 2016, 1, 976–980 CrossRef CAS.
- K. Takechi, Y. Kato and Y. Hase, Adv. Mater., 2015, 27, 2501–2506 CrossRef CAS PubMed.
- X. Wei, W. Xu, M. Vijayakumar, L. Cosimbescu, T. Liu, V. Sprenkle and W. Wang, Adv. Mater., 2014, 26, 7649–7653 CrossRef CAS PubMed.
- J. D. Milshtein, J. L. Barton, R. M. Darling and F. R. Brushett, J. Power Sources, 2016, 327, 151–159 CrossRef CAS.
- T. Liu, X. Wei, Z. Nie, V. Sprenkle and W. Wang, Adv. Energy Mater., 2016, 6, 1501449 CrossRef.
- J. Winsberg, T. Janoschka, S. Morgenstern, T. Hagemann, S. Muench, G. Hauffman, J.-F. Gohy, M. D. Hager and U. S. Schubert, Adv. Mater., 2016, 28, 2238–2243 CrossRef CAS PubMed.
- T. Janoschka, S. Morgenstern, H. Hiller, C. Friebe, K. Wolkersdorfer, B. Haupler, M. D. Hager and U. S. Schubert, Polym. Chem., 2015, 6, 7801–7811 RSC.
- Z. Zhang, L. Zhang, J. A. Schlueter, P. C. Redfern, L. Curtiss and K. Amine, J. Power Sources, 2010, 195, 4957–4962 CrossRef CAS.
- L. Zhang, Z. Zhang, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2012, 5, 8204–8207 CAS.
- J. Huang, L. Cheng, R. S. Assary, P. Wang, Z. Xue, A. K. Burrell, L. A. Curtiss and L. Zhang, Adv. Energy Mater., 2015, 5, 1401782 CrossRef.
- X. Wei, W. Xu, J. Huang, L. Zhang, E. Walter, C. Lawrence, M. Vijayakumar, W. A. Henderson, T. Liu, L. Cosimbescu, B. Li, V. Sprenkle and W. Wang, Angew. Chem., Int. Ed., 2015, 54, 8684–8687 CrossRef CAS PubMed.
- J. Huang, L. Su, J. A. Kowalski, J. L. Barton, M. Ferrandon, A. K. Burrell, F. R. Brushett and L. Zhang, J. Mater. Chem. A, 2015, 3, 14971–14976 CAS.
- J. Huang, B. Pan, W. Duan, X. Wei, R. S. Assary, L. Su, F. R. Brushett, L. Cheng, C. Liao, M. S. Ferrandon, W. Wang, Z. Zhang, A. K. Burrell, L. A. Curtiss, I. A. Shkrob, J. S. Moore and L. Zhang, Sci. Rep., 2016, 6, 32102 CrossRef CAS PubMed.
- C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49–82 RSC.
- K. I. Priyadarsini, M. F. Dennis, M. A. Naylor, M. R. L. Stratford and P. Wardman, J. Am. Chem. Soc., 1996, 118, 5648–5654 CrossRef CAS.
- V. W.-h. Lau, D. Klose, H. Kasap, F. Podjaski, M.-C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef CAS PubMed.
- R. J. Mortimer, Electrochim. Acta, 1999, 44, 2971–2981 CrossRef CAS.
- G. Nagarjuna, J. Hui, K. J. Cheng, T. Lichtenstein, M. Shen, J. S. Moore and J. Rodríguez-López, J. Am. Chem. Soc., 2014, 136, 16309–16316 CrossRef CAS PubMed.
- C. S. Sevov, R. E. M. Brooner, E. Chénard, R. S. Assary, J. S. Moore, J. Rodríguez-López and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 14465–14472 CrossRef CAS PubMed.
- R. Wang, R. Ramaraj, T. Okajima, F. Kitamura, N. Matsumoto, T. Thiemann, S. Mataka and T. Ohsaka, J. Electroanal. Chem., 2004, 567, 85–94 CrossRef CAS.
- F. R. Brushett, J. T. Vaughey and A. N. Jansen, Adv. Energy Mater., 2012, 2, 1390–1396 CrossRef CAS.
- J. D. Milshtein, L. Su, C. Liou, A. F. Badel and F. R. Brushett, Electrochim. Acta, 2015, 180, 695–704 CrossRef CAS.
- A. P. Kaur, S. Ergun, C. F. Elliott and S. A. Odom, J. Mater. Chem. A, 2014, 2, 18190–18193 CAS.
- A. P. Kaur, N. E. Holubowitch, S. Ergun, C. F. Elliott and S. A. Odom, Energy Technol., 2015, 3, 476–480 CrossRef CAS.
- J. D. Milshtein, A. P. Kaur, M. D. Casselman, J. A. Kowalski, S. Modekrutti, P. L. Zhang, N. Harsha Attanayake, C. F. Elliott, S. R. Parkin, C. Risko, F. R. Brushett and S. A. Odom, Energy Environ. Sci., 2016, 9, 3531–3543 CAS.
- S. C. Chieng, M. Kazacos and M. Skyllas-Kazacos, J. Membr. Sci., 1992, 75, 81–91 CrossRef CAS.
- B. Tian, C. W. Yan and F. H. Wang, J. Membr. Sci., 2004, 234, 51–54 CrossRef CAS.
- X. Li, H. Zhang, Z. Mai, H. Zhang and I. Vankelecom, Energy Environ. Sci., 2011, 4, 1147–1160 CAS.
- P. Prakash, D. Hoskins and A. K. SenGupta, J. Membr. Sci., 2004, 237, 131–144 CrossRef CAS.
- R. Liu, L. Wu, J. Pan, C. Jiang and T. Xu, J. Membr. Sci., 2014, 451, 18–23 CrossRef CAS.
- Y. Zhao, S. Si and C. Liao, J. Power Sources, 2013, 241, 449–453 CrossRef CAS.
- S. H. Oh, C. W. Lee, D. H. Chun, J. D. Jeon, J. Shim, K. H. Shin and J. H. Yang, J. Mater. Chem. A, 2014, 2, 19994–19998 CAS.
- T. Sukegawa, I. Masuko, K. Oyaizu and H. Nishide, Macromolecules, 2014, 47, 8611–8617 CrossRef CAS.
- J. Winsberg, S. Muench, T. Hagemann, S. Morgenstern, T. Janoschka, M. Billing, F. H. Schacher, G. Hauffman, J.-F. Gohy, S. Hoeppener, M. D. Hager and U. S. Schubert, Polym. Chem., 2016, 7, 1711–1718 RSC.
- J. Winsberg, T. Hagemann, S. Muench, C. Friebe, B. Häupler, T. Janoschka, S. Morgenstern, M. D. Hager and U. S. Schubert, Chem. Mater., 2016, 28, 3401–3405 CrossRef CAS.
- E. C. Montoto, G. Nagarjuna, J. S. Moore and J. Rodríguez-López, J. Electrochem. Soc., 2017, 164, A1688–A1694 CrossRef CAS.
- Y. S. Kwon, J. Lim, H.-J. Yun, Y.-H. Kim and T. Park, Energy Environ. Sci., 2014, 7, 1454–1460 CAS.
- S. Zhu, Y. Song, J. Shao, X. Zhao and B. Yang, Angew. Chem., Int. Ed., 2015, 54, 14626–14637 CrossRef CAS PubMed.
- Y. Shi, L. Peng, Y. Ding, Y. Zhao and G. Yu, Chem. Soc. Rev., 2015, 44, 6684–6696 RSC.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef.
- D. R. Nevers, F. R. Brushett and D. R. Wheeler, J. Power Sources, 2017, 352, 226–244 CrossRef CAS.
- P. Xie, M. Z. Rong and M. Q. Zhang, Angew. Chem., Int. Ed., 2016, 55, 1805–1809 CrossRef CAS PubMed.
- M. Beidaghi and Y. Gogotsi, Energy Environ. Sci., 2014, 7, 867–884 CAS.
- J. B. Flanagan, S. Margel, A. J. Bard and F. C. Anson, J. Am. Chem. Soc., 1978, 100, 4248–4253 CrossRef CAS.
- T. Suga, S. Takeuchi and H. Nishide, Adv. Mater., 2011, 23, 5545–5549 CrossRef CAS PubMed.
- E. C. Montoto, G. Nagarjuna, J. Hui, M. Burgess, N. M. Sekerak, K. Hernández-Burgos, T.-S. Wei, M. Kneer, J. Grolman, K. J. Cheng, J. A. Lewis, J. S. Moore and J. Rodríguez-López, J. Am. Chem. Soc., 2016, 138, 13230–13237 CrossRef CAS PubMed.
- M. Burgess, E. Chénard, K. Hernández-Burgos, G. Nagarjuna, R. S. Assary, J. Hui, J. S. Moore and J. Rodríguez-López, Chem. Mater., 2016, 28, 7362–7374 CrossRef CAS.
- T. Hagemann, J. Winsberg, B. Haupler, T. Janoschka, J. J. Gruber, A. Wild and U. S. Schubert, NPG Asia Mater., 2017, 9, e340 CrossRef CAS.
- I. Esnal, I. Valois-Escamilla, C. F. A. Gómez-Durán, A. Urías-Benavides, M. L. Betancourt-Mendiola, I. López-Arbeloa, J. Bañuelos, I. García-Moreno, A. Costela and E. Peña-Cabrera, ChemPhysChem, 2013, 14, 4134–4142 CrossRef CAS PubMed.
- E. López, G. Lonzi and L. A. López, Organometallics, 2014, 33, 5924–5927 CrossRef.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- P. Machata, P. Herich, K. Lušpai, L. Bucinsky, S. Šoralová, M. Breza, J. Kozisek and P. Rapta, Organometallics, 2014, 33, 4846–4859 CrossRef CAS.
- J. K. Kochi, Angew. Chem., Int. Ed., 1988, 27, 1227–1266 CrossRef.
- K. M. Davies and J. E. Earley, Inorg. Chem., 1976, 15, 1074–1080 CrossRef CAS.
- Y. Yamaguchi, W. Ding, C. T. Sanderson, M. L. Borden, M. J. Morgan and C. Kutal, Coord. Chem. Rev., 2007, 251, 515–524 CrossRef CAS.
- P. J. Cappillino, H. D. Pratt, N. S. Hudak, N. C. Tomson, T. M. Anderson and M. R. Anstey, Adv. Energy Mater., 2014, 4, 201300566 Search PubMed.
- J. Mun, M.-J. Lee, J.-W. Park, D.-J. Oh, D.-Y. Lee and S.-G. Doo, Electrochem. Solid-State Lett., 2012, 15, A80–A82 CrossRef CAS.
- J. G. Ibanez, C. S. Choi and R. S. Becker, J. Electrochem. Soc., 1987, 134, 3083–3089 CrossRef CAS.
- K. Gong, F. Xu, J. B. Grunewald, X. Ma, Y. Zhao, S. Gu and Y. Yan, ACS Energy Lett., 2016, 89–93, DOI:10.1021/acsenergylett.6b00049.
- Q. Liu, A. E. S. Sleightholme, A. A. Shinkle, Y. Li and L. T. Thompson, Electrochem. Commun., 2009, 11, 2312–2315 CrossRef CAS.
- A. A. Shinkle, A. E. S. Sleightholme, L. T. Thompson and C. W. Monroe, J. Appl. Electrochem., 2011, 41, 1191–1199 CrossRef CAS.
- M. O. Bamgbopa and S. Almheiri, J. Power Sources, 2017, 342, 371–381 CrossRef CAS.
- A. A. Shinkle, T. J. Pomaville, A. E. S. Sleightholme, L. T. Thompson and C. W. Monroe, J. Power Sources, 2014, 248, 1299–1305 CrossRef CAS.
- A. A. Shinkle, A. E. S. Sleightholme, L. D. Griffith, L. T. Thompson and C. W. Monroe, J. Power Sources, 2012, 206, 490–496 CrossRef CAS.
- J.-H. Kim, K. J. Kim, M.-S. Park, N. J. Lee, U. Hwang, H. Kim and Y.-J. Kim, Electrochem. Commun., 2011, 13, 997–1000 CrossRef CAS.
- M. H. Chakrabarti, R. A. W. Dryfe and E. P. L. Roberts, Electrochim. Acta, 2007, 52, 2189–2195 CrossRef CAS.
- M.-S. Park, N.-J. Lee, S.-W. Lee, K. J. Kim, D.-J. Oh and Y.-J. Kim, ACS Appl. Mater. Interfaces, 2014, 6, 10729–10735 CAS.
- A. E. S. Sleightholme, A. A. Shinkle, Q. Liu, Y. Li, C. W. Monroe and L. T. Thompson, J. Power Sources, 2011, 196, 5742–5745 CrossRef CAS.
- Q. Liu, A. A. Shinkle, Y. Li, C. W. Monroe, L. T. Thompson and A. E. S. Sleightholme, Electrochem. Commun., 2010, 12, 1634–1637 CrossRef CAS.
- B. Hwang, M.-S. Park and K. Kim, ChemSusChem, 2015, 8, 310–314 CrossRef CAS PubMed.
- X. Xing, Y. Zhao and Y. Li, J. Power Sources, 2015, 293, 778–783 CrossRef CAS.
- J. A. Suttil, J. F. Kucharyson, I. L. Escalante-Garcia, P. J. Cabrera, B. R. James, R. F. Savinell, M. S. Sanford and L. T. Thompson, J. Mater. Chem. A, 2015, 3, 7929–7938 CAS.
- D. Zhang, H. Lan and Y. Li, J. Power Sources, 2012, 217, 199–203 CrossRef CAS.
- N. Arroyo-Currás, J. W. Hall, J. E. Dick, R. A. Jones and A. J. Bard, J. Electrochem. Soc., 2015, 162, A378–A383 CrossRef.
- C. S. Sevov, S. L. Fisher, L. T. Thompson and M. S. Sanford, J. Am. Chem. Soc., 2016, 138, 15378–15384 CrossRef CAS PubMed.
- W. Ren, E. Zhou, B. Fang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Angew. Chem., Int. Ed., 2014, 53, 11310–11314 CrossRef CAS PubMed.
- Y. Ishikawa, K. Yamamoto and T. Murahashi, Angew. Chem., Int. Ed., 2017, 56, 1346–1350 CrossRef CAS PubMed.
- Y. Zhao, Y. Ding, J. Song, G. Li, G. Dong, J. B. Goodenough and G. Yu, Angew. Chem., Int. Ed., 2014, 53, 11036–11040 CrossRef CAS PubMed.
- Y. Ding, Y. Zhao and G. Yu, Nano Lett., 2015, 15, 4108–4113 CrossRef CAS PubMed.
- M. N. Golovin, D. P. Wilkinson, J. T. Dudley, D. Holonko and S. Woo, J. Electrochem. Soc., 1992, 139, 5–10 CrossRef CAS.
- X. Wei, L. Cosimbescu, W. Xu, J. Z. Hu, M. Vijayakumar, J. Feng, M. Y. Hu, X. Deng, J. Xiao, J. Liu, V. Sprenkle and W. Wang, Adv. Energy Mater., 2015, 5, 1400678 CrossRef.
- L. Cosimbescu, X. Wei, M. Vijayakumar, W. Xu, M. L. Helm, S. D. Burton, C. M. Sorensen, J. Liu, V. Sprenkle and W. Wang, Sci. Rep., 2015, 5, 14117 CrossRef CAS PubMed.
- Y. Ding and G. Yu, Angew. Chem., Int. Ed., 2017, 56, 8614–8616 CrossRef CAS PubMed.
- E. S. Beh, D. De Porcellinis, R. L. Gracia, K. T. Xia, R. G. Gordon and M. J. Aziz, ACS Energy Lett., 2017, 2, 639–644 CrossRef CAS.
- G. Cong, Y. Zhou, Z. Li and Y.-C. Lu, ACS Energy Lett., 2017, 2, 869–875 CrossRef CAS.
- K. Park, J. H. Cho, K. Shanmuganathan, J. Song, J. Peng, M. Gobet, S. Greenbaum, C. J. Ellison and J. B. Goodenough, J. Power Sources, 2014, 263, 52–58 CrossRef CAS.
- Q. Huang, H. Li, M. Gratzel and Q. Wang, Phys. Chem. Chem. Phys., 2013, 15, 1793–1797 RSC.
- H.-s. Kim, T. Yoon, Y. Kim, S. Hwang, J. H. Ryu and S. M. Oh, Electrochem. Commun., 2016, 69, 72–75 CrossRef CAS.
- Y. Ding, Y. Zhao, Y. Li, J. B. Goodenough and G. Yu, Energy Environ. Sci., 2017, 10, 491–497 CAS.
- J. A. Kowalski, L. Su, J. D. Milshtein and F. R. Brushett, Curr. Opin. Chem. Eng., 2016, 13, 45–52 CrossRef.
- F. Pan and Q. Wang, Molecules, 2015, 20, 19711 Search PubMed.
- B. Yang, L. Hoober-Burkhardt, S. Krishnamoorthy, A. Murali, G. K. S. Prakash and S. R. Narayanan, J. Electrochem. Soc., 2016, 163, A1442–A1449 CrossRef CAS.
- C. Qu, Y. Cheng, F. Zhang, Y. Zhang, H. Zhang, X. Li and H. Zhang, Int. J. Hydrogen Energy, 2015, 40, 16429–16433 CrossRef CAS.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- Y. Wang and H. Zhou, Energy Environ. Sci., 2016, 9, 2267–2272 CAS.
- C. Zhang, Y. Ding, L. Zhang, X. Wang, Y. Zhao, X. Zhang and G. Yu, Angew. Chem., Int. Ed., 2017, 56, 7454–7459 CrossRef CAS PubMed.
- L. Zhang, C. Zhang, Y. Ding, K. Ramirez-Meyers and G. Yu, Joule, 2017 DOI:10.1016/j.joule.2017.08.013.
- G.-J. Cheng, X. Zhang, L. W. Chung, L. Xu and Y.-D. Wu, J. Am. Chem. Soc., 2015, 137, 1706–1725 CrossRef CAS PubMed.
- W. Lu, Z. Yuan, Y. Zhao, H. Zhang, H. Zhang and X. Li, Chem. Soc. Rev., 2017, 46, 2199–2236 RSC.
- C. Jia, F. Pan, Y. G. Zhu, Q. Huang, L. Lu and Q. Wang, Sci. Adv., 2015, 1, e1500886 Search PubMed.
- W. Li, H.-C. Fu, L. Li, M. Cabán-Acevedo, J.-H. He and S. Jin, Angew. Chem., Int. Ed., 2016, 55, 13104–13108 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2018 |