
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface enhanced hyper Raman scattering (SEHRS) and its applications
Fani
Madzharova
,
Zsuzsanna
Heiner
and
Janina
Kneipp
 *
*
Humboldt-Universität zu Berlin, Department of Chemistry, Brook-Taylor-Str. 2, 12489 Berlin, Germany. E-mail: janina.kneipp@chemie.hu-berlin.de
First published on 22nd May 2017
Abstract
Surface enhanced hyper Raman scattering (SEHRS) is the spontaneous, two-photon excited Raman scattering that occurs for molecules residing in high local optical fields of plasmonic nanostructures. Being regarded as a non-linear analogue of surface enhanced Raman scattering (SERS), SEHRS shares most of its properties, but also has additional characteristics. They include complementary spectroscopic information resulting from different selection rules and a stronger enhancement due to the non-linearity in excitation. In practical spectroscopy, this can translate to advantages, which include a high selectivity when probing molecule–surface interactions, the possibility of probing molecules at low concentrations due to the strong enhancement, and the advantages that come with excitation in the near-infrared. In this review, we give examples of the wealth of vibrational spectroscopic information that can be obtained by SEHRS and discuss work that has contributed to understanding the effect and that therefore provides directions for SEHRS spectroscopy. Future applications could range from biophotonics to materials research.
 Fani Madzharova | Fani Madzharova studied chemistry at Humboldt-Universität zu Berlin, where she obtained her MSc degree in 2015, with thesis work on surface enhanced hyper Raman scattering (SEHRS) of biological molecules. She is currently working on her doctoral thesis in the group of Janina Kneipp as a Chemiefonds (FCI) fellow. Her research interests include applications of SEHRS, as well as the development and characterization of plasmonic nanostructures for use in surface-enhanced spectroscopies. |
 Zsuzsanna Heiner | Zsuzsanna Heiner received an MSc as Physics Teacher and an MSc in Astronomy in 2005, as well as a PhD in Physics in 2013 at the Department of Optics and Quantum Electronics at the University of Szeged, Hungary. As a PhD student on a Young Researcher Scholarship and as a Junior Research Associate of the Hungarian Academy of Sciences, she pursued research at the Institute of Biophysics in the area of femtosecond time-resolved spectroscopy and its application to biomolecules. In 2013, she joined the Kneipp Group at Humboldt Universität zu Berlin as a postdoctoral fellow, working on developments of SEHRS and other non-linear spectroscopies. In 2015, she was appointed as a Julia Lermontova fellow in optical microspectroscopy at the School of Analytical Sciences Adlershof (DFG GSC1013). Her current research involves combination of different linear and nonlinear micro-spectroscopic methods and their application to biomolecules and complex biological systems. |
 Janina Kneipp | Janina Kneipp received her diploma and doctoral degrees at Freie Universität Berlin. After her dissertation work she was a postdoc at Erasmus Universiteit Rotterdam and Princeton University’s Chemistry Department. She has been conducting research in optical biospectroscopy in the Chemistry Department of Humboldt-Universität zu Berlin since 2008, first as an assistant professor of Analytical Chemistry, and since 2012 as a full professor of Physical Chemistry. The focus of her research lies in the development and application of sensitive vibrational approaches for studies of complex microstructured materials and in plasmon-supported nanospectroscopy, including SEHRS and SERS. |
1. Introduction
The discovery of surface enhanced Raman scattering (SERS)1–3 has sparked growing interest in the examination of the optical properties of nanostructured metal surfaces over the last few decades, which can increase weak Raman scattering (RS) signals by many orders of magnitude even at the single molecule level.4,5 Apart from a so-called chemical contribution, the enormous enhancements were attributed mostly to the highly confined local optical fields of the nanostructures,6,7 arising from resonances of their localized surface plasmons (oscillations of the conductive electrons relative to the metal nuclei) with both incident and Raman scattered light.8In general, all incoherent and also coherent optical effects can benefit from enhanced local optical fields, among them several other vibrational spectroscopies, such as IR absorption,9 hyper Raman scattering (HRS),10,11 coherent anti-Stokes Raman scattering (CARS),12,13 femtosecond stimulated Raman scattering (fs SRS),14 and second hyper Raman scattering.15 This led to the development of several related surface enhanced spectroscopic methods. In this review article, we focus on the enhancement of the incoherent, spontaneous two-photon excited Raman process of hyper Raman scattering (HRS) (Fig. 1). In HRS, the energy of the scattered photons is shifted relative to the second harmonic of the excitation wavelength. HRS is a rather weak process, resulting in intensities that are many orders of magnitude lower than those in normal Raman scattering.16
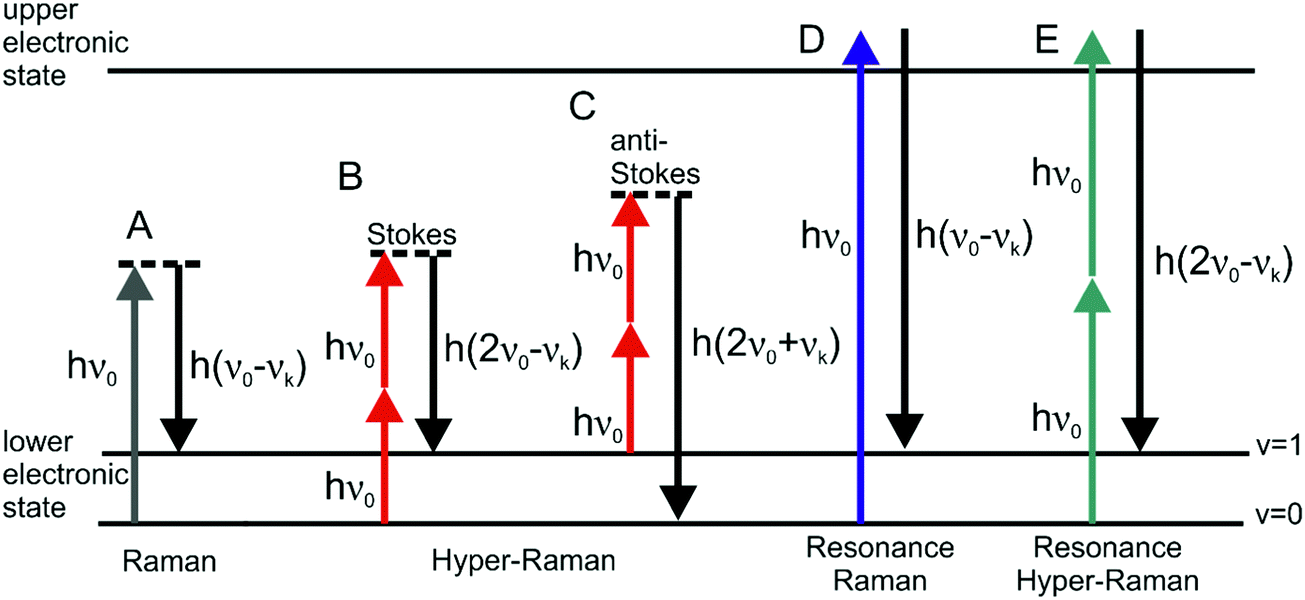 | ||
| Fig. 1 Schematic representation of vibrational: (A) linear Stokes Raman scattering, (B) Stokes hyper Raman scattering, (C) anti-Stokes hyper Raman scattering, (D) Stokes resonant Raman scattering, and (E) Stokes resonant hyper Raman scattering. In each of the cases the molecular system undergoes a vibrational transition from the initial state (v = 0 for Stokes and v = 1 for anti-Stokes) to the final state (v = 1 for Stokes and v = 0 for anti-Stokes), associated with the normal mode k with corresponding frequency νk. Both vibrational states belong to the electronic ground state. The diagram in (E) depicts one possible resonance condition; for more information about resonances in HRS please refer to the literature.25,93,110 | ||
Surface enhanced hyper Raman scattering (SEHRS) is the two-photon excited analogue of SERS and has been proposed very early after the discovery of SERS.10,11 As will be reviewed here, in SEHRS, the non-linear HRS is enhanced to a greater extent than linear RS is enhanced in SERS. Effective SEHRS cross sections up to the order of 10−45 cm4 s that were reported17 are similar to or higher than those of two-photon excited fluorescence. Here, we illustrate that SEHRS can excellently address the need of spectroscopic applications regarding molecular structural sensitivity, detection sensitivity, and spectroscopic imaging. While coherent Raman processes such as (SE-)CARS or (SE-)SRS are restricted to selected Raman lines that match the frequency difference of two excitation lasers, SEHRS is excited by two photons from the same laser and gives information about the entire vibrational spectrum. SEHRS presents several advantages, mostly relying on the multiphoton excitation, which other vibrational spectroscopies do not have. Specifically, HRS is governed by different selection rules than RS, and therefore offers complementary vibrational information. Depending on the molecular symmetry, HRS may probe IR active modes or in addition so-called silent modes, which are seen neither in Raman nor in IR spectra. In a SEHRS experiment, the excitation with light in the near infrared, typically convenient for biological samples, is combined with the desirable detection in the visible spectral range. Besides, the two-photon excitation is favorable for microscopic applications due to the increased penetration depth and limited probed volume,18 resulting in an improved resolution for imaging. Last but not least, SEHRS has the potential to provide better insight into the structure and interaction of molecules on surfaces, as it is much more sensitive than SERS with respect to surface environmental changes.19–24
After starting with a brief introduction to HRS and SEHRS, we will highlight recent advances in SEHRS spectroscopy. In particular, we want to discuss different aspects of experiments and applications of non-resonant and resonant SEHRS. We will focus on currently arising applications in biosensing and microscopy and demonstrate the great potential of SEHRS for vibrational spectroscopic probing in different fields.
2. Hyper Raman scattering
The interaction of materials with incident light can result in absorption, emission or scattering, involving transitions between the molecular (electronic, vibrational, rotational) states of the system. The vibrational Raman effect refers to light scattering phenomena associated with vibrational transitions within the electronic (ground) state of the molecule. While in normal Raman scattering (RS), the scattered photons are Stokes or anti-Stokes shifted by the energy difference between the initial and final vibrational states (Fig. 1A), in the two-photon excited process of hyper Raman scattering (HRS), the scattered radiation occurs near the second harmonic of the excitation wavelength (Fig. 1B and C). HRS as a non-linear process becomes significant when a sufficiently intense electric field is applied.The number of hyper Raman Stokes photons per second nHRS resulting from the annihilation of two photons at frequency ν0 and the creation of a photon at frequency 2ν0 − νk (Fig. 1B) can be expressed as
| nHRS = σHRSn02 | (1a) |
| σHRS ∝ 〈|βif|2〉 | (1b) |
The simulation of HRS spectra involves the determination of the vibrational frequencies and normal modes, the calculation of the hyperpolarizability and of its derivatives (e.g. by finite differentiation), and appropriate orientation averaging according to the illumination–observation geometry (see, e.g., ref. 29). An overview and description of theoretical approaches for obtaining β and HRS simulations, mainly for excitation wavelengths that are off-resonance with electronic transitions in the molecule, can be found in ref. 21 and 29–32.
In the case of resonant HRS (Fig. 1E), calculations of dynamic hyperpolarizabilities are highly desirable to account for resonant enhancements of selected modes. Recently, two methods were implemented and employed to calculate the HRS spectra of rhodamine 6G33 and crystal violet34 under near-resonance conditions, and the results matched very well the experimental data.
3. Enhancement in surface enhanced hyper Raman scattering (SEHRS)
3.1 Electromagnetic and chemical enhancement
Surface enhanced hyper Raman scattering spectroscopy is concerned with the observation of very strong hyper Raman signals, when the scattering molecule is in close proximity to rough metal surfaces or nanoparticles. Apart from the normal HRS, resulting from the interaction of the molecule with the external electromagnetic field of the excitation light, as described in the previous section, the enhancement of the HRS, which occurs due to the presence of a plasmonic metal nanostructure or nanostructured surface, must be discussed. This surface enhancement includes contributions from both metal–light and metal–molecule interactions, giving rise to the so-called electromagnetic (EM) and chemical enhancement mechanisms, respectively.Localized surface plasmons account for the optical properties of metal nanoparticles and result in strong amplification of the electromagnetic fields near the plasmonic nanostructure. These highly confined local optical fields are considered responsible for the electromagnetic enhancement. In a scattering process, as is the case in SERS, plasmonic enhancement is operative in both the incident and the scattered field. SEHRS, as a non-linear effect, benefits particularly from enhanced local fields due to its quadratic dependence on the excitation intensity.
In contrast to the electromagnetic field enhancement that is determined by the plasmonic properties of the metal nanostructure, the chemical enhancement is highly molecule specific and refers to the properties of the metal–molecule system, which can be different from those of the free molecule. The SEHRS spectrum is governed by the hyperpolarizability of the metal–molecule complex. The chemical mechanism can be further divided into contributions arising from (1) metal–molecule ground state interactions and from (2) resonances of the excitation wavelength with metal–molecule charge-transfer transitions or simply with electronic transitions of the adsorbed molecule,35,36 where the electronic states can be altered by the presence of the metal. It is important to mention here that theoretical studies show that the chemical contribution to the overall enhancement in SEHRS can be larger than the corresponding chemical enhancement for SERS.19,21,29,32 Nevertheless, numerous studies show that the main contributions to the very high SEHRS enhancement is related to enhanced local fields of plasmonic structures.
Considering electromagnetic and chemical enhancement mechanisms and including them in eqn (1), the number of SEHRS photons nSEHRS can be written as:
| nSEHRS = σSEHRSn02 | (2) |
| σSEHRS = σHRSads|A(ν0)|4|A|(νHRS)|2 | (2a) |
| σHRSads ∝ 〈|βadsif|2〉 | (3) |
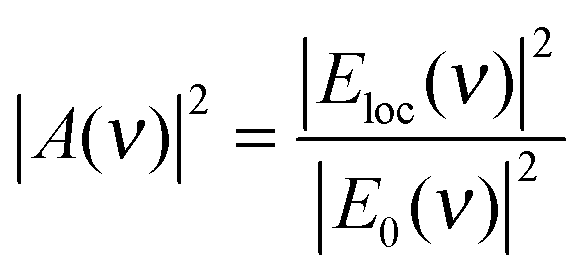 | (4) |
Note that, since HRS depends on the square of the incident radiation intensity (eqn (1)), the enhancement factor for the incident light in SEHRS scales with Eloc4, while for the excitation in SERS it scales only with Eloc2. This shows that HRS can benefit even more than RS from electromagnetic enhancement, if the incident and scattered frequencies are both in resonance with the localized surface plasmons. The EM mechanism, related to the properties of the nanostructure, has been extensively investigated, and more detailed information about the origin and methods for calculation of the field enhancement, as well as its dependence on the geometry of nanostructures can be found, for example, in ref. 35 and 37–41.
Pyridine is the first small organic molecule, for which SEHRS and SERS spectra under non-resonant excitation conditions were reported,1–3,19 and it has remained one of the most extensively investigated model systems for understanding both effects.19,21,29,31,32,42,43 In the different experimental data, the overall SEHRS intensities obtained on electrodes were much greater than those obtained from silver nanoparticle aggregates, indicating strong differences in the enhancement for pyridine, generated by the two types of surfaces. Theoretical work was involved to discuss the enhancement in SEHRS, as it is very difficult to obtain normal HRS spectra under non-resonant excitation conditions.19,29,43 The combination of experimental SERS and SEHRS intensities together with calculated intensities from normal HRS and RS spectra was used to estimate the overall SEHRS enhancement factors.19 Depending on the level of theory used in the calculations and the experimental setup, the values for the enhancement on electrodes ranged from 1010 to 1013,19,29,43 which is much greater than the corresponding overall SERS enhancement factor of 105–107. In other studies, the chemical enhancement in SEHRS was modeled by considering the pyridine molecule on a small silver cluster.21,32 It was found that the chemical contribution (excluding resonance effects) is on the order of 102–104 depending on the adsorption geometry, while for SERS the chemical enhancement is found to be only 101–102.21 Combining the molecule on a small silver cluster model with classical electrodynamics theory for a single silver nanoparticle to account for electromagnetic contributions, the resulting SEHRS enhancement factor was determined to be 109.32 Including aggregated clusters of nanoparticles instead of an isolated nanosphere in the model would possibly result in even larger enhancement factors, reaching the estimated experimental values of 1010 to 1013.
An approach to estimate enhancement factors in SEHRS exploits a direct comparison between SEHRS and SERS signals measured from the same sample. Knowing the excitation intensities and the number of molecules contributing to SERS and SEHRS (note the different scattering volumes for the one- and two-photon excited process), a comparison between SEHRS and SERS signals allows estimation of a ratio of effective cross sections of SEHRS and SERS, i.e. the ratio enhancement factors for both effects (see also the next subsection). Despite very different signal levels of normal Raman and hyper Raman scattering, SERS and SEHRS spectra can appear at comparable signal levels and, for example, can be measured in the same spectrum using the first and second diffraction order of a double grating spectrometer, respectively.44 This implies that hyper Raman scattering performed on silver nanoaggregates must experience an enhancement level six orders of magnitude higher than Raman scattering. Given an enhancement factor for non-resonant SERS performed on such silver nanostructures on the order of 1014,4 the total enhancement factor for SEHRS was reported to be on the order of 1020.17,45 High cross sections in SEHRS enable measuring two-photon excited vibrational spectra down to the limit of single-molecules45–47 and collecting hyper Raman spectra also at the anti-Stokes side (Fig. 2).17,48
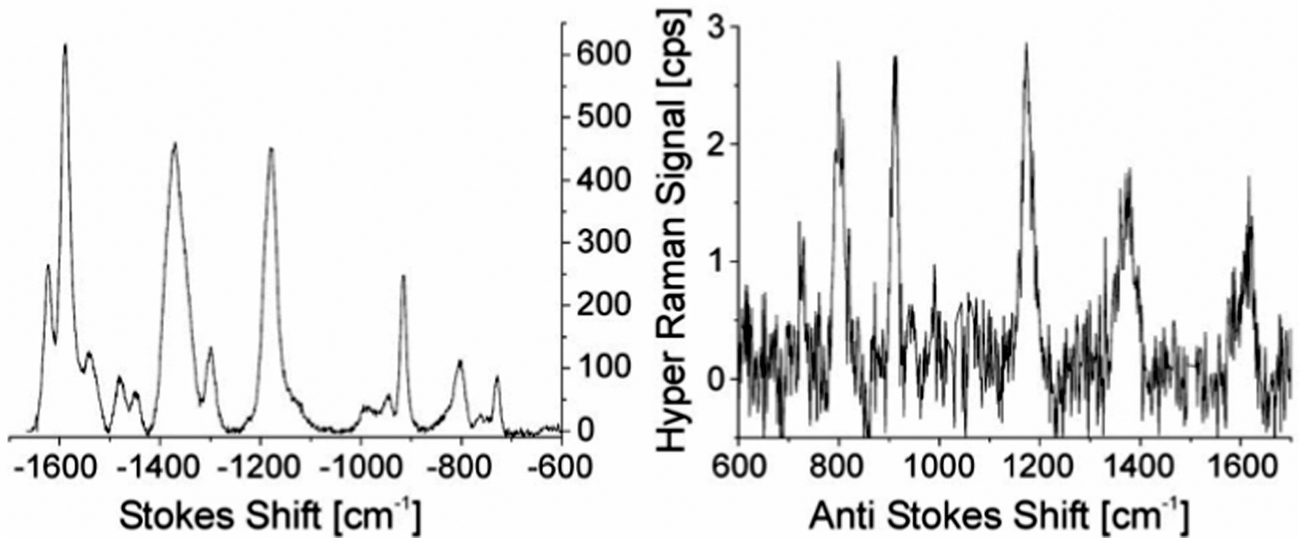 | ||
| Fig. 2 SEHRS Stokes and anti-Stokes spectra of 10−7 M of the dye molecule crystal violet attached to silver nanoaggregates in aqueous solution. The spectra were measured using 1064 nm mode locked ps pulses of average power 40 mW (peak intensity 2 × 1025 photons cm−2 s−1) and collection times of 10 and 60 seconds, respectively. Anti-Stokes scattering appears at a much lower signal level than Stokes scattering, as the number of molecules in the excited vibrational state is much smaller than those in the vibrational ground state, but here anti-Stokes HRS can be observed because of the extremely high effective cross section of SEHRS. Reproduced with permission from ref. 17. Copyright 2006 National Academy of Sciences, USA. | ||
3.2 Understanding the overall SEHRS enhancement in experiments
In an experiment it is hard to distinguish between the chemical and electromagnetic contributions to the SEHRS spectra because both are always included. However, the ratio of the electromagnetic enhancement of one-photon excited RS and HRS, EFEM(SERS)/EFEM(SEHRS), can be estimated for a given absorbance spectrum of the plasmonic sample. Such estimations were made to, e.g., explain the origin of the difference between the SERS and SEHRS enhancement factors for non-resonant spectra of the dye crystal violet44 and for para-mercaptobenzoic acid (pMBA).22 In analogy to the empirical relationship between the electromagnetic SERS enhancement factor and the surface plasmon absorption spectrum,49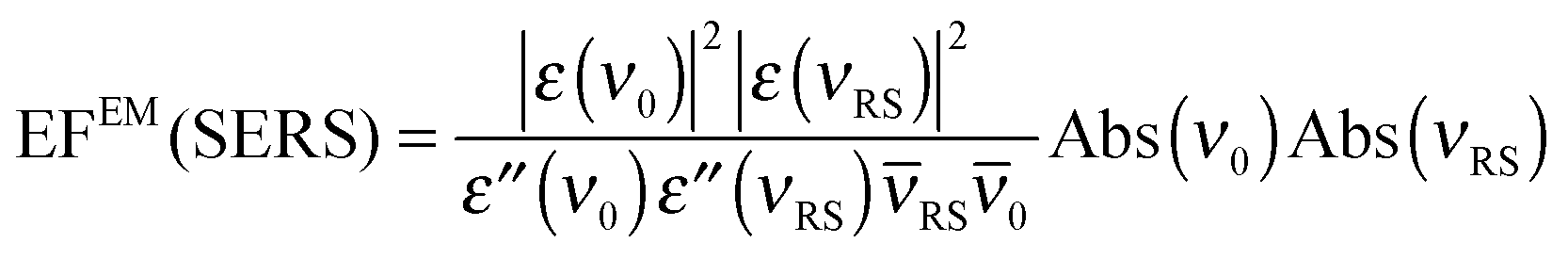 | (5) |
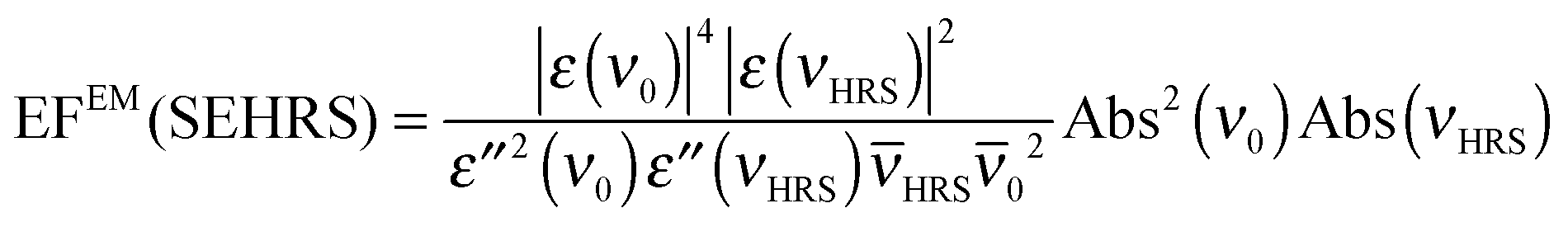 | (6) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) HRS, RS and 0 are the corresponding wavenumbers. Nevertheless, it should be noted that this empirical model does not include possible plasmon resonance effects arising from modes that cannot be observed in the far field UV-vis absorbance spectrum (cf. next section).8,40,50,51
HRS, RS and 0 are the corresponding wavenumbers. Nevertheless, it should be noted that this empirical model does not include possible plasmon resonance effects arising from modes that cannot be observed in the far field UV-vis absorbance spectrum (cf. next section).8,40,50,51
Ratios EFEM(SERS)/EFEM(SEHRS) obtained in this way from absorbance spectra were compared with the ratio of EF(SERS)/EF(SEHRS) determined in Raman experiments that give the total enhancement including electromagnetic and chemical contributions.22,44 Using SEHRS and SERS spectra of crystal violet and pMBA, as well as the corresponding absorbance data, EF(SERS)/EF(SEHRS) and EFEM(SERS)/EFEM(SEHRS) that were in good agreement with each other were obtained, ranging from ∼10−5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 to ∼10−6,44 depending on the respective molecule–nanoparticle system. In both cases, a high similarity of EF(SERS)/EF(SEHRS) and EFEM(SERS)/EFEM(SEHRS) showed that the different EF of SEHRS and SERS can be explained by the difference between the corresponding EFEM.22,44
22 to ∼10−6,44 depending on the respective molecule–nanoparticle system. In both cases, a high similarity of EF(SERS)/EF(SEHRS) and EFEM(SERS)/EFEM(SEHRS) showed that the different EF of SEHRS and SERS can be explained by the difference between the corresponding EFEM.22,44
In the SERS and SEHRS spectra, the scattering powers PSERS and PSEHRS, respectively, are
| PSERS = N0σSERSn0 | (7) |
| PSEHRS = N0σSEHRSn02 | (8) |
Despite the fact that the main contribution to SEHRS is related to EM enhancement, a deeper understanding of the chemical contribution to SEHRS is of great interest for harnessing SEHRS as a sensitive tool for investigations of the interaction of molecules and surfaces.
As the example of pyridine above shows, theoretical simulations can provide deeper insights into the qualitative changes and increased intensities of the HRS spectrum, resulting from adsorption of the molecule on a metal surface. Modeling SEHRS is a challenging task, although significant improvements have been made in this direction recently.21,32,52 Early SEHRS studies, which do not explicitly include surface atoms, assumed a fixed orientation of the molecule with respect to the surface, resulting in different symmetries for the hyper Raman calculations.19,29,31,53 Such simulations account for changes in relative peak intensities between normal HRS and SEHRS spectra, since the model relies on different orientation averaging for both cases, although the vibrational frequencies and hyperpolarizability tensor components are the same for the free and adsorbed molecule. Further improvements include a more accurate evaluation of the chemical enhancement by modeling the HRS spectrum of the molecule in the presence of a small metal cluster,21 which accounts for shifts of vibrational frequencies due to adsorption and yields increased peak intensities in SEHRS compared to HRS. Combined quantum mechanics/classical electrodynamics methods have been employed to estimate both the chemical and the EM enhancement contributions.32 Recently, it was shown that the electromagnetic enhancement can also be evaluated using an atomistic electrodynamics model of the metal nanoparticle.52 In addition, theories going beyond the dipole approximation, which include dipole and quadrupole light–molecule and light–metal interactions, as well as gradients of the local electric field, have been developed and used for the interpretation of SEHRS spectra from small molecules.36,52,54–59 Field gradient effects are of particular interest, as they can contribute not only to the enhancements but also to the selection rules in SEHRS spectra.20,52
4. Instrumentation and experimental aspects in SEHRS
4.1 General considerations
The experimental parameters as well as technical problems in SEHRS experiments can vary greatly. They depend on the optical properties of the plasmonic structures and the required resonant or non-resonant excitation of the molecules of interest, the amount of sample, and the necessary spectral and lateral resolution.As will be discussed in more detail in the next subsection, currently, mostly mode-locked, high repetition rate, few-picosecond, near-infrared lasers are used for excitation of SEHRS. As examples, please refer to the references given in Table 1. Using NIR lasers for two-photon interaction moves the detection range to the convenient visible region, where very sensitive CCDs are available. In the case of SEHRS, excitation with smaller photon energy (compared with SERS in the same detection range) helps in reducing photodamage of the sample and can allow background-free, i.e. fluorescence-free, detection. A further practical advantage of NIR excitation is the deeper penetration into samples. The two-photon excitation being limited to the laser focus leads to reduced interaction volumes and to high spatial resolution.
| Wavelength (nm) | Pulse width | Repetition rate | Intensity (W cm−2) | Ref. |
|---|---|---|---|---|
| a Calculated intensity based on data provided in the papers. | ||||
| 1064 | CW | CW | 3 × 107 | 71–73 |
| 1064 | 200 ns | 5–10 kHz | 106 | 10, 62, 74 and 75 |
| 1064 | 10–15 ns | 10–20 Hz | ∼108 | 11 and 76 |
| 1064 | 80–100 ps | 82 MHz | ∼107 | 19, 20, 42, 53, 60, 77 and 78 |
| 1053 | 50 ps | 76 MHz | ∼107 | 31, 43, 79 and 80 |
| 1064 | 2–7 ps | 76–82 MHz | 107–1010 | 17, 22–24, 64, 68, 70 and 81–85 |
| 750–1050 | 1–5 ps | 80–82 MHz | 107–109a |
33, 44, 45, 47, 61, 65–67, 69, 83 and 86–89 |
| 1550–1570 | 5–6 ps | 80 MHz | ∼5 × 107a |
34 and 90 |
| 1800 | 5 ps | 80 MHz | ∼3 × 107a |
90 |
| 915 | 6 ps | 80 MHz | ∼1 × 108a |
34 |
To observe the two-photon excited Raman spectra of molecules enhanced with plasmonic nanostructures or on electrodes, typical dispersive Raman spectrometers have been used.11,19,22,44,48 While in the 1980s, the reported SEHRS spectra were collected using double or triple monochromators with a photomultiplier or with intensified diode arrays, for examples see ref. 11, 19, 42 and 60, nowadays new generation deep-depletion CCDs, mainly N2 cooled, back-illuminated, Si-based 2D cameras, are suitable for the relatively long integration times required, because of their extremely low dark noise and their high sensitivity in the whole wavelength range of interest.
SEHRS spectra are typically accumulated in the range of a few seconds to a few minutes, depending on the enhancement by the plasmonic structures, cross sections, excitation conditions, detection frequency range and instrument response functions of optical elements. Different authors have reported typical spectral resolutions of 2–6 cm−1 in the full spectral range.22,23,31,48,61 In experiments where the detection frequency range is identical in the one- and two-photon excited case, SERS and SEHRS spectra can be collected quasi-parallel in the same experimental set-up.20,23,44,53,62–64 Simultaneous detection of SEHRS and SERS when using the same excitation wavelength for both one- and two-photon SERS was also reported.44,65
For collecting the scattered light, both forward and backward directions are used.22,33 In forward detection, e.g., using an inverted microscope, optics are easy to handle, and anti-reflection coated lenses or microscope objectives, e.g. NIR coating for excitation and visible-coating for detection,33,63 are commercially available. In contrast, backward detection can pose some challenges, since the focusing and collecting optical components (lenses and dichroic mirrors) must provide appropriate figures of merit regarding both excitation and detection frequencies. As in other Raman experiments, the accessible notch filters needed to clear the signal from the excitation and Rayleigh-scattered light can limit flexibility with respect to the excitation wavelength. For wavelength scanned SEHRS using continuously tuned excitation frequencies in the wavelength range between 780 and 1000 nm, an inverted microscope and special dichroic filters were demonstrated.66,67
The investigated samples, mostly in the liquid phase on electrodes or with nanoparticles, can be measured in cuvettes or micro-containers.10,22–24,42,44,60,68 A few groups report SEHRS spectra on immobilized or lithographically fabricated nanostructures.64,69 As a first real biological application, SEHRS spectra were collected to analyze intrinsic biochemical information from nanometer-scaled volumes in dry-fixed individual culture cells.17,70 Furthermore, using a CW laser at 1064 nm (∼300 mW average power) SEHRS was combined with the optical trapping of nanoaggregates, and spectra of dye molecules were obtained under resonant conditions.46,71
4.2 Laser sources
The continuous development of laser technology that has been taking place since the development of the first laser made it possible more than four decades ago to reach focal intensities ≥108 W cm−2, high enough to drive a broad range of non-linear processes, including SEHRS. Significant effort has been spent on shortening the laser pulse duration, which makes it possible to reach high intensities even at moderate pulse energies. A typical SEHRS experiment requires near-infrared intensities in excess of 107 W cm−2, which is accessible to pulsed lasers or very tightly focused CW lasers.19,46,65 Since the first SEHRS signal observed 35 years ago,10 a vastly expanded selection of lasers has become available at the experimenter’s disposal. At present, mostly mode-locked, high repetition rate, few-picosecond, near-infrared lasers with an average power of 2 × 10−4 to ∼1 W and focused intensities between 107 and 1010 W cm−2 are being used. The minimum intensity in the focus is an approximately constant threshold, irrespective of the operation mode of the laser (CW, Q-switched, or mode-locked). In Table 1, we summarize the experimental parameters in different reports of SEHRS experiments. The following sub-section discusses different excitation pulse parameters in two-photon excited surface enhanced Raman microspectroscopy.4.3 Excitation pulse parameters
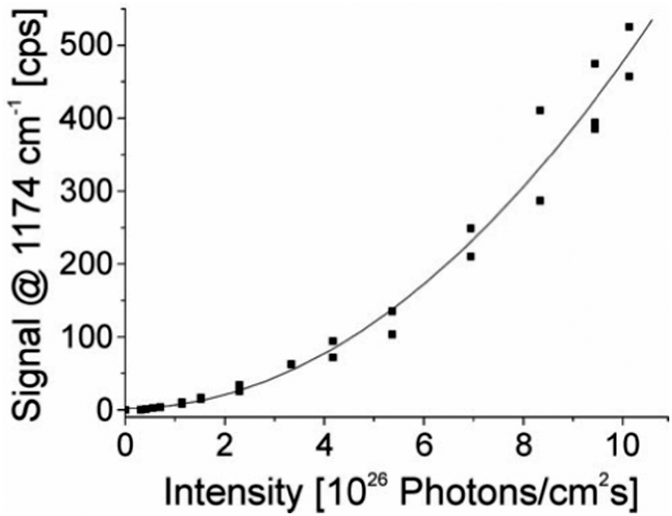 | ||
| Fig. 3 The square-law dependence of the hyper Raman scattering signal on the excitation intensity verifies the two photon process. The signal of a band at 1174 cm−1 in the SEHRS spectra of 10−7 M of the dye molecule crystal violet attached to silver nanoaggregates in aqueous solution is shown as a function of the excitation intensity. The spectra were measured using 1064 nm mode locked ps pulses and collection times of 10 seconds. Reproduced with permission from ref. 17. Copyright 2006, National Academy of Sciences, USA. | ||
As was shown earlier, the photon flux density allows a convenient way to estimate the ratio of one- and two-photon Raman scattering cross sections from scattering powers PSERS and PSEHRS determined in experiments. As an example, photon flux densities of 1026–1029 photons cm−2 s−1 were used to collect SERS and SEHRS spectra of several dyes and of biologically relevant molecules under resonant and non-resonant conditions, respectively.17,22–24,64,84,85 These photon flux densities are in agreement with the above intensity values (see in Table 1) for the resonant (107 W cm−2) and off-resonant (1010 W cm−2) case.
4.4 Focal volumes and amount of probed sample
While the focal spot sizes of the excitation beam can be determined from the Rayleigh criterion, the two-photon interaction volume V can be approximated as a three-dimensional Gaussian volume,18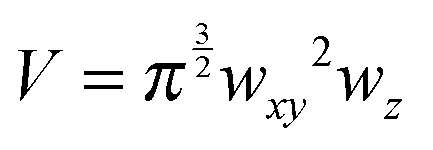 | (9) |
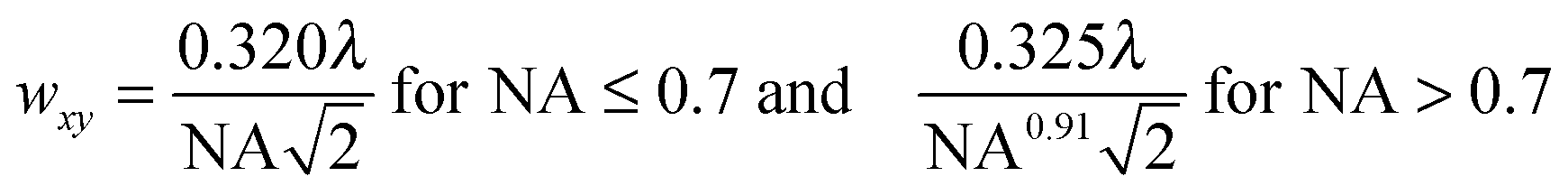 | (10) |
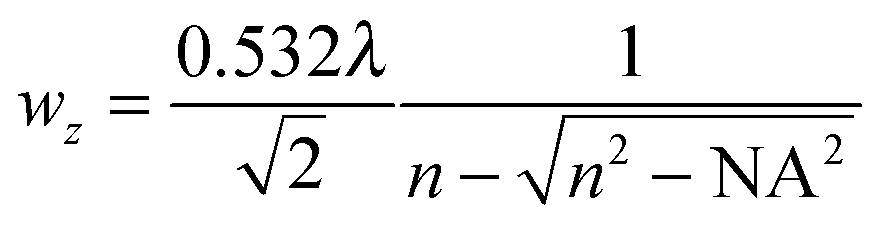 | (11) |
Knowing the interaction volume, the number of nanostructures and number of molecules taking part in a SEHRS experiment can be estimated. For example, several experiments with silver nanoparticle solutions have employed focal volumes of tens of μm3 down to less than a femtoliter, which, at typical nanoparticle concentrations, contain very few silver nanoparticles.22 While the statistical number of nanoparticles in the above defined V volume can be determined using their concentration, specifying the number of molecules in this volume is more difficult. If the required space for a given molecule at the surface of a nanostructure is known, we can determine the number of molecules interacting with the surface of the nanostructures that can participate in SEHRS. This is a more fundamental experimental parameter than molecule concentration. The interaction of molecules with the surface of nanostructures will be discussed in more detail in the following sections. The small interaction volume is also of interest in the development for microspectroscopic imaging using SEHRS.
5. Enhancing nanostructures for SEHRS
5.1 Requirements
SEHRS experiments require nanostructures providing high enhancement factors, so that the low cross sections of hyper Raman scattering (HRS) on the order of 10−65 cm4 s can be overcome. In general, SEHRS experiments have typically been carried out with nanostructures similar to or the same as those used in SERS. While the first observation of SEHRS was made on silver micro powder,11 subsequent investigations employed electrodes19,29,43 and colloidal solutions of metal nanoparticles,10,42,62 which have remained the main two types of enhancing nanostructures for SEHRS.It is well known from SERS theory and experiments that hot spots with extremely high field enhancement occur in the spaces between particles in fractal structures such as nanoaggregates.8,83,91,92 In SEHRS, the effect of aggregation is even more important because aggregated nanoparticles can support plasmon resonances in a wide spectral range from 400 nm to 1200 nm (Fig. 4A and B), although they are not always visible in the far field scattering spectrum (see for example ref. 8, 40, 50 and 51). According to the electromagnetic theory for SEHRS (eqn (2)), this allows enhancing both the incident and scattered fields, whose frequencies are widely separated, and thus optimizing the total enhancement.
 | ||
| Fig. 4 Examples for enhancing nanomaterials used in SEHRS: (A) UV-vis absorbance spectrum of hydroxylamine reduced silver nanoparticles before and after aggregation with magnesium sulfate, and (B) transmission electron micrograph of the silver colloid in (A) used in SEHRS experiments with biomolecules.24 In this experiment, it was possible to collect SEHRS spectra of amino acids only with the aggregated nanoparticles, while SERS spectra could also be obtained with the non-aggregated nanoparticles, thus demonstrating the importance of colloid aggregation in SEHRS. Adapted with permission from ref. 24. Copyright 2017 American Chemical Society. (C) SEM image of immobilized citrate reduced silver nanoparticles on glass slides used for SEHRS hyperspectral mapping.64 Adapted from ref. 64 with permission from the PCCP Owner Societies. | ||
So far, few studies compare SEHRS enhancements of different types of nanostructured materials. Recently, silver nanoaggregates formed by four nanoparticle solutions with silver nanostructures of different size and stabilizing species were compared, and ratios between SERS and SEHRS enhancement factors were estimated from Raman measurements as well as from the absorbance spectra using eqn (5) and (6).22 Earlier investigations compared the overall SEHRS intensities from measurements on roughened electrodes and nanoaggregates.79 Interestingly, it was found that the enhancements depend very much on the specific molecule under study.
As mentioned above, typical plasmonic nanostructures for SERS have been directly transferred to SEHRS experiments. As indicated by more recent results, it is not necessarily an optimum strategy to directly draw conclusions about the enhancement in SEHRS from the enhancement yielded with a particular type of nanostructure in SERS. On the one hand, this is due to different requirements regarding the plasmonic properties. In particular, the design of nanostructures with multiple plasmon resonances to match the incident and scattering frequencies in HRS, active in the same physical region of space, has been proposed.93–95 On the other hand, the surface chemistry of the nanoparticles, which both determines the formation of nanoaggregates (i.e., the plasmonic properties) and at the same time influences the interaction of the molecules with the metal surface, plays an important role in the enhancement. For example, nanoaggregates formed by citrate reduced silver nanoparticles yield higher overall SEHRS intensities than nanoaggregates from hydroxylamine reduced silver nanoparticles, although exactly the opposite is found for SERS.23 In silver nanoaggregates, enhancement was discussed for individual nanostructures, assessing the plasmonic properties as a function of polarization.72,73
Such experiments provide evidence that it is important to design suitable plasmonic materials for SEHRS, and to further understand the factors that influence SEHRS enhancement in the experiments. First theoretical discussions aiming at the optimization of plasmonic nanostructures for SEHRS, e.g. by considering different geometries including double resonant antennas in the visible and UV, and silver heptamers supporting Fano resonances,95 were reported. Further research focusing on the development of SEHRS active nanostructures providing uniform and high enhancement should be the key for obtaining comprehensive vibrational information by means of SEHRS.
A very important feature of both aggregated nanoparticles and electrochemically roughened electrodes is that their structure is neither well defined, nor that it is clear if similar hot spots are equally distributed throughout the sample. There have been several attempts to improve the homogeneity of SEHRS enhancement in nanostructured materials. For example, silver nanocrystal-modified silicon nanowires were found to provide a more spatially uniform response than silver nanoparticles deposited as a film on glass.87 In particular, chemically immobilized silver nanoparticles on glass slides (Fig. 4C), well known for their homogeneous SERS enhancement,96 have proven to be well-suited for SEHRS hyperspectral mapping experiments.64 In addition, fabrication of well-defined structures with nanoscale gaps, commonly used in SERS,97–99 has also been implemented in SEHRS.69 Anisotropic silver dimer arrays were the first artificially designed nanostructures employed in SEHRS measurements.69 However, relatively little work has been done in this direction for SEHRS and further optimization of the electromagnetic enhancement should be possible.
5.2 SEHRS with nanoparticles and nanoaggregates
In recent work, the majority of SEHRS experiments have been carried out using aggregates of metal nanoparticles as enhancing structures. Such nanoparticles can be easily prepared by standardized wet chemical methods, although their reproducibility is often not optimum. As an example, silver nanoparticles with diameters in the range of 50–100 nm obtained by reduction of silver nitrate with sodium citrate100 have been most frequently used, for example by the groups in ref. 17, 22, 42, 44, 66, 68, 71, 78 and 86. Fewer reports employ other preparation techniques for silver nanoparticle solutions resulting in nanostructures with diameters below 50 nm and different surface chemistry, specifically the reduction with hydroxylamine,22–24 borohydride,22 tannic acid,46 hydrazine81 or hydrogen gas.62 Also, silver nanoparticles prepared by green synthesis in situ in intact onion epidermal cells were shown to have sufficient SEHRS activity,84 highlighting the potential of such an approach for exploiting SEHRS in bioanalysis. For the latter purpose, gold nanoparticles, which have also been used for SEHRS,17,60 though less frequently than silver nanostructures so far, would be ideally suited.The aggregation of nanoparticles is often induced by addition of sodium chloride, for examples see ref. 66, 68, 73, 74, 82 and 86, by sodium bromide,47,88,89 potassium chloride42,76,81 or other electrolytes in the nanoparticle solutions. Each type of electrolyte at a given concentration can produce a unique aggregate morphology and surface coverage with ions, which can result in differences between the SEHRS spectra of the same compound.88,89 Nevertheless, it is very difficult to distinguish between chemical effects induced by co-adsorbed ions and effects due to different fields and field gradients that can arise from different gap sizes or aggregate geometry as a reason for the observed spectral changes, since both can be simultaneously varied during the aggregation process. SEHRS is extremely sensitive with respect to changes in surface potential and small changes in the interaction of the molecules with the metal surfaces,20 and depending on the molecule, the chemical contribution to the enhancement can be quite strong as well. Furthermore, as will also be discussed in the next section, the interaction of the molecule with the nanostructure – which is the main prerequisite for the molecule for taking part in SEHRS (and also SERS) – depends critically on the surface charge and charge of the molecule.23,101 As an example, in spectra of para-mercaptobenzoic acid, measured with hydroxylamine-stabilized silver nanoparticles, a reduction in SEHRS enhancement by a factor of 2–3 at very low and very high pH was observed.22 In 532 nm excited SERS spectra with the same samples, the effect was not present.22 Apart from altered interaction of the molecule with the metal nanoparticle surface, the molecules that are used to stabilize the silver nanoparticles, both citrate and hydroxylamine/hydroxide, contain functional groups that can be protonated or deprotonated, depending on the surrounding pH. This leads to changes in surface charge and possible aggregation of the nanostructures. For example, a negative surface charge of the silver particles decreases with decreasing pH by protonation, leading to aggregation and minor changes in gap sizes in the nanoaggregates and therefore to a lower electromagnetic enhancement at very acidic pH.102,103 At this point it must also be mentioned that SEHRS spectra can display strong background contributions, which were discussed in terms of the nanoaggregate properties.22,73,81
5.3 SEHRS with electrochemical electrodes
Electrochemical SEHRS measurements on silver electrodes were found to be convenient for the investigation of small organic molecules such as pyridine, pyrazine, phenazine and related derivatives under non-resonant excitation conditions19,20,31,43,53,79,80 and also to study the dependence of the SEHRS signals on surface potential.19,20,31,43,80 To provide high local field enhancement, the electrodes are usually roughened in oxidation reduction cycles (ORC), which results in modification of the surface morphology at the nanometer scale.19,43,79,80 To improve homogeneity and reproducibility of the surface roughness, modification of the electrode surface by dispersing chemically synthesized metal nanoparticles onto it was proposed, which allowed reversibly obtaining reproducible SEHRS spectra in a wider potential range than with an electrochemically roughened electrode.31 The concept of silver film over nanosphere electrodes, which have more homogeneous and reproducible nanostructure morphology, has also been introduced into SEHRS.20,53 In this type of electrode the surface roughness is produced by vapor deposition of silver on top of a nanosphere monolayer. An important advantage of this system over ORC roughened electrodes is that the concentration and chemical nature of the supporting electrolyte in an electrochemical SEHRS experiment can be varied without altering the surface morphology and thus the enhancement associated with it. In particular, this allows investigation of the effect of co-adsorbed ions on SEHRS spectra,20 which is still not possible for measurements in nanoparticle solutions.6. Non-resonant SEHRS reveals the interaction of molecules with surfaces
6.1 SEHRS spectra provide complementary vibrational information
The selection rules for non-resonantly excited SEHRS have been discussed by comparing SERS and SEHRS spectra from small organic molecules, including pyridine,31,43 pyrazine,31trans-1,2-bis(4-pyridyl)ethylene (BPE),20,53 benzene,31para-mercaptobenzoic acid,22,70 as well as data from nucleobases23,82 and amino acids,24 and recently also the dye crystal violet excited at 1570 nm.34In the case of centrosymmetric molecules such as pyrazine, benzene and BPE, the Raman and hyper Raman active modes are expected to be complementary. For benzene, indeed the SERS and SEHRS spectra do not show any common bands, and the SEHRS spectra are very similar to the IR absorption data.31 Also for pyrazine and BPE “new” Raman-inactive modes appear in the SEHRS data that are not observed in SERS, in addition to some SERS-active bands.20,43 Although the adsorption of molecules can reduce their symmetry, so that Raman-active modes become visible in the hyper Raman spectrum as well, the detailed examination of the SERS and SEHRS spectra for these two molecules showed that little or no symmetry reduction occurs upon adsorption.20,43
The SEHRS and SERS spectra from non-centrosymmetric molecules such as pyridine, nucleobases and aromatic amino acids display bands mostly at the same positions, although with quite different relative intensities.19,24,82 For example, in the SEHRS and IR spectra of pyridine, Golab et al. reported less pronounced signals of the ring breathing modes.19 Also the signal of the symmetric ring breathing mode of all five nucleobases differs between SERS and SEHRS spectra. While in SERS it is often used to estimate the adsorbate orientation with respect to the surface, this signal is weak or medium compared to other bands in SEHRS.23,82 This mode is also very strong in the Raman but medium in the IR absorption spectra of the solid compounds. Fig. 5 (top trace) shows the SEHRS and SERS spectrum of the nucleobase adenine obtained with silver nanostructures. Independent of a varied surface composition (stabilization) of the silver nanoparticles,17,23,82 the differences between the SEHRS and the SERS spectrum of adenine are consistent and obvious in all reports. In the SEHRS spectrum (Fig. 5A, top trace) the signal of the symmetric ring breathing mode at 734 cm−1 is very similar to all other bands, while it dominates the SERS spectrum (Fig. 5B, top trace). Furthermore, the SEHRS data display several strong bands associated with NH2 and N–H deformation modes, specifically the NH2-rocking band at 1026 cm−1, in-plane NH2 scissoring vibrations at 1487 cm−1, 1554 cm−1 and 1653 cm−1, and the bands at 564 cm−1, 1141 cm−1 and 1600 cm−1, which can be associated with N–H bending modes.23
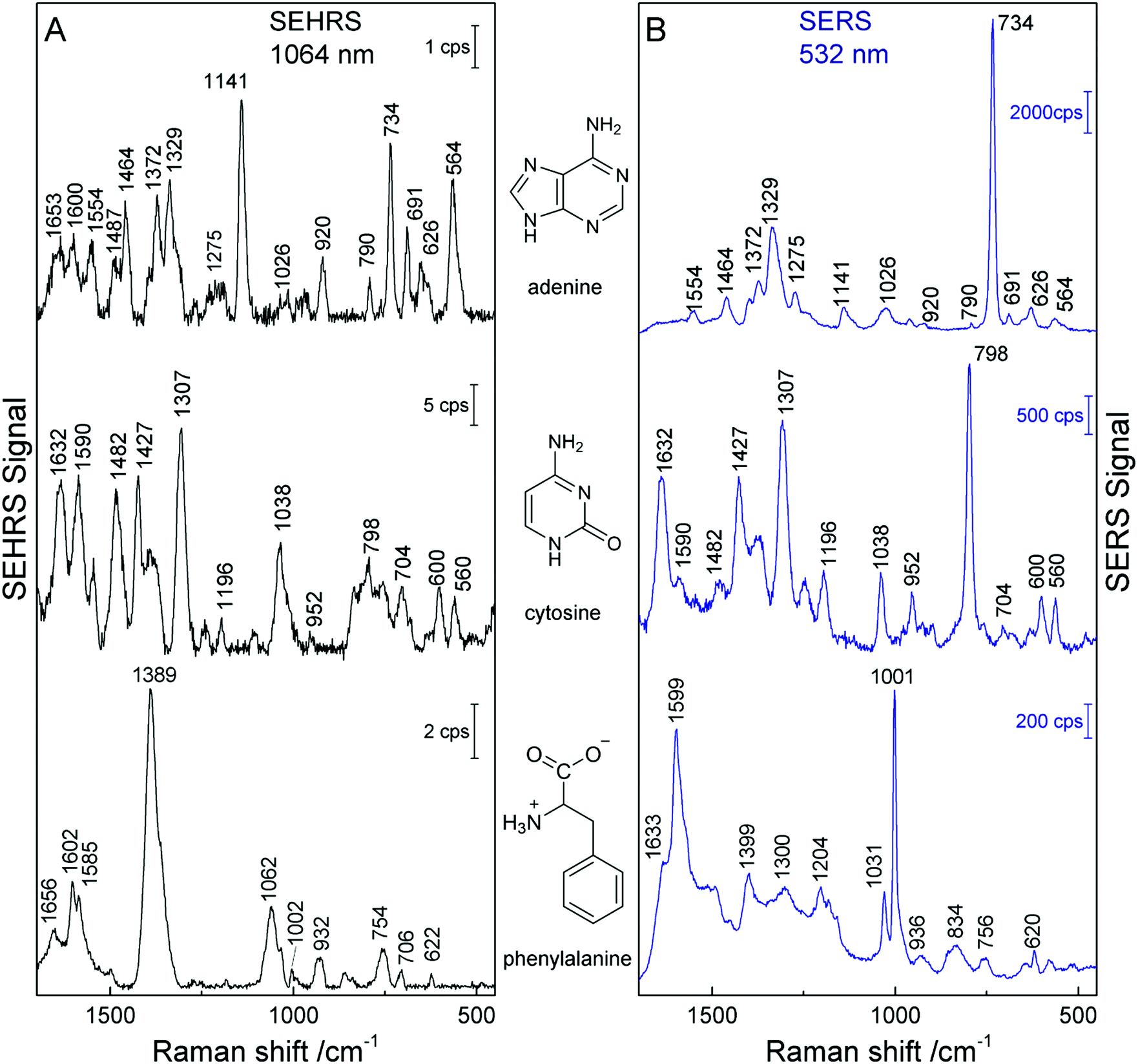 | ||
| Fig. 5 SEHRS spectra (A) and SERS spectra (B) of adenine, cytosine, and phenylalanine. The spectra of adenine and phenylalanine were obtained using nanoaggregates from hydroxylamine reduced silver nanoparticles, and the spectra of cytosine with citrate reduced silver nanoparticles. Experimental parameters in (A): excitation, 1064 nm, peak photon flux density 5 × 1028 photons cm−2 s−1 (adenine and cytosine) and 3 × 1028 photons cm−2 s−1 (phenylalanine), acquisition time, 60 s (adenine and phenylalanine) and 40 s (cytosine); in (B): excitation, 532 nm, peak photon flux density 1 × 1027 photons cm−2 s−1 (adenine and cytosine) and 6 × 1023 photons cm−2 s−1 (phenylalanine), acquisition time, 1 s (adenine and cytosine) and 5 s (phenylalanine), concentration 5 × 10−5 M (adenine and cytosine) and 2 × 10−3 M (phenylalanine). The figure has been reproduced using the data in ref. 23 and 24. | ||
In cytosine, the most obvious differences between the SEHRS (Fig. 5A, middle trace) and the SERS spectrum (Fig. 5B, middle trace) are the pronounced signals of the C–N stretching mode at 1482 cm−1 and a relatively strong NH2 bending mode around 1590 cm−1 in SEHRS. Also here, the ring breathing mode at 798 cm−1 shows a much smaller relative intensity than in the SERS spectrum. The SERS spectrum of cytosine shown in Fig. 5 also has some contributions from the citrate at the surface of the silver nanoparticles, around 952 cm−1. In contrast, in the SEHRS spectrum, these bands are almost absent. The weak citrate signals were discussed as an additional advantage of SEHRS over SERS regarding the selective characterization of analyte molecules.23 As seen for the spectra of uracil and thymine with silver nanostructures obtained at alkaline pH, SEHRS can provide additional information about the interaction of the molecules with the nanoparticle surfaces. For example, a specific deprotonated uracil tautomer could be identified due to a higher enhancement of a characteristic vibration of this molecule in SEHRS compared to SERS.23
In the SEHRS spectra of the amino acids tryptophan, tyrosine, histidine, and phenylalanine, the bands from the ring breathing modes are strongly diminished or absent as well.24 Instead, as can be seen from the intense signal at 1389 cm−1 in the spectrum of phenylalanine in the bottom trace of Fig. 5A, the SEHRS spectra of these biomolecules are dominated by the symmetric COO− stretching vibration.24 At alkaline pH, which was employed in these experiments, the COO− group probably plays an important role in the binding of the amino acids to the silver surface. In contrast, the SERS spectrum of the molecule displays very strong signals of the marker bands at 1599 cm−1 and 1001 cm−1, assigned to the ring stretching (1599 cm−1) and ring breathing (1001 cm−1) (Fig. 5B, bottom trace).
6.2 The sensitivity of SEHRS to local environment and surface orientation
To understand the influence of the interaction of the molecule and the metal surface on the qualitative features in a SEHRS spectrum, many theoretical studies, mainly considering the model of pyridine on a silver surface, were performed.19,21,29,31,32 Assuming perpendicular orientation of the pyridine molecule on the surface, SERS and SEHRS spectra were modeled at various levels of theory. Valley et al.21 and Mullin et al.,32 who also compared their calculated spectra with the experimental work by Golab et al.19 and Li et al.,31 respectively (Fig. 6), showed that the normal HRS spectrum of pyridine is dramatically different from its SEHRS spectrum (see Fig. 6), while the differences between the calculated RS and SERS spectra were much less pronounced.19,21,32 Other simulations have shown that slight tilting of the molecule with respect to the surface normal results in more pronounced changes in the SEHRS spectrum than in SERS, suggesting that SEHRS is more sensitive to adsorbate orientation.19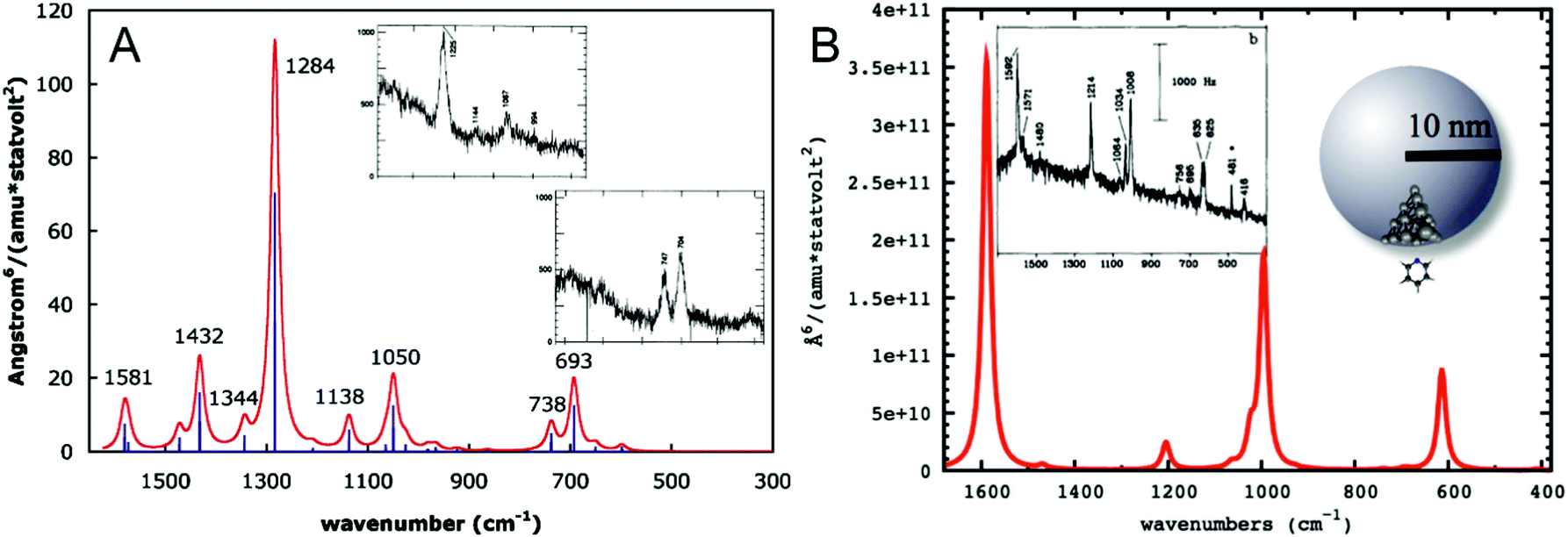 | ||
| Fig. 6 (A) Simulated gas phase HRS spectrum of pyridine reproduced with permission from ref. 21. Copyright 2010 AIP Publishing. (B) Simulated SEHRS spectrum of pyridine reproduced with permission from ref. 32. Copyright 2012 American Chemical Society. Surface effects in (B) were modeled as follows: pyridine–Ag20 cluster electronic structure calculation was used to determine the HRS spectrum and the chemical enhancement factor in the static limit, and then electrodynamics calculation for the nanoparticle was used to obtain the electromagnetic enhancement, which was multiplied with the chemical contribution to obtain the final SEHRS intensities. The insets in (A and B) contain experimental HRS and SEHRS spectra of pyridine from ref. 128 and 19, respectively. | ||
The higher sensitivity of SEHRS with respect to adsorbate geometry and small surface environmental changes has been reported for several species.20,22,24 The SEHRS spectra from BPE on silver electrodes displayed significant variations when the concentration and type of co-adsorbed counterions were changed.20 SEHRS experiments performed at slightly varied concentration of the amino acids histidine and tyrosine with silver nanoaggregates yielded spectra with several differences, while the SERS spectra of the same samples were almost unchanged.24 The changes in the SEHRS spectra indicated that a different interaction with the silver nanostructures took place when the concentration of the molecules was lowered slightly. From the aspect of probing the interaction of amino acids with the silver nanostructures, different selectivity leads to probing of different molecular adsorbate species in SEHRS and SERS.22 In addition to a stronger electromagnetic enhancement effect of HRS due to non-linear dependence on the enhanced local excitation field,94 also the chemical enhancement in SEHRS can greatly differ from that in SERS.21 Furthermore, in a nanostructure providing high SEHRS signals, a specific surface chemistry (e.g., surface coverage with magnesium and sulfate ions) can lead to different concentration dependence of the interaction of the molecules than in aggregates that provide high SERS enhancement. The possibility of probing different sites of interaction between the amino acid molecules and parts of the silver nanostructures in combined SEHRS and SERS experiments will help in understanding the formation and composition of biomolecular coronae around nanoparticles in biological environments, which is mainly determined by adsorption of proteins and amino acids.104,105
6.3 Application to pH sensing
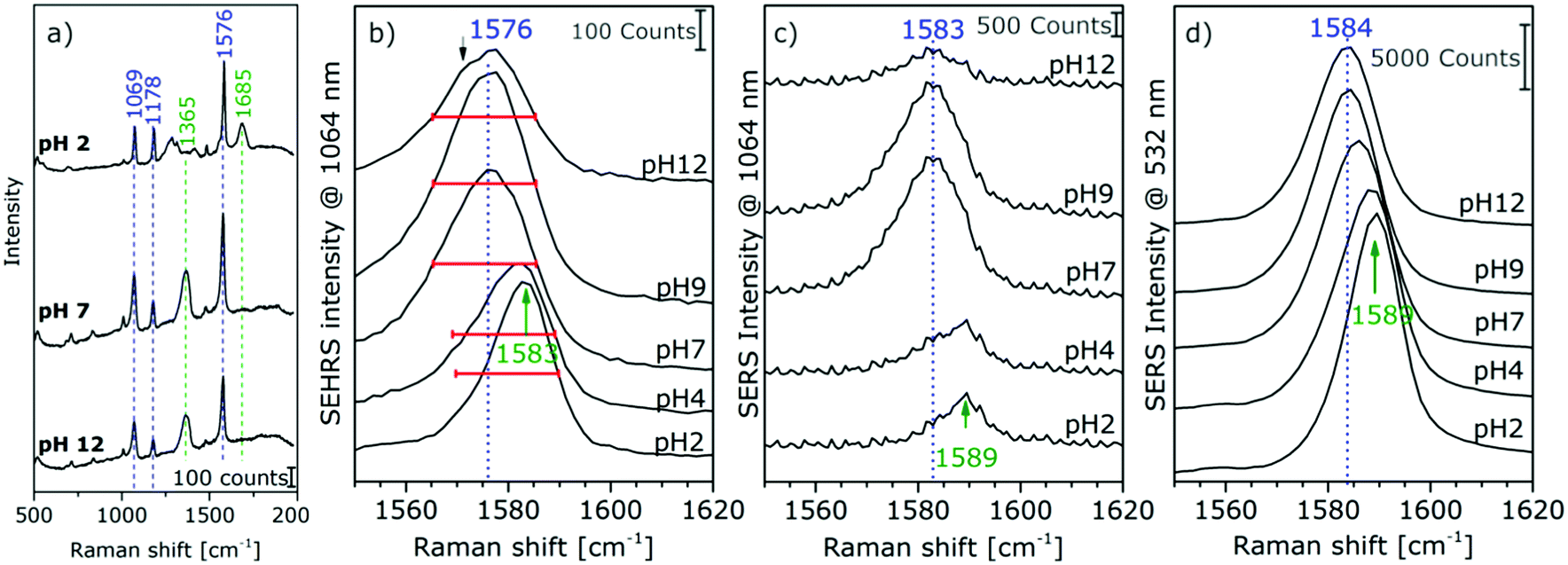
The high sensitivity of SEHRS with respect to surface charge and changes in the local environment of molecules can be applied for vibrational sensing. Additionally, as a two-photon method, SEHRS spectra are collected from smaller volumes than SERS spectra. It was shown that the SEHRS spectrum, similar to the SERS spectrum106 of para-mercaptobenzoic acid (pMBA), changes upon protonation and deprotonation of the molecule.22,70Fig. 7a shows spectra of pMBA measured at different pH values using citrate-stabilized silver nanoparticles from ref. 22. When pH increases, the relative band intensities of vibrations associated with the carboxyl group (marked in green in Fig. 7a) change relative to those of the aromatic ring (marked in blue). Upon deprotonation, the intensity of the COO− band at 1365 cm−1 increases, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration at 1685 cm−1 decreases. Since the relative signal strengths of the bands in the SEHRS spectrum are changed compared to those of the SERS spectrum,22,70 vibrational modes that are useful for pH measurements appear at a higher signal level in the SEHRS spectrum and can therefore be followed down to lower pH values. This enables measurements of pH values over a wide range, from pH 2 to pH 8, using SEHRS nanosensors that were also applied in cells.70 For comparison, other optical pH sensors based on one- or two-photon excited fluorescence, in most cases, require the application of multiple probes to cover such a pH range.
O stretching vibration at 1685 cm−1 decreases. Since the relative signal strengths of the bands in the SEHRS spectrum are changed compared to those of the SERS spectrum,22,70 vibrational modes that are useful for pH measurements appear at a higher signal level in the SEHRS spectrum and can therefore be followed down to lower pH values. This enables measurements of pH values over a wide range, from pH 2 to pH 8, using SEHRS nanosensors that were also applied in cells.70 For comparison, other optical pH sensors based on one- or two-photon excited fluorescence, in most cases, require the application of multiple probes to cover such a pH range.
 | ||
| Fig. 7 (a) Surface enhanced hyper Raman spectra of pMBA in the local field of citrate-stabilized silver nanostructures. Excitation: 1064 nm, photon flux density: 2 × 1025 photons cm−2 s−1, acquisition time: 2 s, pMBA concentration: 9 × 10−7 M, averages of 30 spectra. Extracts of (b) SEHRS and (c and d) SERS spectra of pMBA with Ag (citrate) nanoparticles at an excitation wavelength of 1064 nm (c) and 532 nm (d). Modified with permission from ref. 22. Copyright 2015, Published by the PCCP Owner Societies. | ||
Recently, a better understanding of the two-photon excited SEHRS spectrum of pMBA was obtained, and SEHRS spectra of pMBA, acquired between pH 2 and 12 under non-resonant conditions, together with the visible (532 nm) and near-infrared (1064 nm) excited SERS spectra were discussed.22 Investigating four different types of silver nanoparticles, it was noticed that Raman shifts smaller by 5–15 cm−1 for several bands in SEHRS were very reproducible.22 It was suggested, that these bands indicate probing of different vibrations in SERS and in SEHRS due to the different selection rules,20 and that the shifts are in fact indicative of different adsorption species upon very local changes in the interaction of the molecule when pH changes.22 For high pH values, new band components appear, such as asymmetric broadening and new shoulders, supporting the conclusion that different adsorption species are probed in SERS and SEHRS. Fig. 7b displays this for the example of the ring stretching vibration at 1585 cm−1 in the SEHRS spectra, while the band in the SERS data is only shifted and shows no second component (Fig. 7c and d). The appearance of a low-frequency shoulder in the SEHRS spectrum can be assigned to the non-totally symmetric ring stretching vibration, whereas the high-frequency component corresponds to the totally symmetric ring stretching vibration. From this information it was concluded that at high pH a larger fraction of the molecules must be adsorbed on the surface in a flat orientation and can be specifically probed by SEHRS (Fig. 7a). The flat orientation at neutral and basic pH was also evidenced by a higher intensity of bands from out-of-plane vibrations of the phenyl ring at 684 cm−1 and 710 cm−1 in both the SEHRS and SERS spectra.106–108 Using SERS spectra of pMBA excited with 1064 nm by the same laser, quasi-simultaneous one-photon and two-photon excited measurements of pH were demonstrated in one microspectroscopic setup that were more sensitive than one-photon SERS sensing with excitation in the visible range alone.22 The possibility of sensitive pH measurements in the acidic and neutral pH ranges in small focal volumes enables a variety of applications for sensing in complex microstructured environments.
7. SEHRS in resonance with electronic transitions
7.1 Two-photon resonance enhanced SEHRS
The second harmonic of the NIR wavelengths that are typically used for excitation have resonances in many of the molecules that were investigated so far by SEHRS. A possible transition of a Stokes resonance hyper Raman process responsible for strong two-photon resonant SEHRS signals is shown in Fig. 1E, for more details on resonant hyper Raman scattering (RHRS) please refer to, e.g., ref. 109 by Myers Kelley and references therein.The addition of resonance Raman enhancement by two-photon resonant transitions in the molecule to the surface enhancement makes SEHRS experiments even more sensitive, and enables measurements at low (nanomolar) concentration,82 spectra from single molecules,45–47 as well as SEHRS experiments at low pulse energies86 or the use of CW laser excitation.71
In one of the first reports of SEHRS, differences between SERS spectra excited at 532 nm and the SEHRS spectra excited at 1064 nm of the dye molecules crystal violet (CV) and rhodamine 6G (R6G) were measured.10 In particular there, the relatively high similarity of the SER(R)S and SEHRS spectrum of CV was discussed as being the result of lowering of the high symmetry in the adsorbed CV molecules.10 Both CV and R6G have since then been used to understand the resonance enhancement and surface enhancement in SEHRS both in experiment65,67,86 and theory.33,66 In R6G, different types of resonance enhancement through both the A-term and the B-term in the RHRS93,110 lead to a selective enhancement of some bands in the SEHRS spectra that are not enhanced in the SERS spectra, and therefore both spectra appear very different.33,67 The results give evidence that electronic “dark” states that are not allowed in one-photon absorption can be probed in SEHRS.67
In contrast, the resonant SEHRS and the SERRS spectra of many other dyes can also look very similar, indicating that the same electronic transition is responsible in both cases.111 Among others, they include the spectra of CV,10,82,86 substituted stilbene molecules,61 and malachite green.64,78Fig. 8 shows SEHRS and SERS spectra of both CV and MG from ref. 64. However, as expected, and recently shown for the case of CV, the high similarity vanishes when both SERS and SEHRS spectra are excited off-resonance, at 785 nm and 1570 nm, respectively.34 For β-carotene, the (non-surface enhanced) HRS spectra and the RRS spectra are very different from each other, due to the different selection rules that become very evident for molecules with high symmetry.85,112 Nevertheless, very high similarity of the SEHRS (Fig. 9A) and SERRS (Fig. 9A) spectra is reported, since the interaction with the silver surface lowers the symmetry of the adsorbed molecules.85
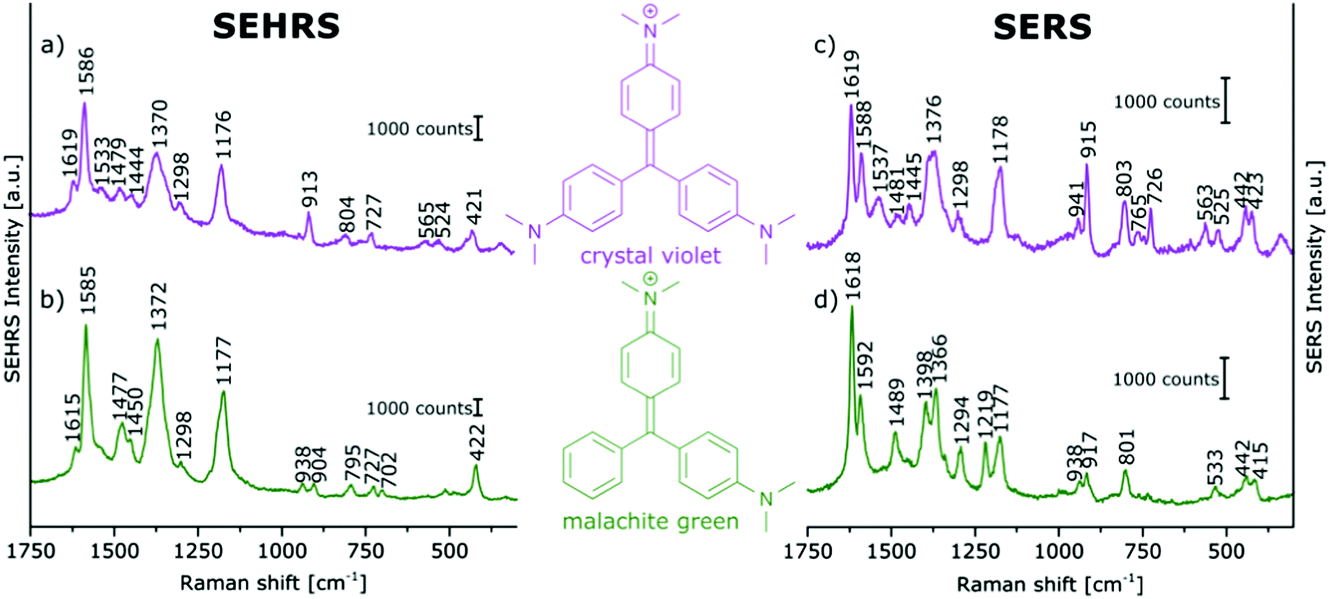 | ||
| Fig. 8 (a and b) SEHRS and (c and d) SERS spectra of aqueous solutions of (a and c) crystal violet, and (b and d) malachite green on immobilized silver nanoparticles. Analyte concentrations: in (a and b) 10−5 M, in (c and d) 10−6 M. Excitation: (a and b) 1064 nm, peak intensity: 1010 W cm−2, 30 s; (c and d) 532 nm, peak intensity: 3 × 109 W cm−2, 100 ms. Modified with permission from ref. 64. Copyright 2016, Published by the PCCP Owner Societies. | ||
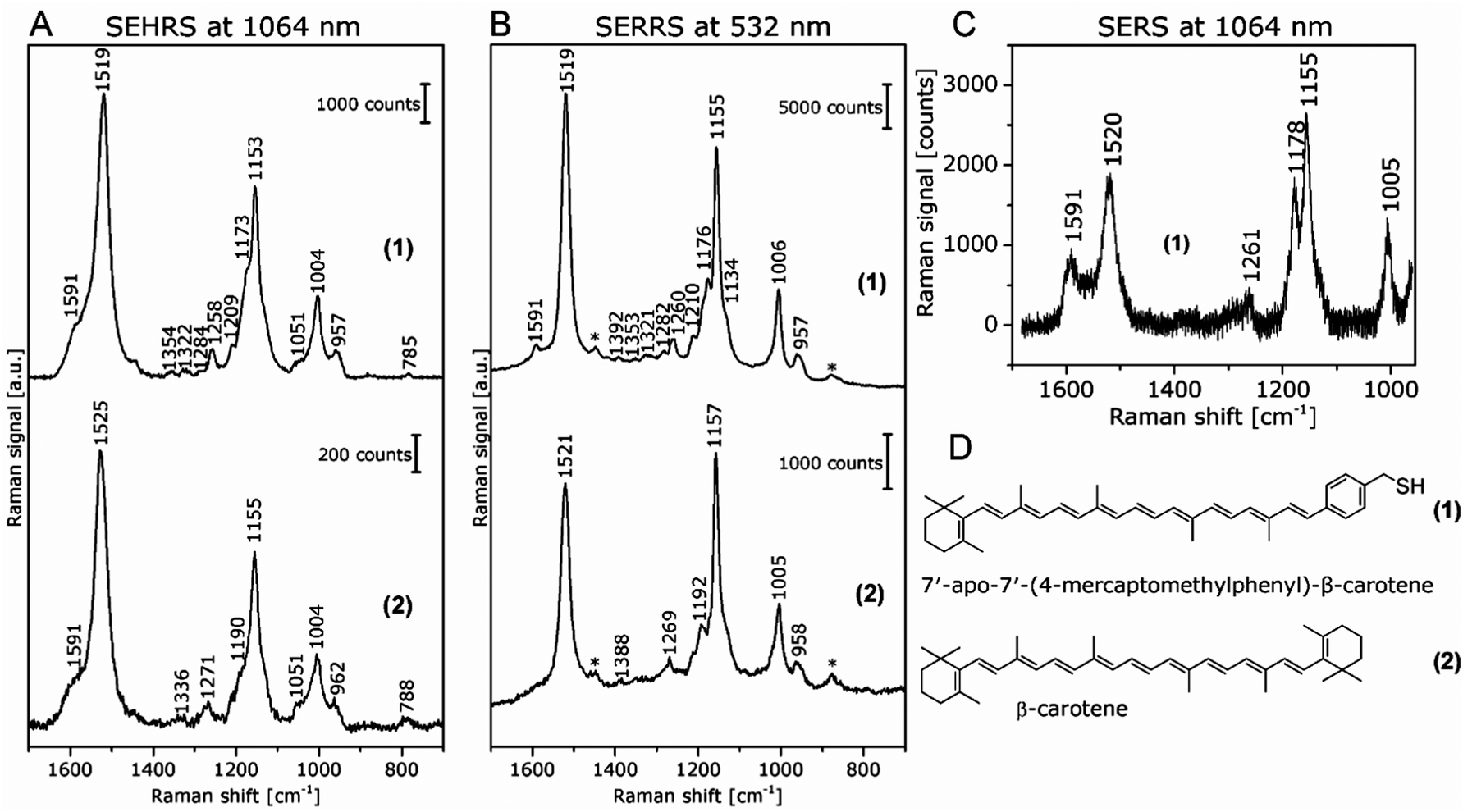 | ||
| Fig. 9 (A) SEHRS spectra excited at 1064 nm and (B) SERRS spectra excited at 532 nm of 7′-apo-7′-(4-mercaptomethylphenyl)-β-carotene (upper spectra in both panels) and of the non-functionalized β-carotene (lower spectra in both panels). Final concentrations of the carotenes in the sample solutions: 3 × 10−7 M; excitation: (A) 1064 nm, peak photon flux density: 6 × 1028 photons cm−2 s−1, acquisition time: 120 s, (B) 532 nm, 7 × 1023 photons cm−2 s−1, acquisition time: 100 ms. The spectra in (A) are averages of 10 baseline corrected spectra. Bands marked with an asterisk in (B) are due to the ethanol in the solvent. (C) SERS spectrum of 7′-apo-7′-(4-mercaptomethylphenyl)-β-carotene; final concentration of the carotenes in the sample solutions: 3 × 10−6 M; excitation: 1064 nm, peak photon flux density: 3 × 1028 photons cm−2 s−1, acquisition time: 30 s. For the spectrum in (C), a total of 100 spectra from 5 different samples were averaged after baseline correction. (D) Structure of 7′-apo-7′-(4-mercaptomethylphenyl)-β-carotene (1) and β-carotene (2). Reproduced with permission from ref. 85. Copyright 2016 American Chemical Society. | ||
The strong contribution by resonance enhancement is obvious in several reports comparing HRS with resonant SEHRS spectra, for example for CV86 and R6G.66,86Fig. 10 shows an HRS and a SEHRS spectrum of CV.
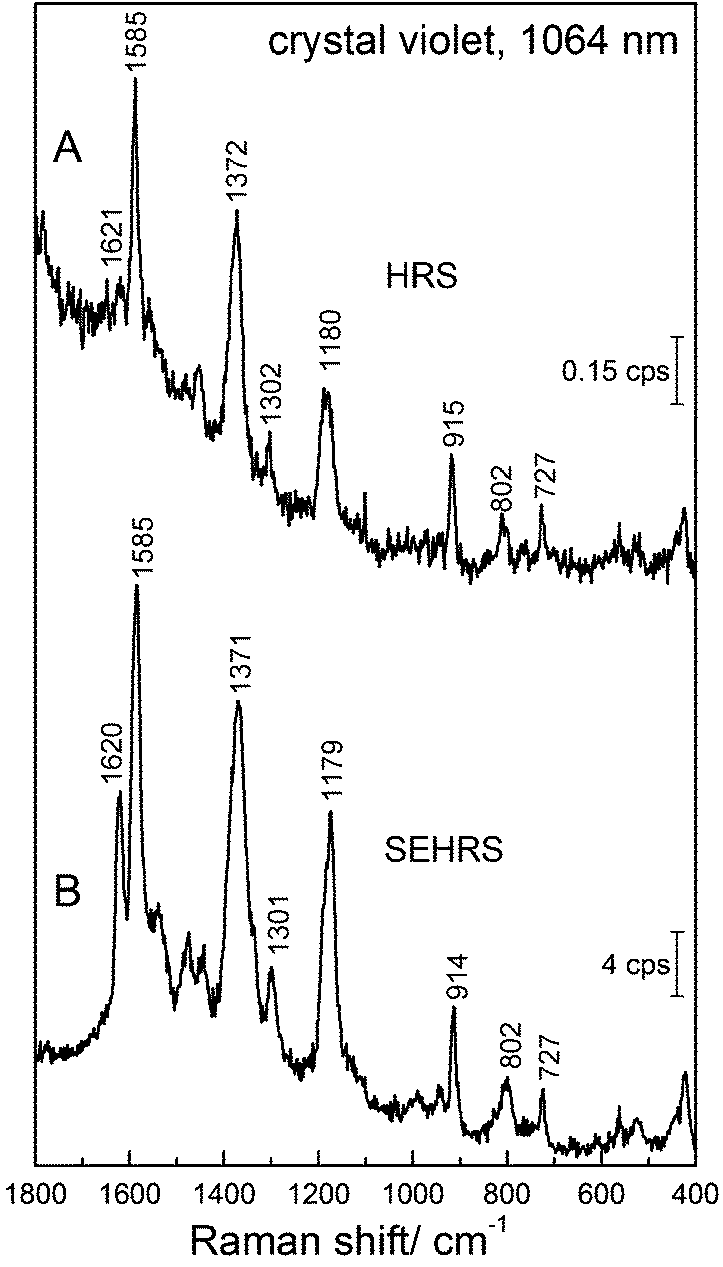 | ||
| Fig. 10 (A) Hyper Raman spectrum of crystal violet dye and (B) surface enhanced hyper Raman spectrum of crystal violet in the local field of silver nanostructures. Excitation: 1064 nm (A and B), photon flux density: 6.8 × 1027 photons cm−2 s−1 (A) and 1.4 × 1027 photons cm−2 s−1 (B), acquisition time: 8 min (A) and 1 min (B), crystal violet concentration: 1 × 10−3 M (A) and 1 × 10−5 M (B). | ||
In these particular data, the SEHRS signals are found to be on the order of 104–105 stronger than the HRS signals (in this case not correcting for the actual number of molecules that participate in the surface enhancement and neglecting reabsorption effects). Leng and Kelley published enhancement factors for different dye molecules, including CV on the order of ∼103.86 Taking into account the actual number of molecules, and considering the contributions of only the hottest hot spots of the SEHRS substrate, K. Kneipp estimated enhancement of ∼1020 for CV on silver nanoaggregates.45
With surface enhancement enabling the acquisition of SEHRS spectral information, a very high sensitivity with respect to very small differences in molecular structure can be used to compare and differentiate the spectra of very similar molecules. In Fig. 8, SEHRS of CV und MG,64,78 which differ only in one dimethylamino group, and their SERS spectra are shown. Several differences between SEHRS and SERS are observed, and in the SEHRS spectra, some differences show more clearly. As an example, the in-plane phenyl deformation mode113 is found at 913 cm−1 for CV and at 904 cm−1 for MG with different intensities compared to the dimethylamino stretching band113 at 938 cm−1 (Fig. 8a and b). In the SERS spectra, the shift of this band is not significant (Fig. 8c and d).
As another example, SEHRS spectra of carotene molecules and thiol-modified carotene, 7′-apo-7′-(4-mercaptomethylphenyl)-β-carotene (Fig. 9A, for structures see Fig. 9D), excited at 1064 nm, together with SERRS spectra measured at 532 nm (Fig. 9B) and non-resonant SERS spectra measured at an excitation wavelength of 1064 nm (Fig. 9C) were used to study the interaction with silver nanoparticles.85 It was discussed that the interaction with the silver surface occurs via the π-electrons of the polyene chain for both functionalized and non-functionalized β-carotene, but only the stronger, functionalization induced interaction enables the acquisition of non-resonant SERS spectra of β-carotene at low concentrations (Fig. 9C). Even though the resonant SEHRS and SERS spectra (Fig. 9A and B) are very similar, the SEHRS spectra contain additional bands of infrared-active modes of carotene, namely a shoulder band at 1051 cm−1 due to a vibration of the ionone ring, and a shoulder at 1591 cm−1, with contributions from an IR-active but Raman-inactive CC stretching mode at 1569 cm−1.85
7.2 Relative intensities in SEHRS, SERS, and SERRS spectra enable discussion of resonance contribution
The experiments with β-carotene at the two different excitation wavelengths (532 nm/1064 nm), providing SERRS, normal Raman, and SEHRS spectra for the resonant case, as well as non-resonant SERS, enabled a discussion of the contributions of the (molecular) resonant Raman enhancement and the surface enhancement.85 The functionalization of carotene, which leads to a stronger interaction with the silver surface gave the possibility of obtaining non-resonant SERS spectra in solution at low concentration. The SEHRS spectra (Fig. 9A) show relatively strong contributions from bands that experience a high SERS enhancement but low resonance enhancement, see, e.g., the bands at 1591 cm−1 and at 1178 cm−1 in Fig. 9A with B. (These bands can be identified with the help of the non-resonant SERS spectrum in Fig. 9C.) In turn, as, relative to these intensities, the intensity of the bands that experience strong resonance enhancement, e.g., at 1520 cm−1 and 1155 cm−1, decreases, the SEHRS data (Fig. 9A) support the theoretical consideration and SERRS experimental observation (see also Fig. 9B) of decreased resonance enhancement due to damping by electromagnetic coupling of the molecules and the metal.114,115In a similar fashion, the SEHRS spectra of the dye Rose Bengal, measured in resonance at 1064 nm excitation resembled very strongly a non-resonant SERS spectrum obtained at 785 nm of the same sample.17 Also here, a strong interaction between plasmons and electronic levels of the molecules, leading to high plasmonic enhancement at the cost of a lower resonance enhancement in SEHRS, could give rise to a decrease in the resonance enhancement in SEHRS, and to similar non-resonant SEHRS and SERS spectra for the low-symmetry molecule.
8. Future applications of SEHRS
8.1 Towards combined intracellular SEHRS/SERS nanoprobing
There is a variety of potential applications of non-resonant and resonant SEHRS in bioanalysis and biophysics, as well as in hybrid materials research, many of which are related to the advantages that are coming with the use of NIR wavelengths for excitation, such as deeper penetration into micro-structured materials and less photodamage, and with the decreased sampling volumes that result from the non-linearity of the effect.The high sensitivity with respect to changes at the surface of plasmonic nanostructures makes SEHRS ideal to probe the interaction of nanostructures with intrinsic biomolecular species in biological samples, analogous to previous reports using one-photon excited SERS.116,117 SEHRS spectra obtained from macrophage cells that incorporated gold nanoparticles show, e.g., pronounced amide II signals of proteins, usually detected by IR absorption spectroscopy. In SEHRS, they can be observed at high lateral resolution. Interestingly, SEHRS also observes spectral bands above 1800 cm−1, which are likely combination modes. The spectral region between 1800 and 2700 cm−1 is almost featureless in other vibrational (Raman and IR) spectra of cells and tissues.17 Better understanding the SEHRS spectra of typical biomolecules, in particular nucleobases17,23,82 and amino acids,24 will allow assessing the spectral information obtained from SEHRS nanosensors in cells. Furthermore, the concept of hybrid nanoprobes, providing SEHRS spectra of a reporter molecule and from the cellular environment, will benefit from the possible combination of SEHRS and SERS. SEHRS labels using the biocompatible reporter molecule Rose Bengal or the dye CV with both resonant SEHRS and non-resonant SERS excitation in the infrared were characterized,7 and used in SERS experiments with cells,118 in principle enabling such experiments in SEHRS as well.
The possibilities of combining SEHRS with SERS for more sensitive measurements of pH using NIR excitation,22 as discussed in Section 6, suggest this combination for the determination of micro-environmental pH (e.g., in biomaterials). SERS/SEHRS pH nanosensors perform pH measurements using the relative signals of spectrally narrow pairs of Raman lines in the same spectrum. This allows quantitative measurement without any correction regarding emission or absorption background signals, a particular advantage for applications in complex biological environments. A combined SEHRS/SERS pH-sensor which uses pMBA would have widespread applications, in endosomal sensing,70 or application in other acidic environments.
8.2 SEHRS hyperspectral mapping
Even though the probing of molecular structure and interaction in SEHRS occurs at the nanoscale and covers the corresponding structural heterogeneity, the combination of SEHRS spectra with microstructural information is highly desirable, for example with applications in cells, tissues, or other micro-structured materials such as microfluidic systems in mind. In hyperspectral mapping and characterization, including SERS mapping,119,120 principal components analysis (PCA), a multivariate technique with wide application in pattern recognition, image compression, and sensing routines121–123 has been applied. In PCA, a data matrix is projected into a variance-weighted coordinate system. This orthogonal linear transformation (or eigenvalue problem) transforms a number of possibly correlated variables of the spectral data set into a smaller number of uncorrelated variables such that the greatest variance by any projection of the data comes to lie on the first coordinate, see, e.g., ref. 123 for recent explanations from the analytical context.Thereby, in a set of spectra, the different types of variance that occur, e.g., due to fluctuation of signals, changes in signal to noise ratio, background, and others, can be separated from the molecular information. In the case of SERS, the full exploitation of the multiplexing capabilities inherent to such data was shown, using spectra of specific molecules and also of molecular mixtures, such as those contained in biological samples.124–127
Multivariate mapping with SEHRS data of large areas in microscopy was recently demonstrated to be feasible64 using silane-immobilized silver nanoparticles on glass slides that had yielded a homogeneous distribution of enhancement of SERS96 on the microscopic scale. In these experiments, the SEHRS spectra were excited in resonance with two well-characterized reporter molecules, crystal violet (CV) and malachite green (MG) at 1064 nm. The surfaces containing both dyes were prepared by immersing slides with immobilized silver nanoparticles first in MG and then in CV solution, following the schematics shown in Fig. 11. The region marked with the rectangle in the center of the slide is at the border between pure MG (green region) and a region where a mixture of MG and CV exists (grey region) because this end of the slide was dipped into both MG and CV. Fig. 11 shows a magnification and indicates several grids of data points (in black, red, green, and blue) that were separated from each other by several millimeters. The microscopic heterogeneity of this (macroscopic) border region was analyzed by collecting grids of SEHRS spectra, each from a nanoscopic sampling volume as discussed in Section 4.
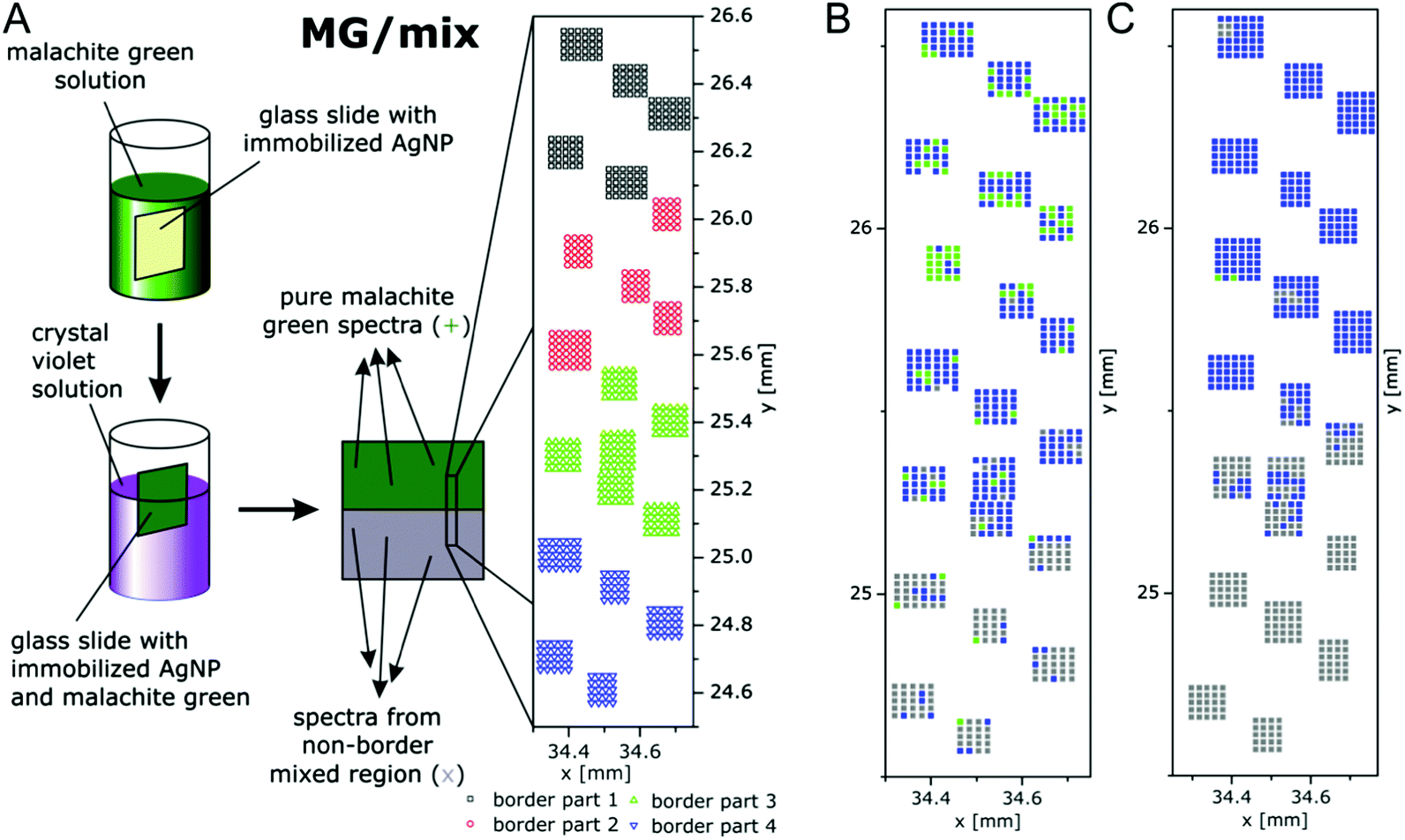 | ||
| Fig. 11 (A) Schematic of the preparation of macroscopic samples that contain a border region of microscopic heterogeneity in the distribution of malachite green (MG) and crystal violet (CV) on immobilized silver nanoparticles. False-color (B) SEHRS and (C) SERS map of the border region of the MG/mix sample shown schematically in (A). The maps are based on the scores of the respective first principles component of the PCA of the SEHRS spectra and of the SERS spectra. The x and y axes of the maps display the absolute coordinates of the x,y-stage that was used for the SEHRS and SERS mapping experiments in order to demonstrate the overlap of the microscopic regions that were probed by SEHRS and SERS. Color code: assignment according to the 1st PC score. Green: MG, blue: between mix and MG, grey: mix. x and y scales show absolute positions in millimeter of the x,y-stage for comparison of the two maps. Modified with permission from ref. 64. Copyright 2016, Published by the PCCP Owner Societies. | ||
The SEHRS spectra of MG and CV on silver nanostructures were discussed before in Fig. 8. For hyperspectral mapping, all SEHRS spectra were analyzed in a PCA. As discussed in ref. 64, analyzing the loadings (eigenspectra) in the PCA, main differences within the data sets of the border regions were found around 914 cm−1, 1587 cm−1 and 1175 cm−1, the latter being assigned to the intense phenyl bands.64 In the variance-weighted principal component space, spectra that are very dissimilar are separated by a large distance, similar spectra are close to one another. Therefore, using the principal component (PC) scores, maps can be constructed for example by plotting an absolute score value of or relative distance between spectra,119 or by defining thresholds for the score value. The latter was used with the mix of CV and MG SEHRS mapping data, and thresholds were defined relative to score values of spectra that were unequivocally assigned to either of the two molecules: along the first PC, three ranges and a corresponding color scale were set, and the score value of each spectrum was combined with its lateral coordinate. A similar analysis was also done with SERS spectra measured from the same areas excited at 523 nm. Both maps are shown in Fig. 11B and C. In the SEHRS map, many spectra resemble pure MG in the upper part (green pixels in Fig. 11B), while most of the SERS spectra (Fig. 11C) are sorted in one group as being spectra from a mix of MG and CV. This is an example that by SEHRS, discrimination between slightly varying molecular structure and composition within a mixture could be more sensitive than in SERS and that in this case, SEHRS provides complementary information.
The separation of the score values, i.e., the generation of image contrast in this example was based on the hyperspectral information of two defined molecules. In other experiments, the number of principal components, offsets and thresholds must be adapted to the questions of the respective experiment. These first results by Gühlke et al.64 indicate that SEHRS microspectroscopic imaging, specifically in combination with SERS microspectroscopy, holds great promise to exploit the combined chemical information from both methods. The ability to obtain SEHRS microspectra from complex samples such as intrinsic biomolecules in cells,17 or of reporter molecules as part of nanosensors70 will help SEHRS hyperspectral imaging to become an important new direction in future nanobiophotonics.
9. Conclusions
To conclude, we have collected here work that highlights the various advantages of surface enhanced hyper Raman scattering (SEHRS) for vibrational probing. There are several reasons that illustrate the importance to further develop probing with SEHRS:(i) From the viewpoint of vibrational spectroscopy, SEHRS provides us with the possibility of using non-resonant (and resonant) hyper Raman scattering (HRS) of molecules in solution. Due to the different selection rules of the two-photon process (HRS can access vibrations that are forbidden in both Raman scattering and also in infrared absorption), we can get different insights into molecular structure and comprehensive vibrational information.
(ii) The high sensitivity of SEHRS with respect to the interaction and orientation of the molecules at the surfaces enables probing of very local surface environment using vibrational information that is inaccessible by SERS. This will help improve our understanding of molecule–nanostructure interactions, and of consequent phenomena, including SERS itself. Vice versa, combined SEHRS and SERS experiments will provide better understanding of SEHRS as well.
(iii) In line with items (i) and (ii), the work reported on non-resonant SEHRS of nucleobases and amino acids illustrates that spectroscopic information obtained by SEHRS can be used to gain new insight into the interaction of biomolecules with nanostructures. Great benefits are expected from a combination of SEHRS with SERS for such structural studies, specifically with excitation in the NIR, which enables in situ spectra from complex biomaterials.
(iv) Recent discussion of two-photon resonant SEHRS spectra shows the great potential of resonant SEHRS to characterize the electronic states of molecules, since electronic states that are not allowed in one-photon absorption can become evident.
(v) The strong enhancement operative in SEHRS can yield two-photon excited vibrational spectra from molecules at very low concentrations. As it takes place in the high local optical fields of nanostructures, SEHRS can act as a sensitive probe of such high fields, and of the plasmonic properties of the nanostructures.
(vi) The advantages of two-photon excitation, leading to relatively high signals, together with the strong confinement of the effect to nanoscopic volumes, suggest SEHRS for vibrational imaging and microscopy, in particular in combination with SERS. This will be important to assess the microscopic heterogeneity of many different kinds of complex biological and hybrid materials.
Acknowledgements
We thank our former colleague Marina Gühlke who actively contributed to several papers on SEHRS that we have discussed here and Katrin Kneipp for critical reading of parts of the manuscript. F. M. acknowledges funding by a Chemiefonds Fellowship of FCI (Fonds der Chemischen Industrie), Z. H. is grateful for a Julia Lermontova postdoctoral fellowship provided by DFG GSC 1013 SALSA, and J. K. acknowledges funding by ERC Grant No. 259432 MULTIBIOPHOT.References
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102–1106 CrossRef CAS PubMed.
- R. R. Chance, A. Prock and R. Silbey, J. Chem. Phys., 1974, 60, 2744–2748 CrossRef CAS.
- K. Kneipp, H. Kneipp and J. Kneipp, Acc. Chem. Res., 2006, 39, 443–450 CrossRef CAS PubMed.
- M. I. Stockman, Opt. Express, 2011, 19, 22029–22106 CrossRef PubMed.
- A. Hartstein, J. R. Kirtley and J. C. Tsang, Phys. Rev. Lett., 1980, 45, 201–204 CrossRef CAS.
- A. V. Baranov and Y. S. Bobovich, JETP Lett., 1982, 36, 339–343 Search PubMed.
- D. V. Murphy, K. U. Vonraben, R. K. Chang and P. B. Dorain, Chem. Phys. Lett., 1982, 85, 43–47 CrossRef CAS.
- E. J. Liang, A. Weippert, J. M. Funk, A. Materny and W. Kiefer, Chem. Phys. Lett., 1994, 227, 115–120 CrossRef CAS.
- Y. Zhang, Y.-R. Zhen, O. Neumann, J. K. Day, P. Nordlander and N. J. Halas, Nat. Commun., 2014, 5, 4424 CAS.
- R. R. Frontiera, A.-I. Henry, N. L. Gruenke and R. P. Van Duyne, J. Phys. Chem. Lett., 2011, 2, 1199–1203 CrossRef CAS PubMed.
- P. D. Simmons, H. K. Turley, D. W. Silverstein, L. Jensen and J. P. Camden, J. Phys. Chem. Lett., 2015, 6, 5067–5071 CrossRef CAS PubMed.
- V. N. Denisov, B. N. Mavrin and V. B. Podobedov, Phys. Rep., 1987, 151, 1–92 CrossRef.
- J. Kneipp, H. Kneipp and K. Kneipp, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17149–17153 CrossRef CAS PubMed.
- W. R. Zipfel, R. M. Williams and W. W. Webb, Nat. Biotechnol., 2003, 21, 1369–1377 CrossRef CAS PubMed.
- J. T. Golab, J. R. Sprague, K. T. Carron, G. C. Schatz and R. P. V. Duyne, J. Chem. Phys., 1988, 88, 7942–7951 CrossRef CAS.
- J. C. Hulteen, M. A. Young and R. P. Van Duyne, Langmuir, 2006, 22, 10354–10364 CrossRef CAS PubMed.
- N. Valley, L. Jensen, J. Autschbach and G. C. Schatz, J. Chem. Phys., 2010, 133, 054103 CrossRef PubMed.
- M. Guhlke, Z. Heiner and J. Kneipp, Phys. Chem. Chem. Phys., 2015, 17, 26093–26100 RSC.
- F. Madzharova, Z. Heiner, M. Gühlke and J. Kneipp, J. Phys. Chem. C, 2016, 120, 15415–15423 CAS.
- F. Madzharova, Z. Heiner and J. Kneipp, J. Phys. Chem. C, 2017, 121, 1235–1242 CAS.
- D. A. Long and L. Stanton, Proc. R. Soc. London, Ser. A, 1970, 318, 441–457 CrossRef.
- S. J. Cyvin, J. E. Rauch and J. C. Decius, J. Chem. Phys., 1965, 43, 4083–4095 CrossRef CAS.
- D. A. Long, The Raman effect – a unified treatment of the theory of Raman scattering by molecules, John Wiley & Sons, Ltd, 2002 Search PubMed.
- J. H. Christie and D. J. Lockwood, J. Chem. Phys., 1971, 54, 1141–1154 CrossRef CAS.
- W. H. Yang and G. C. Schatz, J. Chem. Phys., 1992, 97, 3831–3845 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- X.-Y. Li, Q.-J. Huang, V. I. Petrov, Y.-T. Xie, Q. Luo, X. Yu and Y.-J. Yan, J. Raman Spectrosc., 2005, 36, 555–573 CrossRef CAS.
- J. Mullin, N. Valley, M. G. Blaber and G. C. Schatz, J. Phys. Chem. A, 2012, 116, 9574–9581 CrossRef CAS PubMed.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, J. Phys. Chem. C, 2013, 117, 3046–3054 CAS.
- H. K. Turley, Z. Hu, D. W. Silverstein, D. A. Cooper, L. Jensen and J. P. Camden, J. Phys. Chem. C, 2016, 120, 20936–20942 CAS.
- L. Jensen, C. M. Aikens and G. C. Schatz, Chem. Soc. Rev., 2008, 37, 1061–1073 RSC.
- D. V. Chulhai, Z. Hu, J. E. Moore, X. Chen and L. Jensen, Annu. Rev. Phys. Chem., 2016, 67, 541–564 CrossRef CAS PubMed.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783–826 CrossRef CAS.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. Van Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- J. Zhao, A. O. Pinchuk, J. M. McMahon, S. Li, L. K. Ausman, A. L. Atkinson and G. C. Schatz, Acc. Chem. Res., 2008, 41, 1710–1720 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913–3961 CrossRef CAS PubMed.
- E. J. Zeman and G. C. Schatz, J. Phys. Chem., 1987, 91, 634–643 CrossRef CAS.
- C. K. Johnson and S. A. Soper, J. Phys. Chem., 1989, 93, 7281–7285 CrossRef CAS.
- W.-H. Li, X.-Y. Li and N.-T. Yu, Chem. Phys. Lett., 1999, 305, 303–310 CrossRef CAS.
- H. Kneipp, K. Kneipp and F. Seifert, Chem. Phys. Lett., 1993, 212, 374–378 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Phys., 1999, 247, 155–162 CrossRef CAS.
- H. Yoshikawa, T. Adachi, G. Sazaki, T. Matsui, K. Nakajima and H. Masuhara, J. Opt. A: Pure Appl. Opt., 2007, 9, S164 CrossRef CAS.
- C. B. Milojevich, B. K. Mandrell, H. K. Turley, V. Iberi, M. D. Best and J. P. Camden, J. Phys. Chem. Lett., 2013, 4, 3420–3423 CrossRef CAS.
- S. Nie, L. A. Lipscomb and N.-T. Yu, Appl. Spectrosc. Rev., 1991, 26, 203–276 CrossRef.
- D. A. Weitz, S. Garoff and T. J. Gramila, Opt. Lett., 1982, 7, 168–170 CrossRef CAS PubMed.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- K. Kneipp, H. Kneipp and J. Kneipp, Chem. Sci., 2015, 6, 2721–2726 RSC.
- Z. Hu, D. V. Chulhai and L. Jensen, J. Chem. Theory Comput., 2016, 12, 5968–5978 CrossRef CAS PubMed.
- W. H. Yang, J. Hulteen, G. C. Schatz and R. P. V. Duyne, J. Chem. Phys., 1996, 104, 4313–4323 CrossRef CAS.
- A. M. Polubotko, Opt. Spectrosc., 2010, 109, 510–520 CrossRef CAS.
- A. M. Polubotko and V. P. Smirnov, J. Raman Spectrosc., 2012, 43, 380–388 CrossRef CAS.
- A. M. Polubotko and V. P. Chelibanov, J. Opt. Technol., 2013, 80, 723–730 CrossRef CAS.
- V. P. Chelibanov and A. M. Polubotko, Chem. Phys. Lett., 2014, 608, 344–350 CrossRef CAS.
- A. V. Golovin, A. M. Polubotko and V. P. Chelibanov, Opt. Spectrosc., 2016, 121, 677–688 CrossRef CAS.
- A. M. Polubotko and V. P. Chelibanov, Opt. Spectrosc., 2016, 120, 86–108 CrossRef CAS.
- L. A. Lipscomb, S. Nie, S. Feng and N.-T. Yu, Chem. Phys. Lett., 1990, 170, 457–461 CrossRef CAS.
- W. Leng, H. Y. Woo, D. Vak, G. C. Bazan and A. Myers Kelley, J. Raman Spectrosc., 2006, 37, 132–141 CrossRef CAS.
- A. V. Baranov, Y. S. Bobovich and V. K. Petrov, J. Exp. Theor. Phys., 1985, 61, 435–439 Search PubMed.
- D. W. Silverstein, C. B. Milojevich, J. P. Camden and L. Jensen, J. Phys. Chem. C, 2013, 117, 20855–20866 CAS.
- M. Guhlke, Z. Heiner and J. Kneipp, Phys. Chem. Chem. Phys., 2016, 18, 14228–14233 RSC.
- K. Kneipp, H. Kneipp and F. Seifert, Chem. Phys. Lett., 1995, 233, 519–524 CrossRef CAS.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, J. Am. Chem. Soc., 2011, 133, 14590–14592 CrossRef CAS PubMed.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, ChemPhysChem, 2011, 12, 101–103 CrossRef CAS PubMed.
- S. Nie, L. A. Lipscomb, S. Feng and N.-T. Yu, Chem. Phys. Lett., 1990, 167, 35–40 CrossRef CAS.
- K. Ikeda, M. Takase, Y. Sawai, H. Nabika, K. Murakoshi and K. Uosaki, J. Chem. Phys., 2007, 127, 111103 CrossRef PubMed.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 CrossRef CAS PubMed.
- T. Itoh, Y. Ozaki, H. Yoshikawa, T. Ihama and H. Masuhara, Appl. Phys. Lett., 2006, 88, 084102 CrossRef.
- T. Itoh, H. Yoshikawa, K. Yoshida, V. Biju and M. Ishikawa, J. Chem. Phys., 2009, 130, 214706 CrossRef PubMed.
- T. Itoh, H. Yoshikawa, K.-i. Yoshida, V. Biju and M. Ishikawa, J. Chem. Phys., 2010, 133, 124704 CrossRef PubMed.
- A. V. Baranov and I. R. Nabiev, Proc. SPIE, 1991, 1403, 128–131 CrossRef CAS.
- A. V. Baranov, Y. S. Bobovich and V. I. Petrov, J. Raman Spectrosc., 1993, 24, 695–697 CrossRef CAS.
- Z. P.-X. Pan Duo-Hai, L. Xiang-Sheng, M. Run-Cai and H. Yi-Xian, Chin. Phys. B, 1993, 2, 925–929 Search PubMed.
- N.-T. Yu, S. Nie and L. A. Lipscomb, J. Raman Spectrosc., 1990, 21, 797–802 CrossRef CAS.
- S. Schneider, G. Brehm and P. Freunscht, Phys. Status Solidi B, 1995, 189, 37–42 CrossRef CAS.
- W.-H. Li, X.-Y. Li and N.-T. Yu, Chem. Phys. Lett., 1999, 312, 28–36 CrossRef CAS.
- W.-H. Li, X.-Y. Li and N.-T. Yu, Chem. Phys. Lett., 2000, 327, 153–161 CrossRef CAS.
- G. Brehm, G. Sauer, N. Fritz, S. Schneider and S. Zaitsev, J. Mol. Struct., 2005, 735–736, 85–102 CrossRef CAS.
- H. Kneipp and K. Kneipp, J. Raman Spectrosc., 2005, 36, 551–554 CrossRef CAS.
- S. Kadkhodazadeh, J. B. Wagner, V. Joseph, J. Kneipp, H. Kneipp and K. Kneipp, Plasmonics, 2013, 8, 763–767 CrossRef CAS.
- M. Espina Palanco, K. Bo Mogensen, M. Gühlke, Z. Heiner, J. Kneipp and K. Kneipp, Beilstein J. Nanotechnol., 2016, 7, 834–840 CrossRef PubMed.
- M. Gühlke, Z. Heiner and J. Kneipp, J. Phys. Chem. C, 2016, 120, 20702–20709 Search PubMed.
- W. Leng and A. M. Kelley, J. Am. Chem. Soc., 2006, 128, 3492–3493 CrossRef CAS PubMed.
- W. Leng, A. A. Yasseri, S. Sharma, Z. Li, H. Y. Woo, D. Vak, G. C. Bazan and A. M. Kelley, Anal. Chem., 2006, 78, 6279–6282 CrossRef CAS PubMed.
- P. Almar, I. Taro, V. Prabhat, I. Yasushi and K. Satoshi, Appl. Phys. Express, 2008, 1, 092401 CrossRef.
- A. Palonpon, T. Ichimura, P. Verma, Y. Inouye and S. Kawata, J. Raman Spectrosc., 2009, 40, 119–120 CrossRef CAS.
- H. K. Turley and J. P. Camden, Chem. Commun., 2014, 50, 1472–1474 RSC.
- C. G. Blatchford, J. R. Campbell and J. A. Creighton, Surf. Sci., 1982, 120, 435–455 CrossRef CAS.
- J. M. McMahon, S. K. Gray and G. C. Schatz, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83 Search PubMed.
- A. M. Kelley, Annu. Rev. Phys. Chem., 2010, 61, 41–61 CrossRef PubMed.
- K. Kneipp, H. Kneipp and J. Kneipp, in Plasmonics Theory and Applications, ed. T. V. Shahbazyan and M. I. Stockman, Springer Netherlands, Dordrecht, 2013, pp. 103–124, DOI: 10.1007/978-94-007-7805-4_2 Search PubMed.
- J. Butet and O. J. F. Martin, J. Phys. Chem. C, 2015, 119, 15547–15556 CAS.
- V. Joseph, M. Gensler, S. Seifert, U. Gernert, J. P. Rabe and J. Kneipp, J. Phys. Chem. C, 2012, 116, 6859–6865 CAS.
- X. Zhang, C. R. Yonzon and R. P. Van Duyne, J. Mater. Res., 2006, 21, 1083–1092 CrossRef CAS.
- N. Guillot and M. L. de la Chapelle, Nano, 2012, 6, 064506 Search PubMed.
- A. Shiohara, Y. Wang and L. M. Liz-Marzán, J. Photochem. Photobiol., C, 2014, 21, 2–25 CrossRef CAS.
- P. C. Lee and D. Meisel, J. Phys. Chem., 1982, 86, 3391–3395 CrossRef CAS.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Phys. Chem. A, 2005, 109, 7405–7410 CrossRef CAS PubMed.
- R. A. Alvarez-Puebla, E. Arceo, P. J. G. Goulet, J. J. Garrido and R. F. Aroca, J. Phys. Chem. B, 2005, 109, 3787–3792 CrossRef CAS PubMed.
- M. Kazanci, J. P. Schulte, C. Douglas, P. Fratzl, D. Pink and T. Smith-Palmer, Appl. Spectrosc., 2009, 63, 214–223 CrossRef CAS PubMed.
- D. Drescher, P. Guttmann, T. Buchner, S. Werner, G. Laube, A. Hornemann, B. Tarek, G. Schneider and J. Kneipp, Nanoscale, 2013, 5, 9193–9198 RSC.
- I. Lynch, T. Cedervall, M. Lundqvist, C. Cabaleiro-Lago, S. Linse and K. A. Dawson, Adv. Colloid Interface Sci., 2007, 134–135, 167–174 CrossRef CAS PubMed.
- A. Michota and J. Bukowska, J. Raman Spectrosc., 2003, 34, 21–25 CrossRef CAS.
- S. B. Lee, K. Kim and M. S. Kim, J. Raman Spectrosc., 1991, 22, 811–817 CrossRef CAS.
- Y. Wang, W. Ji, H. M. Sui, Y. Kitahama, W. D. Ruan, Y. Ozaki and B. Zhao, J. Phys. Chem. C, 2014, 118, 10191–10197 CAS.
- A. Myers Kelley, J. Phys. Chem. A, 2008, 112, 11975–11991 CrossRef CAS PubMed.
- Y. C. Chung and L. D. Ziegler, J. Chem. Phys., 1988, 88, 7287–7294 CrossRef CAS.
- A. M. Kelley, W. N. Leng and M. Blanchard-Desce, J. Am. Chem. Soc., 2003, 125, 10520–10521 CrossRef CAS PubMed.
- R. Shimada and H. O. Hamaguchi, J. Chem. Phys., 2011, 134 Search PubMed.
- H. B. Lueck, D. C. Daniel and J. L. McHale, J. Raman Spectrosc., 1993, 24, 363–370 CrossRef CAS.
- D. A. Weitz, S. Garoff, J. I. Gersten and A. Nitzan, J. Chem. Phys., 1983, 78, 5324–5338 CrossRef CAS.
- K. Kneipp and D. Fassler, Chem. Phys. Lett., 1984, 106, 498–502 CrossRef CAS.
- L. A. Austin, B. Kang and M. A. El-Sayed, Nano Today, 2015, 10, 542–558 CrossRef CAS.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 214–226 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, A. Rajaduraj, R. W. Redmond and K. Kneipp, J. Raman Spectrosc., 2009, 40, 1–5 CrossRef CAS.
- A. Matschulat, D. Drescher and J. Kneipp, ACS Nano, 2010, 4, 3259–3269 CrossRef CAS PubMed.
- M. B. Mamian-Lopez and R. J. Poppi, Microchem. J., 2015, 123, 243–251 CrossRef CAS.
- C. Clausen and H. Wechsler, Pattern Recogn., 2000, 33, 1555–1560 CrossRef.
- R. G. Bereton, Chemometrics: data analysis for the laboratory and chemical plant, John Wiley & Sons Ltd, Chichester, UK, 2006, pp. 183–269 Search PubMed.
- S. Stewart, M. A. Ivy and E. V. Anslyn, Chem. Soc. Rev., 2014, 43, 70–84 RSC.
- S. Seifert, V. Merk and J. Kneipp, J. Biophotonics, 2016, 9, 181–189 CrossRef CAS PubMed.
- J. Lin, R. Chen, S. Feng, J. Pan, Y. Li, G. Chen, M. Cheng, Z. Huang, Y. Yu and H. Zeng, Nanomed. Nanotechnol. Biol. Med., 2011, 7, 655–663 CrossRef CAS PubMed.
- K. E. Stephen, D. Homrighausen, G. DePalma, C. H. Nakatsu and J. Irudayaraj, Analyst, 2012, 137, 4280–4286 RSC.
- F. Pozzi, S. Porcinai, J. R. Lombardi and M. Leona, Anal. Methods, 2013, 5, 4205–4212 RSC.
- J. P. Neddersen, S. A. Mounter, J. M. Bostick and C. K. Johnson, J. Chem. Phys., 1989, 90, 4719–4726 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |