
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium-ion batteries: present and future
Jang-Yeon
Hwang†
 a,
Seung-Taek
Myung†
b and
Yang-Kook
Sun
*a
a,
Seung-Taek
Myung†
b and
Yang-Kook
Sun
*a
aDepartment of Energy Engineering, Hanyang University, Seoul, 04763, South Korea. E-mail: yksun@hanyang.ac.kr; Fax: +82 2 2282 7329; Tel: +82 2 2220 0524
bDepartment of Nanotechnology and Advanced Materials Engineering, Sejong University, Seoul 05006, South Korea
First published on 28th March 2017
Abstract
Energy production and storage technologies have attracted a great deal of attention for day-to-day applications. In recent decades, advances in lithium-ion battery (LIB) technology have improved living conditions around the globe. LIBs are used in most mobile electronic devices as well as in zero-emission electronic vehicles. However, there are increasing concerns regarding load leveling of renewable energy sources and the smart grid as well as the sustainability of lithium sources due to their limited availability and consequent expected price increase. Therefore, whether LIBs alone can satisfy the rising demand for small- and/or mid-to-large-format energy storage applications remains unclear. To mitigate these issues, recent research has focused on alternative energy storage systems. Sodium-ion batteries (SIBs) are considered as the best candidate power sources because sodium is widely available and exhibits similar chemistry to that of LIBs; therefore, SIBs are promising next-generation alternatives. Recently, sodiated layer transition metal oxides, phosphates and organic compounds have been introduced as cathode materials for SIBs. Simultaneously, recent developments have been facilitated by the use of select carbonaceous materials, transition metal oxides (or sulfides), and intermetallic and organic compounds as anodes for SIBs. Apart from electrode materials, suitable electrolytes, additives, and binders are equally important for the development of practical SIBs. Despite developments in electrode materials and other components, there remain several challenges, including cell design and electrode balancing, in the application of sodium ion cells. In this article, we summarize and discuss current research on materials and propose future directions for SIBs. This will provide important insights into scientific and practical issues in the development of SIBs.
 Jang-Yeon Hwang | Jang-Yeon Hwang received his BS degree from the Department of Chemical Engineering of Hanyang University in 2012. He is presently a PhD candidate in the Department of Energy Engineering at Hanyang University, Korea, under the supervision of Professor Yang-Kook Sun. His research focuses on materials development in the fields of energy conversion and storage, such as cathode, anode and electrolyte materials for sodium-ion batteries. |
 Seung-Taek Myung | Seung-Taek Myung is a Professor of Nano Engineering at Sejong University, South Korea. He received his PhD degree in Chemical Engineering from Iwate University, Japan, in 2003. His research interests embrace development of electro-active materials and corrosion of current collectors of rechargeable lithium and sodium batteries. |
 Yang-Kook Sun | Yang-Kook Sun received his PhD degree from Seoul National University, Korea. He was group leader at Samsung Advanced Institute of Technology and contributed to the commercialization of the lithium polymer battery. He has worked at the Hanyang University in Korea as a professor since 2000. His research interests are the synthesis of new electrode materials for lithium-ion batteries, Na ion batteries, Li–S batteries, and Li–air batteries. |
1. Introduction
Fossil fuels are the most widely used energy resource worldwide. Risks related to resource depletion, environmental pollution, and political unrest with regard to fossil fuel production have led to the rapid emergence of a variety of intermittent renewable and cleaner energy sources such as wind, solar and wave. In order to integrate these renewable energies into the electrical grid, a large-scale energy storage system (ESS) is vital to peak shift operation.1 Among various energy storage technologies, using an electrochemical secondary battery is a promising method for large-scale storage of electricity due to its flexibility, high energy conversion efficiency, and simple maintenance.1,2 LIBs, which have become common power sources in the portable electronic market since their first commercialization by Sony in the early 1990s,3 are the primary candidates for ESSs. The introduction of LIBs into the automotive market as the battery of choice for powering hybrid electric vehicles (HEVs), plug-in hybrid electric vehicles (PHEVs) and electric vehicles (EVs) could reduce dependence on fossil fuels. Lithium, the primary ingredient in LIBs, is non-uniformly distributed within the Earth's crust. As a result, the Andean states have been dubbed the ‘new Middle-East’.4 However, the increasing demand for lithium associated with these new and large-scale applications is expected to skyrocket the price of lithium, affecting reserves as well, as it is not a naturally abundant element. Based on the calculations, overall global Li consumption in 2008 was nearly 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 280 tons; hence, present mineable resources could be sustained for approximately 65 years at most at an average growth rate of 5% per year,2,5 making the implementation of the above-mentioned applications difficult and very costly.
280 tons; hence, present mineable resources could be sustained for approximately 65 years at most at an average growth rate of 5% per year,2,5 making the implementation of the above-mentioned applications difficult and very costly.
Sodium, the fourth most abundant element on earth, has a seemingly unlimited distribution.6 Supplies of sodium-containing precursors are vast, with 23 billion tons of soda ash located in the United States alone. The abundance of resources and the much lower cost of trona (about $135–165 per ton), from which sodium carbonate is produced, compared to lithium carbonate (about $5000 per ton in 2010), provide a compelling rationale for the development of SIBs to be used as alternatives to LIBs.7,8 Because an alternative to lithium is needed to realize large-scale applications, SIBs have attracted considerable research attention in recent years. SIBs were initially studied when the development of LIBs began in the 1970s and 1980s, but due to rapid advances in the development and success of commercial applications of LIBs, SIBs were largely abandoned.9–15 Moreover, during those years, the overall quality of materials, electrolytes and glove boxes was insufficient for handling sodium, making it difficult to observe electrode performance. In the 1980s, prior to the commercialization of LIBS, a few US and Japanese companies developed SIBs in full cell configurations where a sodium-lead alloy composite and a P2-type NaxCoO2 were used as the anode and cathode, respectively. Despite the remarkable cyclability over 300 cycles, the average discharge voltages were lower than 3.0 V, which did not attract much attention against carbon//LiCoO2 cells exhibiting an average discharge voltage of 3.7 V.16–18 The battery components and the electrical storage mechanism of SIBs and LIBs are basically the same except for their ion carriers. In terms of cathode materials, the intercalation chemistry of sodium is very similar to that of lithium, making it possible to use similar compounds for both systems. However, there are some obvious differences between these systems. Na+ ions (1.02 Å) are larger compared to Li+ ions (0.76 Å), which affects the phase stability, transport properties, and interphase formation.9 Sodium is also heavier than lithium (23 g mol−1 compared to 6.9 g mol−1) and has a higher standard electrode potential (−2.71 V vs. SHE as compared to −3.02 V vs. SHE for lithium); thus, SIBs will always fall short in terms of energy density. However, the weight of cyclable Li or Na is a small fraction of the mass of the components, and the capacity is determined primarily by the characteristics of the host structures that serve as electrodes. Hence, in principle, there should be no energy density consequences of the transition from LIBs to SIBs.7 In addition, aluminum undergoes alloy reaction with lithium below 0.1 V vs. Li/Li+, which indicates that aluminum is available as a current collector for anodes in sodium cells. Therefore, aluminum is a cost-effective alternative to copper as an anode current collector for SIBs.
Various cathode materials for SIBs have been reported; for instance, layer and tunnel type transition metal oxides, transition metal sulfides and fluorides, oxyanionic compounds, Prussian blue analogues and polymers. However, the search for an anode with appropriate Na voltage storage, a large reversible capacity, and high structural stability remains an obstacle to development of SIBs. Graphite, which is a common anode material in LIBs, has a moderate Li storage capacity (∼350 mA h g−1) at approximately 0.1 V vs. Li/Li+.2 Recent studies have demonstrated that graphite does not properly intercalate sodium ions.19,20 Non-graphitic anodes, which consist largely of various carbonaceous materials such as carbon black21 and pitch-based carbon-fibers,22 allow insertion of sodium ions. Hard carbons, which are synthesized at high temperatures from carbon-based precursors, have been comprehensively modeled,23,24 characterized,25 and thermally tested26 in Na cells. These non-graphitic carbonaceous materials are considered to be the “first-generation” anodes of choice for SIB systems. SIBs are not fabricated with sodium metal due to dendrite formation, high reactivity, and an unstable passivation layer in the most organic electrolytes at room temperature. The high reactivity of metallic sodium with organic electrolyte solvents and dendrite formation during Na metal deposition are even more problematic than they are in Li metal anodes. The low melting point of sodium at 97.7 °C also presents a safety hazard for devices using Na metal electrodes at ambient temperature.27 Thus, it is important to use a true Na-ion system, where Na ions are exchanged between cathodes and anodes in a ‘rocking-chair’ format.
A new type of electrolyte for SIBs is needed, as the use of organic liquid electrolytes raises practicality and safety issues. The most common electrolyte formulations for SIBs are NaClO4 or NaPF6 salts in carbonate ester solvents, particularly propylene carbonate (PC). Metallic sodium anodes corrode continuously in the presence of these organic electrolytes, rather than forming a stable solid electrolyte interface (SEI). According to XPS and TOF-SIMS analyses performed by Komaba et al.,27 when NaPF6 is used as the electrolyte salt, the SEI film on hard carbon is predominantly an inorganic salt that contains precipitated species such as NaF on the surface.7 Palacin and colleagues28 found that NaClO4 and NaPF6 in an EC:PC solvent mixture represent the best electrolyte for a hard carbon anode. Developing aqueous electrolytes instead of organic electrolytes could be essential to the success of SIBs. Recently, an aqueous rechargeable battery with Na2NiFe(CN)6 and NaTi2(PO4) as the cathode and anode, respectively, demonstrated a good rate and cycle life with a theoretical energy density of 42.5 W h kg−1.29 Thus, it is possible to achieve higher energy density by selecting the appropriate electrode material. Nevertheless, an aqueous electrolyte system is more complicated than an organic system because of the (1) elimination of residual O2 from the electrolyte, (2) maintenance of electrode stability in the aqueous electrolyte, (3) inhibition of H3O+ co-intercalation into the electrode and (4) efficiency of the internal consumption of O2 and H2 produced from the cathode and anode sides when overcharged, overdischarged or improperly operated in a closed aqueous battery system. All of these issues are important for practical applications of aqueous battery systems.30,31
Scheme 1 and Fig. 1 illustrate a schematic SIB image that can adopt several representative candidate materials such as cathode and anode materials, electrolytes, separators, and binders that are discussed in the present paper. Most studies of SIBs explored the electrochemical performance of new electrodes and materials used with Na metal in half-cells, as this field is still in its early stages, and it is thus difficult to create full cells. In that regard, discussion and justification of SIBs are complicated compared to those of LIBs. In addition, experimental conditions such as the purity of Ar gas, electrolyte quality, and the glove box can influence the performance of SIB cells. Hence, comparison of battery performance between studies can be challenging.18
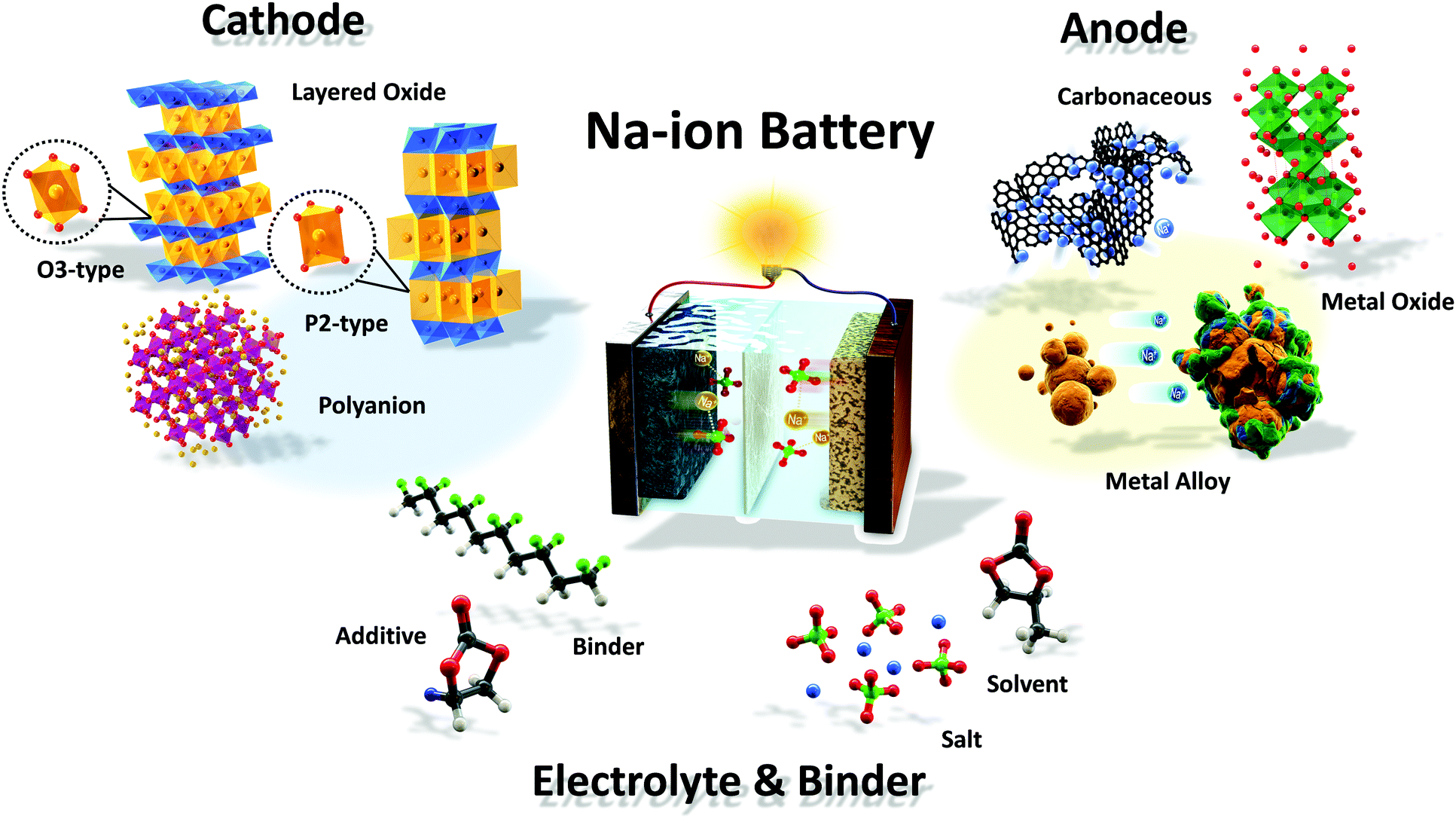 | ||
| Scheme 1 Illustration of a Na-ion battery system. | ||
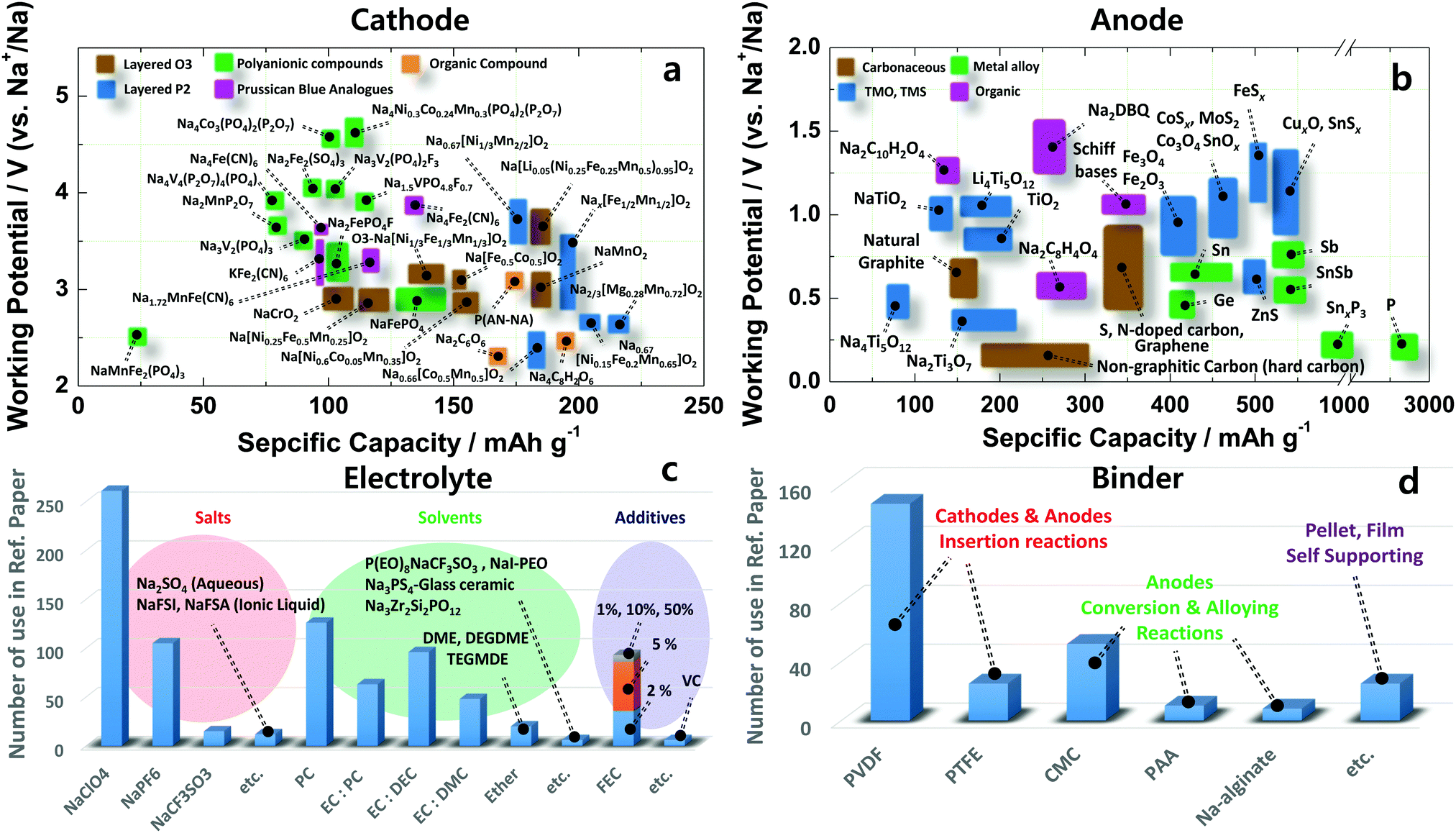 | ||
| Fig. 1 Recent research progress in sodium ion batteries: (a) cathode, (b) anode, (c) electrolyte and (d) binder. | ||
2. Cathode materials
Similar to LIBs, highly reversible cathode materials based on the intercalation reaction, which involves interstitial introduction of a guest species (Na+ in the present context), are needed for high capacity and good cyclability of SIBs. These electrode materials are mainly categorized into oxides, polyanions such as phosphates, pyrophosphates, fluorosulfates, oxychlorides, and NASICON (Na super ionic conductor) types, and organic compounds, which are mentioned in detail in Sections 2 and 3. These cathode materials exhibit a minimal structural change with intercalation, which ensures a reversible intercalation reaction that affects the cycle life. However, continuous structural evolution is inevitable during Na+ ion intercalation into the host structure interaction because of the large Na+ ion size (coordination number 6: 1.02 Å) relative to that of Li+ (coordination number 6: 0.76 Å). Also, sodiated transition metal materials are highly hygroscopic, even with brief exposure to air,32 and caution is necessary to avoid hydration of the material, particularly the surface, which results in formation of NaOH that degrades electrode performance due to its insulating properties. Thus, the preparation of sodiated cathode materials and batteries requires meticulous handling and moisture-free conditions.2.1. Two-dimensional layer transition metal oxides
The above-mentioned prototype compounds are composed of two- and three-dimensional crystal structures. Early investigation of two-dimensional layer oxides was performed by Delmas and Hagenmuller in the early 1980s.13,14,33 They defined the crystal structure of layered compounds depending on the stacking sequence of alkali ions between layers. Sodiated transition metal oxides, Na1−xMO2 (M: transition metal), were representatively classified into two main groups, O3 type and P2 type (Fig. 2), by Delmas et al.33 Those crystal structures comprise sheets of edge-sharing MO6 octahedral layers sandwiched between Na ion layers into which ionic species are inserted in an octahedral (O) or a prismatic (P) environment. The number (O2, O3, P2, and P3) indicates the packing number of Na ion octahedral or prismatic layers within each unit cell. In addition, the prime symbol (′) indicates monoclinic distortion, such that O′3 and P′3 represent monoclinic distortion of the O3 and P3 phases, respectively.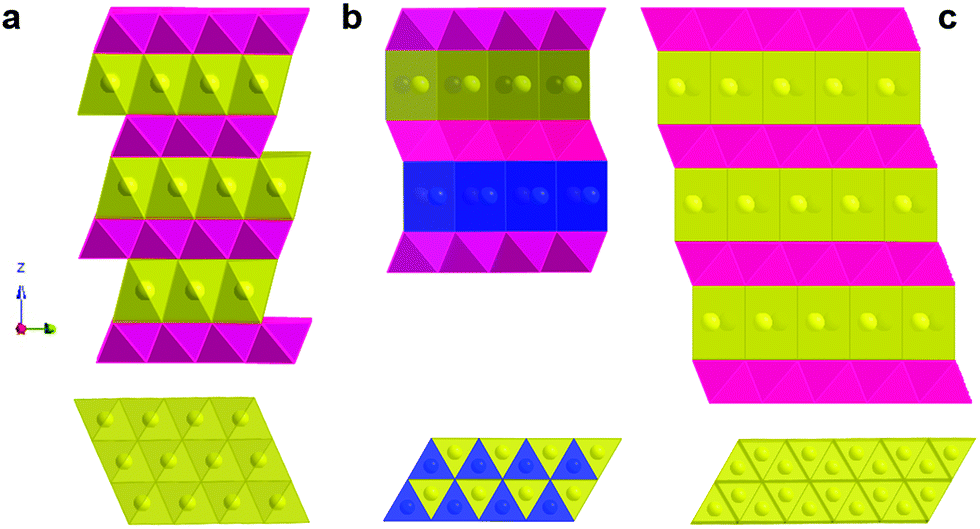 | ||
| Fig. 2 Crystal structures; (a) perspective view (top) and top view of the (00l) layer of the O3 structure, (b) perspective view (top) and top view of the (00l) layer of the P2 structure in which yellow sphere is face shared Na1 and blue one is edge sharing Na2 atoms, and (c) perspective view (top) and top view of the (00l) layer of the P3 structure. | ||
The O3 type is stable when the x value is high in Na1−xMO2 (x is close to 0), in which the average oxidation state of M is close to 3+. Electrochemical de-/sodiation of the O3 structure progresses with reversible structural transformation of O3 ↔ O′3 ↔ P3 ↔ P′3. Na+ ions energetically favor a prismatic environment when Na+ ions are partly extracted from the crystal structure, which creates vacancies. At the same time, this extraction induces strong repulsion of oxygen in the Na layers, and the interlayer distance thus expands. Na+ diffusion occurs faster in the P′3 phase due to the greater interlayer distance compared to O3. These transitions are followed by gliding of the MO2 slab without breakage of M–O bonds, as proposed by Delmas et al.33
The P2 type has a different optimal environment for Na compared to the O3 type. In particular, the structure is stable when the Na content is in the range of 0.3–0.7 in Na1−xMO2, in which the average oxidation state of M is above 3.3+. The presence of vacancies in the structure causes strong repulsion of oxygen in the Na layers, resulting in expansion of the interlayer distance. This leads to Na+ ions occupying prismatic (P) sites due to the large Na ionic size. Na+ ions occupy two different types of trigonal prismatic sites: Naf (Na1) contacts the two MO6 octahedra of the adjacent slabs along its face, whereas Nae (Na2) contacts the six surrounding MO6 octahedra along its edges. Adjacent Naf and Nae sites are too close together (considering the Na+ ionic radius) to be occupied simultaneously. The P2 phase is maintained in a wide desodiation range up to Na0.46MO2. Further desodiation shifts the phase toward O2 due to gliding (π/3 rotation) of the MO6 octahedra and contraction of the crystal structure, reducing the interlayer distance. Based on O3 ↔ O′3 ↔ P′3 the phase transitions for the O3 type and P2 ↔ O2 for the P2 type, structural variation from the O3/P3 to P2/O2(OP4) type or vice versa is not possible in Na cells unless the M–O bond breaks. Apart from phase transition, the P3 type can be produced not only via electrochemical desodiation, but also at low temperatures when synthesizing P2 type compounds; P3 and P2 are low and high temperature phases, respectively, indicating that phase transition requires breakage or reformation of M–O bonds via heat treatment.
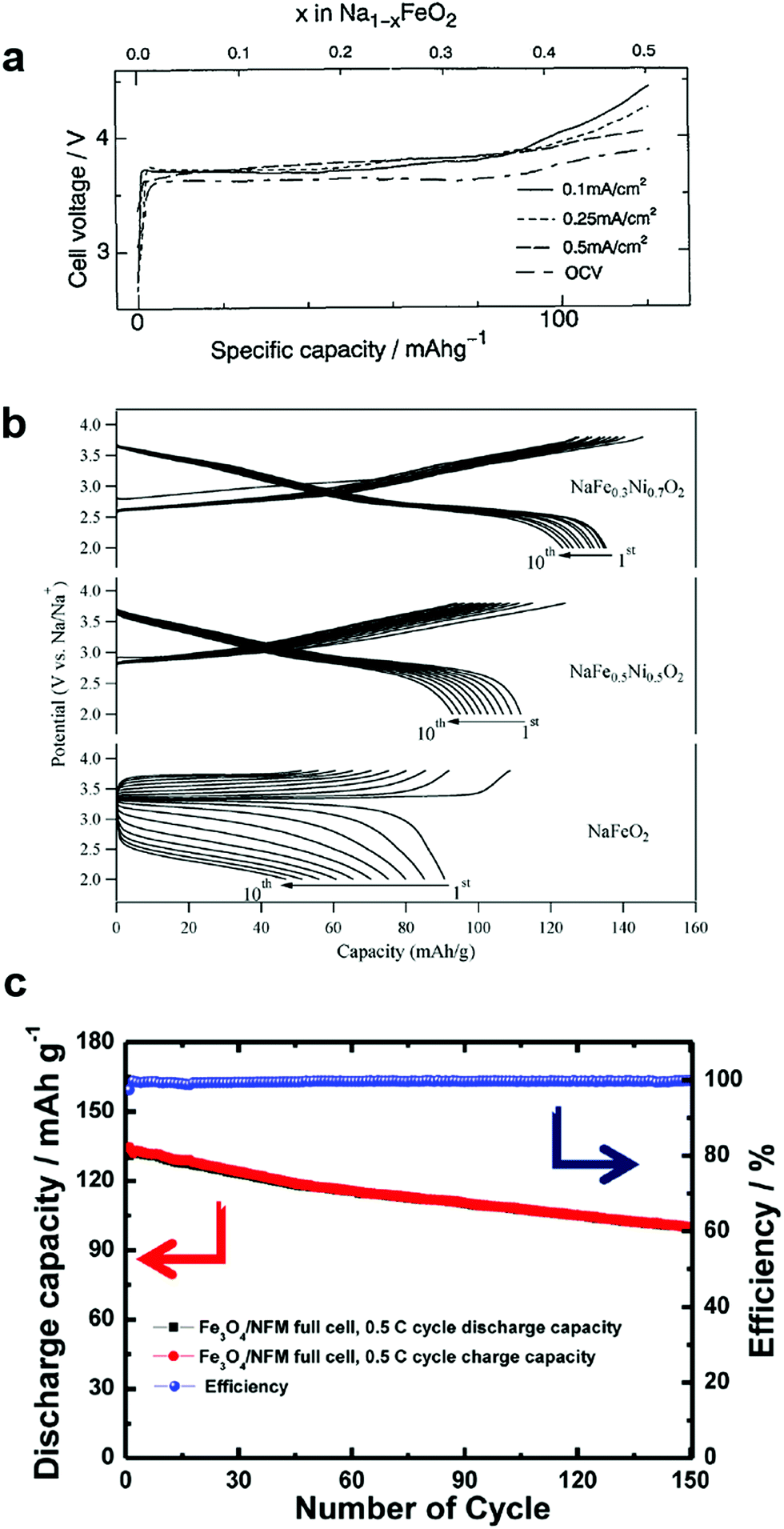 | ||
| Fig. 3 (a) Charge and discharge curves of O3-NaFeO2. (Reprinted from ref. 39, Copyright 1994, with permission from Elsevier.) (b) Charge and discharge curves of O3-Na[Fe1−xNix]O2. (Reproduced with permission from ref. 48, Copyright 2014 American Chemical Society.) (c) Clce performance for the Fe3O4/Na[Ni0.25Fe0.5Mn0.25]O2 full cell. (Reproduced with permission from ref. 51, Copyright 2014 American Chemical Society.) | ||
Partial substitution of Fe sites by Co is interesting in terms of capacity retention and rate performance, NaFe0.5Co0.5O2.47 In particular, the dramatic suppression of the irreversible capacity is attributed to the suppression of iron migration with the help of Co occupation in transition metal sites instead of Fe. Also, the presence of Co enabled an increase in electrical conductivity, such that the electrode was active up to 30 C-rates (102 mA h g−1). Wang et al.48 stabilized the crystal structure of NaFeO2 by forming a solid solution with NaNiO2, NaFe1−xNixO2 (0 ≤ x ≤ 1). The dilution of the Fe concentration, NaFe0.3Ni0.7O2, effectively increased capacity and retention (135 mA h g−1 and 74% retention after 30 cycles) via Fe3+/4+ and Ni3+/4+ redox couples (Fig. 3b). In addition, they also recorded the 57Fe Mössbauer spectra to determine the valence state and investigate the mechanism of the electrochemical reaction of Fe in O3-NaFe1−yNiyO2 (y = 0, 0.5, and 0.7). Recent report by Nanba et al. also well supported the electrochemical reaction mechanism of NaFe1−xNixO2 (0 ≤ x ≤ 1) electrodes.49
Extension of the layer structure toward a three component system, Na[Ni1/3Fe1/3Mn1/3]O2, was suggested by Kim et al.50 For synthesis, they used oxalate coprecipitation, [Ni1/3Fe1/3Mn1/3]C2O4, due to the difficulty of forming hydroxides when Fe is involved. Na[Ni1/3Fe1/3Mn1/3]O2 could deliver a discharge capacity of 120 mA h g−1 in the voltage range of 2–4 V for a half cell. They also tested the cyclability after adopting a hard carbon anode (100 mA h g−1 after 150 cycles). Oh et al. also synthesized spherical Na[Ni0.25Fe0.5Mn0.25]O2 using [Ni0.25Fe0.5Mn0.25](OH)2.51 They found that the electronic states of Ni, Fe, and Mn were 2+, 3+, and 4+, respectively, of which Ni2+/4+ and Fe3+/4+ were responsible for electrochemical activity in the range of 2.1–3.9 V via the X-ray absorption nearest edge spectral (XANES) analysis technique. Mn remained electrochemically inactive, but preserved the crystal structure during the electrochemical reaction. A Fe3O4/Na[Ni0.25Fe0.5Co0.25]O2 full cell with a conversion anode and an insertion cathode delivered a capacity of approximately 130 mA h (g-Na[Ni0.25Fe0.5Mn0.25]O2)−1 with approximately 76.1% retention at the 150th cycle (Fig. 3c).
Despite the good reversibility of Na[Ni0.25Fe0.5Mn0.25]O2, the discharge capacity limit of 140 mA h g−1 in O3 type layer materials needs to be overcome. The voltage limitation to 3.9 V prevents iron migration during charge. Oh et al. suggested another approach to raise the upper voltage cutoff to 4.4 V, in order to offer more capacity in O3 type compounds using the Mn3+/4+ redox, but reducing the concentration of Fe3+ in Na[Li0.05(Ni0.25Fe0.25Mn0.5)0.95]O2.52 As anticipated the compound was active based on the Ni2+/4+, Fe3+/4+, and Mn3+/4+ redox reactions, resulting in a large capacity of approximately 180 mA h g−1 at a 0.1C-rate in the voltage range of 1.7–4.4 V. Additionally, the presence of a stronger Li–O bond relative to that of Ni–O and Mn–O in the transition metal layer was responsible for stabilization of the crystal structure, enabling better capacity retention during cycling.
Na0.7FeO2 is not stable in P2 type layer compounds because of the intrinsic instability of tetravalent Fe in an oxide framework, as described in Section 2.1.1. Yabuuchi and Komaba et al. introduced a P2-Nax[Fe0.5Mn0.5]O2 layer compound that delivers 190 mA h g−1 of reversible capacity (Fig. 4a).46 Partial substitution of Fe by Mn made it possible to utilize the electrochemical oxidization of Fe3+ to Fe4+ reversibly, in particular in a voltage range of 3.8–4.2 V. Upon desodiation, the P2 phase was maintained at 3.8 V by the oxidation of Mn3+/4+, Na0.42[Fe0.5Mn0.5]O2, after which the oxidation of Fe3+/4+ led to phase transformation from P2 to OP4 at 4.2 V (Na0.13[Fe0.5Mn0.5]O2) as shown in Fig. 4b. An in situ XRD study also demonstrated that P2-Na2/3[Fe0.5Mn0.5]O2 undergoes a reversible P2–OP4 phase transition at the end of charge.54 This achievement is remarkable in terms of capacity in comparison to the O3 type Na[Fe0.5Mn0.5]O2 that delivers less than 120 mA h g−1 in early cycles. Despite a high capacity approximating 190 mA h g−1 on the first cycle, capacity fading was inevitable in the P2-Nax[Fe0.5Mn0.5]O2 compound.
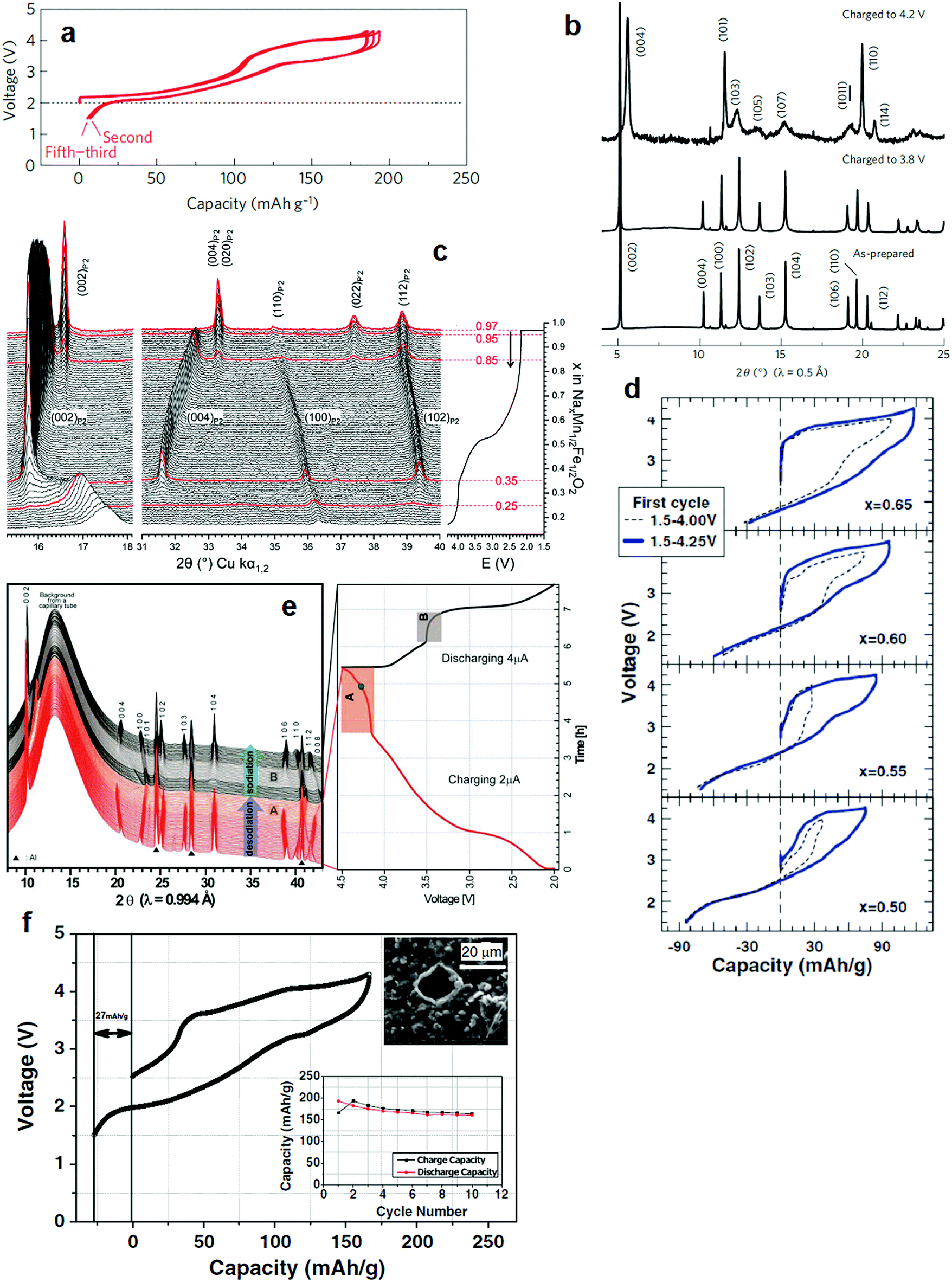 | ||
| Fig. 4 (a) Charge and discharge curves of P2-Na2/3[Fe1/2Mn1/2]O2 and (b) SXRD patterns of P2-Na2/3[Fe1/2Mn1/2]O2 samples charged to 3.8 and 4.2 V in which the P2 phase was transformed into OP4 one. (Reproduced by permission from ref. 46, Nature Publishing Group, Copyright 2012.) (c) Operando in situ XRD patterns of the P2-Nax[Fe1/2Mn1/2]O2 electrode. The red XRD patterns correspond to the limits of the different structural domains: P′2 phase for x = 0.85–0.97 in Nax[Fe1/2Mn1/2]O2 and Z-phase for x = 0.25–0.35 in Nax[Fe1/2Mn1/2]O2. (Reproduced with permission from ref. 55, Copyright 2011 The Royal Society of Chemistry.) (d) First cycle for 1.5–4.0 V (dashed lines) and 1.5–4.25 (solid lines) for selected cells. (Reproduced with permission from ref. 57, Copyright 2013 The Electrochemical Society.) (e) In situ synchrotron XRD patterns of the P2-Na0.7[Fe0.4Mn0.4Co0.2]O2. (Reproduced from ref. 58 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) (f) First charge and discharge curves of the Nax[Fe1/2Mn1/2]O2/NaN3 composite electrode. (Reprinted from ref. 60, Copyright 2013, with permission from Elsevier). | ||
Morternard de Boisse et al. raised another issue regarding the structural change during the first discharge from OCV to 1.5 V.55 The P2 phase, Na0.62[Fe0.5Mn0.5]O2, was transformed into orthorhombic P′2 (space group: Cmcm), Na0.97[Fe0.5Mn0.5]O2, by cooperative Jahn–Teller distortion at the end of discharge. In a P2 type layer structure, Nae and Naf sites are both simultaneously occupied at a ratio close to 2:1. This distribution results from the strong sodium–sodium repulsion interaction, which prevails over sodium–transition metal repulsion. Hence, Nae and Naf cannot be occupied simultaneously in the P2 phase. All Na+ ions are located at one site, and the stoichiometry of Na[Fe0.5Mn0.5]O2 can be stabilized in the orthorhombic structure. Note that the atomic displacement parameter of Na in the P′2 phase (0.7(2) Å2) is much lower than that of the P2 phase (1.9(3) Å2), indicating that Na+ ions are less mobile in the orthorhombic P′2 phase. One interesting finding that contrasts with the results of Yabuuchi and Komaba et al. is that when electrochemical sodiation forms the orthorhombic P′2 phase, the resulting desodiation to 4.3 V leads to the formation of not OP4, but also a new and unindexable “Z” phase (Nax[Fe0.5Mn0.5]O2, x < 0.25) with poor crystallinity (Fig. 4c).55 Talaie et al. revealed phase “Z”, which is a result of migration of Fe3+ into tetrahedral sites in the interlayer space, showing a short range order between two adjacent layers.56 This migration is highly reversible, although it induces polarization of the cell. Addition of Ni instead of Fe was very effective at mitigating migration of Fe3+ and thus improved the cycling performance.
Thorne et al. determined the relationship between the Na and Fe content in Na1−x[Fe1−xMnx]O2 (0 ≤ x ≤ 0.5).57 The Na content is a decisive factor establishing the phase (O3, O′3, P2, etc.). As clearly seen in Fig. 4d, the length of the first desodiation plateau increased with x in Na1−x[Fe1−xMnx]O2. The length of the first sodiation plateau also increased as x decreased due to more sodium vacancies in Na1−x[Fe1−xMnx]O2. The oxidation states in Na1−x[Fe1−xMnx]O2 are Fe3+ and Mn4+, as suggested by Mössbauer spectroscopy. Therefore, desodiation oxidized Fe3+ to Fe4+, while the sodiation of Na1−x[Fe1−xMnx]O2 selectively reduced Fe4+/3+ at high voltages above 3.5 V and reduced Mn4+/3+ oxidation below 3.5 V. This coincides with the results of Yabuuchi and Komaba. Jung et al.58 explored the effects of Co on P2-Na0.7[(Fe0.5Mn0.5)1−xCox]O2 (x = 0–0.2). Note that the partial replacement of Fe by Co in O3-Na[Fe1−xCox]O2 dramatically enhanced capacity and performance.47 Similarly, the addition of Co into the transition metal layers of Na0.7[(Fe0.5Mn0.5)1−xCox]O2 resulted in higher capacity and contributed to stable cycling behavior during cycling, in particular for Na0.7[(Fe0.5Mn0.5)0.8Co0.2]O2. In contrast to P2-Nax[Fe0.5Mn0.5]O2, P2-Na0.7[(Fe0.5Mn0.5)0.8Co0.2]O2 underwent a phase transition toward the O2 phase when desodiated and the O2 phase was transformed into P2 upon sodiation. The P2 to O2 transition was reversibly achieved via the gliding of slabs due to prismatic site instability without Na+ ions. This simple phase transition is related to the addition of Co in the transition metal layers, as a result of structural stabilization. The phase transition from P2 to OP4 is supposed to occur (Fig. 4e); however, the added Co that stabilizes the crystal structure is likely to suppress phase transition towards the OP4 structure because of the suppression of Fe3+ migration to a tetrahedral or octahedral interspace. Therefore, the simple phase transition from P2 to O2 is responsible for the better capacity retention of Co-doped Na0.7[(Fe0.5Mn0.5)1−xCox]O2. In comparison with the O3 layer structure, the simple phase transition during the de-/sodiation process of P2 layer compounds is obviously advantageous in preserving the original structure during cycling.
However, an important issue regarding the Na-deficient P2 structure is that such high capacities can be obtained only after the first cycle with a Na metal counter electrode. Compensation of Na in the synthetic state causes formation of an O3 and/or O′3 structure with a further increase in the Na content of Nax[Fe1/2Mn1/2]O2 (x ≥ 0.8).59 Therefore, the irreversible capacity of the first cycle is an intrinsic issue with P2 type materials. Singh et al. suggested using a NaN3 additive, sacrificial salt, which acts as follows: 2NaN3 → 3N2 + 2Na+ + 2e−.60 This oxidative decomposition was effective in circumventing the irreversible capacity loss in Na2/3[Fe1/2Mn1/2]O2 in the first cycle. NaN3 was used as a source of extra Na+ ions added to the cathode. Hence, NaN3-added P2 Na2/3[Fe1/2Mn1/2]O2 could successfully reduce the irreversible first charge capacity from 58 mA h g−1 to 27 mA h g−1 (Fig. 4f). Taking into account N2 release after oxidative decomposition at high potentials, an appropriate amount of NaN3 is recommended because decomposition can cause swelling of cells. However, degassing is possible in pouch-type cells, such that the addition of sacrificial salts is likely to facilitate full cell configuration of the P2 cathode materials.
Sensitivity in air, in particular uptake of CO2 in air, is a serious problem because of the formation of electrochemical-inactive Mn4+ on the surface of active materials. Ni-doping of the transition metal sites in Nax[Fe0.5Mn0.5]O2 is less prone to react in air.61
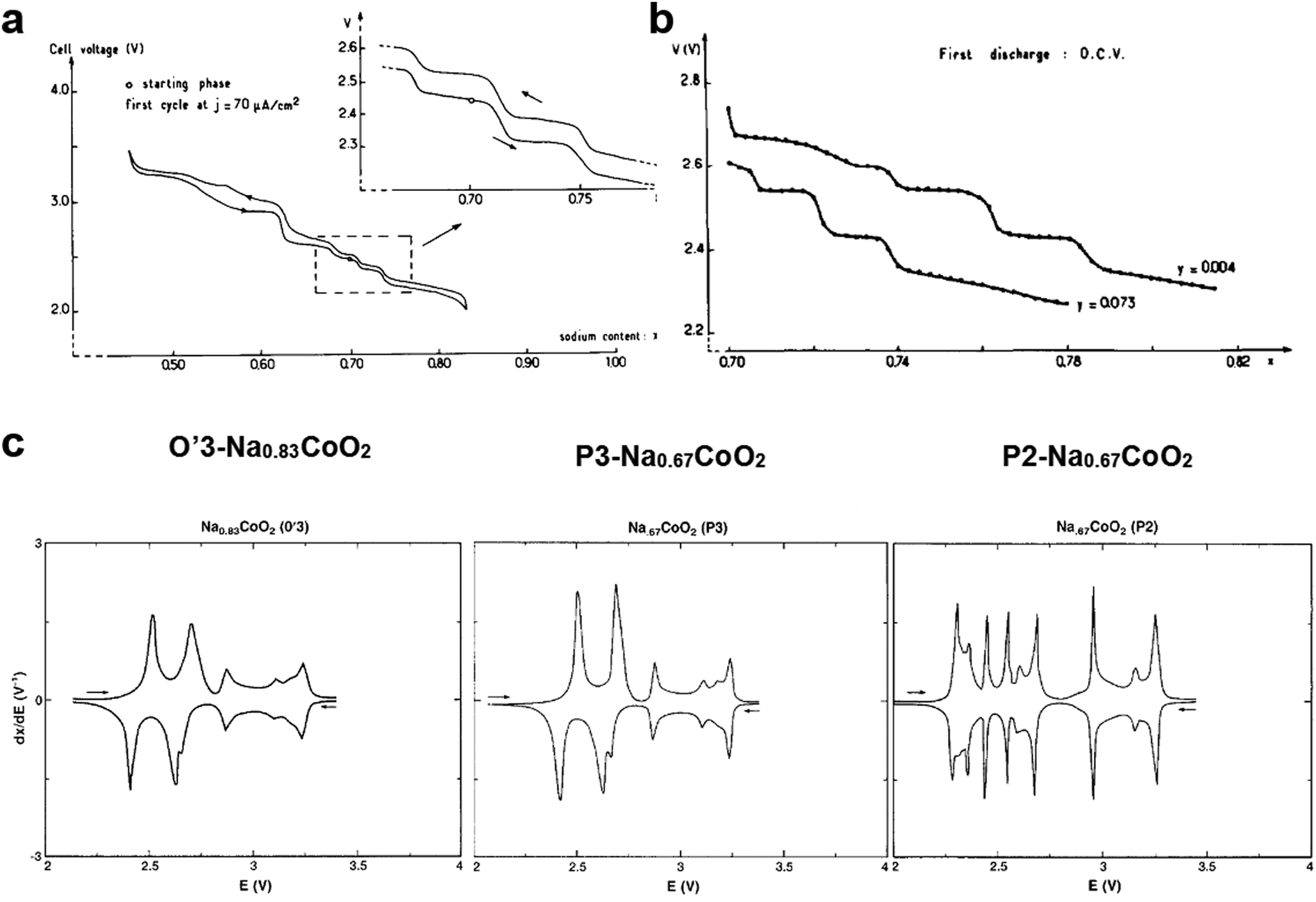 | ||
| Fig. 5 (a) First charge and discharge curves of P2-Na0.7CoO2. (Reprinted from ref. 33, Copyright 1981, with permission from Elsevier.) (b) Comparison of open circuit EMF composition curves obtained from P2-Na0.7CoO2−y. (Reprinted from ref. 69, Copyright 1989, with permission from Elsevier.) (c) Comparison of dQ/dV curves of O′3-Na0.83CoO2 (left), P3-Na0.67CoO2 (center), and P2-Na0.67CoO2 (right). (Reproduced with permission from ref. 71, Copyright 1988 The Electrochemical Society.) | ||
Delmas suggested the presence of several NaxCoO2 structures and Shacklette confirmed that it has four phases.71 In particular, O3, O′3, and P3 layer structures were formed in a temperature range of 400–600 °C, whereas the P2 structure was stable only when the heating temperature was >700 °C in an oxygen atmosphere via the conversion of the P3 phase, which involved rotation of CoO6 octahedra and Co–O bond breakage. The voltage profile, operating voltage, and phase transition are very similar for the O3 and P3 phases during de-/sodiation (Fig. 5c). For the P2 phase, although similar behavior was observed in a voltage range of 2.7–3.5 V, phase transition appeared more complicated below 2.7 V compared with O3 and P3 (Fig. 5c). The difference in Na+ ordering for the P2 and P3 phases could account for the behavior variation below 2.7 V, at which considerable Na+ is included at trigonal sites. Capacity retention was over 80% for 300 cycles for P2-Na0.7CoO2, whereas P3-Na0.7CoO2 exhibited retention of approximately 60% for over 250 cycles. The higher capacity for the P2 phase may be due to its higher crystallinity compared with the P3 material produced at low temperatures. Ma et al. confirmed that P2-Na0.7CoO2 can be used for long-term cycling in a P(EO)8NaCF3SO3 electrolyte at 90 °C.72
Berthelot et al. revisited P2-NaxCoO2 to investigate the phase diagram in a range of 0.45 ≤ x ≤ 0.9, in which nine potential drops were found during the second discharge (Fig. 6a).62 Weak polarization, which is an intrinsic property of P2-Na0.7CoO2 due to its high conductivity, enabled characterization of each voltage drop via in situ XRD. The (008) diffraction peak sometimes splits into two peaks, which is indicative of a two-phase domain when the discharge curve exhibits voltage plateaus, while the voltage drop indicates a strictly fixed peak due to the single phase reaction, demonstrating the presence of ordered phases (Na0.5CoO2, Na0.67CoO2 (No. 3), Na0.72CoO2 (No. 7), Na0.76CoO2 (No. 8), and Na0.79CoO2 (No. 9), although they are present only in very narrow ranges. Between the voltage plateau and the voltage drop, the diffraction peak shifts slightly with solid solution behavior. Na ordering arises from two different repulsive interactions, sodium–sodium and sodium–cobalt. Occupation of Na at the shared edges (Nae) for P2-Na2/3CoO2 was predicted by first-principles calculations63,67 and was experimentally verified.62,68 At the same time, some of the Na ions are located at the shared faces (Naf). For P2-Na0.5CoO2, an ordered phase, half of Na occupies Nae sites while the rest appears at Naf sites, which probably minimizes the repulsive in-plane interaction between Naf and Nae. The distribution of Na+ ions in these two positions depends on the Na content in the compound, which affects the Na+/vacancy-ordered structure.
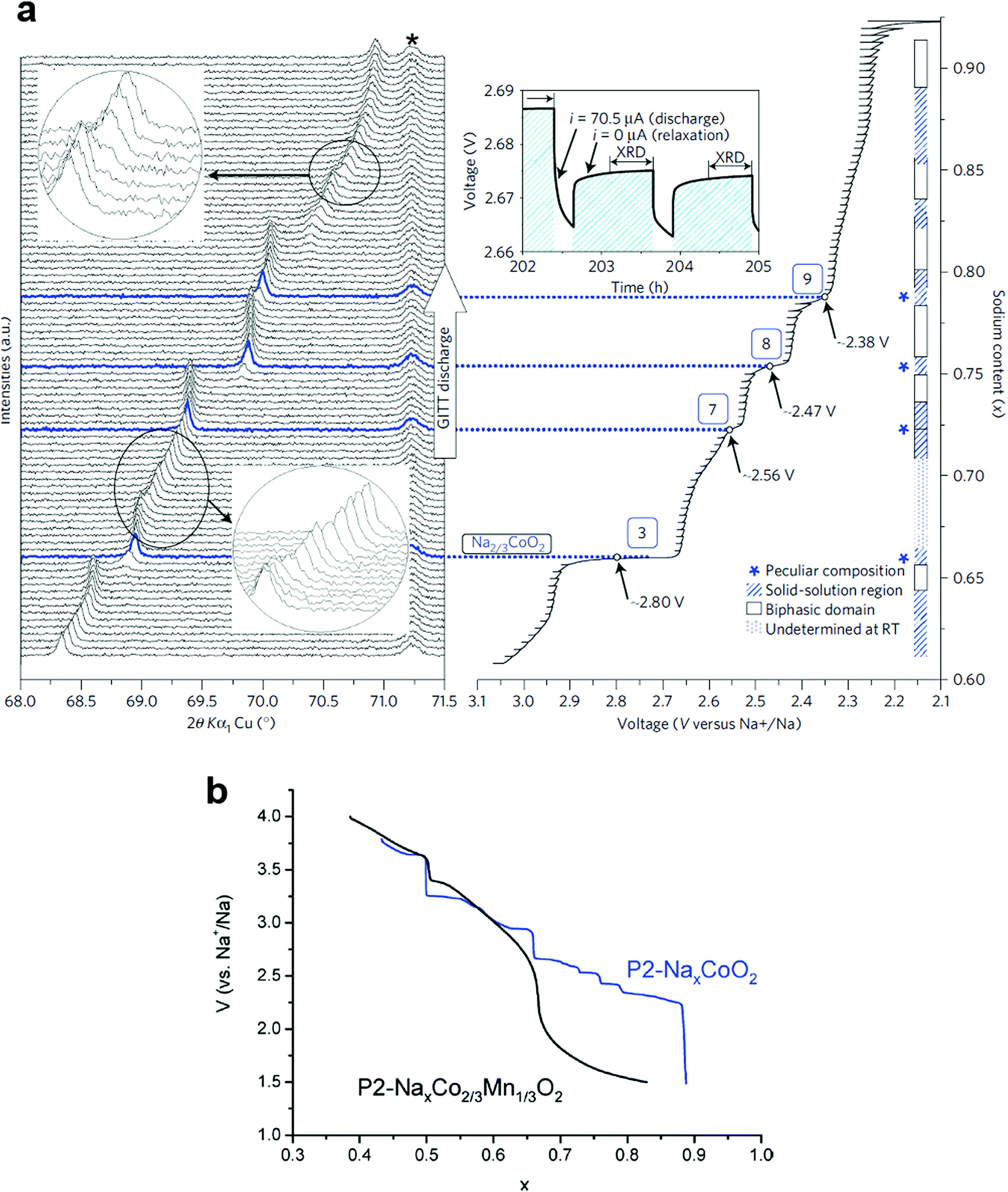 | ||
| Fig. 6 (a) In situ XRD patterns during sodium ion intercalation in P2-NaxCoO2. (Reproduced by permission from ref. 62, Nature Publishing Group, Copyright 2011.) (b) Comparison between the second discharges obtained for a P2-Nax[Co2/3Mn1/3]O2 cell (black curve) and a NaxCoO2 cell (blue curve). (Reproduced with permission from ref. 76, Copyright 2011 The Royal Society of Chemistry.) | ||
Croguennec et al. obtained T#2-Li2/3[Co2/3Mn1/3]O2via ion-exchange from P2-Na2/3[Co2/3Mn1/3]O2, in which the oxidation states of Co and Mn were 3+ and 4+, respectively.73,74 Cheng et al. also confirmed that the oxidation states of Co and Mn for P2-Na2/3[Co2/3Mn1/3]O2 are stabilized at 3+ and 4+, respectively.75 Carlier et al.76 evaluated P2-Na2/3[Co2/3Mn1/3]O2 in the Na cell and Yang et al.77 extended the stability of P2-Na2/3[Co1−xMnx]O2 to x = 0.5. The resulting P2-Na2/3[Co2/3Mn1/3]O2 shows suppression of Na+/vacancy ordering in the structure because the stepwise voltage plateau disappeared in a range of 0.5 ≤ x ≤ 0.83 in Nax[Co2/3Mn2/3]O2 (Fig. 6b). Note that P2-Na0.74CoO2 formed nine distinct phases in a range of 0.5 ≤ x ≤ 0.9 in NaxCoO2 (Fig. 6a). However, the single phase domain for x = 0.5 in NaxCoO2 is still found in Na2/3[Co2/3Mn1/3]O2, although the voltage drop at that composition is lower than in NaxCoO2. The main difference after Mn substitution for Co is abrupt voltage decay in a range of 0.65 ≤ x ≤ 0.83 in Nax[Co2/3Mn1/3]O2. They further revealed that Co3+/2+ and Mn4+/3+ reactions were involved in the low voltage plateau, as similar behavior was found when Li1+x[Ni1−x−yCoxMny]O2 was overlithiated on discharge.
Yang et al.77 successfully stabilized the P2-Na2/3[Co1−xMnx]O2 phase to x = 0.5, although the resulting capacity was limited to 120 mA h g−1. Meanwhile, Co3+/2+ and Mn4+/3+ reactions were more dominant in a voltage range of 1.5–2.1 V. Lowering the synthetic temperature to 700 °C resulted in P2/P3-Na2/3[Co0.5Mn0.5]O2 compound.78 This biphasic compound greatly improved the capacity to 180 mA h g−1 at a rate of 0.1C in a voltage range of 1.5–4.3 V compared with pure P2-Na2/3[Co0.5Mn0.5]O2.77 Rate cycling performance at a rate of 5C was also remarkably high, delivering approximately 125 mA h g−1 with retention of 91% for 100 cycles. Even at high rates, half capacity was delivered in the voltage range of 1.5–2.1 V associated with the Co3+/2+ and Mn4+/3+ reactions. The structural stabilization achieved in the biphasic compound may explain the excellent electrochemical performance. However, the capacity obtained in the low voltage region may decrease the energy density as use these electrode materials for cathodes.
Recently, Matsui et al. explored the possibility of Ca-doping at Na sites to form Na2/3−xCaxCoO2.79 The similarity in the ionic radius of Ca2+ (1.00 Å) versus Na+ (1.02 Å) allows the incorporation of Ca2+ into the Na sites. Although the delivered capacity decreased to some extent due to Ca2+ in the Na layers, Na2/3−xCaxCoO2 could suppress the multiple phase transition during charge and discharge. For example, Na5/8Ca1/24CoO2 could be cycled even at 5 mA cm−1 with very little capacity decay, while capacity fade was inevitable in Ca-free Na2/3−xCoO2. Post-cycled electrodes showed formation of a sodium poor phase of Na2/3CoO2, while Na2/3−xCaxCoO2 suppressed this phase separation. This work emphasizes that stability in the Na environment is another topic of investigation that will contribute to development of a long-term cyclable cathode for Na cells.
Xia et al. tested the reactivity of desodiated Na0.35CoO2 derived from P2-Na0.65CoO2 in the NaPF6-based electrolyte.80 Na0.35CoO2 decomposed to Na0.7CoO2 and Co3O4 with oxygen release from the crystal structure. The NaPF6 salt rapidly reacted with Na0.35CoO2, and NaCoF3 was produced via the exothermic reaction. This reaction has not been reported in a LixCoO2 system thus far. Selection of the electrolytic salt is another important issue to utilize the electrode in Na cells.
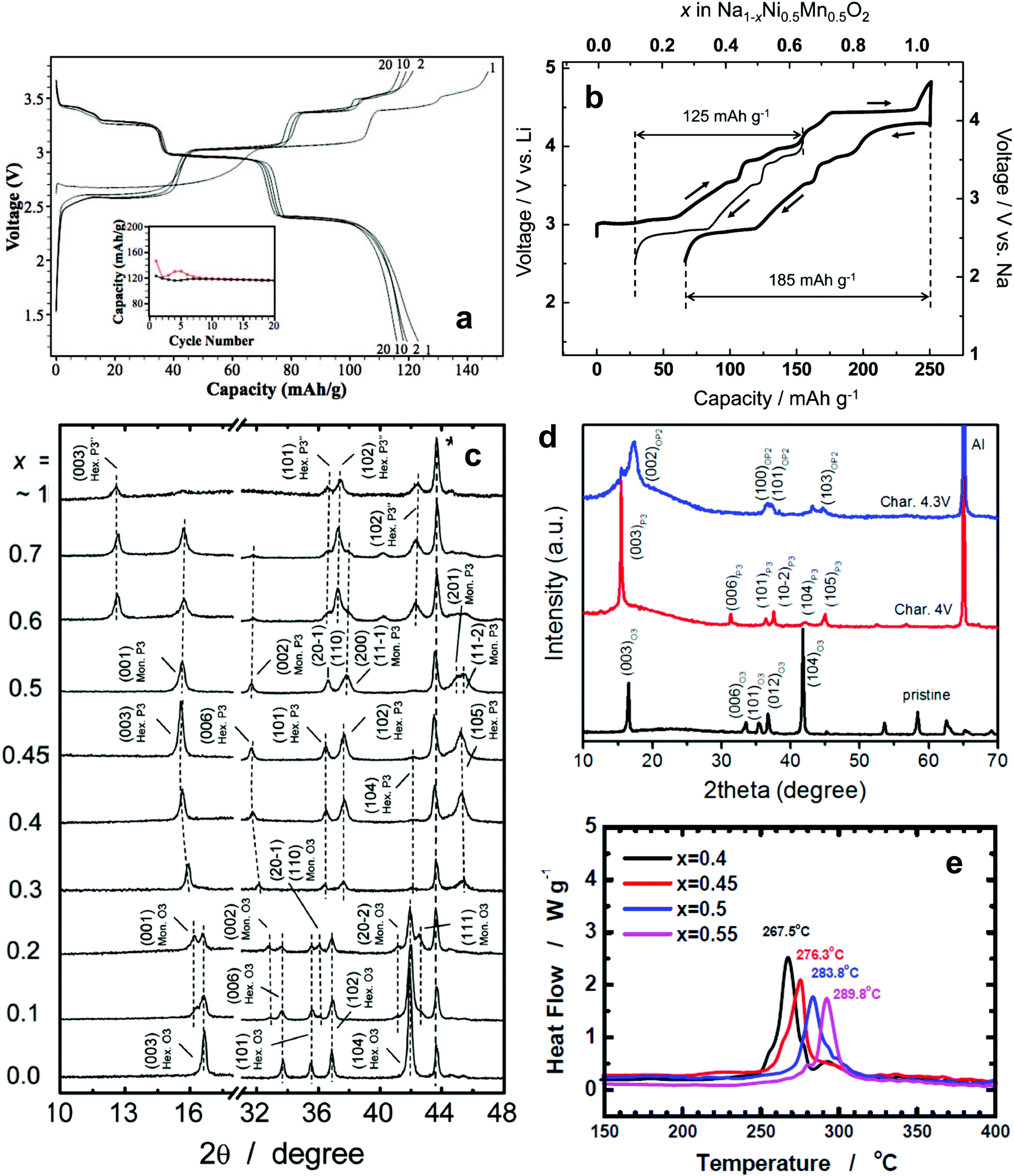 | ||
| Fig. 7 (a) Charge and discharge curves of O′3-NaNiO2. (Reproduced with permission from ref. 81, Copyright 2013 The Electrochemical Society.) (b) The first charge and discharge curves of O3-Na[Ni0.5Mn0.5]O2 and (c) structural evolution of Na1−x[Ni0.5Mn0.5]O2 (x = 0–1). (Reproduced with permission from ref. 83, Copyright 2012 American Chemical Society.) (d) Ex situ XRD patterns of O3-Na[Fe0.2(Ni0.5Mn0.5)0.8]O2 at various charge states. (Reproduced with permission from ref. 84, Copyright 2015 American Chemical Society.) (e) DSC traces of desodiated Na1−d[Ni0.75−xFexMn0.25]O2 obtained after charging to 3.9 V. (Reprinted from ref. 85, Copyright 2016, with permission from Elsevier.) | ||
Komaba et al. applied the findings of research on Li[Ni0.5Mn0.5]O2 to Na[Ni0.5Mn0.5]O2, in which the average oxidation states of Ni and Mn are 2+ and 4+, respectively.83 The material was also crystallized into an O3-type layer structure and could be solely activated by the redox reaction of Ni2+/4+. In comparison with NaNiO2, the presence of tetravalent Mn induced the formation of Ni2+ in Na[Ni0.5Mn0.5]O2. The two-electron reaction led to a high discharge capacity of 185 mA h g−1 in a voltage range of 2.5–4.5 V (Fig. 7b), whereas optimization of the electrode performance limited the upper voltage cutoff to 3.8 V, resulting in a capacity above 100 mA h g−1 over 20 cycles. In contrast to NaNiO2, the O3 ↔ O′3 ↔ P3 ↔ P′3 phase transition was highly reversible in a voltage range of 2.5–3.8 V (Fig. 7c). Fe-substituted Na[Ni0.5Mn0.5]O2, however, underwent different structural transformation during desodiation compared with Na[Ni0.5Mn0.5]O2.84 Phase evolution from P3 to P3′′ was predominant in Na[Ni0.5Mn0.5]O2, whereas Na+ extraction from Fe-substituted Na[Ni0.5Mn0.5]O2 facilitated a gradual evolution from P3 to OP2 due to the migration of Fe3+ ions from the octahedron of the transition metal layers to the interstitial tetrahedron or the octahedron of the Na layers (Fig. 7d). Nonetheless, capacity retention was improved in a voltage range of 2–4.3 V. In particular, a long irreversible reaction due to the formation of P3′′ phase transitioned to the OP2 phase in a voltage range of 4–4.3 V on charge, although the length of the plateau did not appear on discharge. This peculiar behavior seems to be related to the effect of Fe. Recently, Hwang et al. investigated the collation of Ni and Fe contents in Na[Ni0.75−xFexMn0.25]O2 (x = 0.4, 0.45, 0.5, and 0.55).85 The structural and thermal stabilities, which affect cycle retention and rate capability, were dependent on the Fe content. Although the discharge capacity of higher Fe content materials decreased slightly, the improved cycling performance and rate capability compensate for the slightly reduced capacity. The increase in the Fe content seems to improve conductivity, which is associated with low band gap energy (∼2.5 eV for Fe2O3).86 Therefore, enhanced rate performance with increasing Fe content is attributed to the improvement in electric conductivity derived from Fe in the compound. Also, their DSC study revealed that reactive Ni4+ ions in the desodiated host material cause oxygen removal from the crystal structure, and that oxygen evolution can effectively suppress the Fe increase in the crystal structure (Fig. 7e).
They also developed a radially-aligned hierarchical columnar structure in spherical particles with a varied chemical composition from the inner end (Na[Ni0.75Co0.02Mn0.23]O2) to the outer end (Na[Ni0.58Co0.06Mn0.36]O2) of the structure.87 An electrochemical reaction based on Ni2+/3+/4+ had a discharge capacity of 157 mA h (g-oxide)−1 with a capacity retention of 80% (125 mA h g−1) over 300 cycles in combination with a hard carbon anode. The cathode also exhibited good temperature performance, even at −20 °C, which enables the Ni redox reaction.
Ti-Substituted O3-Na[Ni0.5Ti0.5]O2 is also interesting due to its superior cyclability under moderate conditions, with an average operating potential of 3.1 V (vs. Na+/Na) and delivery of a reversible capacity of 121 mA h g−1 at 20 mA g−1. Since the average oxidation states of Ni and Ti are 2+ and 4+, respectively, the Ni2+/4+ redox reaction is responsible for electrochemical activity.88 Increasing the Ni content resulted in higher capacity, while a dramatic degradation in capacity and thermal properties was observed. In contrast, increasing the Fe content improved capacity retention and thermal stability in a highly desodiated state. Hence, it is worth noting that Ni redox such as Ni2+/4+ or Ni3+/4+ contributes to high capacity delivery, while optimization of the Ni content in the transition metal layer is important in ensuring capacity retention and thermal properties.
 | ||
| Fig. 8 (a) Crystal structure of monoclinic O′3-Na0.93MnO2 projected in the a–c-plane and (b) charge and discharge curves of monoclinic O′3-Na0.93MnO2. (Reproduced with permission from ref. 90, Copyright 2011 The Electrochemical Society.) (c) Schematic illustration of orthorhombic P′2-Na0.7MnO2. (Reprinted from ref. 95, Copyright 2010, with permission from Elsevier.) (d) Charge and discharge curves of P2-Na2/3MnO2. (Reproduced with permission from ref. 99, Copyright 2014 American Chemical Society.) (e) Charge and discharge curves of P2-Na2/3[Ni1/3Mn2/3]O2 (Reproduced with permission from ref. 101, Copyright 2001 The Electrochemical Society), in which the inset represents the diffusion paths of P2 (left) and O2 (right) that favors diffusion in the P2 phase (Reproduced with permission from ref. 103, Copyright 2013 The Royal Society of Chemistry) and (f) in situ powder X-ray diffraction of Na2/3[Ni1/3Mn2/3]O2 showing the reversible evolution of P2–O2 phase transition. (Reproduced with permission from ref. 101, Copyright 2001 The Electrochemical Society.) | ||
Ma and Ceder et al. reported that monoclinic O′3-NaMnO2 could deliver charge and discharge capacities of approximately 210 mA h g−1 and 197 mA h g−1, respectively, in a voltage range of 2–3.8 V and at a rate of C/30 (Fig. 8b).90 This behavior differs from an earlier report by Mendiboure and Hagenmuller.91 The charge and discharge curves show many plateaus and voltage drops, with eight and five charge and discharge plateaus, respectively. The hysteresis was reversible even after cycling. A long plateau observed at 2.63 V from Na0.93MnO2 to Na0.7MnO2 is associated with a two-phase reaction. Their ex situ XRD investigation revealed that the second phase was Na0.7MnO2, although this was not consistent with orthorhombic Na0.7MnO2 (space group: Cmcm) or P2-Na0.7MnO2 (space group: P63/mmc). Capacity retention of O′3-NaMnO2 was approximately 74% for 10 cycles without a significant structural change compared to the fresh electrode. In Mn3+-containing cathode materials, capacity fade is usually mentioned with Mn dissolution, which causes disproportionation to Mn2+ and Mn4+. Only a small percentage of Mn was dissolved from the active material (less than 32 μg from 2–3 mg of the active material), such that dissolution was not likely to affect electrode performance. Their successive work demonstrated the readiness of the 180° Na–O–Mn3+–O–Na strip formation in contrast to VNa–O–Mn3+–O–Na, albeit insufficient Na+ ions in Na5/8MnO2, because Na+ ions relax to the highly distorted octahedral sites, where they share the symmetric attraction of two neighboring Jahn–Teller distorted –O–Mn3+–O–Na configurations along the [100] axis.91 This structural imperfection may be the reason for the gradual capacity fade observed during cycling in this system.
High temperature-type β-NaMnO2 has a zigzag layer structure composed of two edge-sharing stacks of the MnO6 octahedra. Between two neighboring sheets, the sodium ions occupy octahedral sites.92 In the Li system, orthorhombic LiMnO2 is directly synthesized, indicating that the orthorhombic structure is energetically favored relative to monoclinic LiMnO2, which is usually produced via ion exchange from α-NaMnO2. In the Na system, however, first principles calculations indicate that the monoclinic α-NaMnO2 is energetically more stable than orthorhombic β-NaMnO2.93,94 β-NaMnO2 is active in a narrow range (0.85 ≤ x ≤ 0.96) in NaxMnO2.91 In this range, a two-phase domain is attributed to β-NaMnO2 and NaxMnO2. Phase transformation towards spinel was possible in β-LiMnO2, because Mn can migrate into tetrahedral Li sites to form the spinel phase. This phase transition is unlikely to occur because tetrahedral sites rarely form in spinel with Na due to the large ionic size of Na+ relative to Li+.
Reducing the Na content to 0.7 induces formation of a different crystal structure: P2-, P3, and orthorhombic (P′2) Na0.7MnO2 (Fig. 8c).95 As mentioned in Section 2.1, P3 is the low temperature type and P2 is the high temperature type (Fig. 2). Stoyanova found that orthorhombic Na0.7MnO2 (space group: Cmcm) is stable at 1000 °C (Na2/3[Mn3+3/2Mn4+1/3]O2).95,96
Among these polymorphs, P2-Na0.7MnO2 and its derivatives have been intensively studied.97–99 An early report by Caballero et al. showed reversible capacity delivery of more than 150 mA h g−1 in a voltage range of 2–3.8 V in Na cells.98 They also suggested that intercalation of Na+ ions occurred in several steps, but the resulting capacity fade progressed upon successive cycling tests. Increasing the synthetic temperature to 900 °C, Yabuuchi et al. could improve the capacity up to 190 mA h g−1 in a voltage range of 1.5–4.3 V during several early cycles (Fig. 8d).99,100 The electrode performance is similar to that of O3′-NaMnO2, although the related phase transition is different.
P2-Na2/3[Ni2+1/3Mn4+2/3]O2 was first reported in 2001 by Lu and Dahn et al.101 This compound is stable in moist air, and hydration via the insertion of water molecules does not occur.102 Although it has a relatively low theoretical capacity (173 mA h g−1), P2-Na2/3[Ni2+1/3Mn4+2/3]O2 showed an average operating voltage of 3.5 V with a Ni2+/4+ redox reaction, delivering approximately 160 mA h g−1 in a voltage range of 2–4.5 V (Fig. 8e). Because of the similarity in the ionic size between Ni2+ and Mn3+, Ni2+ prefers to occupy the Mn3+ sites instead of Mn4+ in Na2/3MnO2. Their in situ XRD study revealed a reversible P2–O2 phase transition stemming from the oxygen shift, in which the O2 phase prevails at a voltage plateau above 4 V on charge and 3.8 V on discharge (Fig. 8f). This is the main difference from P2-Na0.67MnO2; the absence of the Jahn–Teller distortion Mn3+ is responsible for the occurrence of the P2–O2 phase transition. This transition is associated with a large volume change when the O2 phase appears. Recently, Meng et al. revisited P2-Na2/3[Ni1/3Mn2/3]O2 and observed phase transformation from P2 to O2 at 4.2 V.103 The long voltage plateau is the evidence of two forms of Na+ ion ordering: one row of Naf and two rows of Nae in Na1/2[Ni1/3Mn2/3]O2 and Na orders in rows on either Nae or Naf order in Na1/3[Ni1/3Mn2/3]O2, in which the latter corresponds to the region where the O2 phase is dominant. In the P2 phase, the path with the minimum energy passes through a shared face between two neighboring Na prismatic sites. Here, Na+ ions need around 170 meV for diffusion in the P2 phase. In the O2 phase, Na+ ions cross the tetrahedron between two octahedral sites by means of a vacancy mechanism.104 The required energy for Na+ diffusion in the O2 phase is 290 meV, indicating slow Na+ mobility in the O2 phase, as was confirmed experimentally. It is reasonable because the diffusion path of Na+ ions is more spacious in the P2 phase relative to the O2 phase; this leads to a lower activation barrier (Fig. 8e inset). Hence, a cycling test in the voltage range of 2.3–4.1 V, which is associated with the P2 phase reaction, resulted in excellent cycling behavior retaining 96% of the initial capacity (∼80 mA h g−1). They further extended their work to inhibit O2 phase formation in a deeply desodiated state to retain the capacity for a long-term. Surprisingly, Li substitution in the transition metal layer, Na0.80[Li0.12Ni0.22Mn0.66]O2, inhibited P2–O2 transition up to 4.4 V on charge, showing a simple solid solution reaction across the entire range.105 As a result, there was no characteristic voltage plateau in a voltage range of 4.1–4.4 V. This feature ensured excellent capacity retention as high as 91% for 50 cycles (∼120 mA h g−1 at the first discharge).
Substitution of divalent elements such as Mg2+ and Zn2+ in Ni is effective for diluting the effect of the above-mentioned P2–O2 phase transition.106–113 A decrease in the initial charge capacity is natural because the electro-active species Ni2+ was reduced by substitution, as shown in Fig. 9a, which reflects the origination of the capacity drop from the Ni2+/4+ redox reaction. Stepwise voltage plateaus were not dominant throughout the operation range of 2–4.5 V.106,107 Compared with Na2/3[Ni1/3Mn2/3]O2, the capacity drop in Na0.67[Ni0.2Mg0.1Mn0.7]O2 was only 6 mA h g−1 for 50 cycles.107 Using in situ XRD, instead of the P2 phase, a new phase identified as an OP4 phase was found at voltages above 4.2 V, where a short voltage plateau was observed compared to Mg-free Na2/3[Mn1/3Mn2/3]O2 (Fig. 9b). The presence of electro-inactive Mg2+ in transition metal layers facilitates occupation of Na+ ions in prismatic sites, stabilizing the overall charge balance of the compounds. Since the Na+ ions are less extracted from the host structure assisted by Mg2+ in the transition metal layers, the original P2 phase is retained as the major phase, though transition to OP4 because deep desodiation is observed at high voltage as a minor phase. Hence, the P2–O2 transition and the Na+/vacancy ordering in Na2/3[Ni1/3−xMgxMn0.7]O2 are suppressed during cycling via Mg substitution. This P2–OP4 phase transition is highly reversible; the cycling performance demonstrates its superiority compared to the P2–O2 phase transition.101 While Zn-substituted P2-Na2/3[Ni1/3−xZnxMn2/3]O2 induces reversible phase transition during electrochemical reactions.111,112 Although the capacity was still limited to approximately 140 mA h g−1 by the Ni2+/4+ redox reaction due to reductions in electro-active Ni2+, the enhanced structural integrity achieved by Zn2+ in the transition metal layer enabled good cyclability due to readiness for transformation and a less variation in unit volume. Further studies explored partial replacement of Ni sites by Al.113 The substitution displayed sloping charge–discharge curves due to the diminution of the electro-active Ni2+ concentration, suggesting that Ni2+/4+ redox has superior capacity delivery. The substituent stabilized the host structure and the resulting cyclability was significantly improved, in particular for Al-doped P2-Na2/3[Ni1/3Mn2/3]O2, which had 94.8% (from ∼147 mA h g−1) retention over 30 cycles.
 | ||
| Fig. 9 (a) Continuous charge and discharge curves of P2-Na0.67[Mg0.1Ni0.2Mn0.7]O2 and (b) in situ powder X-ray diffraction of P2-Na0.67[Mg0.1Ni0.2Mn0.7]O2 showing the reversible evolution of P2–OP4 phase transition at the end of the charge and beginning of the discharge. (Reproduced with permission from ref. 107, Copyright 2016 American Chemical Society.) (c) Continuous charge and discharge curves of P2-Na0.70[Mn0.60Ni0.30Co0.10]O2 in the voltage range of 1.5–4.3 V and (d) XRD patterns of the material recovered on both sides of the plateau: (a) after charge at 4.0 V and (b) after charge at 4.3 V, where G denotes graphite. (Reproduced with permission from ref. 114, Copyright 2014 The Electrochemical Society.) (e) Initial charge and discharge curves of P2-Na2/3[Mg0.28Mn0.72]O2 and (f) structural changes of P2-Nax[Mg0.28Mn0.72]O2 in the Na cells examined by synchrotron X-ray diffraction (SXRD). Highlighted SXRD patterns (marked by a dotted square) are also shown in the inset. Mg and Mn superlattice ordering is still visible after electrochemical cycle tests. (Reproduced with permission from ref. 120, Copyright 2014 The Royal Society of Chemistry.) | ||
Replacement with a trivalent 3d transition metal element such as Co and Fe is also interesting because they are electrochemically active in the same operating range.114–116 Yoshida et al. investigated the composition of P2-Na0.7[Mn0.6Ni0.3Co0.1]O2, in which the oxidation state of Mn is approximately 3.4+, in a voltage range of 1.5–4.3 V.114 Note that the oxidation state of Mn is 4+ in P2-Na2/3[Ni1/3Mn2/3]O2. As expected, the material exhibited multiple voltage plateaus. In comparison with P2-Na2/3[Ni1/3Mn2/3]O2, the improvement in capacity during the first cycle (approximately 204 mA h g−1) is due to the additional capacity contributed by Mn3+/4+ redox below 4 V (Fig. 9c). The long plateau above 4 V confirmed the validity of the Ni2+/4+ redox reaction. As cycles went by, the length of the upper voltage plateau related to Ni2+/4+ and the lower voltage plateaus related to Mn3+/4+ 2 V decreased. This can be interpreted in two ways: (1) the progressive formation of the O2 phase at high voltage and the effect of the Jahn–Teller distortion in the crystal lattice. This further limited the cycling region to three parts: 1.5–4.3 V, 1.5–4.0 V, and 1.7–4.0 V. It is evident that, although the Jahn–Teller distortion is present in low voltage regions, severe capacity fade occurs in high voltage regions. This explains that the P2–O2 phase transition is not favored when maintaining the capacity because of the large difference in the c-axis, which causes a drop in the capacity (Fig. 9d). Jahn–Teller distortion seems to be less pronounced in capacity retention. In contrast, instead of Co, Yuan et al. suggested that Ni substitution with Fe (P2-Na0.7[Mn0.65Ni0.15Fe0.2]O2) led to an improvement in capacity and retention: 208 mA h g−1 with 71% retention over 50 cycles.115 The phase transition was P2 to OP4 in this case.117
Tetravalent Ti-substituted P2-Na2/3[Ni1/3Mn2/3−xTix]O2 yielded a wide solid solution range of 0 ≤ x ≤ 2/3.118,119 Due to the similarity of both Mn4+ and Ti4+ in the valence and ionic radius, such a solid solution can be formed readily throughout the range. The reason remains unclear, but an increase in the Ti content resulted in a decrease in capacity. In particular, Na2/3[Ni1/3Mn2/3−xTix]O2 (x = 1/6) delivered approximately 127 mA h g−1 with an average operating voltage of approximately 3.7 V on discharge, which corresponds to 470 W h kg−1. More importantly, the stepwise voltage profiles of Na2/3[Ni1/3Mn2/3]O2 were obviously diluted by Ti substitution. XRD suggested that the volume change of the fully charged state was reduced from 23.1% in Ti-free compounds to 12–13% for Ti-substituted compounds, confirming the suppression of Na/vacancy ordering and phase transitions during sodium intercalation.
A new mechanism was introduced in P2-Na2/3[Mg0.28Mn0.72]O2 and P2-Na5/6[Li1/4Mn3/4]O2 by Yabuuchi and Komaba et al.120,121 In this compound, since Mg2+ and Li+ are electrochemically inactive, the only possible reaction should be associated with Mn redox upon de-/sodiation. While the average oxidation states of Mn are 3.84+ for Na2/3[Mg0.28Mn0.72]O2 and 3.88+ for Na5/6[Li1/4Mn3/4]O2. Nonetheless, both materials exhibited large capacities of approximately 220 mA h g−1 for Na2/3[Mg0.28Mn0.72]O2 (Fig. 9e) and 180 mA h g−1 for Na5/6[Li1/4Mn3/4]O2. Similarly, a recent report by Slater et al. also showed a large capacity of 200 mA h g−1 for P2-Na0.85[Li0.17Ni0.21Mn0.64]O2.7 Taking the Mn3+/4+ redox species into account, it is not possible to explain the delivery of high capacity. Indeed, there is almost no delivery of capacity in the voltage cutoff up to 4 V on charge; however, those capacities are delivered above 4 V, which is related to oxidation of oxide ions, resulting in the release of oxygen from the oxide lattice. Oxygen removal causes formation of Mn3+, which simultaneously participates in oxidation on charge and is reduced on discharge. This causes rearrangement of the in-plane cation ordering of the crystal structure, which is similar to a Li-rich Li2MnO3 system.122 P2–O2 transition was also observed in these compounds (Fig. 9f), which is worth mentioning because of the theoretical capacity of 173 mA h g−1 based on the Ni2+/4+ redox reaction. Ni2+ was successfully overcome by the oxygen compensation accompanied by the Mn3+/4+ redox reaction.
Surface modification was performed on P2-Na2/3[Ni1/3Mn2/3]O2via wet milling in an Al-containing aqueous solution.123 Na-containing transition metals react very quickly when moisture is present. Although P2-Na2/3[Ni1/3Mn2/3]O2 is stable in air, ion exchange between Na and H occurs when the reaction progresses in an aqueous medium. Optimized experimental conditions facilitate the formation of Al2O3-coated P2-Na2/3[Ni1/3Mn2/3]O2, although there is some possibility of Al doping in the crystal structure because additional heating after the removal of the aqueous medium was performed at 650 °C for 10 h. Al2O3-coated P2-Na2/3[Ni1/3Mn2/3]O2 had good cycling performance for 300 cycles with approximately 72% retention in a voltage range of 2.5–4.3 V. Although the detailed mechanism related to this high capacity retention remains unclear, structural stabilization of the host material by the coating was suspected.
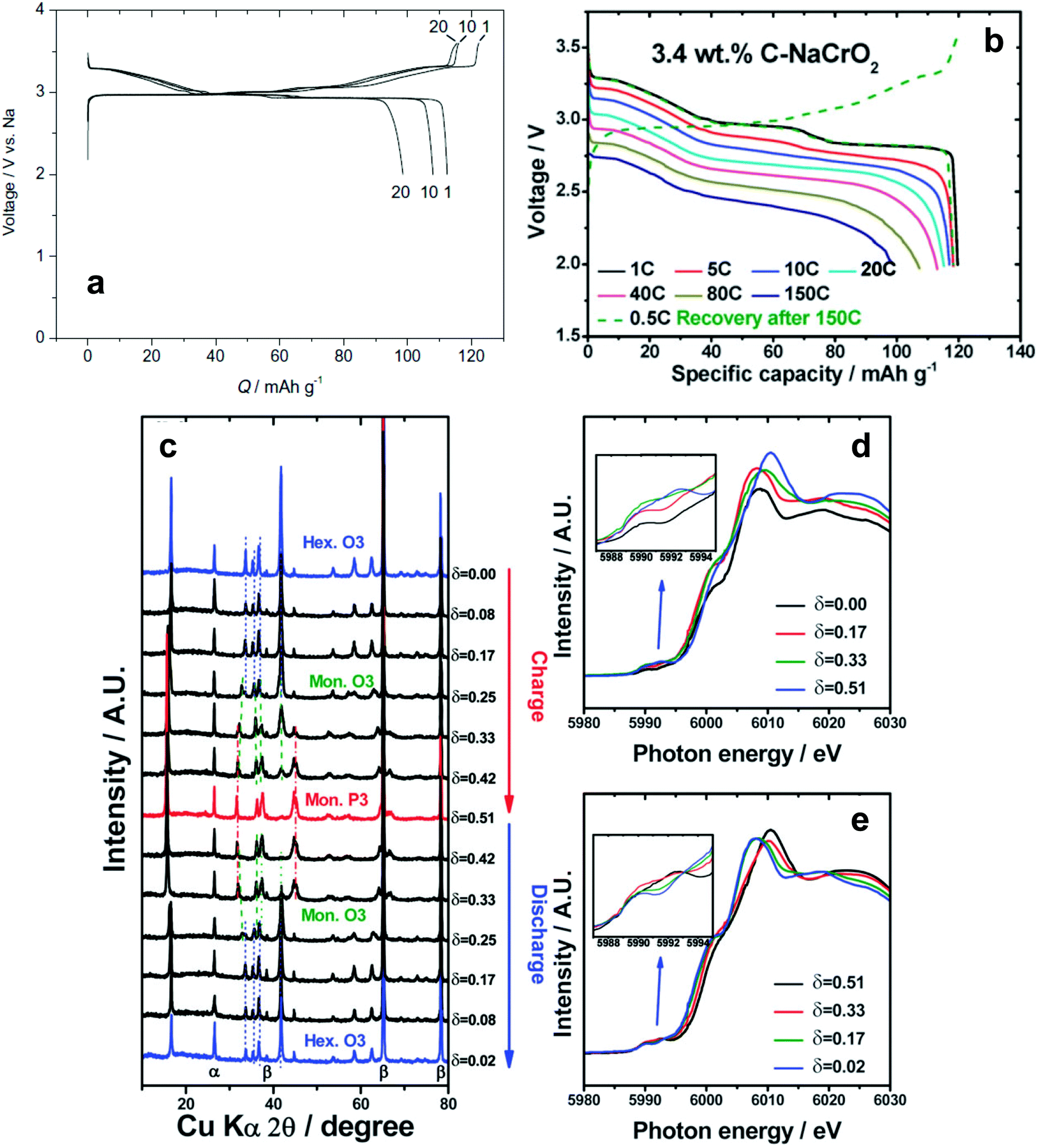 | ||
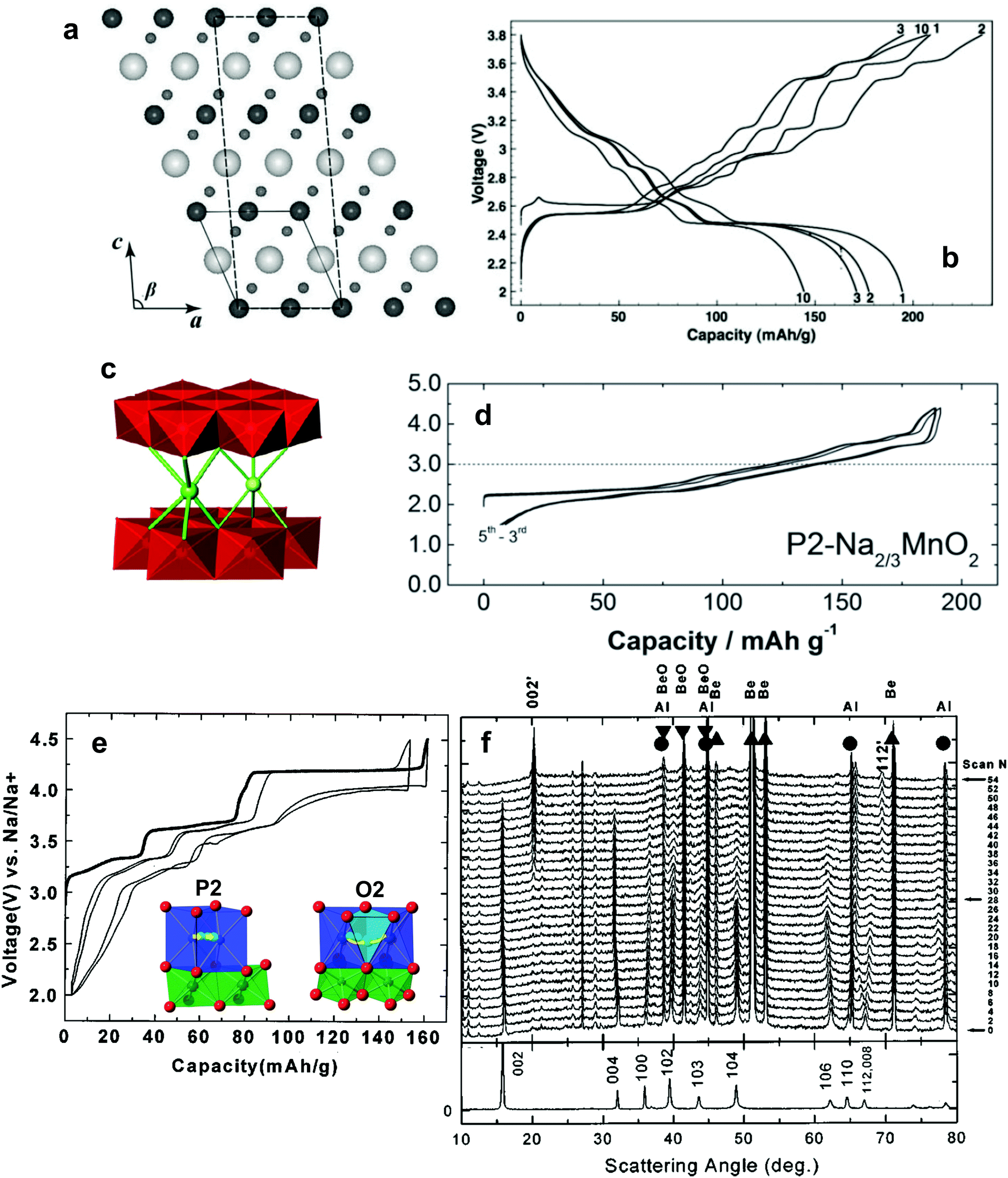
| Fig. 10 (a) Charge and discharge curves of O3-NaCrO2. (Reprinted from ref. 128, Copyright 2010, with permission from Elsevier.) (b) Rate performances of 3.4 wt% carbon-coated O3-NaCrO2. (c) Ex situ XRD patterns obtained during charging and discharging, Cr K-edge XANES spectra obtained during (d) charging and (e) discharging. (Reproduced with permission from ref. 130, Copyright 2015 The Royal Society of Chemistry.) | ||
In consideration of synthetic conditions, NaCrO2 is usually produced in a reducing atmosphere to retain the oxidation state of Cr at 3+. This condition enables carbon coating, which can dramatically improve electrode performance. Ding et al. found that citric acid-assisted carbon coating slightly improved the cycling stability of NaCrO2 electrodes.130 Although the cycling performance of carbon-coated NaCrO2 was improved compared to the bare material, operation at high rates was not possible, presumably due to inhomogeneous or excessively thick carbon layers. The XANES study revealed that Cr3+ is oxidized to a higher chemical state on charge. The above results are not satisfactory for use in practical applications because of rapid capacity fading (ca. 80% in the 50th cycle) and disappointing rate capability. Yu et al. modified the surfaces of the NaCrO2 particles via carbonization of pitch, which improved the electrical conductivity to approximately 10−1 S cm−1.131 The carbon-coated NaCrO2 electrode exhibited excellent cyclability and an ultrafast rate capability of up to a 150C-rate (Fig. 10b). High electrical conductivity successfully promotes reversible insertion and extraction of sodium ions accompanied by a facile complementary redox reaction of the Cr3+/Cr4+ couple as confirmed by XANES (Fig. 10c–e). The NaCrO2 electrode was also sufficiently stable in an intermediate temperature ionic liquid, NaFSA-KFSA, at 363 K.132 Excellent electrochemical performance of these cathodes was demonstrated in both half and full sodium ion cells. Apart from the electrode performance, the carbon coating layer also delays exothermic decomposition by preventing oxygen evaporation from the crystal lattice; specifically, the lower heat generation from the desodiated Na0.5CrO2 was ascribed to the suppression of oxygen evolution during the exothermic reaction, which results from the presence of carbon coating layers.
A major challenge associated with sodiated cathode materials is immediate water absorption after exposure to air. This leads to the formation of NaOH and Na2CO3 on the surface of active materials. The sodium diffuses to the surface, and electrochemically inactive parts are formed within the particles. Even worse, the NaOH and Na2CO3 formed are electrical insulators. The carbon coating can prevent moisture uptake due to its hydrophobic characteristics.
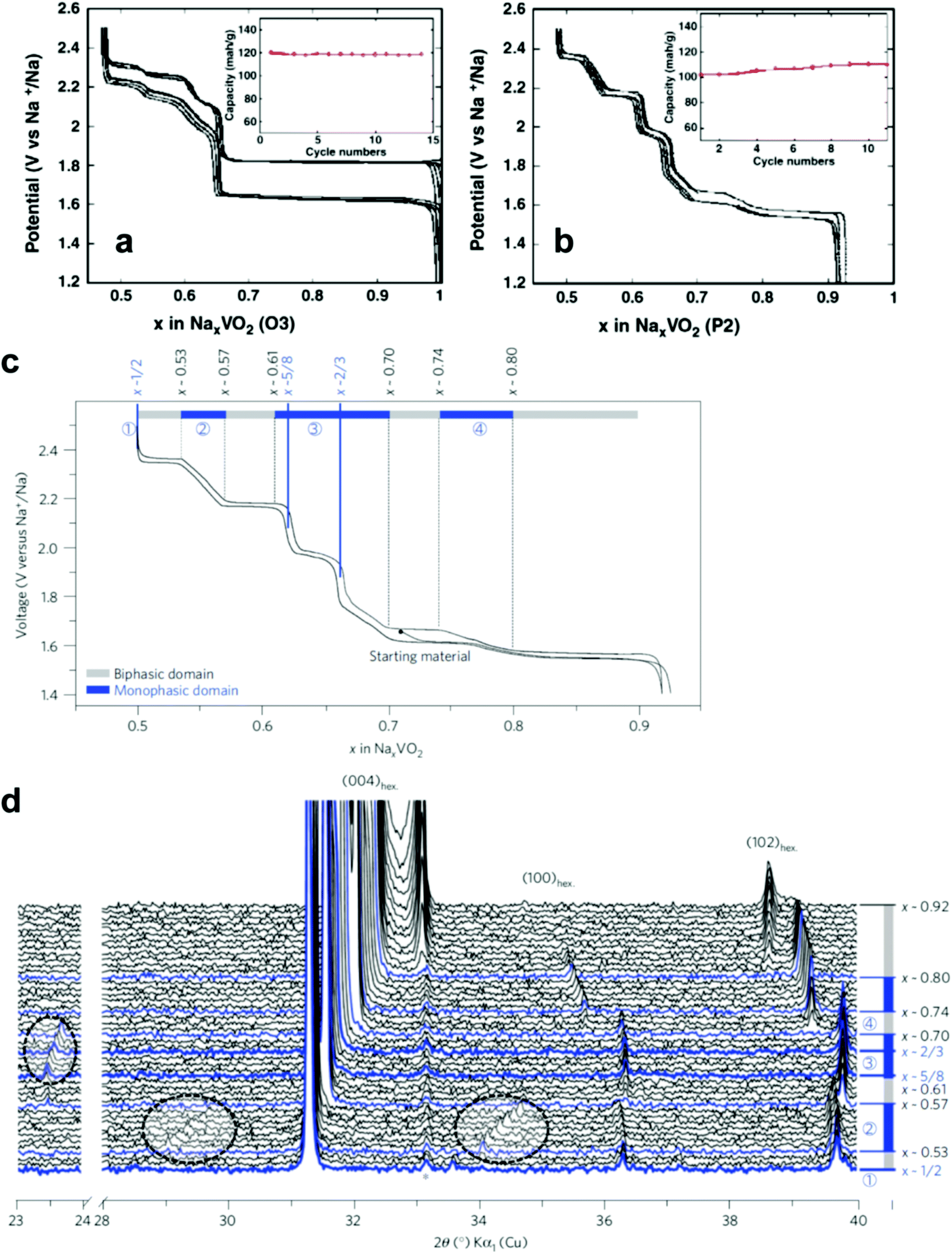 | ||
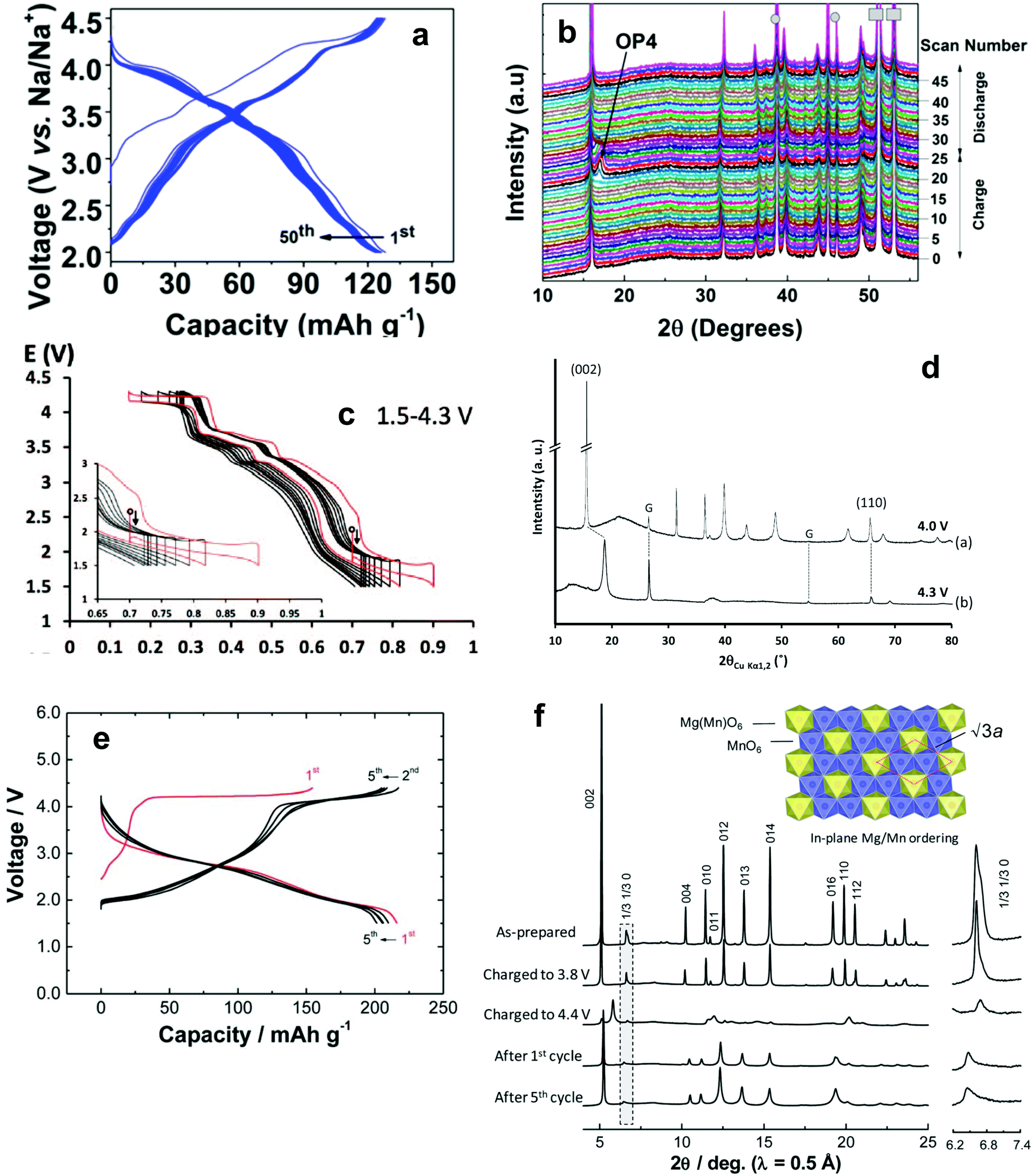
| Fig. 11 Voltage–composition curves of (a) O3-NaVO2 and (b) P2-Na0.7VO2. (Reprinted from ref. 135, Copyright 2011, with permission from Elsevier.) P2-NaxVO2: (c) the limits of the biphasic domains and the solid solutions (d) in situ X-ray diffraction data recorded during the galvanostatic intermittent titration technique experiments. (Reproduced by permission from ref. 136, Nature Publishing Group, Copyright 2013.) | ||
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m without ordering of the superstructure, unlike Li2MnO3. The first discharge capacity was approximately 150 mA h g−1 at the first cycle and this capacity was maintained throughout the cycling test. For Na+ insertion and extraction, the electrochemical reaction proceeded via a two-phase reaction with hex-I and hex-II phases. Although further studies are needed to elucidate the structural evolution during Na+ insertion and extraction, the use of layered Na2MO3 is an important approach for exploring new transition metal oxides.
m without ordering of the superstructure, unlike Li2MnO3. The first discharge capacity was approximately 150 mA h g−1 at the first cycle and this capacity was maintained throughout the cycling test. For Na+ insertion and extraction, the electrochemical reaction proceeded via a two-phase reaction with hex-I and hex-II phases. Although further studies are needed to elucidate the structural evolution during Na+ insertion and extraction, the use of layered Na2MO3 is an important approach for exploring new transition metal oxides.
2.2. Two- or three-dimensional layer transition metal oxides and fluorides
Na-deficiency or Na-free transition metal oxides are interesting because of their reversible Na+ insertion and extraction. These compounds usually have an open structure to allow Na+ ions into their crystal structure. Particularly in three-dimensional structures, Na+ ions can diffuse in x, y, and z directions; fast Na+ migration is possible relative to two-dimensional structures. Many of these compounds are synthesized at low temperatures, such that their large surface area with small particles contributes to unexpectedly high rate performance. Since the ionic size of Na+ is larger than that of Li+, the host structure should be sufficiently rigid or have a large tunnel size to facilitate the entry of large ionic species. Otherwise, the crystal structure could collapse upon repetitive Na+ insertion and extraction. These electrodes are active compared to Na metal; however, the main difficulty is that a sodiated anode is needed to construct a full cell. Recent research introduced low voltage operating P2-Na0.62[Ti0.37Cr0.63]O2 anode materials.138 Plenty of possibilities remain regarding the development of full cells utilizing these transition metal oxides and sodiated anode materials.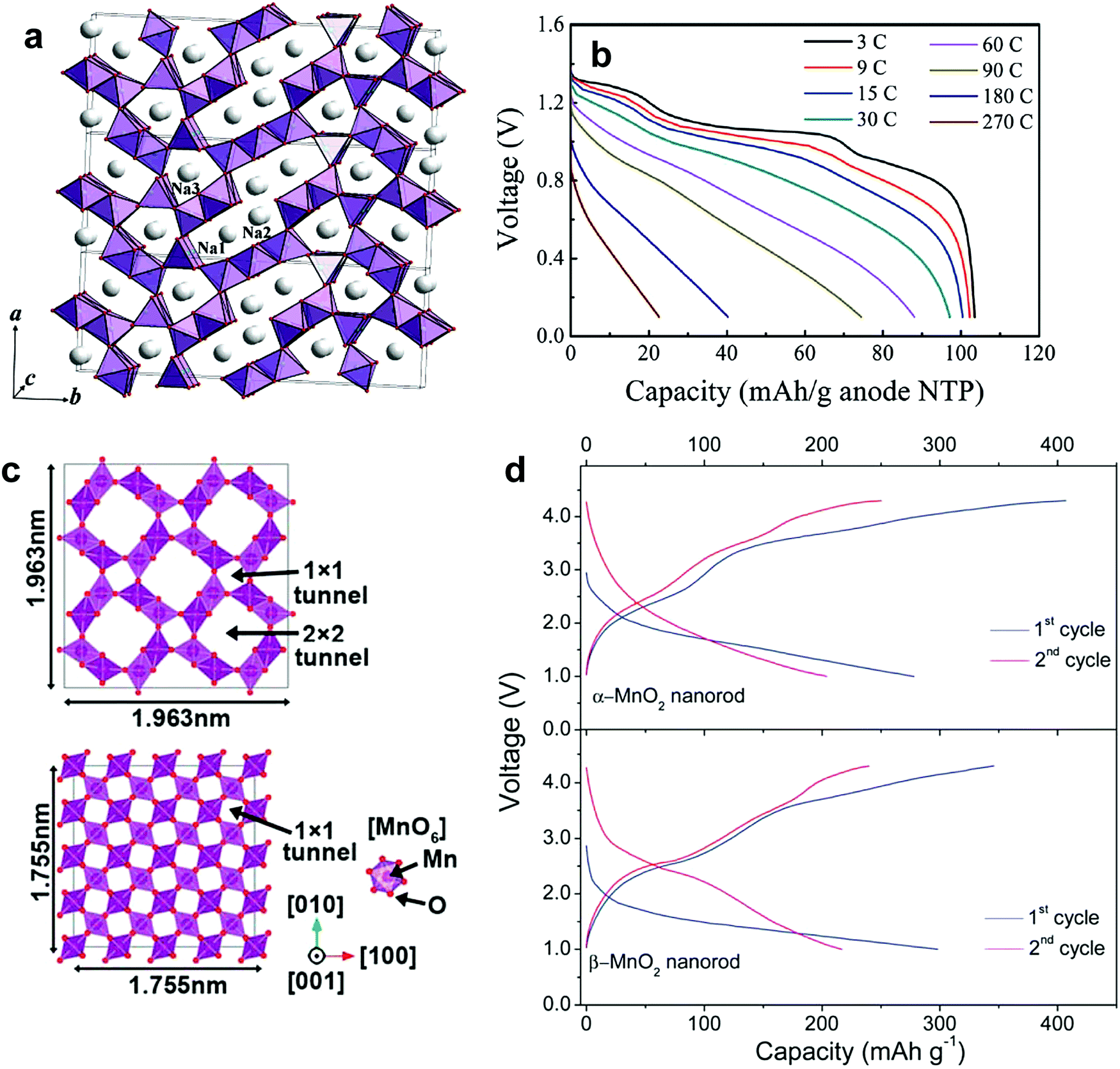 | ||
| Fig. 12 (a) The crystal structure of Na0.44MnO2 perpendicular to the ab–plane. (Reproduced with permission from ref. 141, Copyright 2007 American Chemical Society.) (b) Discharge curves of NTP-C/NMO full cells in two-electrode configuration at different C-rates. (Reproduced from ref. 145 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) (c) The crystal structure of α-MnO2 along the [001] direction (top) and β-MnO2 along the [001] direction (bottom) and (d) the resulting initial charge and discharge curves of α-MnO2 (top) and β-MnO2 (bottom). (Reproduced with permission from ref. 146, Copyright 2013 The Royal Society of Chemistry.) | ||
The application of Na0.44MnO2 electrodes has also been highlighted in aqueous systems. Whitcare et al. documented a full cell with a Na0.44MnO2 cathode and an activated carbon anode in a 1 M NaSO4 aqueous electrolyte.144 Despite a smaller capacity in the aqueous solution relative to the aprotic ones, the full cell could be tested without apparent capacity loss over 1000 cycles. In the NaTi2(PO4)/Na0.44MnO2 system, the cell was capable of operation at over 100C-rates and stable cycling for over 1000 cycles (Fig. 12b).145
Na0.4MnO2 consists of a 2 × 3 tunnel structure called romanechite.89 Ba+ and water molecules have been introduced to stabilize the large tunnel structure. Although the Na+ insertion and extraction mechanism remains unclear, 0.3 mol of the Na+ ion could be inserted into the host structure.
Few studies have explored Na-free MnO2 for Na+ insertion. Because of the relatively large tunnel size of α- and β-MnO2, Na+ insertion is also possible for both α- and β-MnO2. α-MnO2, which is called hollandite, is composed of double chains of edge-sharing MnO6 octahedra that are linked at the corners to form a 2 × 2 and 1 × 1 tunnel structure (Fig. 12c). Because of the large size of the 2 × 2 tunnel, Na+ insertion into the empty tunnel is possible, as suggested by Su et al.146 and Islam et al.147 (Fig. 12d). A relatively large capacity of approximately 280 mA h g−1 was obtained at the first discharge in α-MnO2 nanorods, whereas the retained capacity was only 75 mA h g−1 after 100 cycles. In contrast, rutile-type β-MnO2 shows a 1D channel 1 × 1 tunnels along the c-axis composed of individual chains of the MnO6 octahedral units. In general, insertion of ion species, i.e., Li+, is not easy due to the small size of the 1 × 1 tunnels.148 Su et al. also tested β-MnO2 in Na cells, which delivers approximately 300 mA h g−1 during the first cycle.146 In comparison with α-MnO2, the β-MnO2 nanorods exhibited a higher discharge capacity of approximately 145 mA h g−1 after 100 cycles. They attributed the better electrochemical performance of β-MnO2 to the large number of empty tunnels that accommodate the Na+ ions. Since both α- and β-MnO2 have a different tunnel structure, both materials should exhibit different charge and discharge behavior.149 Nevertheless, the same voltage profiles showing sloppy discharge curves are more likely to be related to pseudo-capacitance behavior. λ-Type MnO2, which is produced by electrochemical delithiation of LiMn2O4, showed discharge of approximately 200 mA h g−1 during the first cycle with reasonable capacity retention.150 The λ-type MnO2 phase was transformed into O′3 NaMnO2via electrochemical cycling and showed stable cycling performance.
Recently, Tapavcevic et al. reintroduced layered α-V2O5 using a nanostructured bilayer concept, of which the interslab distance was approximately 13.5 Å, which is significantly larger than that of conventional V2O5 showing 4.4 Å.152 This was possible because they used electrochemical deposition from aqueous vanadyl sulfate on a Ni foil substrate followed by heat treatment at 120 °C for removal of water molecules present in the interslabs (Fig. 13a). The electrode was activated by a V5+/4+ redox reaction in a voltage range of 1.5–3.8 V, but showed sloping charge–discharge curves. The electrode delivered a reversible capacity of approximately 250 mA h g−1, which is close to the theoretical capacity, with excellent cyclability of 300 cycles (Fig. 13b).
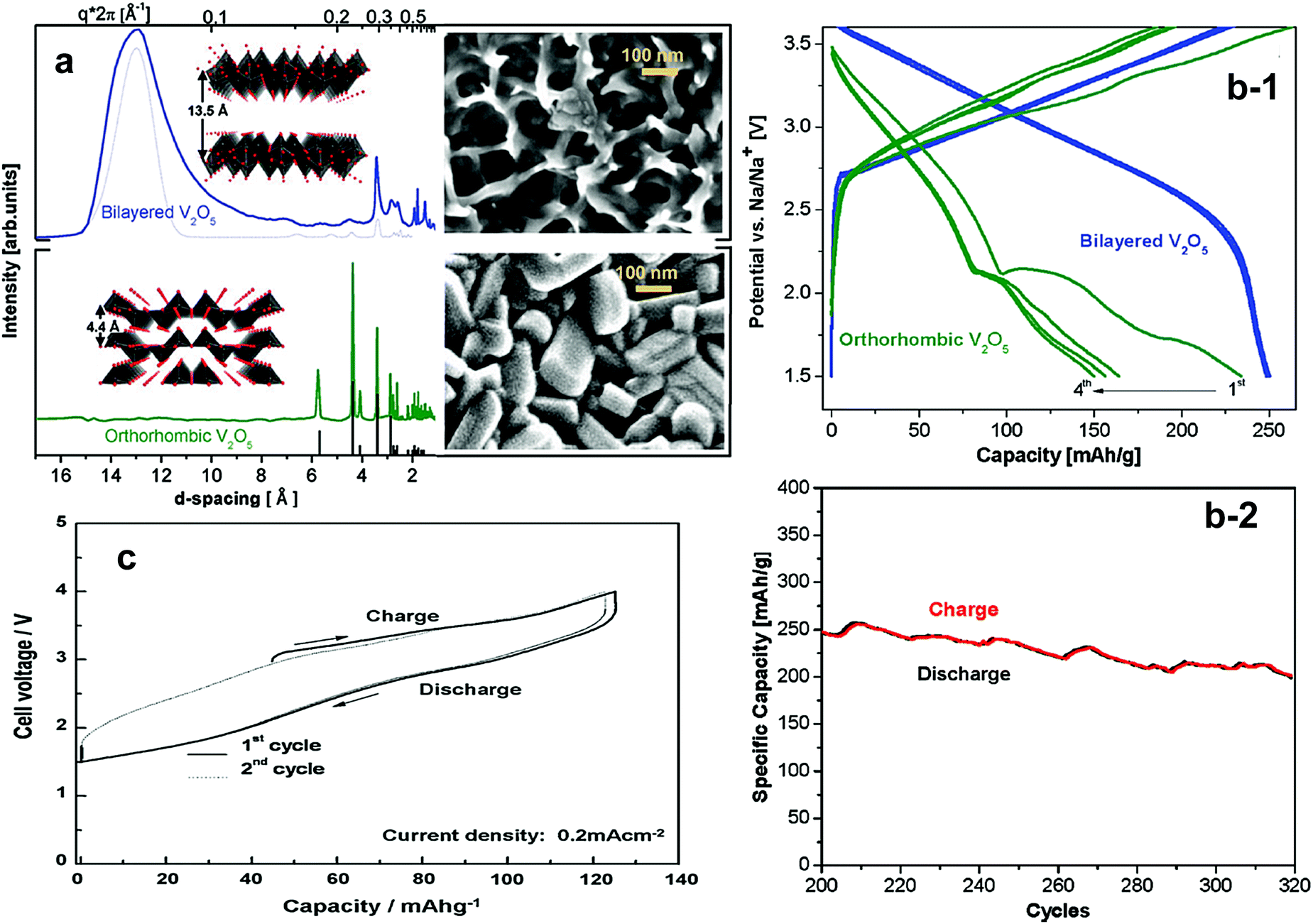 | ||
| Fig. 13 (a) Synchrotron XRD and SEM images of electrodeposited bilayered V2O5 (top) and orthorhombic V2O5 (bottom) and (b-1) first four charge–discharge cycles of bilayered V2O5 and orthorhombic V2O5 and (b-2) the cycle life of bilayered V2O5. (Reproduced with permission from ref. 152, Copyright 2012 American Chemical Society.) (c) Initial charge and discharge curves of NaFeF3 (reprinted from ref. 155, Copyright 2009, with permission from Elsevier.) | ||
Surface modification of VO2(B) using reduced graphene oxide (rGO) substantially improved repetitive Na+ insertion ability.153 Although it has an open bronze structure to accommodate ion species into the empty channels, structural collapse or amorphization was observed in Na cells. The charge and discharge curves were sloppy, delivering approximately 150 mA h g−1, in which the V4+/3+ reaction was related to the electrochemical reaction. Both Na+ insertion into VO2(B) and pseudo-capacitive behavior were confirmed via XRD and XAS studies.
c) have large bottlenecks in diffusion pathways for ion carriers such as Li+ and Na+. Although they have a high theoretical capacity (∼200 mA h g−1), their electrochemical performance is affected by the low electric conductivity of metal fluorides. Success has been achieved through mechanical milling and compositization with nanosized carbons. Okada's group introduced several metal fluorides (metal: Fe, V, Ti, Co, and Mn) including sodiated compounds.154–157 A reversible Fe3+/2+ redox reaction achieved Na+ insertion and extraction, which could deliver 150 mA h g−1 during the first cycle. All MF3 compounds except FeF3 (M: V, Ti, Co, and Mn) exhibited disappointing Na+ storage. They further developed sodiated metal fluoride crystallized into the perovskite structure, NaMF3 (M: Fe, Mn, Ni). Among those materials, NaFeF3 had a relatively a large discharge capacity (128 mA h g−1) with an average cell voltage of 2.7 V (Fig. 13c), while NaNiF3 and NaMnF3 suffered from capacities below 40 mA h g−1 with sloppy voltage decay. Even when the synthesis method was switched from mechano-milling to a solution-based method, the resulting capacity did not exceed the prior report at the same current density (0.2 mA cm−1), but instead reached 180 mA h g−1 at a rate of 0.01C.157 The strong ionic character of the M–F bond must be overcome to achieve a high capacity even at high rates. Homogeneous dispersion of nanosized metal fluoride materials onto a conducting carbon matrix might facilitate high rate performance.
2.3. Three-dimensional polyanion compounds
In comparison with oxide and fluoride systems, transition metal polyanion materials have shown significant thermal stability, which is supported by the presence of covalent bonds such as P and O, in particular cathodes are in a deeply-charged (oxidized) state. Oxygen evolution is common in layered compounds at temperatures above 200 °C;51,85,158–162 however, such behavior is dramatically suppressed by the presence of P–O covalent bonds in the crystal structure. These phenomena prevail in Li cells and also apply to Na systems, since the related material chemistry during electrochemical reactions does not vary significantly from Li systems with the exception of the charge carrier, Na+, in Na cells. Polyanion-based materials usually exhibit lower electric conductivity relative to oxides, such that surface modifications using electro-conducting carbons, which contribute to a dramatic increase in electrical conductivity, improve electrochemical performance. The basic form begins from NaFePO4; interestingly, some factors such as (i) variation in the charge carrier number of Na in Na sites, (ii) partial or full replacement of Fe by the other transition metals, (iii) a mixed anion system with F, OH, CO2, and (iv) extension towards mixed phosphate (PO4)3− and pyrophosphate (P2O7)4− ions also stabilize the crystal structure in the Na system. Unfortunately, because moisture absorption occurs easily in these polyanion systems, avoiding hydration and formation of NaOH on the surfaces of particles can be a challenge. Heterogeneous surfaces may lead to misinterpretation of the electrode performance in Na-containing aprotic electrolytes.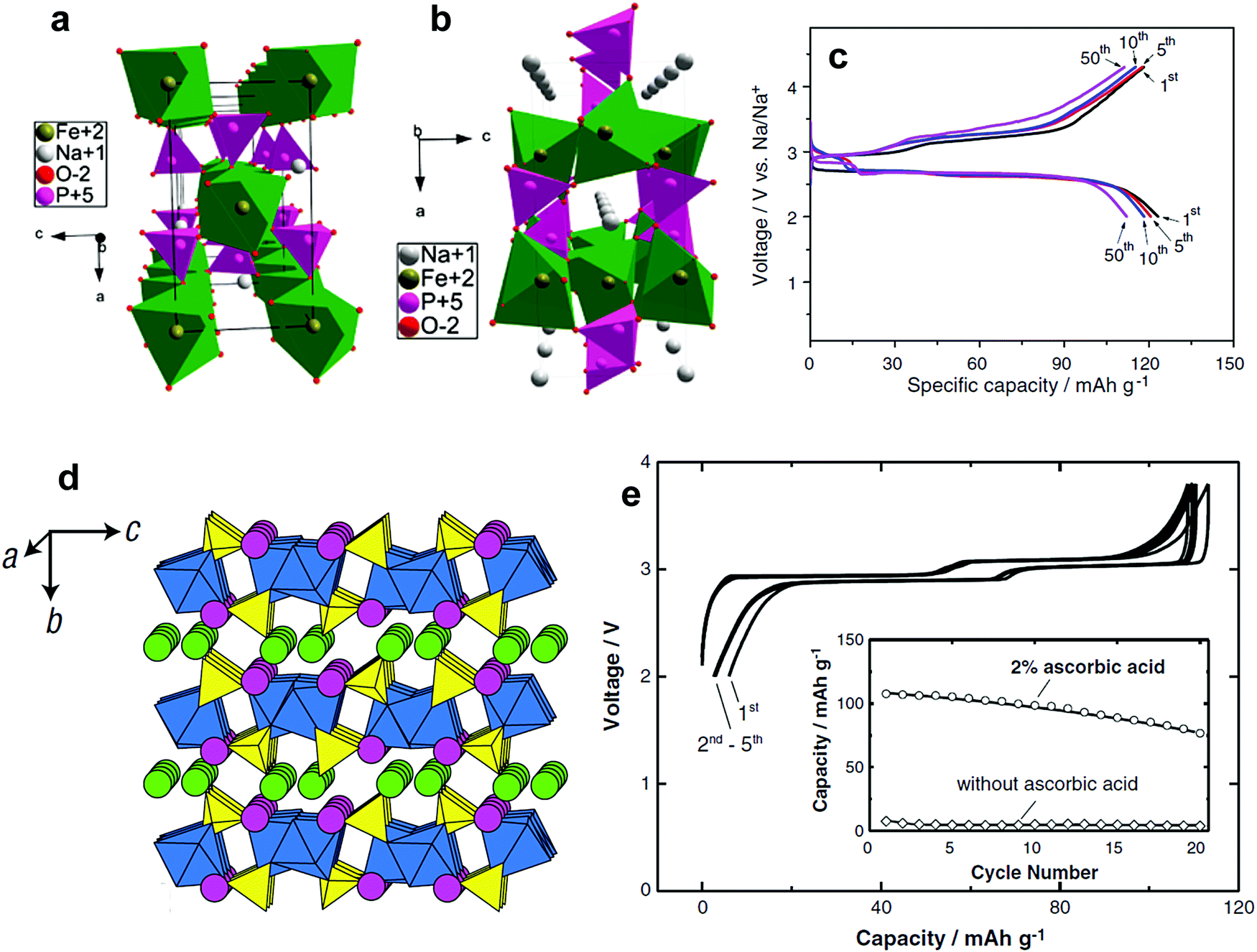 | ||
| Fig. 14 (a) The crystal structure of maricite NaFePO4 and (b) olivine NaFePO4. (Reproduced with permission from ref. 164, Copyright 2010 American Chemical Society.) (c) Charge–discharge profiles of NaFePO4 for 50 cycles. (Reprinted from ref. 169, Copyright 2012, with permission from Elsevier.) (d) Structure of Na2FePO4F along the [100] direction, iron octahedra: blue and the phosphate tetrahedral: yellow, and Na (1) in green and Na (2) in pink. (Reproduced by permission from ref. 176, Nature Publishing Group, Copyright 2007.) (e) Charge/discharge curves of the Na2FePO4F cell cycled at a rate of 6.2 mA g−1. (Reprinted from ref. 178, Copyright 2012, with permission from Elsevier.) | ||
Electrode performance is dependent on the crystal structure. Amorphous NaFePO4 exhibited a high discharge capacity of ∼150 mA h g−1, but operated at a low voltage (2.4 V) with a sloping discharge profile.165 Maricite NaFePO4 is electrochemically inactive, as the structure blocks Na+ extraction and insertion.166 Olivine NaFePO4 shows a relatively high operating voltage (2.8 V) with two distinct voltage plateaus, delivering over 120 mA h g−1 capacity (Fig. 14c).163–173 During the electrochemical reaction, the oxidation state of Fe is systematically altered followed by a redox reaction of Fe3+/2+, as confirmed by the XAS studies performed by Ali et al.173 Carbon coating led to excellent long-term cyclability. An interesting feature of triphylite NaFePO4 is the appearance of intermediate Na0.7FePO4, which has the same crystal structure as the triphylite NaFePO4, during electrochemical testing, whereas this phase is not found in LiFePO4. Moreau et al. suggested that the advent of Na0.7FePO4 could be related to cationic ordering in the crystal structure.164 Similar behavior is also observed in NaxCoO262 in comparison with LixCoO2.174 Casas-Cabanas et al. demonstrated that Na+ ion insertion into FePO4 occurs via the intermediate phases due to large volume mismatches between FePO4 and NaFePO4.170 Therefore, three phases, FePO4, Na0.7FePO4, and NaFePO4, appear simultaneously on discharge, while they are separated into two first-order phase transitions on charge, which may be related to Na+/vacancy ordering in the crystal structure. In comparison with LiFePO4, the charge transfer resistance of NaFePO4 is high, and the diffusion coefficient of Na+ ions is approximately 2 orders of magnitude lower.175 Recent density functional theory calculation revealed that the migration energy of Na+ ions in NaFePO4 is 0.05 eV higher than that of Li+ ions in LiFePO4, which reflects the slow kinetics in NaFePO4.172 The large ion size of Na+ relative to Li+ is associated with slow diffusion of Na+ ions into the crystal structure. An extension of NaFePO4, Na[Fe0.5Mn0.5]PO4, could be synthesized;168 however, the electrode did not show voltage plateaus, but rather a sloping curve due to the high interface strain between the Na-rich and Na-poor phases that was ascribed to the large size of Na+ ions, allowing 0.6 mol Na+ into Nax[Fe0.5Mn0.5]PO4. Since large Na+ ions are associated with migration, minimization of the interface energy is essential to the utilization of the other transition metals such as Co, Mn, and Ni, among others.
The ionicity of fluorides over oxides and sulfides is ascribed to their higher electronegativity, which thus increases the operating voltage of the electrodes. This concept was successfully applied to the discovery of Na2FePO4F, which is applicable to both Li and Na systems.176 This material crystallizes in orthorhombic structures with a Pbcn space group (Fig. 14d). Bioctahedral Fe2O7F2 units comprising face-sharing FeO4F2 octahedra are connected via bridging F atoms to form a chain, and are joined by PO4 tetrahedra to form FePO4F layers. The two Na cations located in the interlayer enable facile two-dimensional migration pathways. However, structural evolution from the layer structure to a tunnel one occurred (space group P21/n) when Fe was substituted for Mn greater than 25%. In the case of the tunnel structure, the Na cations are located within channels.177 Notwithstanding the advantages of high operating voltage relative to NaFePO4, the strong ionicity lowers the intrinsic electric conductivity. Hence, the bare Na2FePO4F delivered almost no capacity. Surprisingly, the carbon-coated Na2FePO4F could deliver approximately 110 mA h g−1, which corresponds to a 90% theoretical capacity.178 Two voltage plateaus are evident with small polarization. As anticipated from the crystal structure, Tripathi et al. predicted Na+ ion conduction in Na2FePO4F, which occurs along the two-dimensional interlayer with low activation energy.179 However, further investigation is needed to improve cycling performance by adding more carbon to provide better electron conduction. Carbon-coated Na2[Fe0.5Mn0.5]PO4F (6 wt% of carbon) also showed a reversible capacity of 110 mA h g−1, confirming the feasibility of Na+ ion migration into/out of the tunnel structure.180 Polarization of the Mn redox is clearly greater than that of the Fe2+/3+ redox, which is similar to those observed in LiFePO4 and LiMnPO4.
In place of divalent transition metal elements, a trivalent element is also available in the form of NaVPO4F. Barker et al. first introduced the material as an electrode material for hybrid-ion cells in 2003.181 During the initial charging, Na+ ions are extracted from the host material while Na+ ions are plated on the surface of the Li metal anode in a Li cell. This process releases an atomically equivalent amount of lithium back into the electrolyte. Therefore, Li+ ions are reinserted into the host material during the first discharge because Na is immobilized on the Li metal surface. NaVPO4F has tetragonal symmetry with a I4/mmm space group, which is an isostructure of α-Na3Fe2(PO4)2F3.182 This structure demonstrates facile Na+ ion diffusion through an extended three-dimensional framework constructed from VO4F2 octahedra and PO4 tetrahedra (Fig. 15a). Na+ ions are present in the empty channel and diffuse along the c-axis. Coupling with a hard carbon anode, NaVPO4F, led to two-step voltage responses (Fig. 15b), while the delivered discharge declined to less than 50% of the initial capacity (approximately 82 mA h g−1). Zhao et al. found another crystal system in this compound: monoclinic (C2/c) as the low temperature form and tetragonal (I4/mmm) as the high temperature form.183 Cr-doping into the V site of NaVPO4F crystallized to the monoclinic phase showed a discharge capacity of 80 mA h g−1 with 91% retention for 20 cycles.184,185 Sauvage et al. reinvestigated Na–V–P–O–F materials because the structural parallelism between NaVPO4F and Na3Al2(PO4)2F phases was not straightforward owing to the difficulties encountered in determining the stoichiometry.186 They found that a 10 wt% excess of NaF could yield a single phase. Reliable refinement results were obtained assuming a crystal structure of Na3V2(PO4)2F, which can be presented as Na1.5VOPO4F0.5 instead of NaVPO4F. In this structure, Na+ ions are disordered across two sites (8h and 8j) and the oxidation state of V is 4+ (Fig. 15c). The crystal structure is composed of a VO5F octahedral and PO4 tetrahedra sharing O vertices parallel to the ab-plane. Along the c-axis direction, VO5F octahedra are connected via F vertices located in the same ab-plane as the disordered Na atoms. This structure could accommodate 0.56 Na+ (approximately 87 mA h g−1) per formula unit in Na1.5VOPO4F0.5 with two different voltage plateaus, 3.6 and 4 V (Fig. 15d). In consideration of the theoretical value (156 mA h g−1) assuming a V4+/5+ reaction, the delivered capacity can be improved further with electro-conducting coating layers. Due to structural and electrolytic instability, the capacity retention in the upper voltage region is not promising compared to the capacity delivered at the lower voltage plateau. These results, including capacity and retention, agree with an earlier report by Barker et al.181 The oxidation state of V was lowered to 3.8+ in Na1.5VPO4.8F0.7.187 In comparison with Na1.5VOPO4F0.5,188 the oxygen content was reduced slightly, but the equivalent content of F increased. Although structural refinement was performed based on space group P42/mnm, the XRD pattern, including the Bragg peak position, is the same for both compositions. Na1.5VPO4.8F0.7 is comprised of tetrahedral PO4 and VO5F/VO4F2 octahedral units that share corners, which yields a three-dimensional open framework with Na+ ions located at interstitial sites (Fig. 15e). Na1.5VPO4.8F0.7 provided a discharge capacity of approximately 137 mA h g−1 based on the V3.8+/5+ redox reaction (1.2 mol Na+ per formula unit in Na1.5VPO4.8F0.7) with excellent cyclability for 500 cycles (Fig. 15f).
 | ||
| Fig. 15 (a) The crystal structure of α-Na3Al2(PO4)2F3 along the a-axis. (Reprinted from ref. 182, Copyright 1999, with permission from Elsevier.) (b) Electrochemical performance data for a typical hard carbon//NaVPO4F cell. (Reproduced with permission from ref. 181, Copyright 2003 The Electrochemical Society.) (c) The crystal structure of Na1.5VOPO4F0.5 in the (ac) plane and (d) the P.I.T.T. curve of the composite Na1.5VOPO4F0.5/C recorded at a 1Na/100 h rate. (Reprinted from ref. 186, Copyright 2006, with permission from Elsevier.) (e) The crystal structure of Na1.5VPO4.8F0.7 and two different local environments for V4+ and V3+ ions (i.e., VO5F and VO4F2 octahedra, respectively): Na (yellow), V cyan), P (purple), O (red), and F (green) atoms and Na layers denoted as dashed boxes and (f) charge–discharge profile at a C/10 rate for the Na1.5VPO4.8F0.7 cathode (voltage window: 2.0–4.5 V vs. Na+/Na) appeared with the average voltage (3.8 V vs. Na+/Na) as a horizontal dotted line. (Reproduced with permission from ref. 187, Copyright 2013 American Chemical Society.) (g) Synchrotron radiation diffraction data in the P42/mnm space group (top) and in the low-symmetry orthorhombic group Amam (bottom). (Reproduced with permission from ref. 191, Copyright 2014 American Chemical Society.) and (h) first charge and discharge curves for Na3V2(PO4)2F3 a rate of 0.2 mA cm−2. (Reprinted from ref. 190, Copyright 2013, with permission from Elsevier.) | ||
To further lower the average oxidation state of V efforts were made employing Na3V2(PO4)3F3 with space group P42/mnm.188–190 Very recently, Bianchini et al. revisited the Na3V2(PO4)3F3 compound to clarify its crystal structure, although the structure was successfully refined by Le Mine et al. in 1988.191 They claimed that structural discrepancies in the literature originated from the possible existence of F and O mixing in the structure.182,188–190 A high-resolution diffraction study using synchrotron radiation revealed a suitable orthorhombic structure with space group Amam rather than a tetragonal structure with space group P42/mnm (Fig. 15g). Interestingly, when F was absent, the structure was crystallized to NASICON-type rhombohedral Na3V2(PO4)3 with space group Rc, which will be addressed in Section 2.3.4. Similar to Na1.5VOPO4F0.5186 and Na1.5VPO4.8F0.7,187 the presence of fluorine with phosphorus ions raises the operating voltage originating from the strong inductive effects of the anion group, even though the V3+/4+ redox couple is available for delivery of high capacity (approximately 120 mA h g−1) in Na3V2(PO4)3F3 compounds, with two voltage plateaus at 3.7 and 4.2 V (Fig. 15h). Computational calculations revealed that Na+ ions located at the Na2 site are more readily extracted due to the lower diffusion barrier. Compared with two earlier reports by Barker et al.181 and Sauvage et al.,186 this work is notable because variation in the anion configuration and the content of oxygen fluorine dramatically improved electrochemical performance, even at high rates.
In 2008, Adam et al.193 introduced the crystal structure of Li2MnP2O7, and it was confirmed using Li+ intercalation cathode materials.194,195 Recent developments in SIBs have motivated the exploration of facile Na+ intercalation materials towards pyrophosphates. Na2FeP2O7 with triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) was first studied by Honma et al.196 and Barpanda et al.,197 and the crystal structure was stabilized into the triclinic P phase (Fig. 16a). The structure was comprised of corner-sharing FeO6 octahedra creating Fe2O11 dimers, which are interconnected via corner-sharing and edge-sharing with P2O7 pyrophosphate groups. FeO6 octahedra and PO4 tetrahedra are connected in a staggered fashion, thus creating large tunnels along the [110] direction, which accommodate Na atoms at four distinct sites. However, these materials are not competitive cathode materials because of the two phosphate groups per transition metal in terms of capacity (theoretical capacity approaching 97 mAh g−1) even though the related material chemistries are interesting. The appearance of stepwise voltage plateaus (roughly at 2.5 and 3 V) may indicate the presence of Na+/vacancy ordering in the structure (Fig. 16b). An ex situ study suggested that a two-phase reaction prevails in the upper voltage plateau and a single phase reaction is dominant in the lower voltage region.198 According to calculation by Clark et al.,199 low activation energy was found for long-range diffusion in all crystallographic directions in Na2FeP2O7 (M: Fe and Mn), indicating three-dimensional Na+ ion diffusion. Therefore, electrochemical studies indicated moderate performance at high rates, which is related to the availability of three-dimensionality and low migration energy for Na+ ion diffusion in Na2FeP2O7. The good electrode performances of Na2FeP2O7 were further evidenced in an inorganic ionic liquid NaFSA-KFSA (FSA: bis(fluorosulfonyl)amide) at 363 K.200
was first studied by Honma et al.196 and Barpanda et al.,197 and the crystal structure was stabilized into the triclinic P phase (Fig. 16a). The structure was comprised of corner-sharing FeO6 octahedra creating Fe2O11 dimers, which are interconnected via corner-sharing and edge-sharing with P2O7 pyrophosphate groups. FeO6 octahedra and PO4 tetrahedra are connected in a staggered fashion, thus creating large tunnels along the [110] direction, which accommodate Na atoms at four distinct sites. However, these materials are not competitive cathode materials because of the two phosphate groups per transition metal in terms of capacity (theoretical capacity approaching 97 mAh g−1) even though the related material chemistries are interesting. The appearance of stepwise voltage plateaus (roughly at 2.5 and 3 V) may indicate the presence of Na+/vacancy ordering in the structure (Fig. 16b). An ex situ study suggested that a two-phase reaction prevails in the upper voltage plateau and a single phase reaction is dominant in the lower voltage region.198 According to calculation by Clark et al.,199 low activation energy was found for long-range diffusion in all crystallographic directions in Na2FeP2O7 (M: Fe and Mn), indicating three-dimensional Na+ ion diffusion. Therefore, electrochemical studies indicated moderate performance at high rates, which is related to the availability of three-dimensionality and low migration energy for Na+ ion diffusion in Na2FeP2O7. The good electrode performances of Na2FeP2O7 were further evidenced in an inorganic ionic liquid NaFSA-KFSA (FSA: bis(fluorosulfonyl)amide) at 363 K.200
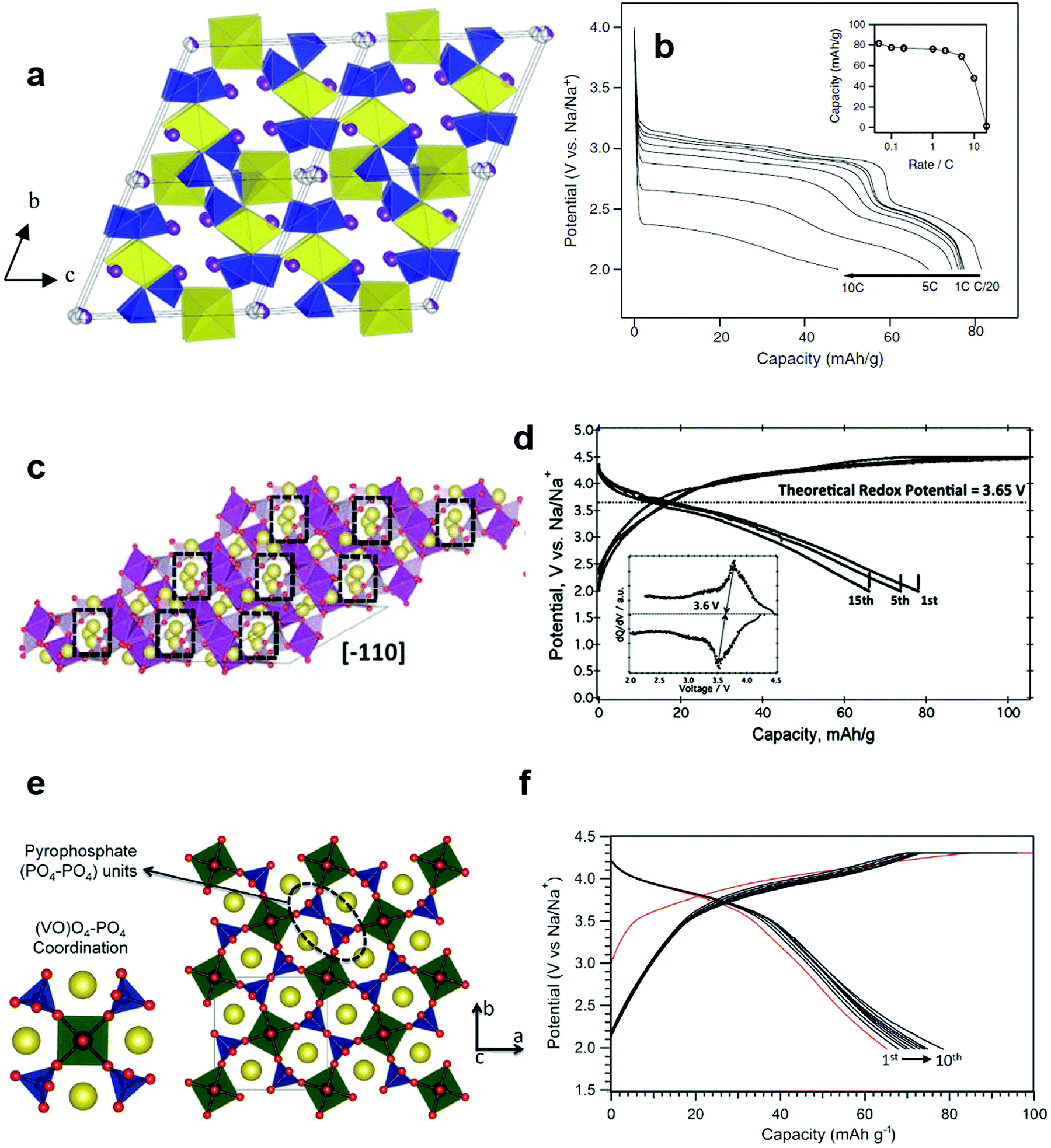 | ||
| Fig. 16 (a) The crystal structure of Na2MP2O7. (Reproduced with permission from ref. 199, Copyright 2014 The Royal Society of Chemistry.) (b) Discharge capacity of Na2FeP2O7 as various current densities. (Reprinted from ref. 197, Copyright 2012, with permission from Elsevier.) (c) The crystal structure of β-Na2MnP2O7 polymorph consisting of MnO6 octahedra (pink), PO4 tetrahedra (light purple) and Na atoms (yellow) along [−110] projection in which the possible sodium migration tunnels are highlighted by black dotted rectangles and (d) charge and discharge profiles of the β-Na2MnP2O7 cathode at a rate of C/20. (Reproduced with permission from ref. 202, Copyright 2013 The Royal Society of Chemistry.) (e) The crystal structure of Na2(VO)P2O7: corner-sharing between (VO)O4 and PO4 units (top) and the side and top view of the (VO)O4 square pyramid (bottom) and (f) the resulting charge and discharge curves of Na2(VO)P2O7 at a rate of C/20. (Reproduced from ref. 203 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Ha et al. extended the composition to Na3.32Fe2.34(P2O7)2, which can be represented as Na1.66Fe1.17P2O7, of which the crystal structure is identical to Na2FeP2O7, but the new composition is able to increase the theoretical capacity to approximately 110 mA h g−1.201 The first irreversible capacity was negligible and the lower voltage was shortened. The electrode performance was superior to that of Na2FeP2O7 in terms of capacity retention and rate capability up to a rate of 10C. However, Na3.32[Fe0.5Mn0.5]2.34(P2O7)2 and Na3.32Mn2.34(P2O7)2 had very low capacities, which was a controversial result of the computational study of Clark et al.199 The poor electrochemical activity seems to be similar to that of LiMnPO4, which requires strategies for nanosizing and doping with the other divalent elements to improve electric conductivity.
Recent work of Barpanda et al. identified a new polymorph of β-Na2MnP2O7, which was also stabilized into a triclinic structure, P.202 The structure consists of distorted MnO6 octahedral and PO4 tetrahedral blocks creating tunnels accommodating Na atoms along the [001] direction. The structure has corner-sharing MnO6–MnO6 (Mn2O11) dimers, which are in turn connected by PO4–PO4 (P2O7) diphosphate units in a mixed-edge and corner-sharing fashion (Fig. 16c). A galvanostatic test of the electrode provides a reversible discharge capacity of 80 mA h g−1 associated with an average operating voltage of 3.6 V (Fig. 16d). This coincided with earlier DFT calculations suggesting that this compound can undergo a Mn2+/3+ redox reaction at around 3.65 V.199 The sloppy discharge curve is the signature of β-Na2MnP2O7, which is different from Na2FeP2O7 operating with two distinct voltage plateaus. Structural investigation is further required to elucidate the reaction mechanism. They also suggest a new pyrophosphate oxyanionic framework compound, tetragonal Na2(VO)P2O7 with space group P4bm.202 This material belongs to the fresnoite family consisting of a VO5 or (VO)O4 square pyramid and PO4 tetrahedral units (Fig. 16e). Each VO5 pyramid is connected to four independent PO tetrahedra in a corner-sharing fashion, thus forming [VP2O11] units. Also, each PO4 tetrahedron is connected to two VO5 units and one PO4 unit that share corners. The delivered capacity was approximately 80 mA h g−1, which is close to the theoretical capacity (93 mA h g−1) (Fig. 16f). The discharge curve was sloppy with the V5+/4+ redox reaction showing an average operating voltage of 3.8 V. Apart from the above-mentioned compounds, the other pyrophosphates with Ni(II), Cu(II), Ti(II), Co(II) and higher transition metal oxidation states have not yet been explored as host materials for Na+ insertion.
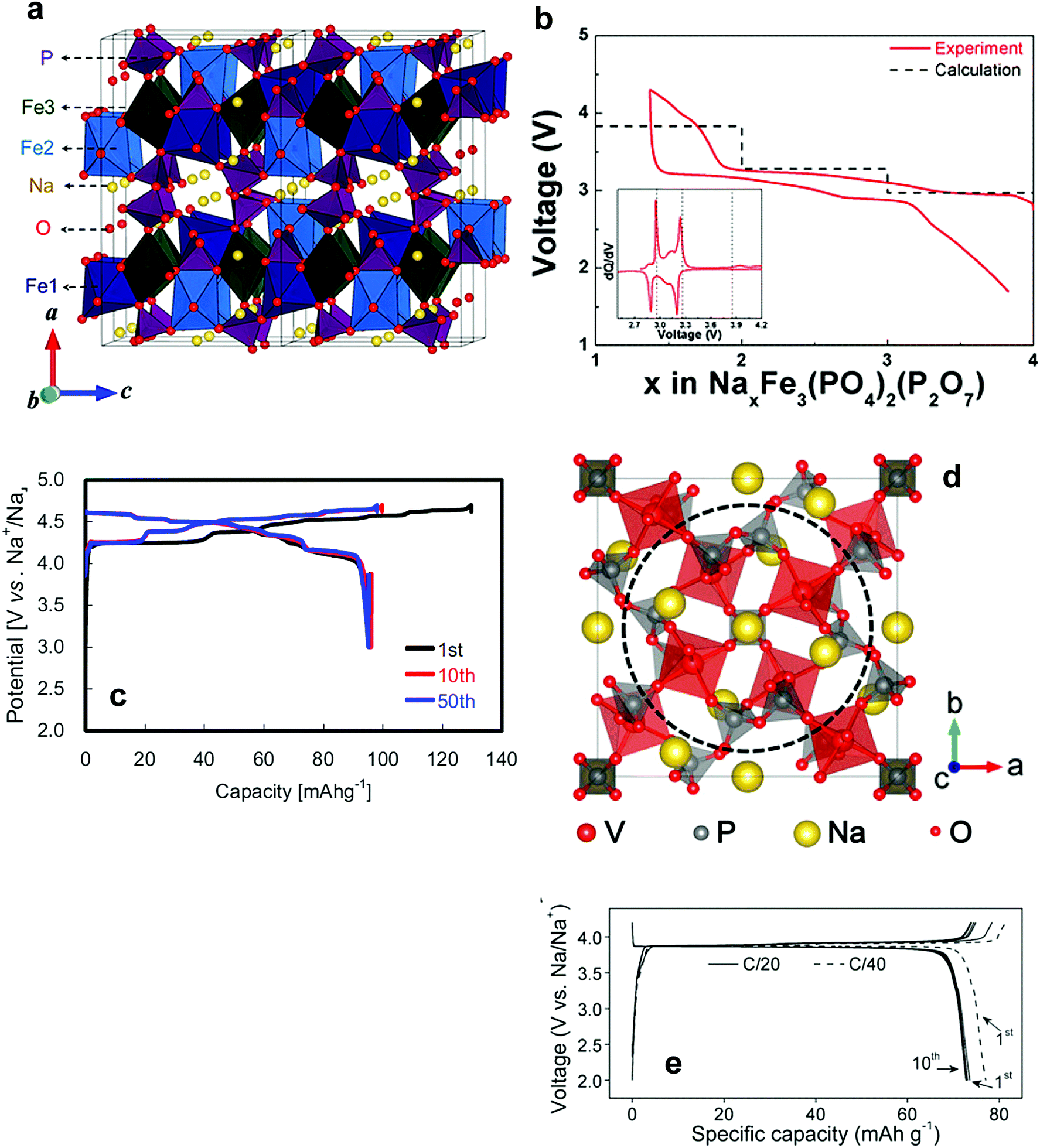 | ||
| Fig. 17 (a) Schematic representation of Na4Fe3(PO4)2(P2O7) and (b) galvanostatic charge/discharge profiles of Na4Fe3(PO4)2(P2O7) under a C/40 rate and the calculated average voltage at each region, where the inset shows the dQ/dV curve of initial charge/discharge profiles. (Reproduced with permission from ref. 205, Copyright 2012 American Chemical Society.) (c) Galvanostatic charge/discharge curves at 1st, 10th and 50th cycles of Na4Co3(PO4)2P2O7. (Reprinted from ref. 206, Copyright 2012, with permission from Elsevier.) (d) The projected crystal structure along the c-axis: gray tetrahedrons, PO4; red octahedrons, VO6; yellow spheres, Na atoms. The (VP2O7)4PO4 unit is denoted as a dashed circle and (e) galvanostatic profiles of Na7V4(P2O7)4PO4 measured at C/20 and C/40 in the first cycles. (Reproduced with permission from ref. 208, Copyright 2014 PNAS.) | ||
Another interesting compound, Na7V4(P2O7)PO4, was suggested by Lim et al.208 This material has a crystallized tetragonal structure with a P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c space group. The crystal structure has a basic unit, (VP2O7)4PO4, which is composed of a central tetrahedron [PO4] that shares corners with four [VO6] octahedra, and each diphosphate group [P2O7] bridges the two adjacent [VO6] octahedra with shared corners. The interconnected (VP2O7)4PO4 units comprise a three-dimensional channel for Na+ ion diffusion (Fig. 17d). Based on the V3+/4+ redox reaction, Na7V4(P2O7)PO4 exhibited a flat plateau at 3.88 V associated with a biphasic reaction governed by the first-order transition, delivering approximately 90 mA h g−1 (Fig. 17e). Addition of rGO further extended its cycle life to 1000 cycles with 78% capacity retention.
21c space group. The crystal structure has a basic unit, (VP2O7)4PO4, which is composed of a central tetrahedron [PO4] that shares corners with four [VO6] octahedra, and each diphosphate group [P2O7] bridges the two adjacent [VO6] octahedra with shared corners. The interconnected (VP2O7)4PO4 units comprise a three-dimensional channel for Na+ ion diffusion (Fig. 17d). Based on the V3+/4+ redox reaction, Na7V4(P2O7)PO4 exhibited a flat plateau at 3.88 V associated with a biphasic reaction governed by the first-order transition, delivering approximately 90 mA h g−1 (Fig. 17e). Addition of rGO further extended its cycle life to 1000 cycles with 78% capacity retention.
c space group, has a NASICON-type framework built up from isolated VO6 octahedra and PO4 tetrahedral units interlinked via corners to form a framework anion, [V2(PO4)3]3−. Two different Na atoms are located in the empty channels of the framework with two different oxygen environments (Fig. 18a).209 Early work by Gopalakrishnan et al. demonstrated the possibility of Na+ extraction to form V2(PO4)3 using chlorine in CHCl2 at room temperature.210 Electrochemical Na+ extraction was investigated by Masquelier211 and Plashnitsa et al.212 and two voltage plateaus were found at 3.4 and 1.6 V. The latter work also tested a full symmetric cell, adopting Na3V2(PO4)3 for both cathodes, but the performance was unsatisfactory. Jian et al. modified the surface of Na3V2(PO4)3 using electro-conducting carbons derived from sugar.209 The Na3V2(PO4)3/C electrode exhibited improved capacity in both the upper and lower voltage regions compared with earlier works (Fig. 18b). They further reduced the irreversible capacity, changing the conventional electrolyte to NaFSI/PC from the PC-based compound. The new combination electrolyte increased the initial Coulombic efficiency above 98%.213 Saravanan et al. employed a solution-based template method using CTAB as a cationic surfactant that left approximately 6 wt% of carbon after synthesis in a reducing environment.214 They demonstrated stable long-term cyclability for over 30000 cycles at a rate of 40C (4.68 A g−1) with superior rate capability (Fig. 18c). Their symmetrical cell also evidenced the cycling stability for over 5000 cycles. Jian et al. further analyzed the crystal structure during the electrochemical reaction.215 As mentioned in Fig. 18a, Na atoms are localized to two different sites (6b, M1 and 18e, M2) in Na3V2(PO4)3. The flat curve during desodiation of Na3V2(PO4)3 indicates a biphasic reaction based on the V3+/4+ redox reaction as a result of Na+ extraction at the M2 site, which is related to the formation of NaV2(PO4), in which only one Na (6b, M1) resides in the crystal structure. NMR further revealed that, in Na3V2(PO4)3, the Na atoms were not randomly distributed at M2 sites, but appeared in an ordered arrangement locally; an Na+ ion at the M2 sites in Na3V2(PO4)3 is mobile, whereas an Na+ ion at the M1 site is immobile, corresponding to 2 mol Na+ per formula unit in Na3V2(PO4)3, while 1 mol Na+ per formula unit in Na3V2(PO4)3 can retain the framework during the electrochemical reaction.
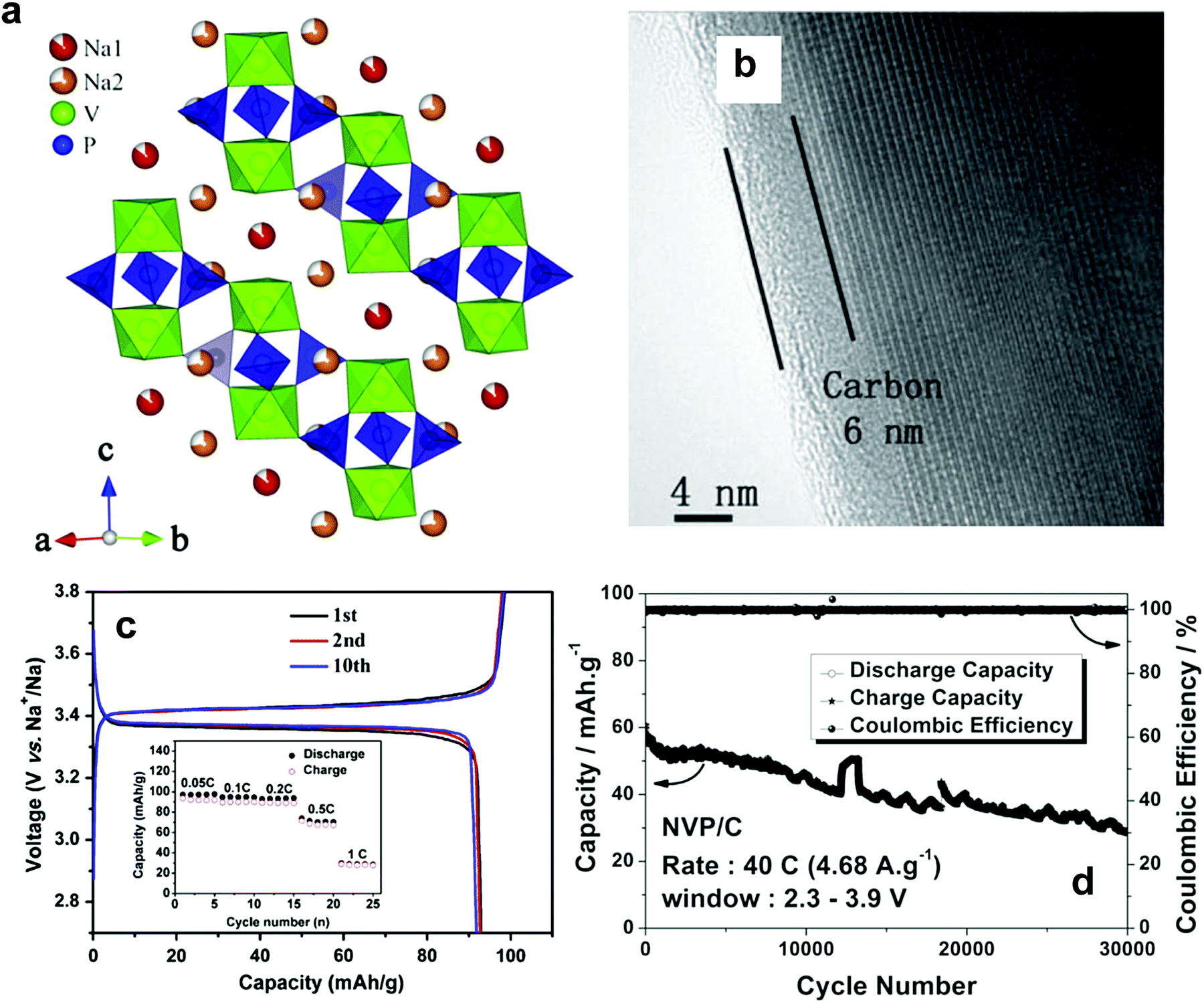 | ||
| Fig. 18 (a) The crystal structure of Na3V2(PO4)3. (Reprinted from ref. 209, Copyright 2012, with permission from Elsevier.) (b) TEM bright-field image of carbon-coated Na3V2(PO4)3. (Reproduced from ref. 213 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) (c) Charge and discharge curves in a voltage range of 1.0 to 3.8 V for carbon-coated Na3V2(PO4)3. (Reprinted from ref. 209, Copyright 2012, with permission from Elsevier.) (d) Long term cycle life and Coulombic efficiency for 30000 cycles at a rate of 40C for carbon-coated Na3V2(PO4)3. (Reproduced from ref. 214 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
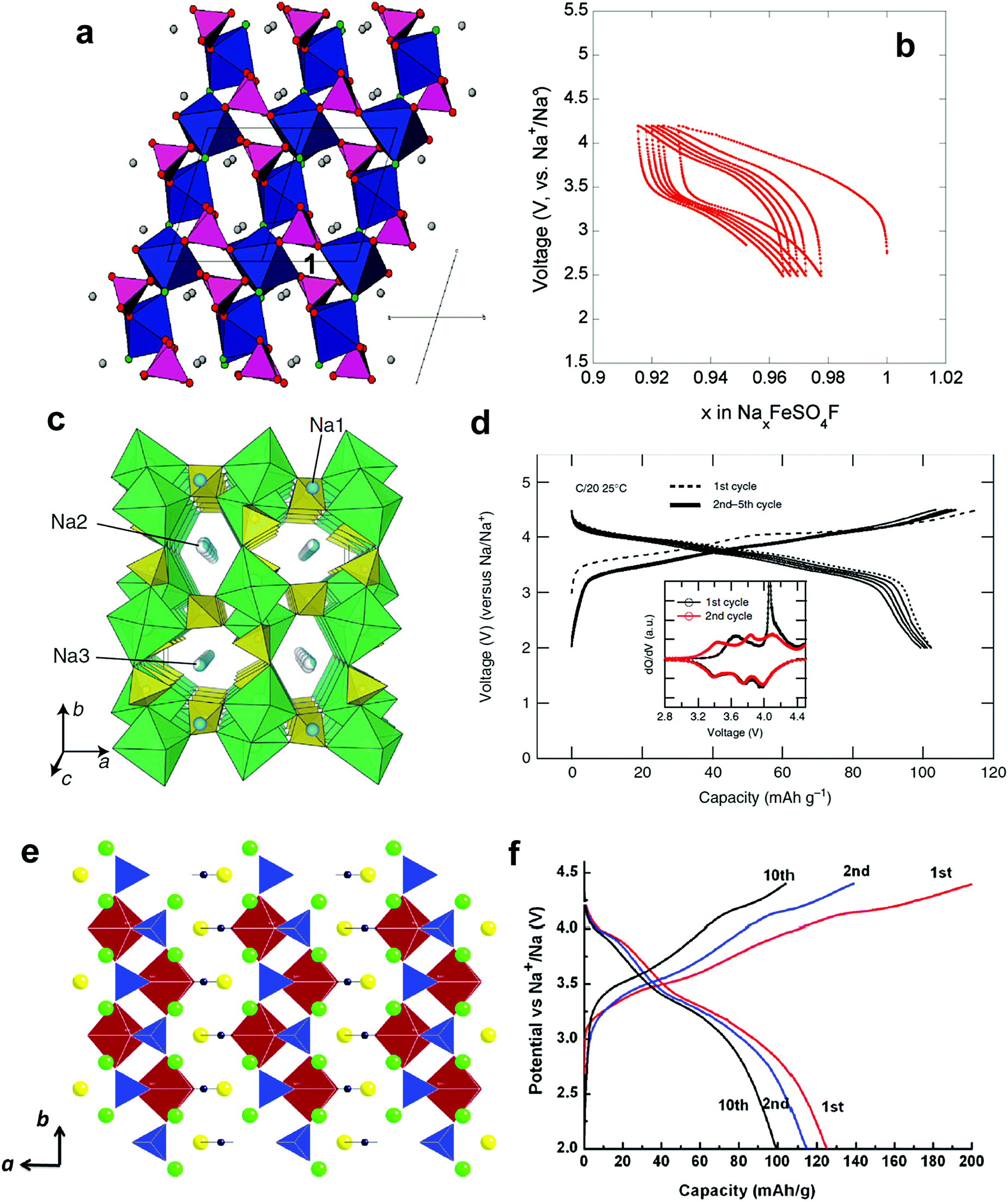 | ||
| Fig. 19 (a) Crystal structure of Monoclinic sodium-based metal fluorosulphates and (b) the electrochemical voltage profile of NaFeSO4F cycled in a sodium cell with a metallic Na anode and a 1 M Na-TFSI:PC electrolyte at a rate of C/20. (Reproduced with permission from ref. 217, Copyright 2010 American Chemical Society.) (c) Crystal structure of Na2Fe2(SO4)3 projected along the c-axis and (d) galvanostatic charging and discharging profiles of the Na2−xFe2(SO4)3 cathode cycled between 2.0 and 4.5 V at a rate of C/20 (2 Na in 20 h) at 25 °C. First (1st) cycle is shown in dashed black line, and 2nd–5th cycle in solid black lines. (inset) The differential galvanostatic profiles (dQ/dV) of the Na2Fe2(SO4)3 cathode. (Reproduced by permission from ref. 219, Nature Publishing Group, Copyright 2014.) (e) The structure of Na3MnPO4CO3 viewed along [001]: Mn octahedra, brown; PO4 tetrahedra, blue; CO3 triangular planar, black; sodium, green (Na1 site) and yellow (Na2site) spheres and (f) the resulting voltage curves of Na3MnPO4CO3 at the first, second, and 10th cycles with a C/100 rate. (Reproduced with permission from ref. 221, Copyright 2013 American Chemical Society.) | ||
Sodiated metal carbonophosphates, such as sidorenkite Na3MPO4CO3 (M: Mg, Mn, Fe, Co, Ni, and Cu), were recently introduced as a new intercalation cathode by Chen et al.220,221 Two different series of (PO4)3- and (CO2)2-ions have bonds with M in Na3MPO4CO3 compounds, which are stable with a sidorenkite structure and a P21/m space group (Fig. 19e). In Na3MnPO4CO3, the MnO6 octahedra are connected by a PO4 tetrahedra to form a double layer. The CO3 group shares an oxygen edge with MnO6. The two-dimensional interslab accommodates two Na atoms at two different interstitial sites, where Na1 and Na2 coordinate with seven and six O atoms, respectively. As anticipated, their computations suggested that the sidorenkite structure may have good intercalation properties.222 Carbophosphate showed high discharge (125 mA h g−1) (Fig. 19f), which represented 66% of the theoretical capacity (191 mA h g−1). It is interesting that Na3MPO4CO3 underwent a topotactic solid solution reaction during cycling, activated by a two-electron electrochemical reaction of Mn2+/3+ and Mn3+/4+ redox couples.
m).229 The double-perovskite ordering of both the anion orientations and the cations introduces a rhombohedral site symmetry along each of the four [111] axes that would stabilize alkali-ion displacement along a cubic [111] axis toward the more octahedral-site complex. The four-fold degeneracy of these displacements keeps the cubic phase at higher temperature and lower Na concentration, but a cooperative Na displacement at higher Na concentration reduces the crystal symmetry to rhombohedral, stabilizing the rhombohedral phase (Fig. 20a). Note that the larger is the concentration of Na+ ions in the compound, the higher is the temperature of transition from rhombohedral to cubic symmetry. Electrochemical reactions occurring at low and high voltage are the redox reactions of low-spin Fe3+/2+ and high-spin Mn3+/2+, respectively. The post-cycled electrode did not show a notable structural change after 50 cycles. Na1.72MFe[CN]6 reacted at a rate of 40C (Fig. 20b). Replacement of Mn with Fe, FeFe[CN]6, lowered the operation voltage of the lower voltage plateau to 3.1 V on charge and 2.8 V on discharge, delivering a discharge capacity of approximately 120 mA h g−1.230 The electrode was substantially stable over 600 cycles with excellent capacity retention. Na4Fe[CN]6 was electrochemically active in Na cells, showing approximately 90 mA h g−1 on discharge with a flat voltage plateau at 3.4 V.231 The lower capacity is due to the one-electron reaction by the low-spin Fe3+/2+ redox.
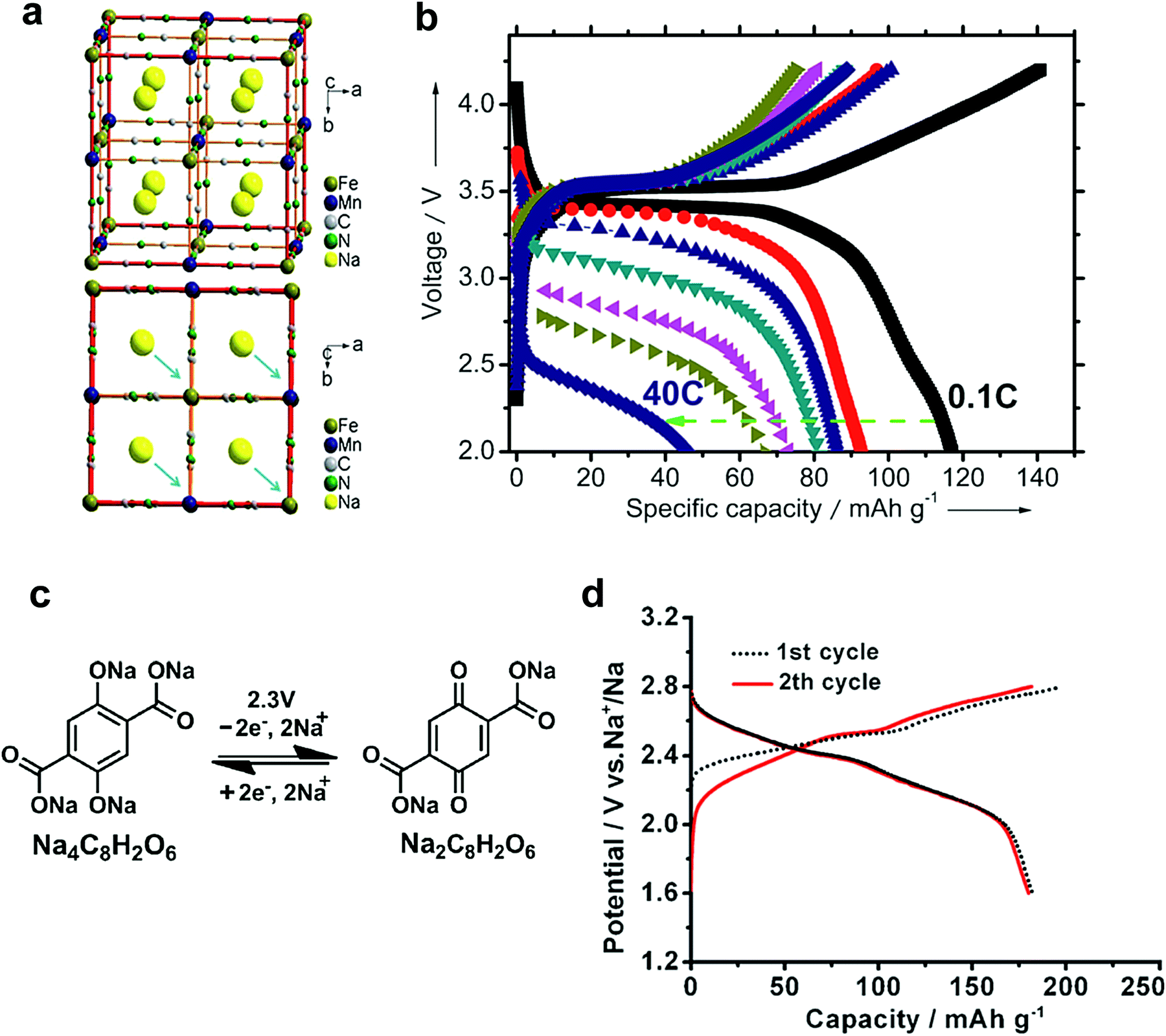 | ||
| Fig. 20 (a) Crystal structures of cubic Na1.4MnFe[CN]6 (upper) and the alkali-ion displacement along a [111] resulting in rhombohedral symmetry of Na1.72MnFe[CN]6 (lower) and (b) charge/discharge curves of a Na1.4MnFe[CN]6/Na half-cell at various current densities (follow the arrow by order of 0.1, 0.85, 3.5, 8.5, 17, 26, and 40C, 120 mA g−1 at 1C). (Reproduced from ref. 229 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) (c) Electrochemical redox reaction mechanism of Na+ ions with Na2C8H2O6/Na4C8H2O6 at potentials of 2.3 V and (d) the resulting initial discharge/charge profiles of Na4C8H2O6/Na half cells at a rate of C/10 (19 mA g−1). (Reproduced from ref. 236 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Organic molecules can undergo a reversible electrochemical redox reaction. The advantages of such a reaction are structural diversity, flexibility, molecular level controllability, and resource renewability.232–235 Recently, Wang et al. reported the electrochemical performance of 2,5-dihydroxyterephthalic acid (Na4DHTPA), Na2C8H2O6, with enolate and carboxylate groups as the cathode material for room temperature SIBs (Fig. 20c).236 They demonstrated that two reversible Na+ ion electrochemical reactions occurred with redox couples of Na2C8H2O6/Na4C8H2O6 as the cathode at 2.3 V, with a capacity of 180 mA h g−1 and excellent cyclability (Fig. 20d). Na2C6O6237 and aniline–nitroaniline copolymer238 also exhibited high rechargeable capacity over 170 mA h g−1 with good cyclability.
3. Anode materials
As mentioned in Section 2, several platforms of cathode materials have been introduced for SIBs.239 Simultaneously, as the cathode counterpart, the recent development of anodes for SIBs has been achieved by the use of selected carbonaceous materials, transition metal oxides (or sulfides) and intermetallic and organic compounds, as anodes.240 Research on the above anode materials for SIBs has been developed in three main categories, based on the reaction mechanism during sodiation/desodiation processes: (1) the insertion reaction, (2) the conversion reaction, and (3) the alloying reaction. Carbonaceous materials and titanium-based oxides have been introduced as insertion anode materials.241,242 Recent efforts expand the applications towards transition metal oxides (TMOs) or transition metal sulfides (TMSs) that are driven by a conversion reaction.243 Also, Na–Me (Me = metal) alloying compounds of group 14 or 15 elements motivated by the alloying reaction are recommended as possible candidate materials with the help of an electro-conducting carbon matrix to improve their specific capacity and cyclability.244,245 Such conversion and alloying reaction materials are known to deliver high capacities but suffer from huge volume expansion of the host materials due to the continuous self-pulverization of the electrode materials.240,243 More seriously, to compete with lithium ion batteries, a great challenge to overcome is the sluggish reaction kinetics from the large ionic size of Na+ ions (1.02 Å), which impedes fast Na+ storage.99,240,246 Therefore, scientists should make more efforts to improve the electrode performances. In this section, we discuss present research progress in anode materials for SIBs.3.1. Insertion materials
Based on an insertion reaction, carbonaceous and titanium-based oxides have been extensively studied as anodes for SIBs. Several carbon-based materials such as graphitic and non-graphitic carbons have been investigated for Na+ storage.241 These carbon materials are widely accepted because of their ability to accommodate Na+ ions into their structure. In particular, hard carbon is interesting because of its reasonable capacity of ∼300 mA h g−1 and low operating potential (almost zero, ∼0 V vs. Na+/Na).22,23,247 However, the Na+ storage mechanism in a disordered carbon structure is still controversial.247–251 On the other hand, titanium-based oxide compounds have been widely studied because of their low operation voltage and cost as well.242 Analogous to LIBs, titanium-based oxide anodes, including various polymorphs titanium dioxide (TiO2), spinel-lithium titanate (Li4Ti5O12) and sodium titanate (NaxTiyOz), were reported as promising anode materials.3.1.1.1. Graphitic carbon (graphite). During the electrochemical reduction, Li+ ions are inserted between graphene layers, and Li-graphite intercalation compounds (Li-GIC) are formed through stage transformations in Li cells.252,253 Since the 1980s, the electrochemical behavior of graphite with Na+ ions has been studied based on the graphite/polyethylene oxide NaCF3SO3/Na cell254 (Fig. 21a). However, Na+ insertion into graphite is significantly impeded and degradation of electrolyte and/or electrode materials was observed255 (Fig. 21b). First-principles calculation results of the formation energy for Na-GIC exhibited that Na hardly intercalates into graphite because of the energetic instability of the Na-GICs. Graphite is stressed when some Na+ intercalates into graphite because of the thermodynamic instability of binary Na-intercalated GICs (b-GIC), which is assumed to be the result of an unfavorable mismatch between the graphite structure and the size of the Na ion.256 Namely, due to the unfavorable formation of NaC6 and NaC8 that are thermodynamically unstable at the first stage of Na-graphite intercalation compound (Na-GIC) formation in SIBs, the capacity of natural graphite is limited.256–258 As a recent breakthrough, Jache et al. facilitated Na-ion storage in graphite using solvated-Na-ion intercalation, forming ternary GICs. That is the intercalation of solvated alkali ions “co-intercalation” by reduction of graphite according to the following equation: Cn + e− + A+ + y
solv ↔ A+(solv)yCn−.258 Zhu et al. also confirmed that solvated Na+ ions intercalate into graphite via a stage-evolution process, forming a set of ternary graphite intercalation compounds.259 Ether-based electrolytes with a high donor number can form stable Na+ solvated species with non-polar characteristics for co-intercalation into natural graphite.260–262 Kim et al. reported a reversible phase transition during intercalation and deintercalation processes of solvated-Na-ions in natural graphite via an operando X-ray diffraction analysis261 (Fig. 21c). They investigated the unusual Na storage behavior in natural graphite through Na+-solvent co-intercalation using different solvents (EC/DEC, DME, DEGDME and TEGDME) and Na salts (NaPF6, NaClO4, NaCF3SO3). Ether-based electrolytes could suppress electrolyte decomposition, resulting in the formation of a negligible SEI film on the graphite surface, enabling Na+-solvent transport to the graphite lattice. In contrast, carbonate-based electrolytes (EC/DEC) form relatively thick insulating SEI layers on the graphite surface, which block Na+-solvent transport262 (Fig. 21d). As a result, under the limited conditions using ether-based electrolytes, the natural graphite delivered a capacity of 150 mA h g−1 with reasonable retention for 2500 cycles, and produced over 75 mA h g−1 at 10 A g−1 in the DEGDME electrolyte containing NaPF6 salt. On the other hand, Wen et al. proposed expanded graphite with an enlarged interlayer lattice distance of 4.3 Å. Note that graphite has a typical interlayer space of 3.4 Å.263 Their in situ TEM study revealed reversible insertion and extraction of Na+ ions during the electrochemical reaction (Fig. 21e). As a result, the expanded graphite could exhibit a moderate capacity of approximately 284 mA h g−1 at a current of 20 mA g−1 with good capacity retention over 2000 cycles (Fig. 21f). Recently, Kang et al. reported the sodium ion intercalation behavior of expanded graphite oxide (GO) as an anode material. According to their report, the electrochemical properties of GO strongly depend on the amounts and ratios of different functional groups.264
 | ||
| Fig. 21 (a) Graphite current–potential curve in the graphite/PEO-NaCF3SO3/Na cell at 82 °C. (Reprinted from ref. 254, Copyright 1988, with permission from Elsevier.) (b) Li+ and Na+ insertion mechanisms in graphene and graphite. (Reproduced by permission from ref. 255, Nature Publishing Group, Copyright 2015.) (c) In operando synchrotron X-ray diffraction analysis of the structural evolution of the ternary Na–ether–graphite system. (Reproduced with permission from ref. 261, Copyright 2015 The Royal Society of Chemistry.) (d) Different Na storage mechanisms in natural graphite between in NaPF6 in EC/DEC (top) and NaPF6 in DEGDME (bottom). (Reproduced from ref. 262 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) (e) In situ TEM investigation of the sodium storage mechanism in the EG-1h sample. (f) Electrochemical performances of PG, GO, EG-1h and EG-5h in organic 1.0 M NaClO4 in a polycarbonate solvent liquid electrolyte. (Reproduced by permission from ref. 263, Nature Publishing Group, Copyright 2014.) | ||
3.1.1.2. Non graphitic carbon (hard carbon). In 1993, Doeff and co-workers first reported the Na storage performance of disordered soft carbon prepared by pyrolysis of petroleum coke251 (Fig. 22a). They demonstrated the extent of the Na+ insertion/extraction reaction into the soft carbon and discussed the possibility of its application for SIBs. Stevens and Dahn reported the insertion mechanism of Na+ ions into disordered hard carbon (Fig. 22b).22,23,247 The suggested mechanism was the “house of cards” for Na+ ion storage, which is composed of the two domains in a disordered hard carbon structure without staging transition. First, Na+ ions are inserted between parallel graphene sheets (in the sloping voltage region) upon increasing the interlayer space. Second, Na+ ions fill the nanopores (in the plateau region) of the disordered carbon structure.22,23,249 The hard carbon prepared by carbonization of glucose showed a reversible electrochemical reaction in Na cells and it delivered a high specific capacity of approximately 300 mA h g−1 with a low operating potential of about ∼0 V. Later, Komaba and co-workers performed a systematic study on electrochemical sodium insertion into hard carbon to understand the related structural change.27 Upon reduction to 0.2 V (in the sloping region), the resulting XRD peaks shifted to lower angles, indicating that the interlayer spacing between the graphene sheets was expanded due to the sodium insertion (Fig. 22c). They also confirmed the reversible Na+ insertion process into the nanopores via small angle X-ray scattering measurements (SAXS) in the voltage range of 0.2 V to 0 V (in the plateau region) upon reduction. From these results, they suggested that the enlarged interlayer spacing (d-spacing = 3.8 Å) or the smaller crystallite size of the parallel graphene within hard-carbon in comparison to those of the graphite (d-spacing = 3.354 Å) is responsible for the sodium insertion. In addition, the 23Na NMR study demonstrated the reversible electrochemical reaction mechanism of Na+ ions with hard carbon: (i) two resonance peaks at 9.9 ppm and 5.2 ppm were ascribed to reversible sodium present between disordered graphene sheets in hard carbon and (ii) one broader resonance peak between −9 and −16 ppm was ascribed to the appearance of Na+ ions in heterogeneously distributed closed nanopores in hard carbon.265 In 2011, Wenzel and co-works demonstrated high capacity carbon anode materials with an excellent rate capability of 100 mA h g−1 at C/5 at room temperature for the first time266 (Fig. 22f). They prepared templated carbon with hierarchical porosity via the nanocasting route by using pitch carbon and investigated the co-relationships between the physico-chemical properties and the electrochemical properties with commercially available porous and non-porous carbon materials (porous carbon: Timrex 100, 300, 500 and activated carbon, non-porous carbon: graphite). The highest surface area (1041 m2 g−1) of activated carbon and the highest pore volume (1.008 cm3 g−1) of Timrex 500 show the low reversibility with Na+ ions. Meanwhile, their templated carbon controlled by microstructural approaches minimized the diffusion lengths within the electrode (surface area: 346 m2 g−1, pore volume: 0.798 cm3 g−1). As a result, the carbon matrix showed excellent electrochemical activity for Na+ ions. Their results imply that high capacity and rate capabilities cannot be simply achieved by enlarging the surface area or by increasing the porosity. On the other hand, Tsai et al. further confirmed the reaction process through an ab initio study showing that a larger interlayer distance is not the only factor that helps Na intercalation. Additionally, the vacancy defects (MV: mono-vacancy and DV: di-vacancy) in hard carbon can greatly enhance the Na+ ion intercalation because of the strong ionic binding energy between the Na+ ions and the defects, which effectively overcomes the van der Waals interaction.248 Further, Bommier et al. discussed the new storage mechanism in the sloping region (as a function of the charge–discharge curve), which can be explained through Na+ ion storage at defect sites.250 They also confirmed that Na+ ions can be intercalated into the hard carbon lattice particularly in the low voltage plateau region. These findings suggest that the Na+ storage is related to a three-step process (Fig. 22d) rather than the two-step storage mechanism proposed in the card-house model (Fig. 22e); (i) Na+ ions are adsorbed at defective sites in the slope-voltage region, (ii) Na+ ions are intercalated in the hard carbon lattice, and (iii) Na+ ions are adsorbed at the pore surface in the plateau region. Such research results show that the Na+ insertion mechanism into a disordered structure is still controversial. Therefore, further theoretical and experimental investigation should be conducted to clarify the related reaction mechanism for development of hard carbon materials.
 | ||
| Fig. 22 (a) First cycle of the Na/DME, NaClO4/(ground) petroleum coke cell. (Reproduced with permission from ref. 251, Copyright 1993 The Electrochemical Society.) (b) Sodium potential profiles of glucose pyrolyzed to 1000 °C. The insets show details of the low-potential region. (Reproduced with permission from ref. 247, Copyright 2000 The Electrochemical Society.) (c) Ex situ XRD patterns of hard-carbon electrodes: (1) pristine electrode, galvanostatically reduced to (2) 0.40 V, (3) 0.20 V (4) 0.10 V, (5) 0.00 V, and (6) oxidized to 2.00 V after reduction to 0.00 V (• PVdF binder, ? unknown). (Reproduced from ref. 27 with permission, Copyright 2011 Wiley-VCH Verlag GmbH & Co. KGaA.) (d) Potentiogram and schematic of the proposed Na-ion three part storage mechanism. (Reproduced with permission from ref. 250, Copyright 2015 American Chemical Society.) (e) Typical potential vs. capacity profile of hard carbon when tested against sodium metal counter electrodes. (Reproduced with permission from ref. 249, Copyright 2015 The Electrochemical Society.) (f) Specific capacities (C/5, Na-insertion) upon cycling for different carbons and Coulombic efficiency for the templated carbon at room temperature. (Inset image: the SEM image of templated carbon.) (Reproduced with permission from ref. 266, Copyright 2012 The Royal Society of Chemistry.) | ||
To date, hard carbons have been the most widely used carbon source of SIB anodes and have been proved to significantly improve the electrochemical performance of SIBs. However, there are a lot of challenges facing the development of hard carbon anodes for practical applications. The reversibility of the hard carbon was found to depend on the carbon precursor, particle sizes and manufacturing processes. And, the appropriate low pore volumes and surface areas can achieve the higher reversible capacities. The suitable additives and electrolytes are highly desirable for reversibility of Na+ storage. Therefore, based on a number of previous studies, we should consider some factors, including particle sizes, additives, electrolytes, vacancy defects and porosity measurements, to develop the high irreversible capacity and high rate capability in hard carbons. Furthermore, we should investigate the relationship between the electrochemical characteristics of hard carbon and solid electrolyte interphase (SEI) layer formation. At the same time, the computational works such as DFT calculation and ab initio calculation should be conducted to predict and support the experimental results.
3.1.1.3. Non graphitic carbon (graphene). Graphene, unique two-dimensional carbon materials, has great physical and chemical advantages such as a large surface area, superior electronic conductivity, and chemical stability.267 Such advantages of graphene can bring shorter paths for fast ion diffusion and produce a large exposed surface offering more ion insertion channels.268 Therefore, for LIBs and SIBs, graphene can be considered as an anode material for energy-storage systems255,269–274 (Fig. 21b). Moreover, graphene or reduced graphene oxide (RGO) help to improving the Li and Na storage capability in its composite anodes. Recently, Wang et al. demonstrated the reversible sodium ion storage performances in reduced graphene oxide (RGO).274 RGO possesses higher electrical conductivity and active sites with large interlayer distances and provides a disordered structure enabling it to store a larger amount of Na+ ions. In their study, RGO anodes exhibited a moderate specific capacity of 141 mA h g−1 at 40 mA g−1 with stable cycle retentions for over 1000 cycles. Ding et al. synthesised different kinds of few-layer graphene at different carbonization temperatures (600–1400 °C) and investigated the Na+ ion storage mechanism in their graphene structure.275 At a higher carbonization temperature of 1100 °C, they obtained an optimized highly ordered pseudographic structure having a large interlayer spacing of 0.388 nm, which exhibited promising Na+ insertion performances. On the other hand, Datta et al. reported that the presence of defects enhances the adsorption of Na atoms in graphene sheets.276 However, such defects of graphene represent a serious drawback such as low Coulombic efficiency that may cause Na metal plating on the rGO surfaces.255
3.1.1.4. Heteroatom doping. As an effective strategy to enhance the electrochemical properties of carbonaceous materials as anodes, heteroatom (such as N, B, S and P) doped hard carbon and graphene (or graphene liked materials) are introduced.277–291 Hetero atom doping into a carbon structure tends to create a defect site to absorb Na+ ions and improves the electrode–electrolyte interaction by functionalizing the carbon surface. Commonly, nitrogen doping into a carbon structure significantly improves ion transport and charge-transfer processes.279,280 Wang et al. proposed a 3D interconnected structure of free-standing flexible films composed of nitrogen-doped porous nanofibers that exhibited 212 mA h g−1 at 5 A g−1 with stable capacity retentions of 99% after 7000 cycles282 (Fig. 23a). In a recent work, sulfur-doped disordered carbon was proposed as an anode material for SIBs. Sulfur doping into the carbon structure provided additional reaction sites for accommodation of Na+ and/or contributing to facile ion diffusion by enlarging the interlayer distance. Li et al. synthesized a sulfur-doped disordered carbon that has a high sulfur doping level (∼26.9%) and a unique 3D coral-like structure. The as-prepared sulfur-doped carbon exhibited a high reversible capacity of 516 mA h g−1 with excellent rate capability as well as superior cyclability for 1000 cycles288 (Fig. 23b).
 | ||
| Fig. 23 (a) HAADF image and the elemental mapping of N-CNF (left). Long term cycling performance of N-CNF at a current density of 5 A g−1 (right). (Reproduced from ref. 282 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) (b) HR-TEM image and EDS elemental mapping of DC-S (left). Electrochemical performances of DC-S (right). (Reproduced with permission from ref. 288, Copyright 2015 The Royal Society of Chemistry.) (c) The preparation process of NDCS derived from okara (left). High rate capability and long cycling stability of the NDCS at 4.5C for 2000 cycles (right). (Reproduced from ref. 296 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
3.1.1.5. Biomass derivatives. As mentioned above, disordered carbon appears to be the most suitable anode material for SIBs. Biomass-derived disordered carbon materials have been widely investigated due to their low production cost and low energy consumption during the synthesis procedures.275,292–297 Wood carbon anodes were derived from natural woods via a simple carbonization process, which functions as a binder-free and current collector-free anode material.292 This material exhibited a high areal capacity of 13.6 mA h cm−2 with a high mass loading of 55 mg cm−2. A recent work, reported by Yang et al., showed a sustainable route using a biomass byproduct, okara, as a natural nitrogen fertilizer possessing high content N-doped carbon sheets.296 Okara was carbonized and exfoliated to expand between carbon sheets that led to a high specific surface area. This benefits facile intercalation and deintercalation of Na+ ions into the carbon structure upon cycling. This unique structure and large surface area were responsible for the high capacity of approximately 292.2 mA h g−1 with stable cycling performances for over 2000 cycles (Fig. 23c). From the green and recycle points of view, using biomass (bio-waste) is of great significance for the future direction of material development.
Such works present that carbonaceous materials exhibit promising Na+ ion storage performances as anodes for SIBs. Notwithstanding, the initial irreversible capacity, limited capacity, poor rate capability, and low mass loading should be resolved for cell balancing for practical battery applications. Herein, we have mentioned few relevant ones and suggest the possible propositions as follows. The irreversible capacity upon the initial cycle is the most critical issue as it severely hindered the practical cell energy density achievable. Attaining an attractive rate capability is another important factor for a practical energy storage system. In addition, tap density is also a crucial parameter determining the volumetric energy density of the cells. The appropriate low pore volumes and surface areas were required for higher reversible capacities. Using suitable electrolytes, additives, and binders is also an effective approach for decreasing the initial irreversible capacity. And morphology and size control is a substantial strategy to facilitate mass transport and storage, which can significantly improve the rate capability. Most of all, for developing practical SIBs using high mass loading electrodes, a comprehensive study of the impact of the electrode thickness on the rate capability, energy and power density and long-term cycling behavior is required.298
3.1.2.1. Titanium dioxides. The several TiO2 polymorphs, including anatase-TiO2,301–314,316–323 rutile-TiO2,324–328 brookite-TiO2,329,330,332 and bronze-TiO2333 have been investigated as anode materials for SIBs294–296 (Fig. 24a–d). Among them, most research results were reported using anatase TiO2 because the activation barrier for Na+ insertion into the anatase lattice is comparable to that of lithium, which is rather remarkable considering its significantly larger ionic radius.301–303 Xiong et al. reported the feasibility of electrochemically-grown amorphous titanium dioxide nanotube electrodes in Na cells for the first time304 (Fig. 25a). On the other hand, Mattsson et al. claimed that high crystalline and/or micronized TiO2 cannot easily support Na+ insertion because of the ionic size of Na+ and a much higher sodium diffusion barrier compared with Li.305 Recently, high electrochemical activity of TiO2 with Na+ ions was achieved by reducing the particles to the nanometer size for shortening of the migration length for Na+ insertion. Xu et al. used anatase nanocrystalline TiO2 for Na+ storage.306 Wu et al. interpreted the reaction process during Na+ insertion and extraction, highlighting the substantial influence of the electrolyte composition (salt and solvent) and cut-off potential307 (Fig. 25b). Through NMR and electrochemical studies, Gonzalez et al. explained the irreversible process (electrolyte decomposition) up to 0.3 V observed at the first Na+ insertion process in the TiO2 electrode.308 Kim et al. observed that anatase TiO2 nanorods can be stored in the host structure through the Na+ insertion and extraction reaction coupled with the Ti4+/Ti3+ redox reaction via ex situ XRD and XAS studies309 (Fig. 25c and d). Their carbon coating on the anatase TiO2 nanorod surface played an important role in improving the capacity and rate capability. Furthermore, they also suggested that the presodiation technique was an effective way to minimize the initial irreversible reaction of the TiO2 anode. Recently, Passerini's group found that only the (de-)insertion of Na+ ions in the newly formed amorphous sodium titanate phase appears to be reversible (uptake/release 0.41 Na per TiO2), while all the other processes (metallic titanium, sodium superoxide, and oxygen evolution) appear to be irreversible (Fig. 25e and f).310 Usui et al. investigated rutile-TiO2 that showed the reversible reaction of Na+ insertion and extraction into and from the crystal lattice of rutile TiO2326 (Fig. 26a). In addition, the appropriate amount of Nb doping (0.06 mol%) on rutile TiO2 materials could substantially improve the electronic conductivity. As a result, the Ti0.94Nb0.06O2 electrode exhibited excellent cycling performance with a reversible capacity of 160 mA h g−1 at the 50th cycle. Later, Zhang et al. proposed rutile TiO2 microspheres anchored by nanoneedle clusters, which exhibited a capacity retention of 83.1% after 200 cycles at a current density of 0.1C-rate.327 Huang et al. found that one of the polymorphs, the low crystalline brookite-TiO2 was available for Na+ ion storage.329 Pioneer works of unusual TiO2 polymorphs, including the brookite-phase, the bronze-phase and their mixture phase, are recently investigated.330–333
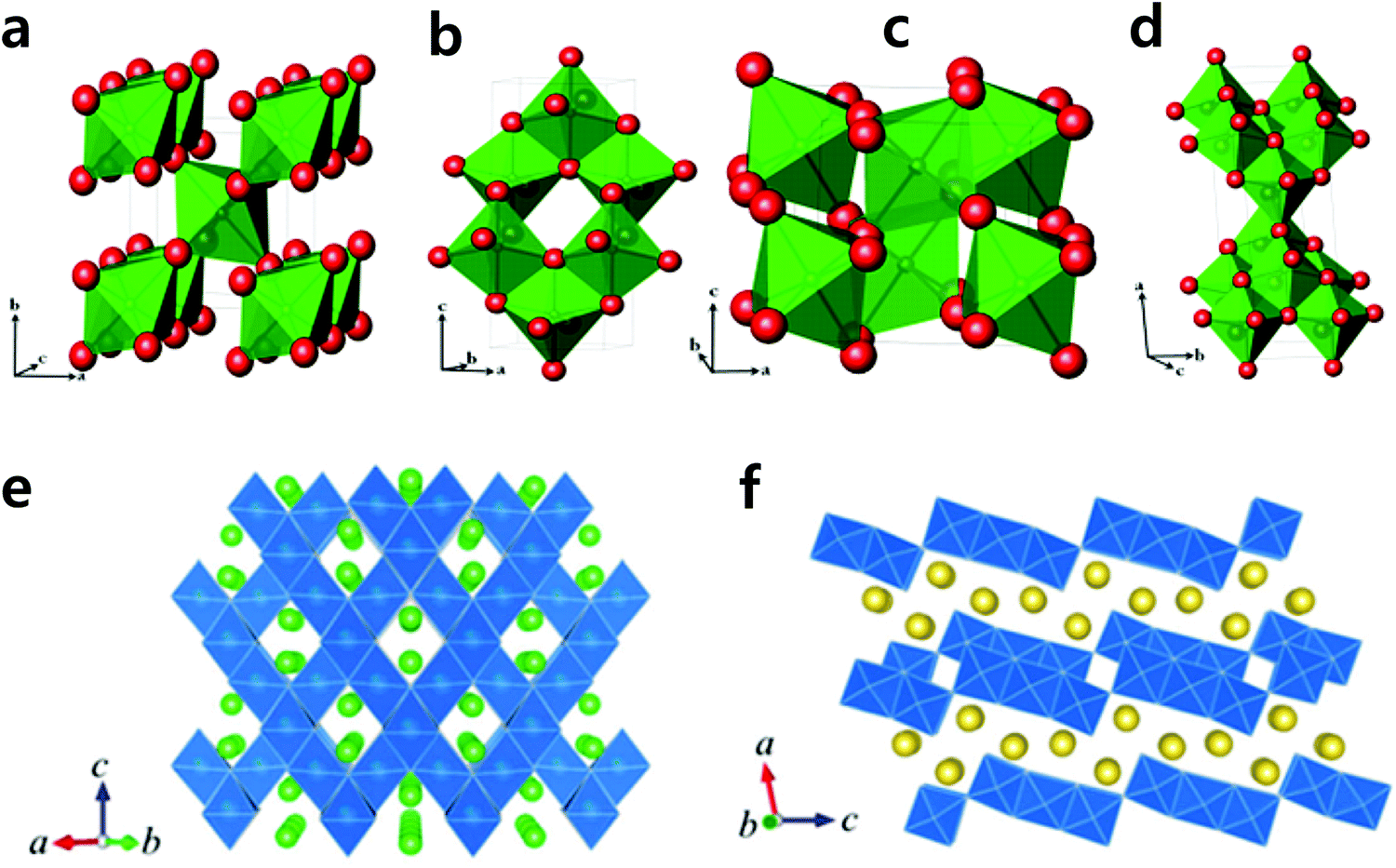 | ||
| Fig. 24 Crystal structures of TiO2. (a) Rutile, (b) anatase, (c) bronze, (d) brookite. (Reprinted from ref. 299, Copyright 2015, with permission from Elsevier.) Crystal structures of (e) spinel-type Li4Ti5O12 and (f) Na2Ti3O7. Li and Na atoms are represented by green and yellow spheres, respectively. (Reproduced with permission from ref. 300, Copyright 2016 The Royal Society of Chemistry.) | ||
 | ||
| Fig. 25 (a) Charge/discharge galvanostatic curves of amorphous 80 nm I.D. TiO2NT in Na half cell (red for discharge and black for charge). (Reproduced with permission from ref. 304, Copyright 2011 American Chemical Society.) (b) Galvanostatic investigation of the influence of the lower cut-off potential based on Coulombic efficiency (dis-)charge capacities. (Reprinted from ref. 307, Copyright 2014, with permission from Elsevier.) (c) Ex situ XRD patterns and (d) XANES K-edge spectra of 2.9 wt% carbon-coated anatase nanorod TiO2. (Reproduced with permission from ref. 309, Copyright 2014 American Chemical Society.) (e) In situ XRD analysis of the (de-)sodiation mechanism of anatase TiO2 nanoparticles, (f) scheme of the reaction mechanism. (Reproduced from ref. 310 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
 | ||
| Fig. 26 (a) Cycling performances of thick-film electrodes consisting of Ti1−xNbxO2 with rutile and anatase structure in Na cell. (Reproduced with permission from ref. 326, Copyright 2015 American Chemical Society.) (b) Long-term cycling performance of G–TiO2(B) electrode at a current density of 500 mA g−1. Inset image represent the illustration of partially bonded graphene–TiO2-B (001) interface. (Reproduced by permission from ref. 331, Nature Publishing Group, Copyright 2015.) (c) Charge–discharge voltage curves of TiO2, TiO2-F, and TiO2-F on CNT at 0.1C-rate. (Reprinted from ref. 312, Copyright 2015, with permission from Elsevier.) (d) Rate capability of TiO2 and S-TiO2 at various C-rate. (Reproduced from ref. 323 with permission, Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
However, sodium storage of TiO2 suffers from the sluggish sodium kinetics due to the larger ionic size of Na+ ions. To overcome this drawback, strategies such as the nano-architecture modified by metallic Ti and high conductivity carbon additives were introduced.309,311,328,331 Kim et al. fabricated pitch carbon-coated anatase TiO2 nanorods, which exhibited a high capacity of 193 mA h g−1 and superior rate capability.309 Recently, Zhang et al. proposed graphene-rich wrapped petal-like rutile TiO2 tuned by carbon dots, which exhibited the outstanding sodium-storage performances.328 Chen et al. proposed a chemically bonded graphene (G)–TiO2(B) composite as a long-cycle life material. Kinetics analysis reveals Na+ intercalation pseudo-capacitive behaviour in the G–TiO2 sodium cell, which is highly beneficial to fast charge storage and long-term cyclability331 (Fig. 26b).
Doping to reduce the average oxidation state of Ti is another interesting strategy to enhance the reversible Na storage performances of TiO2.312,313,315,323 Hwang et al. reported ultrafast sodium storage performance in fluorine-doped anatase TiO2 nanoparticles embedded on carbon nanotubes312 (Fig. 26c). Nanosized anatase TiO2 partially doped with fluorine (TiO2−δFδ) to form electro-conducting trivalent Ti3+ led to facile Na+ insertion into an anatase TiO2 structure. In addition, TiO2−δFδ was modified by electro-conducting carbon nanotubes (CNTs) to further enhance the electric conductivity. Boron doping can also enhance the photocatalytic activity of TiO2 due to or partly due to the formation of Ti3+ ions induced by oxygen vacancies which can increase the conductivity of TiO2.315 Recently, Wang et al. prepared B-doped TiO2via the facile hydrothermal method and demonstrated a high reversible capacity of 150 mA h g−1 at a high current rate of 2C with stable cycling performance for over 400 cycles.313 Ni et al. introduced self-supported S-doped TiO2 nanotubes with high electronic conductivity323 (Fig. 26d). When S is incorporated into TiO2, the S 3p states will be partially delocalized. The S 3p states can contribute to the formation of the valence band, and thus increases the width of the valence band, resulting in a decreased band gap energy (S-TiO2: 2.6 eV, TiO2: 3.0 eV).334 The high conductivity assisted in decreasing the band gap energy, which enabled delivery of a high capacity of 320 mA h g−1 at 33.5 mA g−1 with a stable capacity retention of 91% for over 4400 cycles at a high current density of 3.35 A g−1.
3.1.2.2. Lithium titanate. A spinel Li4Ti5O12 has been extensively studied as one of the most promising anode materials for long-life stationary LIBs because it has a flat and high potential around 1.5 V (vs. Li/Li+) during charge and discharge and an excellent cycle life due to the negligible volume change.335 Recently, Zhao et al. revisited spinel Li4Ti5O12 as a Na+ insertion host material for SIBs300,336 (Fig. 24e). A reversible capacity of 145 mA h g−1 was delivered with a relatively low insertion/extraction potential of 1.0 V. Also, they suggested an unexpected mechanism with a mixture of LiNa6Ti5O12 and Li7Ti5O12 as the final product after Na+ insertion. Later, Sun et al. precisely interpreted the Na+ insertion behavior via DFT calculations and advanced electron microscopy337 (Fig. 27a and b). They also improved the electrochemical properties of Li4Ti5O12 by varying the electrolyte and binders. Usually, for LIBs, Li+ insertion into the Li4Ti5O12 anode occurs through a two-phase reaction between spinel Li4Ti5O12 and rock-salt Li7Ti5O12. In Na cells, Na+ insertion into Li4Ti5O12 results in a three-phase reaction. Due to the different ionic sizes of Li+ and Na+, the Na+ ions are favorably occupied in the 16c site of the Li4Ti5O12 lattice, and they simultaneously induce phase separation into two rock-salt phases of LiNa6Ti5O12 and Li7Ti5O12 as follows: 2Li4Ti5O12 + 6Na+ + 6e− ↔ Li7Ti5O12 + Na6LiTi5O12. Later, Kim et al. observed the structural evolution and the chemical state of Ti at the initial cycle based on ex situ XRD and XPS measurements339 (Fig. 27c–e). Yu et al. observed three phase transition behavior of Li4Ti5O12 during Na+ insertion through in situ X-ray diffraction. They also investigated a size-dependent sodium storage mechanism in Li4Ti5O12.340 For 440 nm size Li4Ti5O12, only a small amount of Na can be inserted (0.27Na+ per formula unit of Li4Ti5O12), which corresponds to a specific capacity of 16 mA h g−1. While the degree of the reversible Na+ insertion into 44 nm size Li4Ti5O12 was greatly increased (3Na+ ion per formula unit of Li4Ti5O12) and nearly reached its theoretical capacity of 175 mA h g−1 (Fig. 27f). They claimed that downsizing the Na+ host structure of Li4Ti5O12 is a crucial factor to improve the sluggish Na+ ion diffusion kinetics. Analogous to this scenario, Hasegawa et al. reported nano-sized Li4Ti5O12 materials with hierarchically porous structures and flower-like morphologies.341 According to their reports, Na+ insertion/extraction capability is strongly dependent on their nanoarchitectural design and calcination temperature. These nanostructured Li4Ti5O12 electrodes calcined at 700 °C displayed a flower-like porous structure and exhibited a remarkably high rate performance of 146 mA h g−1 and 105 mA h g−1 at 10C and 30C-rates, respectively, even without carbon-coating. On the other hand, Yu et al. reported Na+ ion transport kinetics and coupled pseudocapacitive charge in thin film Li4Ti5O12 electrodes.342 The synergistic effect of typical Na+ insertion and the extra pseudocapacitive charge storage produced an unexpected high capacity of 225 mA h g−1 in thin film Li4Ti5O12 electrodes. They claimed that the pseudocapacitance effect on a typical insertion electrode is a potential solution to overcome the capacity limit for Na+ insertion anodes.
 | ||
| Fig. 27 (a) Crystal structure of spinel Li4Ti5O12 viewed from the [110] crystallographic direction showing separated Ti and O columns. (b) STEM imaging of a three-phase coexistence region. HAADF, ABF images and ABF line profile of Li4Ti5O12 (Li4), Li7Ti5O12 (Li7) and Na6LiTi5O12 (Na6Li) phase. (Reproduced by permission from ref. 337, Nature Publishing Group, Copyright 2013.) (c) First discharge–charge curve of carbon-coated Li4Ti5O12 and it indicating the places where ex situ XRD and XPS measurements were carried out; (d) ex situ XRD patterns (left) and (e) XPS profiles of carbon-coated Li4Ti5O12 (right). (Reprinted from ref. 339, Copyright 2015, with permission from Elsevier.) (f) The charge–discharge curve of Na storage into the crystallite size distribution of nanosized (r = 44 nm, r = 120 nm) and submicrosized (r = 440 nm) Li4Ti5O12. (Reproduced with permission from ref. 340, Copyright 2013 American Chemical Society.) (g) TEM, HR-TEM images of G-PLTO (grapheme-wrapped porous Li4Ti5O12) and long-term cycling performance of G-PLTO at 3C-rate for 12000 cycles. (Reproduced from ref. 343 with permission, Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Another approach to improve the electrochemical performances is combining carbon additives with Li4Ti5O12 materials. Kim et al. proposed pitch carbon-coated Li4Ti5O12 nanowires, which significantly increased the electronic conductivity and delivered a high capacity of 168 mA h g−1 at 0.2C-rate.339 Chen et al. fabricated porous Li4Ti5O12 nanofibers confined in a highly conductive 3D-interconnected graphene framework for SIB anodes343 (Fig. 27g). This unique structure of porous Li4Ti5O12 nanofibers wrapped with 3D graphene offers not only short pathways for Na+ diffusion and highly conductive networks for electron transport but also abundant Li4Ti5O12–electrolyte (solid–liquid) and Li4Ti5O12–graphene (solid–solid) interfacial sites for Na+ adsorption, giving rise to additional interfacial Na+ storage, a high reversible capacity of 195 mA h g−1 at a 0.2C-rate (exceeding the theoretical capacity based on the Na+ insertion reaction) and a long cycle life of 12000 cycles.
3.1.2.3. Sodium titanate. The Na2Ti3O7 structure has been most investigated as a promising anode with the lowest operating potential for SIBs347–353 (Fig. 24f). Senguttuvan et al. firstly reported that sodium titanate, Na2Ti3O7, can reversibly uptake 2 mol of Na+ ions per formula unit with a low operating potential plateau at 0.3 V vs. Na/Na+
347 (Fig. 28a). A plateau with this voltage can be quite advantageous in a full cell with a suitable cathode in terms of energy density compared to the other oxide type anodes. Typical voltage profiles of carbon black and a composite of Na2Ti3O7 with carbon black indicated an irreversible electrochemical process at ca. 0.7 V vs. Na/Na+, which corresponds to the reaction of carbon black and a reversible plateau around 0.3 V vs. Na/Na+ with concomitant intercalation of additional 2Na+ ions in the structure. Xu et al. reported the computational and experimental results for an in-depth understanding of the sodium storage mechanism of the Na2Ti3O7 structure.348 Based on the calculations of the electrostatic interaction in the crystal structure, 2 mol of Na+ ions are intercalated into the Na2Ti3O7 structure to form Na4Ti3O7 producing strong electrostatic repulsion, leading to structural instability and low operating voltage. Also, this strong electrostatic repulsion in the fully sodiated state induces the self-relaxation phenomena. In addition, they observed the Ti4+/Ti3+ redox couple upon cycling via the XAS technique (Fig. 28b). Furthermore, their carbon-coated Na2Ti3O7 electrodes delivered a theoretical capacity of 177 mA h g−1 (corresponding to uptake of 2 Na+ ions per Na4Ti3O7).
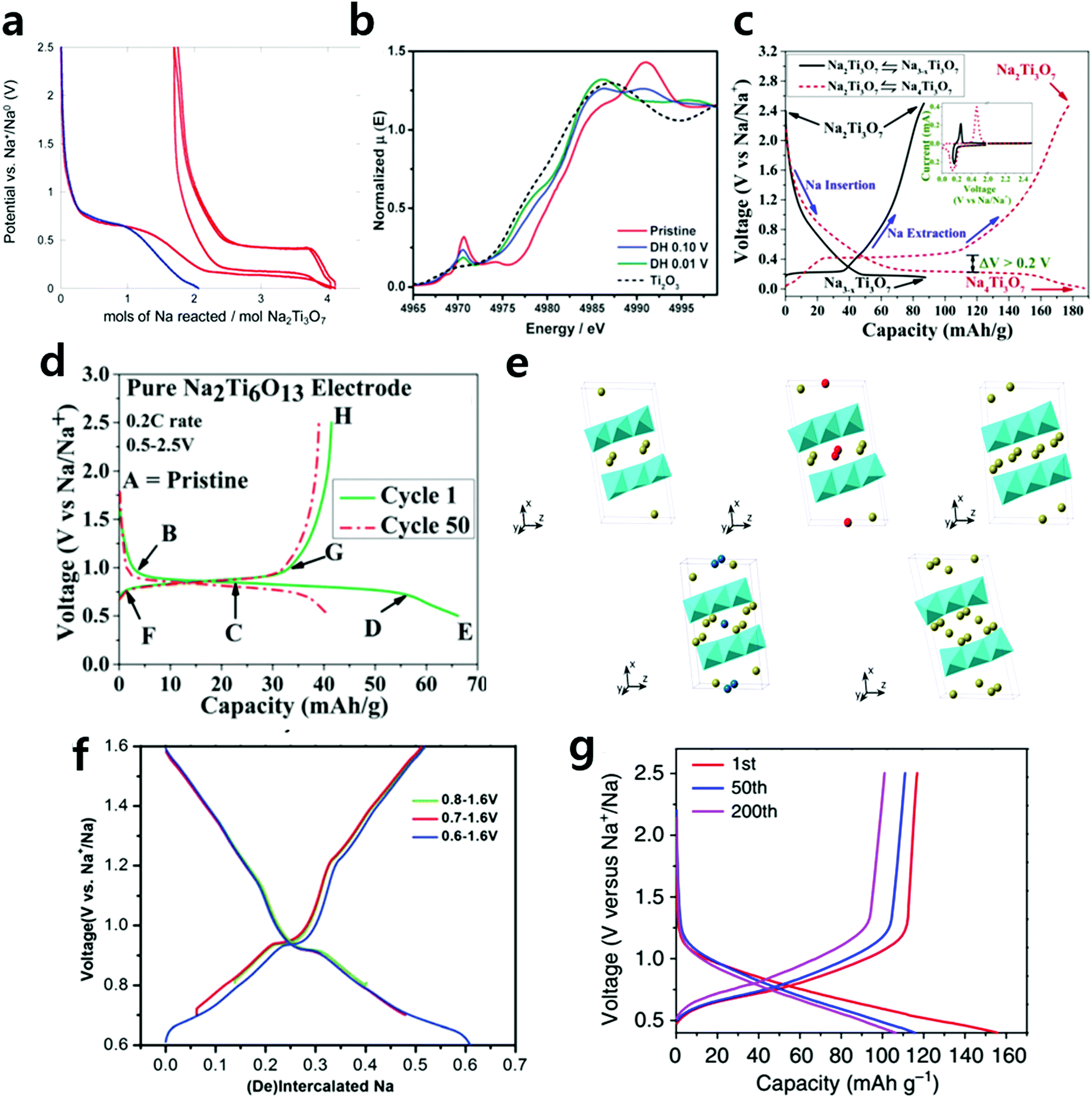 | ||
Fig. 28 (a) Voltage versus composition profile for the electrochemical reduction of carbon black (blue curve) and composite electrodes containing Na2Ti3O7 and 30% carbon black (red curve). (Reproduced with permission from ref. 347, Copyright 2011 American Chemical Society.) (b) Normalized Ti K-edge XANES for Na2Ti3O7 at pristine state (red), after discharged to 0.10 V (blue), and after discharged to 0.01 V (green). (Reproduced with permission from ref. 348, Copyright 2014 The Royal Society of Chemistry.) (c) Representative C/5 cycling of Na2Ti3O7 ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) Na3–xTi3O7 and Na2Ti3O7 Na4Ti3O7 pathways. (Reprinted from ref. 350, Copyright 2015, with permission from Elsevier.) (d) Charge–discharge curves of Na2Ti6O13 at 0.2C-rate in the voltage range of 0.5–2.5 V vs. Na/Na+. (Reproduced with permission from ref. 355, Copyright 2013 The Royal Society of Chemistry.) (e) Crystal structure change at different Na+ insertion into the Na2+4Ti6O13 structure based on the DFT calculations. (Reproduced with permission from ref. 356, Copyright 2015 American Chemical Society.) (f) Comparison of the amount of cycled Na in voltage windows with lower cut-off voltages from 0.6–0.8 V. (Reproduced with permission from ref. 361, Copyright 2015 The Royal Society of Chemistry.) (g) The 1st, 50th and 200th discharge/charge curves at a current rate of C/10 (10.6 mA g−1) in the voltage range of 0.4 and 2.5 V versus Na+/Na. (Reproduced by permission from ref. 362, Nature Publishing Group, Copyright 2013.) Na3–xTi3O7 and Na2Ti3O7 Na4Ti3O7 pathways. (Reprinted from ref. 350, Copyright 2015, with permission from Elsevier.) (d) Charge–discharge curves of Na2Ti6O13 at 0.2C-rate in the voltage range of 0.5–2.5 V vs. Na/Na+. (Reproduced with permission from ref. 355, Copyright 2013 The Royal Society of Chemistry.) (e) Crystal structure change at different Na+ insertion into the Na2+4Ti6O13 structure based on the DFT calculations. (Reproduced with permission from ref. 356, Copyright 2015 American Chemical Society.) (f) Comparison of the amount of cycled Na in voltage windows with lower cut-off voltages from 0.6–0.8 V. (Reproduced with permission from ref. 361, Copyright 2015 The Royal Society of Chemistry.) (g) The 1st, 50th and 200th discharge/charge curves at a current rate of C/10 (10.6 mA g−1) in the voltage range of 0.4 and 2.5 V versus Na+/Na. (Reproduced by permission from ref. 362, Nature Publishing Group, Copyright 2013.) | ||
Rudola et al. investigated the physicochemical and electrochemical properties of Na2Ti3O7 and calculated the diffusion coefficient of Na+ ions in the Na2Ti3O7/CB electrode.349 Later, Rudola et al. observed the intermediate phase of Na3−xTi3O7 during the sodiation/desodiation process350 (Fig. 28c). During the sodiation process, based on the ex situ XRD results, two discharge plateaus revealed the following two phase reactions: Na2Ti3O7 → Na3–xTi3O7 (black curve in Fig. 28c) and Na3–xTi3O7 → Na4Ti3O7 (red curve in Fig. 28c). The lower discharge plateau causes an irreversible transformation that leads to the loss of the sodium storage pathway in subsequent cycles. By controlling the cut-off potential from 0.01–2.5 V (with low plateaus) to 0.155–2.5 V (without low plateaus), the Na2Ti3O7 ⇆ Na3–xTi3O7 pathway has the lowest redox voltage of 0.2 V vs. Na/Na+ with a moderate capacity of 89 mA h g−1. In addition, it showed excellent rate performance up to 80C-rate and a good cycle life for over 1500 cycles. On the other hand, Pan et al. observed that nanosized Na2Ti3O7 intermediate phase is avoided and Na4Ti3O7 directly translates to Na2Ti3O7 during the Na+ extraction process.351 They also discussed two main reasons for the low Coulombic efficiency and continuous capacity fading of the Na2Ti3O7 electrode: (i) partial decomposition on the Na2Ti3O7 particle surface (instability of the SEI layer) and (ii) structural distortion upon Na+ insertion/extraction. Later, Muñoz-Márquez et al. observed the instability of SEI formation during the charge process and precisely investigated the composition and evolution of the solid–electrolyte interphase in Na2Ti3O7 electrodes.352 Although Na2Ti3O7 is considered as a promising anode with the lowest operating potential for Na-ion batteries, poor capacity retention remains the serious drawback. Therefore, further investigation including reducing the catalytic activity of Na+ ions with titanium and application of protective coating of active materials should be conducted to improve the electrochemical performances of Na2Ti3O7 electrodes.
Other types of sodium titanates were also widely studied as potential anodes for SIBs.354–364 Trinh et al. reported reversible electrochemical activity of Na2Ti6O13 in sodium cells with less than half a mole of sodium per formula unit.354 Based on the ex situ XRD measurements in the voltage range of 0.5–2.5 V, Rudola et al. reported the sodium insertion/extraction mechanism of Na2Ti6O13 as follows: Na2Ti6O13 + xNa+ + xe− ↔ Na2+xTi6O13 with x = 0.85.355 Later, Shen et al. show that the capacity of the Na2Ti6O13 anode material can be enhanced from 49.5 mA h g−1 (Na2+1Ti6O13) to 196 mA h g−1 (Na2+4Ti6O13) by lowering the cutoff voltage from 0.3 to 0 V.356 Simultaneously with experimental works, Shen et al. predicted the structure and average voltage change of reduced phases at various compositions of Na2+xTi6O13 (x = 0–4) by using density functional theory (DFT) calculations (Fig. 28e). Shirpour et al. suggested layered sodium titanate, structurally identical to sodium nonatitanate, which was capable of reversibly intercalating Na+ ions at a low potential of about 0.3 V vs. Na/Na+.357 During the discharge process, typical voltage curves of a dehydrated nonatitanate electrode had a reversible capacity of 125 mA h g−1 at 30 mA g−1 accompanied by irreversible decomposition of the electrolyte which includes formation of a SEI layer in the voltage range of 0.3–0.9 V similar to the other titanium-based oxides.
The sodium titanate form of Na4Ti5O12 has been investigated as the two crystal structures of trigonal Na4Ti5O12 (T-Na4Ti5O12) and monoclinic Na4Ti5O12 (M-Na4Ti5O12) by Woo et al. and Naeyaert et al., respectively.358,359 T-Na4Ti5O12 has a tunnel-structured three-dimensional framework, whereas M-Na4Ti5O12 has a quasi-2D layered structure. Both electrodes can incorporate intercalated Na+ ions into their structures, however, 2D channels with partially occupied Na sites, providing broader pathways, can deliver higher reversible capacity than T-Na4Ti5O12. The tunnel structured sodium titanate form of Na2Ti7O15 was also investigated as a possible Na+ insertion host material for SIBs anodes. Li et al. proposed Na2Ti7O15 nanotubes on the Ti net substrate, which exhibited a high reversible capacity of 258 mA h g−1 at 50 mA g−1 and an excellent capacity retention of 96% after 200 cycles at 1.0 A g−1.364
Recently, as promising anode materials, layered structured sodium titanate compounds of O3-NaTiO2 and P2-Na0.66[Li0.22Ti0.78]O2 were introduced.360–362 Through the in situ X-ray diffraction studies, Wu et al. demonstrated the reversible O3–O′3 phase transition of O3-NaTiO2 and proposed the Na+ insertion/extraction mechanism (Fig. 28f). In an optimal voltage window of 0–1.6 V, approximately 0.5 mol of Na+ can be reversibly intercalated in NaTiO2, showing a reversible capacity of 152 mA h g−1 and stable cycle retention after 60 cycles.361 For P2-Na0.66[Li0.22Ti0.78]O2, a reversible capacity of 116 mA h g−1 was delivered at an average storage voltage of 0.75 V362 (Fig. 28g). In addition, it exhibited zero strain characteristics of only ∼0.77% volume change during sodium insertion–extraction, ensuring a potentially long cycle life for over 1200 cycles.
3.2 Conversion materials
Some kinds of transition metal oxide (TMO),365–399 transition metal sulfide (TMS)400–454 and transition metal phosphide (TMP)455–466 compounds can adopt Na+ ions through conversion reactions. Unlike intercalation and alloying reactions, where metal atoms are reversibly shuttled in and out of a host lattice, conversion reactions involve the chemical transformation of one or more of the atomic species into a host lattice to form a new compound.426 Depending on the transition metal, insertion–extraction or alloying–dealloying was combined with conversion reactions. Analogous to the reaction in LIBs, conversion materials have been considered as potential anode materials for SIBs due to their high theoretical specific capacities. However, large volume expansion/contraction upon the sodiation–desodiation process accelerated tremendous damage of electrodes, which led to the loss of electrical contact and subsequently rapid capacity fading. Moreover, sluggish mobility of Na+ ions due to the large ionic size (1.02 Å) is still a challenge to fully utilize their theoretical capacity. In recent years, to deal with such problems, strategies such as advances in the nanotechnology and/or carbon-conducting technique have been introduced, facilitating the development of high performance conversion materials as anodes for SIBs. In this section, the recent research achievements for transition metal oxides, transition metal sulfides and transition metal phosphide as Na conversion hosts are summarized and discussed.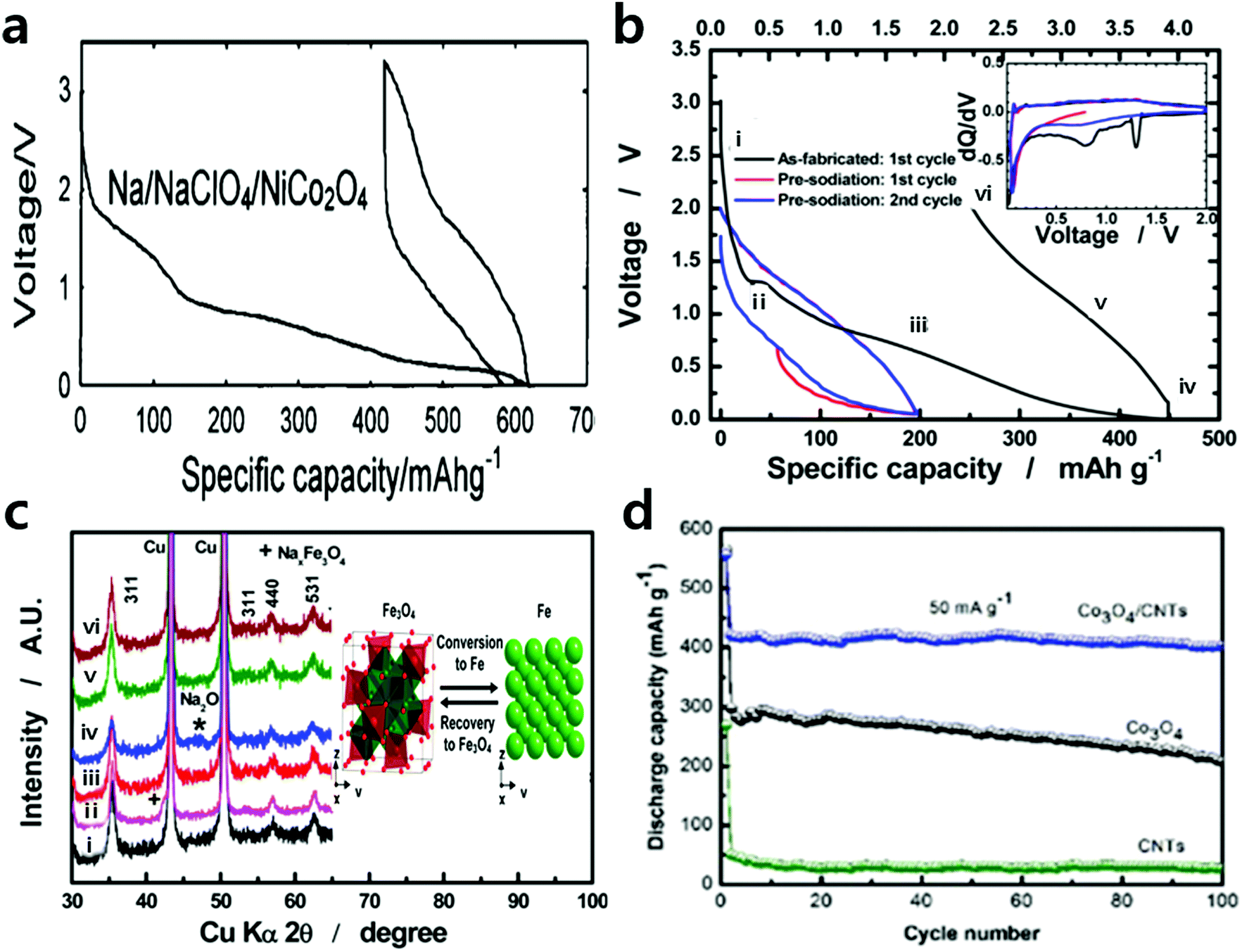 | ||
| Fig. 29 (a) Voltage profile of Na/NaClO4(EC:DMC)/NiCo2O4 cell between 3.3 and 0.01 V. (Reproduced with permission from ref. 365, Copyright 2002 American Chemical Society.) (b) The first charge–discharge cycle of a Na/C–Fe3O4 cell at a constant current density of 20 mA g−1 (0.1C-rate, 0–2 V) and related differential capacity curves (inset). (c) Ex situ XRD analysis of the C-Fe3O4 electrode at different discharging and charging state in (b) and scheme of the electrochemical process of the C-Fe3O4 cell. (Reproduced with permission from ref. 51, Copyright 2014 American Chemical Society.) (d) Cycling performance of CNTs, Co3O4 and Co3O4/CNTs electrodes. (Reproduced with permission from ref. 379, Copyright 2015 The Royal Society of Chemistry.) | ||
3.2.1.1. Iron oxides (Fe3O4, Fe2O3). Komaba et al. reported applicability of Fe3O4 in Na batteries through the insertion reaction in the voltage range of 1.2–4.0 V.366 Recently, Hariharan et al. proposed the possibility of Fe3O4 materials in the conversion reaction mechanism with Na+ ions at a discharge voltage of 0.04 V: Fe3O4 + 8e− + 8Na+ ↔ 3Fe + 4Na2O.367 Through the conversion reaction, a discharge capacity of 643 mA h g−1 was delivered during the initial cycles with a high Coulombic efficiency of 57%. Oh et al. suggested pitch carbon as a coating additive for nano-sized Fe3O4 (Fig. 29b). Oh et al. also observed the conversion reaction of the C/Fe3O4 electrode upon the sodiation–desodiation process51 (Fig. 29c). To further ensure electric conductivity, Park et al. introduced a composite of C/Fe3O4 embedded on carbon nanotubes, which delivered 440 mA h g−1 for the first discharge and 321 mA h g−1 for the first charge with a high Coulombic efficiency of 73%.368 More recently, Liu et al. synthesized extremely small Fe3O4 quantum dots on hybrid carbon nanosheets, which demonstrated a high capacity of 416 mA h g−1 at 0.1 A g−1 and a superior cycle retention of 70% for 1000 cycles at 1.0 A g−1.369 Fe2O3 has also been considered to be a promising electrode material for SIBs due to its excellent chemical stability, high capacity, easy fabrication, low cost and nontoxicity.373–377 Similar to Fe3O4 materials, sodium storage in Fe2O3 is mainly achieved via a reversible conversion reaction, by forming Fe nanoparticles dispersed in the Na2O matrix.
3.2.1.2. Cobalt oxides (Co3O4). Rahman et al. proposed the reversible conversion reaction mechanism of Co3O4 with sodium ion via cyclic voltammogram and ex situ XRD analyses: Co3O4 + 8Na+ + 8e− ↔ 4Na2O + 3Co.378 Based on the XRD results of the first cycle and after 20 cycles, Rahman et al. suggested that the conversion reaction is not completed in the first discharge to 0.01 V, and the conversion reaction tends to continuously occur during further cycling. Following the conversion reaction, a reversible capacity of 447 mA h g−1 and ∼86% capacity retention were demonstrated after 50 cycles. Later, to improve the electrochemical performances of Co3O4, Rahman et al. fabricated a carbon composite architecture of Co3O4/CNTs by combining a molten salt precipitation process and liquid plasma.379 CNTs as carbon additives in this composite anode provided several advantages during the sodiation–desodiation process: (1) significantly decreases the absolute stress/strain, (2) accommodates a large volume change and prevents aggregation, (3) its CNT matrix provides electronic conductivity and stability, and (4) reduces the ion or electron transfer time. The results showed a high capacity of 403 mA h g−1 at 50 mA g−1 and a superior rate capability of 190 mA h g−1 at 3.2 A g−1 (Fig. 29d).
3.2.1.3. Tin oxides. Recently, various nanostructured tin-based oxide (SnO, SnO2) materials and their composites have been proposed.383–390 Gu et al. visualized the sodiation–desodiation process and investigated the failure mechanism of SnO2 nanowire anodes via an in situ TEM technique383 (Fig. 30a). At the initial stage of the Na insertion process into SnO2, a displacement reaction occurs, leading to the formation of amorphous NaxSn nanoparticles dispersed in the Na2O matrix. Further sodiation led to crystallization to Na15Sn4 (x = 3.75) from NaxSn. At this stage, the nanowire experiences a huge volume expansion from 67 to 145 nm in diameter with the sodiation. Upon desodiation, NaxSn transforms into Sn nanoparticles. Associated with dealloying, pores are formed, leading to a structure of Sn particles confined in a hollow matrix of Na2O. These pores greatly increase electric impedance, therefore accounting for the poor cyclability of SnO2. Later, Lu et al. investigated the electrochemical properties and the sodium storage mechanism of SnO and SnO2 materials386 (Fig. 30b). Based on ex situ XRD analysis and SAED pattern results, they confirmed that SnO and SnO2 are able to store sodium in their structure through the combined conversion and alloying reactions: (1) SnO → SnO + 2Na+ + 2e− ↔ Sn + Na2O, Sn + Na2O + xNa+ + xe− ↔ NaxSn + Na2O, (2) SnO2 → SnO2 + 4Na+ + 4e− ↔ Sn + 2Na2O, Sn + 2Na2O + xNa+ + xe− ↔ NaxSn + 2Na2O (Fig. 30c). According to their report, low oxygen contents of SnO showed better electrochemical performances, which implies that the Sn
:O ratio and the respective Sn:Na2O ratio in the converted electrode play important roles in delivering the capacity (Fig. 30d). Recently, a strategy to overcome such failures and maximize the utilization of the high theoretical capacity of SnO2 (∼782 mA h g−1) has been introduced such as composite anodes with porous carbon, MWCNTs, and graphene.384–390 Xie et al. synthesized SnO2 with nitrogen-doped graphene nanohybrids via an in situ hydrothermal method and this composite anode delivered an initial reversible capacity of 339 mA h g−1 at 20 mA g−1 and excellent capacity retentions even at a high current density of 640 mA g−1390 (Fig. 30e).
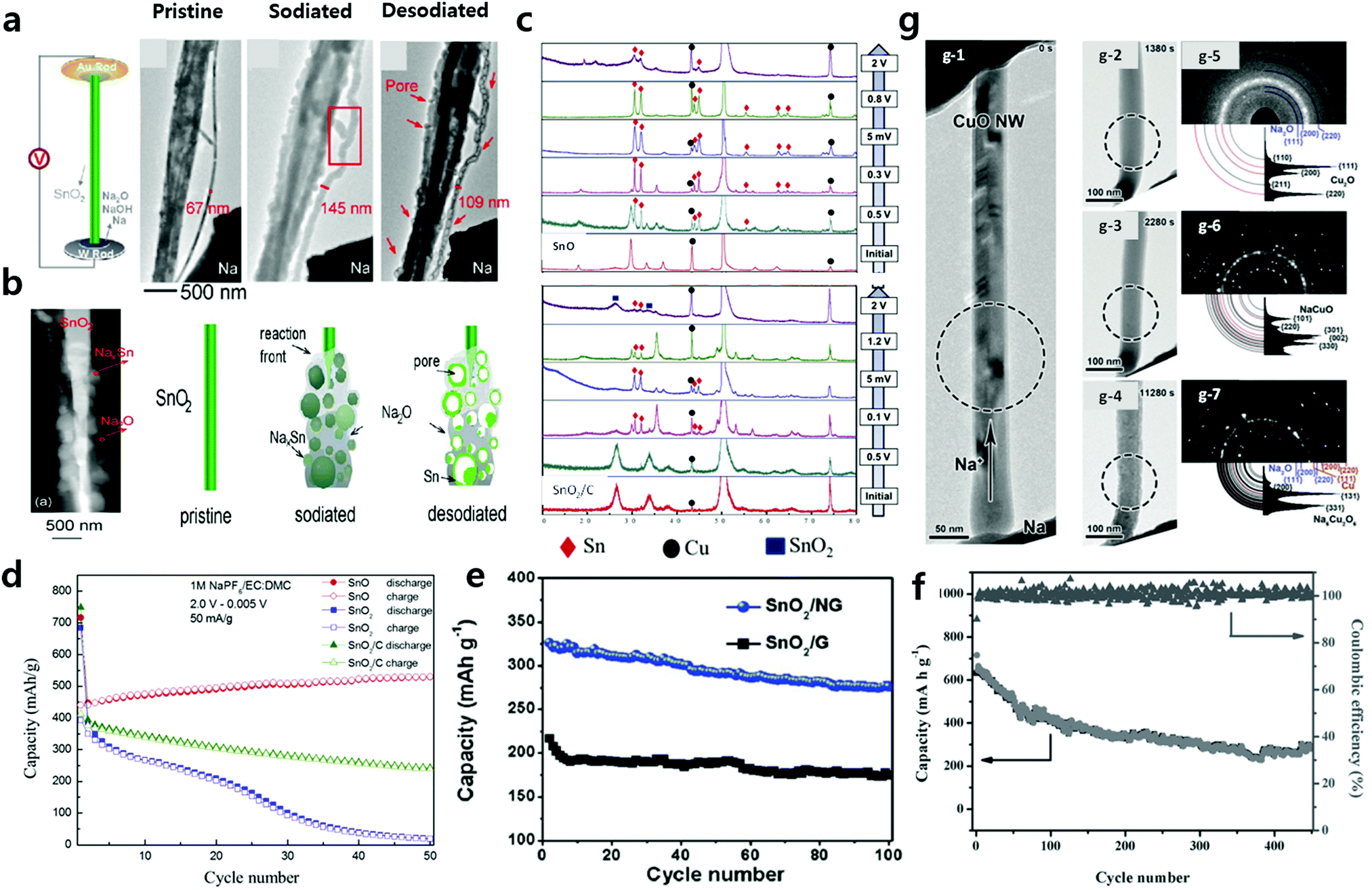 | ||
| Fig. 30 (a) TEM images of pristine, sodiated, and desodiated states of SnO2 nanowire. (b) STEM Z-contrast image showing the reaction front of the SnO2 nanowire (left) and schematic drawing showing the morphology evolution of the SnO2 nanowire upon Na insertion and extraction (right). (Reproduced with permission from ref. 383, Copyright 2013 American Chemical Society.) (c) Ex situ XRD profiles of SnO (top) and SnO2/C (bottom) electrodes over the first charge/discharge cycle. (d) Cycle life of SnO, SnO2, and SnO2/C electrodes. (Reprinted from ref. 386, Copyright 2015, with permission from Elsevier.) (e) Cycling performance of SnO2/NG and SnO2/G composites at a current density of 20 mA g−1. (Reproduced with permission from ref. 390, Copyright 2015 The Royal Society of Chemistry.) (f) Cycling performance of the binder-free CuO nanorod array (CAN) electrode at a high current density of 200 mA g−1. (Reproduced from ref. 392 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) (g) In situ observation of the sodiation process in CuO nanowires using TEM. (Reproduced with permission from ref. 394, Copyright 2015 The Royal Society of Chemistry.) | ||
3.2.1.4. Copper oxide. Copper-based oxide materials have also been proposed due to the Earth's abundant distribution, chemical stability, and high theoretical capacity.391–396 Klein et al. reported that Cu2O can be applicable and thereby exhibited a high capacity of ∼600 mA h g−1 at 0.1C.243 Yuan et al. proposed flexible and porous CuO nanorod arrays by engraving Cu foils. The arrays delivered a high specific capacity of 640 mA h g−1 at 20 mA g−1 and good cycle retentions over 400 cycles392 (Fig. 30f). Liu et al. demonstrated the morphology change and phase transformations in CuO nanowires during the sodiation process. And, based on the in situ TEM results, Liu et al. suggested detailed conversion mechanisms as follows: 2CuO + 2Na+ + 2e− → Cu2O + Na2O, Cu2O + Na2O → NaCuO, 7NaCuO + Na+ + 2e− → Na6Cu2O6 + Na2O + 5Cu394 (Fig. 30g). However, similar to the other metal oxides, Cu-based oxides are still hindered by a large volume change during cycling. Recently, to enhance the electronic conductivity and the accommodation of volume variation upon cycling, Lu et al. proposed the micro-nanostructured CuO/C spheres. The as-prepared CuO/C spheres exhibited a high capacity of 402 mA h g−1 after 600 cycles at a current density of 200 mA g−1 and superior rate capabilities.395
3.2.2.1. Cobalt sulfides. Cobalt sulfide is an interesting metal chalcogenide semiconductor material and has a number of applications.400–403 Recently, superior Li-storage properties of cobalt sulfide were achieved by designing a novel nanoarchitecture and fabricating hybrid nanocomposites with various carbon additives.404–406 Shadike et al. fabricated CoS2 and CoS2–MWNCT materials and investigated their electrochemical performances with a Na storage mechanism in ether-based electrolytes and commonly used carbonate-based electrolytes. They also investigated the evolution processes of CoS2 and CoS2–MWCNT during cycling via ex situ TEM407 (Fig. 31a). During the sodiation/desodiation process, CoS2 can accommodate Na+ ions as follows: CoS2 + 4Na+ + 4e− ↔ Co + 2Na2S. Both CoS2 and CoS2–MWCNT electrodes have a similar sodium storage mechanism, but their electrochemical performances are quite different. Using MWCNTs as carbon additives provides several advantages such as 3D electron conductive networks with a high surface area, which facilitate the fast penetration of sodium ions and diffusion of electrolytes. As a result, the CoS2–MWCNT electrode exhibited a high initial discharge capacity of 826 mA h g−1 with a high Coulombic efficiency of 93%, and a stable cycle life for 100 cycles in an ether-based electrolyte (1 M NaCF3SO3–DGM) (Fig. 31b).
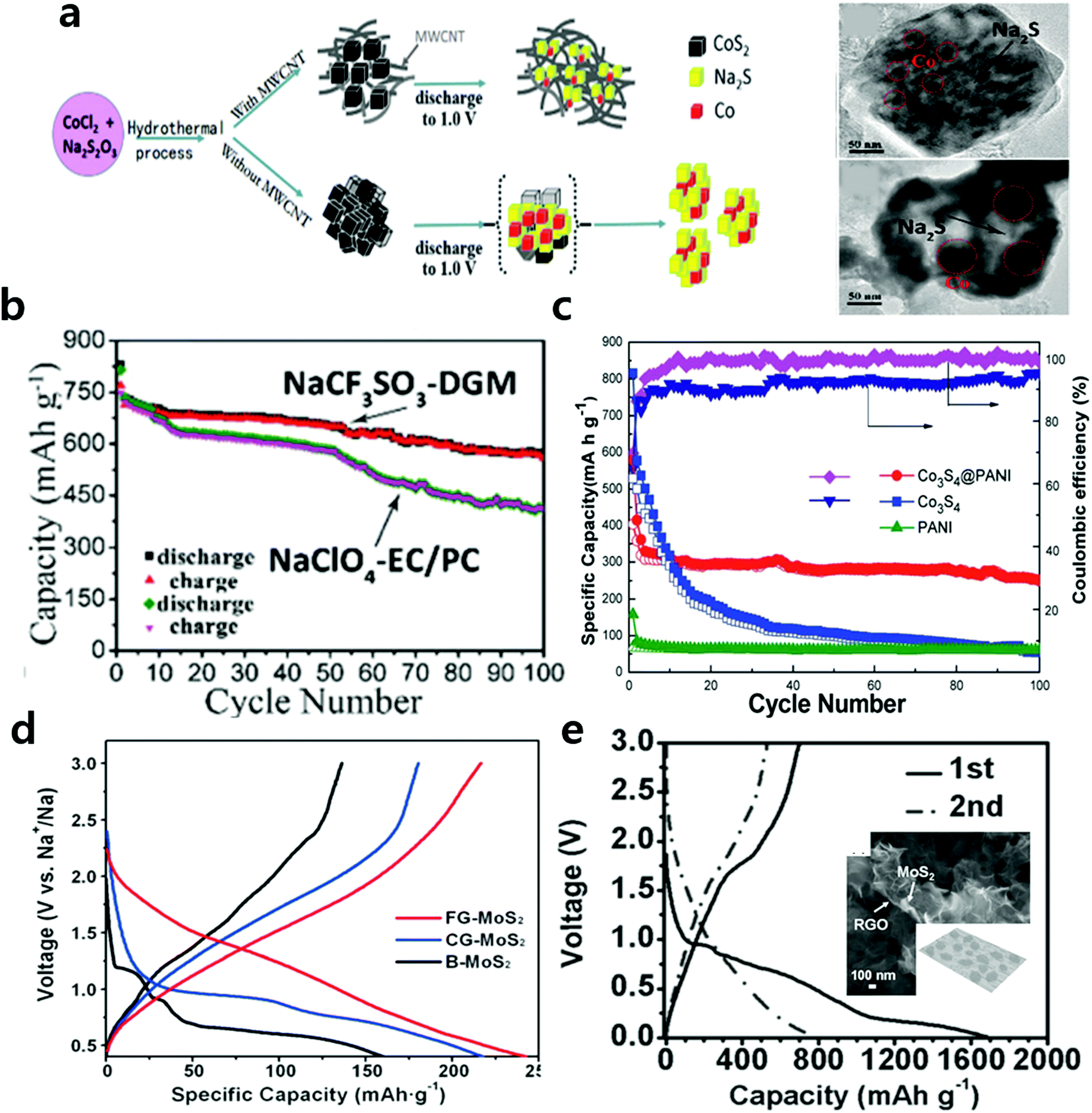 | ||
| Fig. 31 (a) Schematic diagram of the evolution process of the as-prepared CoS2–MWCNT and bare CoS2 electrodes before and after the first discharge to 1.0 V (left). And, ex situ TEM images of sodiated CoS2–MWCNT and bare CoS2 particles (right). (b) CoS2–MWCNT electrodes at a current density of 100 mA g−1 in 1.0 M NaCF3SO3–DGM and 1.0 M NaClO4–EC/PC. (Reproduced with permission from ref. 407, Copyright 2015 The Royal Society of Chemistry.) (c) Cycling performance of Co3S4, PANI, Co3S4@PANI electrodes. (Reproduced with permission from ref. 410, Copyright 2016 The Royal Society of Chemistry.) (d) Charge and discharge curves of the as-prepared FG-MoS2, CG-MoS2, and B-MoS2 at the first cycle. (Reproduced from ref. 413 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) (e) Galvanostatic charge–discharge profiles of MG-3 and SEM and TEM images of MG-3 (inset image). (Reproduced from ref. 419 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Later, Peng et al. proposed the unique hybrid nanocomposite of cobalt sulfide (CoS) nanoplates anchored onto reduced graphene oxide (rGO) sheets and demonstrated the impressive high specific capacity of 540 mA h g−1 at 1 A g−1, excellent rate capability, and superior cycle retention of 88% after 1000 cycles in Na cells.409 More recently, Zhou et al. fabricated Co3S4@PANI (polyaniline) nanotubes, in which polyaniline is uniformly coated on both the exterior and inner surfaces of Co3S4 nanotubes (Fig. 31c).410 Based on the CV results in the potential window of 0.05–2.0 V (vs. Na+/Na), they claimed that Co3S4 electrodes can store Na+ ions through the combined insertion and conversion reaction. In the first scan, a cathodic peak at 0.98 V is commonly assigned to an initial process of the Na+ insertion reaction: Co3S4 + xNa+ + xe− → NaxCo3S4. The peak at 0.72 V is related to the conversion reaction of Co3S4 with Na and the formation of a solid electrolyte interphase: NaxCo3S4 + (8 − x)Na+ + (8 − x)e− → 4Na2S + 3Co. The flexible PANI layers coated on both outer and inner surfaces of Co3S4 nanotubes would form “protective layers”, thus preventing structure collapse and pulverization of the Co3S4 nanotubes during cycling.
3.2.2.2. Molybdenum sulfides. In the case of molybdenum disulfide (MoS2), Mo and S atoms are covalently bonded to form 2D S–Mo–S trilayers and the adjacent planes are stacked by van der Waals interactions, which facilitate intercalation of the large Na+ ion.411,414 According to previous reports, MoS2 can store the Na+ ions through the intercalation and/or conversion reactions depending on the operation voltage window.413,415–417,419 The electrochemical reaction of MoS2 is interpreted as the following two step reactions: MoS2 + xNa+ + xe− → NaxMoS2 (above 0.4 V), NaxMoS2 + (4 − x)Na+ + (4 − x)e− → 2Na2S + Mo (below 0.4 V).413 Hu et al. prepared MoS2 nanoflowers with expanded interlayer spacing of the (002) plane, which exhibited stable electrochemical performances that followed an intercalation reaction by controlling the cut-off voltage to 0.4–3.0 V413 (Fig. 31d). As a result, this material delivered a high discharge capacity of 350 mA h g−1 at 50 mA g−1 and stable cycle retention for over 1500 cycles. On the other hand, the intercalation reaction of MoS2 at a limited voltage above 0.4 V delivered a lower specific capacity in consideration of their theoretical capacity of 668 mA h g−1 when 4Na+ ions reacted with MoS2 through the conversion reaction.417,419 However, a conversion type chemical reaction usually brings about a serious volume change to the electrode materials and sluggish kinetics for reconstruction of the original active materials. Recently, to overcome such problems through the electrochemical conversion reactions of MoS2 (below 0.4 V), high conductivity carbon additives and novel nanoarchitecture design were proposed.414–422 Su et al. prepared few-layer MoS2 nanosheets and conducted the electrochemical test in the voltage range of 0.01–3.0 V.418 The results show that few-layer MoS2 nanosheets exhibited a high capacity of 530 mA h g−1 and a high rate capability. Xie et al. prepared a series of sheet-on-sheet structured MoS2/RGO nanocomposites and investigated the effect of heterointerfacial areas on sodium storage performances.419 Computational calculation and experimental results show that the 2D MoS2/rGO heterointerface can increase the conductivity of MoS2 and capture more Na atoms due to maintaining the high diffusion mobility of Na+ ions on the MoS2 surface and high electron transfer efficiency from Na to MoS2, respectively. As a result, this material delivered a high capacity of 352 mA h g−1 even at a high current density of 640 mA g−1 in the voltage range of 0.01–3.0 V (Fig. 31e).
3.2.2.3. Iron sulfides. Natural and/or synthetic FeS2 materials have been demonstrated to be potential electrode materials among metal sulfides due to their high theoretical capacity of 894 mA h g−1 and environmental friendliness. Ahn and co-works reported a Na/synthetic FeS2 battery for the first time423,424 (Fig. 32a). Later, Hu et al. demonstrated room temperature sodium storage performances of the FeS2 microspheres with only the intercalation reaction by simultaneously selecting a compatible ether-based electrolyte and tuning the cut-off voltage to 0.8 V. The intercalation reaction mechanism between Na+ ions and FeS2 within the operation voltage range of 0.8–3.0 V is as follows: FeS2 + xNa+ + xe → NaxFeS2 (x < 2),429 The resulting exhibited high capacity of 170 mA h g−1 at a high current density of 20 A g−1 and superior long term cyclability over 20
000 cycles (capacity retention of ∼90%) (Fig. 32b). Based on the ex situ XRD and TEM measurements, they also observed the structural evolution of FeS2 during the electrochemical reaction with Na+ ions (Fig. 32c). The XRD patterns collected at the charge-end state after the 50th cycle (VIII) indicates the formation of a layered compound of trigonal NaxFeS2 with x around 1.6. Also, the layered structures of FeS2 could be maintained within the selected potential window of 0.8–3.0 V, enabling the highly reversible intercalation reaction. On the other hand, Walter et al. applied nanocrystalline FeS2 materials within the operation voltage range of 0.02–2.5 V. The conversion reaction mechanism between Na+ ions and FeS2 is as follows: FeS2 + 2Na+ + 2e− → Na2FeS2, Na2FeS2 + 2Na+ + 2e− → 2Na2S + Fe.430 Upon sodiation below 0.8 V, FeS2 nanocrystals led to the formation of Na2S involving most likely only amorphous phases. This amorphous state can effectively reduce mechanical stress upon expansion and contraction during cycling. The FeS2 nanocrystals delivered a high capacity above 500 mA h g−1 for 400 cycles at a current density of 1 A g−1. Compared to pyrite (FeS2), a few studies on ferrous sulfide (FeS) are reported due to poor cyclability and rate capability.431,432 Recently, Wei et al. proposed the flexible and self-supported carbon-coated FeS on carbon cloth films, which display high reversible capacity and superior rate capability.431
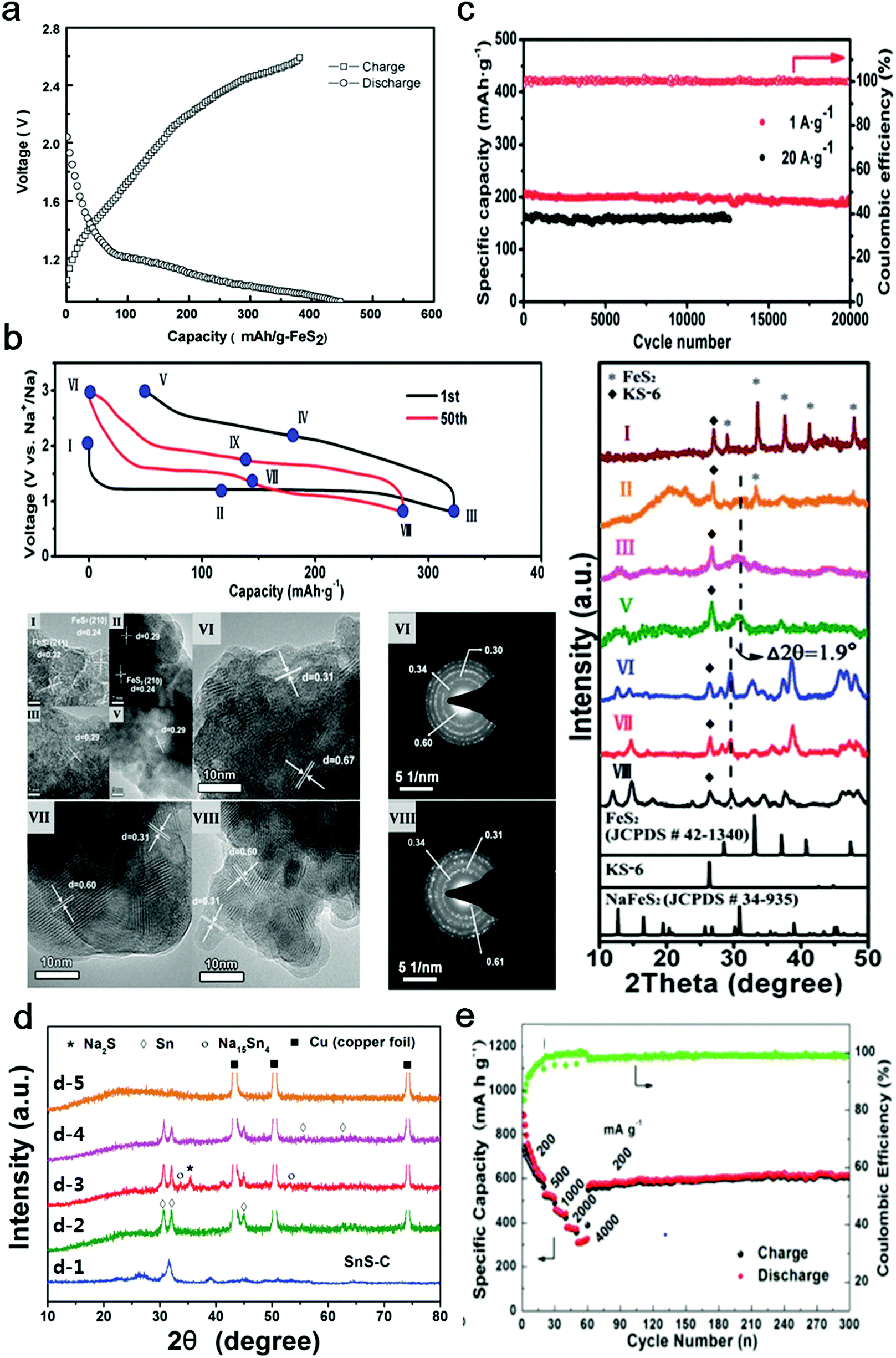 | ||
| Fig. 32 (a) Charge–discharge voltage profiles of the Na/FeS2 cell at room temperature. (Reprinted from ref. 423, Copyright 2008, with permission from Elsevier.) (b) Structural evolution of FeS2 during the electrochemical reaction with sodium: TEM images (left) and ex situ XRD results (right). (c) Cyclic performance of FeS2 microspheres. (Reproduced with permission from ref. 429, Copyright 2015 The Royal Society of Chemistry.) (d) Ex situ XRD patterns of the SnS–C electrode at different discharging and charging states. (d-1: fresh electrode; d-2: after 1st discharging to 0.6 V; d-3 after 1st discharging to 0.01 V; d-4 after 1st charging to 0.8 V; d-5 after 1st charging to 2.0 V.) (Reproduced with permission from ref. 433, Copyright 2014 The Royal Society of Chemistry.) (e) Rate capability and long-term cyclability with Coulombic efficiency of the SnS2/G-20 electrode. (Reproduced with permission from ref. 442, Copyright 2015 The Royal Society of Chemistry.) | ||
3.2.2.4. Tin sulfides. Tin-based sulfide (SnS, SnS2) compounds have attracted considerable attention due to their high theoretical capacity with combined conversion and alloying electrochemical reactions.433–437 Wu et al. proposed tin-sulfide nanocomposite (SnS–C) anode materials based on combined conversion and alloying reactions: SnS + 2Na+ + 2e− ↔ 5Na2S + Sn (conversion reaction), Sn + 3.75Na+ + 3.75e− ↔ Na3.75Sn (alloying reaction)433 (Fig. 32d). The prepared SnS–C composite has a small crystalline size of SnS and good carbon coating, which synergistically facilitates electrochemical utilization and maintains the structural integrity. As a result, the prepared SnS–C composite exhibited a high Na storage ability delivering a capacity of 568 mA h g−1 at 20 mA g−1 and an excellent cycling stability of 97.8% after 80 cycles as well as a high-rate capability. Later, Zhu et al. proposed a 3D porous interconnected metal sulfide/carbon nanocomposite by the ESD technique without adding carbonaceous materials such as carbon nanotubes and graphene.436 SnS2 has also been applied as an anode material for SIBs.438–448 SnS2 has a sandwich structure that consists of covalently bonded S–Sn–S trilayers separated by a relatively large van der Waals interaction.438 A large interlayer d-spacing of 5.90 Å can effectively accommodate Na+ ions.439–441 In addition, intermediate products of amorphous NaS2 suppress the pulverization and aggregation during Na–Sn alloying reactions.440 Liu et al. synthesized exfoliated-SnS2 restacked on graphene that ultrasmall SnS2 nanoplates (with a typical size of 20–50 nm) composed of 2–5 layers are homogeneously decorated on the surface of graphene.442 This unique structure facilitates Na+ ion diffusion and delivers a high capacity of 650 mA h g−1 at 200 mA g−1 with stable cyclability at ∼610 mA h g−1 without notable capacity fading for 300 cycles (Fig. 32e). SnS2 can store the Na+ ions through following three step processes: (1) intercalation reaction: xNa+ + SnS2 + xe− → NaxSnS2, (2) conversion reaction: 4Na+ + SnS2 + 4e− → 2Na2S + Sn, (3) alloying reaction: Sn + 3.75Na+ + 3.75e− → Na3.75Sn. Such improved electrochemical performances of conversion materials are attributed to the electro-conducting carbons. The introduction of carbon additives such as graphene and/or carbon nanotubes into active materials is indispensable to have advantages over conversion materials such as effective stress relief, accommodation of large volume expansion/shrinkage, and facilitation of electron and Na+ ion transport.
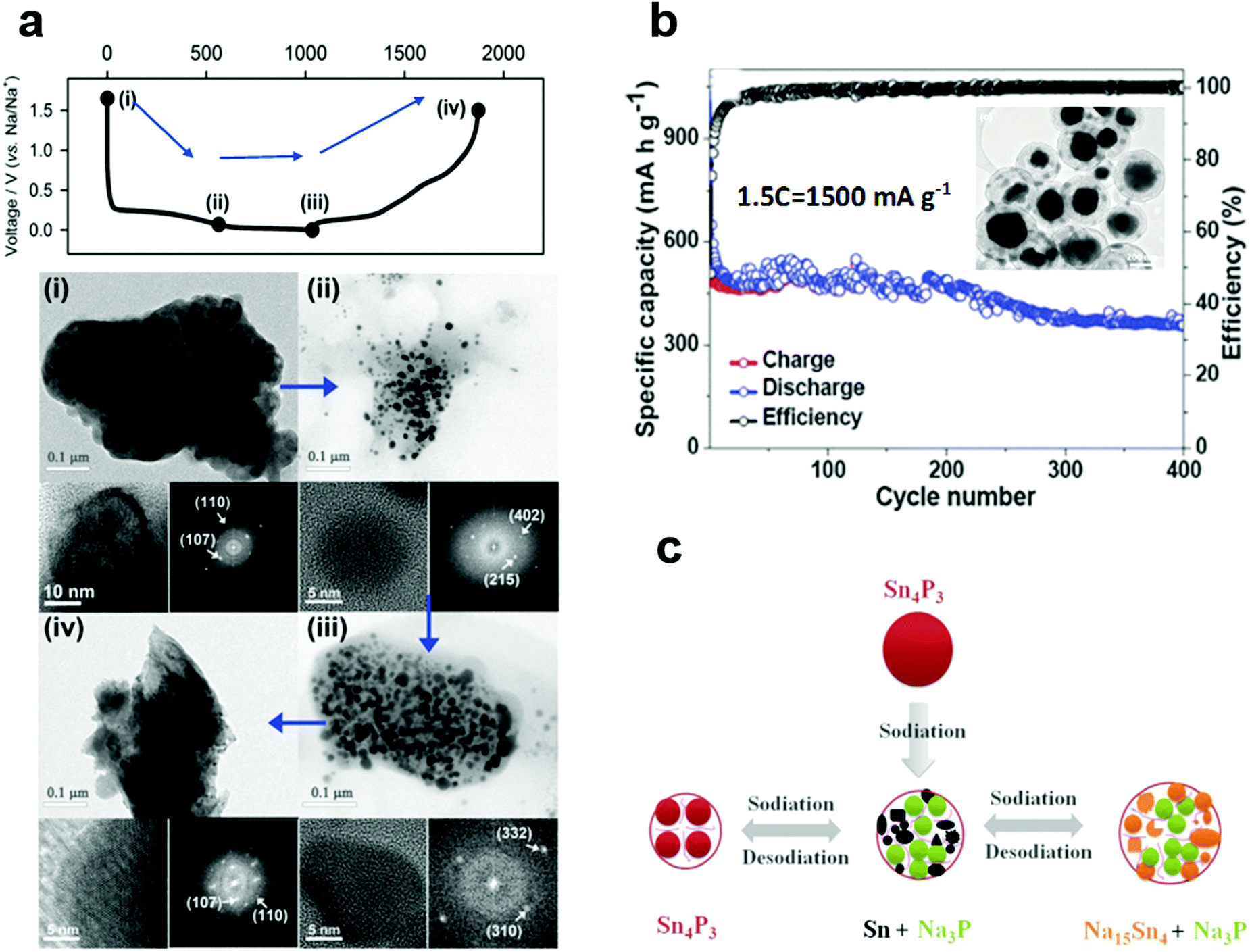 | ||
| Fig. 33 (a) First galvanostatic charge and discharge profiles of the Sn4P3 electrode (top). The ex situ HR-TEM bright-field images, enlarged HR-TEM images and corresponding FFT patters at each point. (Reproduced from ref. 460 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) (b) Long cycling performances of yolk–shell Sn4P3@C nanospheres at 1.5C. (Reproduced with permission from ref. 463, Copyright 2015 The Royal Society of Chemistry.) (c) Self-healing effect of the conversion reaction for the Sn4P3 anode. (Reproduced with permission from ref. 465, Copyright 2016 American Chemical Society.) | ||
3.3 Alloying reaction materials
Na+ insertion materials, such as carbonaceous materials and titanium-based oxide compounds, have been successfully applied as Na storage materials that deliver a reasonable capacity with relatively small volume expansions during the electrochemical insertion/extraction reaction with Na.241,300 However, these materials still suffer from limited capacity utilization due to their intrinsic constraint ascribed to their structures, which lowers the specific energy density of SIBs. Similar to the conversion materials, alloying materials can be suggested as attractive anodes for SIBs because they can store a large number of sodium ions in the host structure with a relatively low operating potential (below 1.0 V).244,245 Multiple reactions with Na per single atom produce a high specific capacity during the alloying–dealloying reactions.99 Metals (Sn, Bi), metalloids (Si, Ge, As, Sb) and polyatomic nonmetal compounds (P) in group 14 (Fig. 34) or 15 elements (Fig. 37) in the periodic table have been widely studied as potential anode materials for SIBs. However, depending on the host materials and electrochemical sodiation levels, the large Na+ ion causes huge volume changes during the alloying–dealloying reaction. This repetitive volume changes under the constraints imposed by the battery packaging give rise to complex mechanical stresses in active particles, ultimately leading to their fracture or pulverization.240 To date, through various experimental works focusing on electrochemical and mechanical responses of these alloys to Na interactions, the desirable architectures and/or enhanced electrode designs have been investigated. In this section, we specifically discussed group 14 and group 15 elements with a focus on their Na alloy reaction mechanisms and summarize various strategies for high performance sodium anode materials.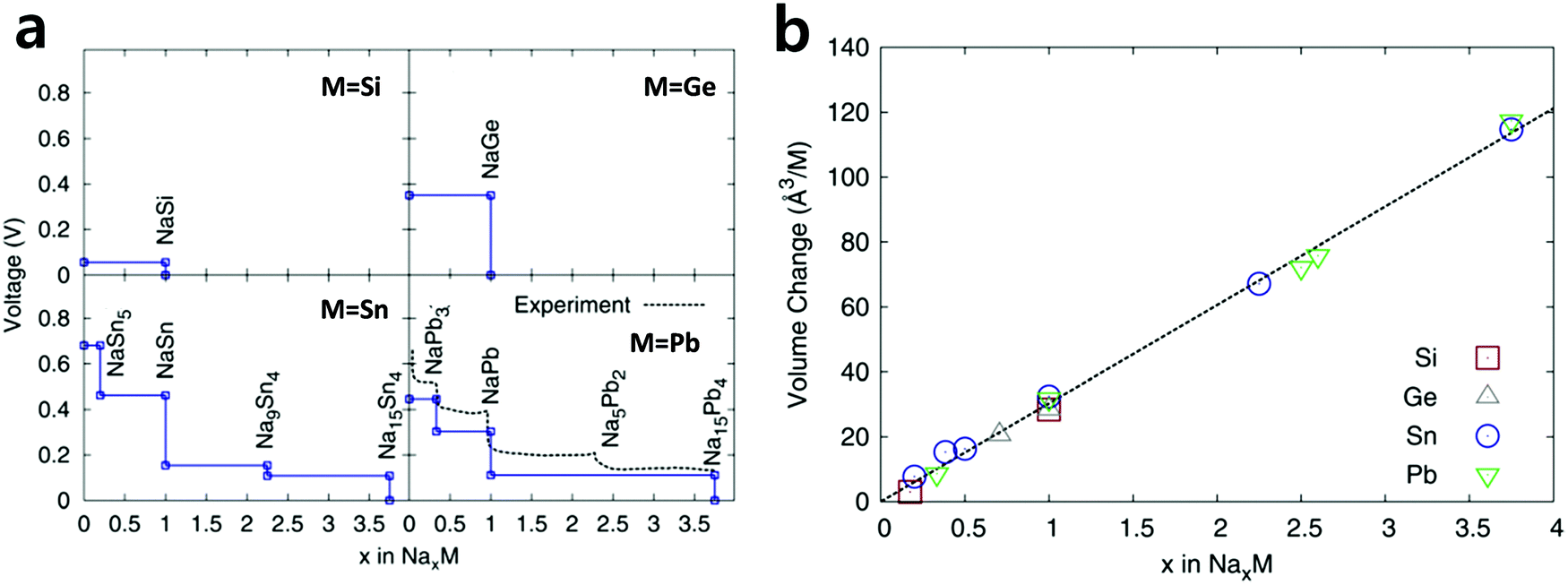 | ||
| Fig. 34 (a) Na–M voltage curves calculated using DFT and (b) the volume changes for group 14 elements in periodic table. (M = Si, Ge, Sn, and Pb.) Experimental values are also given if available. (Reproduced with permission from ref. 244, Copyright 2011 The Electrochemical Society.) | ||
3.3.1.1. Silicon. In recent years, Si-based anode materials were intensively studied for LIBs due to their abundance in Earth's crust and their high specific capacity through electrochemical alloying reactions with Li.467,468 Theoretically, silicon can uptake 4.4 Li+ ions per Si atom and deliver a high specific capacity of 4000 mA h g−1. Morito et al. firstly demonstrated the phase diagram between Na and Si, which indicated the fully sodiated form of Na–Si.469 However, based on a single-atom diffusion model, Morito et al. deduced that bulk Si is not a promising anode material for Na batteries because Si can only uptake 1Na per Si atom and exhibit poor Na diffusion kinetics469 (Fig. 34a). Through the computational calculation, desirable electrode design to facilitate Na+ intercalation and migration into Si was suggested such as structural modification and control of the activation barrier.470–476 Structurally modified Si was predicted to demonstrate better electrochemical performance such as in amorphous Si due to more favorable binding between Na and Si. Reasonable activation barriers for Na+ diffusion were also predicted, where 0.4 eV was required for Na+ migration in amorphous Si.457 Based on theoretical works, Xu et al. experimentally proved reversible electrochemical Na+ ion uptake in Si for the first time475 (Fig. 35a and b). Xu et al. prepared nanoparticles containing both amorphous and crystalline Si, which demonstrated an excellent reversible capacity of 279 mA h g−1 at 10 mA g−1 and a stable capacity retention of 248 mA g−1 after 100 cycles at 20 mA g−1. Also, through the various analysis techniques, they proposed the possible Na storage mechanism as follows: xNa + Si ↔ xNaSi + (1 − x)Si (sodiation), NaSi ↔ Na(1 − x)Si + xNa (desodiation). More recently, Zhang et al. precisely investigated the sodiation/desodiation behavior of microsized and nanosized crystalline-Si, hereafter referred to as c-Si, electrodes during the Na+ insertion/extraction processes via cyclic voltammogram, operando XRD and Raman analysis.476 The operando XRD patterns reveal that no structural conversions or reversible reactions could proceed in the microsized c-Si electrode, and the capacity of the microsized c-Si electrode is negligible. On the other hand, the voltage plateau of the nanosized c-Si electrode below 0.5 V in the discharge profile is consistent with the reduction peak at 0.2 V in the CV curve, which is related to the Na ion uptake and the alloying process with c-Si (Fig. 35c). It means that the electrochemical behavior of c-Si strongly depends on the particle size. The operando Raman analysis results demonstrated that an irreversible crystal structure transformation from c-Si to amorphous-Si takes place during the first sodiation process. This newly generated a-Si is beneficial for the reversible Na+ insertion reaction due to its more disordered crystal structure (Fig. 35d). According to their report, the whole process includes four phases along with a growing degree of sodiation. In stage one, Na ions diffuse along the ion channels and accumulate at tetrahedral sites between (111) planes because this position is the most stable site for Na-ion insertion. In stage two, along with the increase of the concentration of Na+, the bonds of Si–Si are broken, and Na atoms make new bonds with Si atoms in stable positions. In stage three, further sodiation leads to the break of the majority of the Si–Si bonds, and crystalline Si is transformed into an amorphous Na–Si alloy. In stage four, after the desodiation process, the amorphous structure is maintained, leading to the a-Si structure (Fig. 35e).476
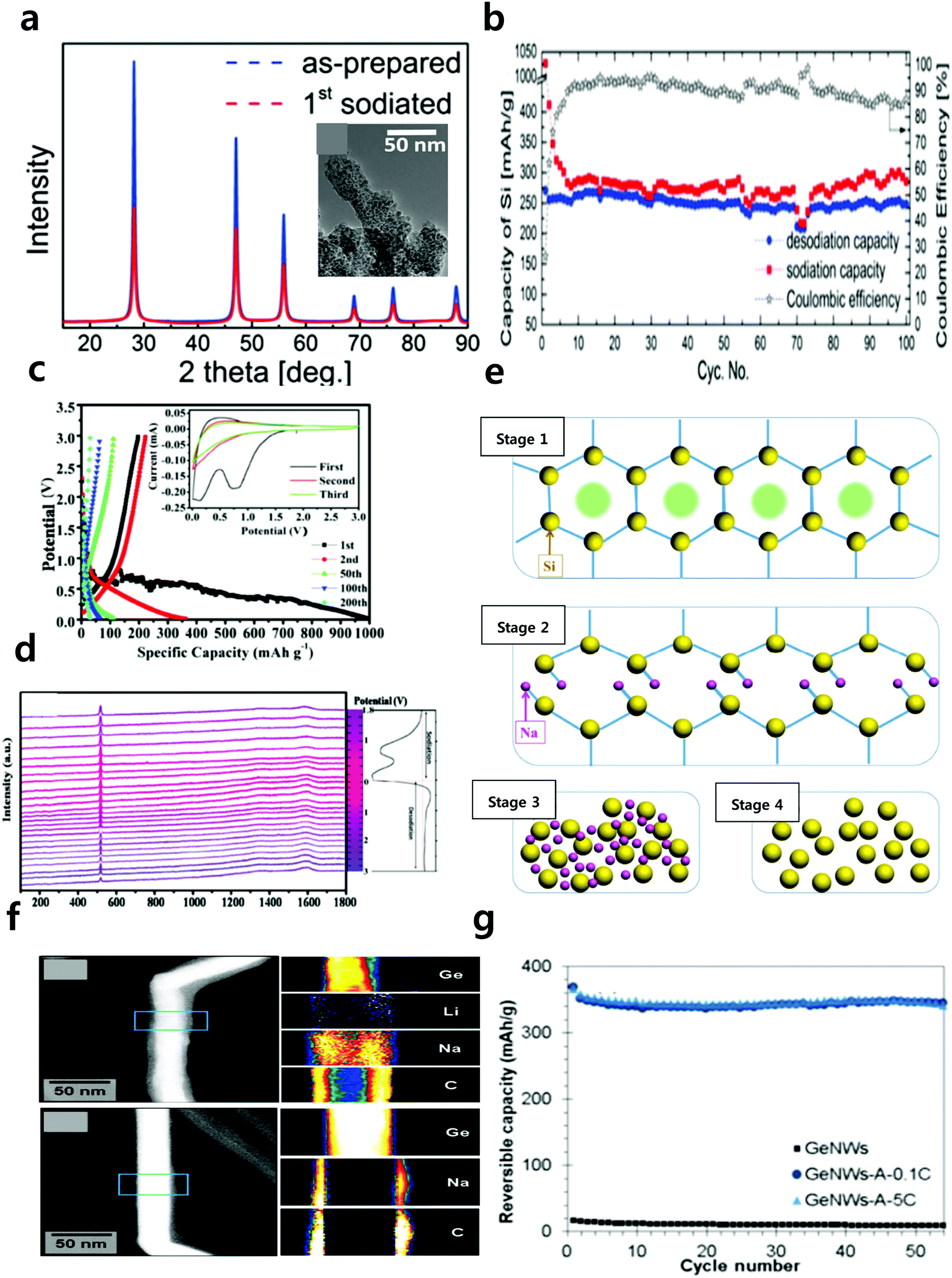 | ||
| Fig. 35 (a) X-ray diffraction patterns on as-synthesized Si nanoparticles and inset image show the morphology of sodiated Si NP electrode after charge/discharge for 100 cycles at 20 mA g−1. (b) Cycling test of Si NP electrodes in the voltage range of 0.01–2.8 V. (Reproduced from ref. 475 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) (c) Charge–discharge profiles for selected cycles at 500 mA g−1 (inset: the first three CV curves for the nanosized c-Si electrode), (d) in operando Raman analysis of the electrochemical behavior towards Na+ for the nanosized c-Si electrode and (e) schematic illustrations explaining the changes in the nanosized c-Si during cycling. (Reproduced from ref. 476 with permission, Copyright 2016 Wiley-VCH Verlag GmbH & Co. KGaA.) (f) TEM analysis of GeNWs-A-0.1C and GeNWs after initial sodiation process and HAADF micrograph and EELS elemental maps of Ge, Na, Li, and C. (g) Reversible capacity as a function of cycle number for GeNWs and GeNWs-A electrodes. (Reproduced with permission from ref. 481, Copyright 2014 American Chemical Society.) | ||
3.3.1.2. Germanium. Germanium has a similar chemistry to silicon, bonding with a maximum of one Na atom.477,478 The theoretical calculation predicted that sodium could alloy with germanium to form Na–Ge compounds which delivers a theoretical capacity of 369 mA h g−1.479 However, similar to silicon, germanium is also theoretically predicted not to be capable of storing Na in its crystalline structure because the large ionic size of sodium compared to lithium results in much higher activation energy for hopping between interstitial sites in the lattice (0.51 eV for lithium vs. 1.5 eV for sodium).479 Therefore, to overcome the sluggish kinetics of Na+ in germanium, a novel nanostructure and/or electrode design was introduced such as amorphous germanium in thin film and nanowire form. Baggetto et al. prepared a germanium thin film electrode and demonstrated the reversible reaction with a high discharge capacity of 350 mA h g−1, which is close to the theoretical value.480 The shapes of the sodiation and desodiation voltage profiles are quite flat which indicates that the sodiation reaction proceeds via a two-phase reaction mechanism: a reaction front separating the sodium-rich and sodium-poor phases propagates through the material as the reaction progresses. Recently, Kohandehghan et al. proposed amorphous germanium nanowires and thin films with a single lithiation–delithiation activation process.481 Activation with Li induces amorphization in germanium nanowires and thin films, which reduces the barrier for nucleation of the NaxGe phase. Also, a dense distribution of nanopores provides an additional Na diffusion path during the sodiation–desodiation process. The TEM and EELS mapping results clearly show that activated germanium nanowires (GeNWs-A) uniformly take up more Na+ ions at the sodiated state compared to unactivated germanium nanowires (GeNWs). For the case of GeNWs, the Na+ ion is primarily present on its surface at the sodiated state, being associated with both the SEI layer and the irreversibly formed sodium oxide (Fig. 35f) As a result, GeNWs-A delivered a high reversible capacity of 355 mA h g−1 at a 1C-rate and superior rate capabilities (Fig. 35g).
3.3.1.3. Tin. Sn has been regarded as one of the most promising anode materials owing to its high theoretical specific capacity of 847 mA h g−1, based on the full sodiation state of Na15Sn4.482–495 Based on the theoretical calculations, Chevrier and Ceder et al. proposed a voltage profile for sodium insertion into tin compounds244 (Fig. 34a). The phase diagram of Na–Sn indicated that the sodiation of Sn progressed in a series of steps: Sn → NaSn5 → NaSn → Na9Sn4 → Na15Sn4.482–484 Komaba et al. experimentally demonstrated that Sn undergoes a reversible electrochemical redox reaction to reversibly form Sn–Na intermetallic phases.482 Based on the DFT calculations and in situ X-ray diffraction results, Ellis et al. suggested that electrochemical sodiation of Na with Sn proceeds through the following reaction steps: plateau 1: Na + Sn → NaSn3*, plateau 2: Na + NaSn3* → a-NaSn, plateau 3: 5Na + 4(a-NaSn) → Na9Sn4, plateau 4: 6Na + Na9Sn4* → Na15Sn4 (a-amorphous, *-new crystalline phase)483 (Fig. 36a). Huang's group investigated the microstructural evolution and phase transformation with volumetric expansions of tin nanoparticles during electrochemical sodiation via the in situ transmission microscopy technique484 (Fig. 36b). According to their report, Sn undergoes a two-step sodiation process to form amorphous NaSn2 (56% expansion) in the first step and sequentially to form amorphous Na9Sn4, Na3Sn (336% expansion), and crystalline Na15Sn4 (420% expansion) phases in the second step. Later, Wang et al. visualized the 3D structural/chemical evolution of the Sn electrode during multi-electrochemical cycles (sodiation–desodiation) via in situ synchrotron hard X-ray nanotomography485 (Fig. 36c). They also suggest two important critical sizes of 0.5 μm for low complexity and 1.6 μm for high complexity, which provide new insights into the failure mechanism and materials fracture in SIBs (Fig. 36d). Such various strategies have been tested to clarify the electrochemical sodiation mechanism of Sn; however, there remains disparity between computational and experimental works.486 And the extremely high volumetric expansion–contraction during the uptake–release process of 3.75 Na atoms in Sn is the main obstacle for full use of the tin-based anode. Because it will lead to cumulative pulverization, leading to the loss of electrical contact and accelerating the capacity decay during cycling. Therefore, most works focused on dealing with the serious volume change during alloying–dealloying reactions. Carbon-conducting techniques such as carbon coating and/or formation of composites with a 2D or 3D carbon matrix are very effective for buffering the volume strain and creating the electric conducting pathways.487–495 Xie et al. suggested a unique free standing electrode configuration with 3D architectures. They prepared a free-standing electrode composed of core–shell structured Sn@CNT nanopillar arrays on a carbon paper (Sn@CNT-CP).490 This 3D architecture was able to provide buffering capability against mechanical strain during NaxSn alloy formation and efficient 3D transport pathways for both Na+ ions and electrons. As a result, the as-prepared Sn@CNT-CP electrode exhibited promising electrochemical performance, including high capacity and good cyclability extending to 100 cycles. The 2D graphene-backboned matrix was also introduced to function as a physical barrier to buffer the drastic volume change and impede the aggregation of Sn nanoparticles during Na–Sn alloying–dealloying reactions. Recently, Sn@C composites with ultrasmall tin nano particles (1–8 nm) are introduced by Liu et al.491,492 Later, Luo et al. fabricated a hierarchical tin@carbon composite composed of a graphene carbonaceous matrix and well-confined tin nanoparticles with a typical size of ∼15 nm as anodes for Na storage493 (Fig. 36e). This composite delivered a high specific capacity of 413 mA h g−1 and reversible sodium storage properties with negligible capacity fading after100 cycles (Fig. 36f).
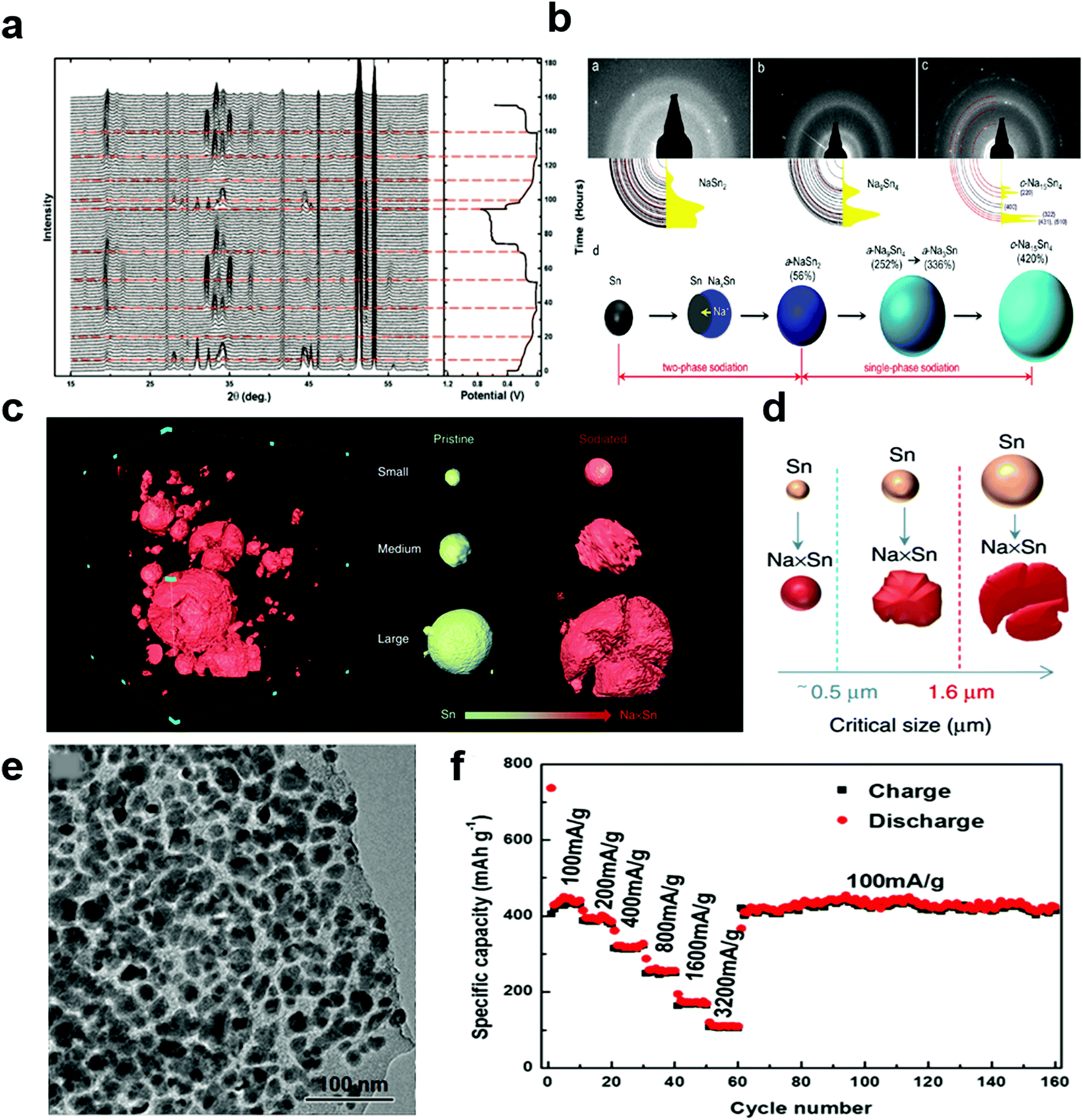 | ||
| Fig. 36 (a) In situ XRD data and the corresponding voltage curve. Dashed lines indicated the separating two-phase regions. (Reproduced with permission from ref. 483, Copyright 2012 The Electrochemical Society.) (b) Three a-NaxSn phases in the single-phase sodiation and schematic illustration of the structural evolution of Sn NPs during the sodiation. (Reproduced with permission from ref. 484, Copyright 2014 American Chemical Society.) (c) 3D morphologies of the sodiated electrode and selected three particles with different sizes and fracture and (d) schematic illustration of two critical sizes for Sn fracture in NIB. (Reproduced by permission from ref. 485, Nature Publishing Group, Copyright 2015.) (e) TEM images of F-G/Sn@C composites and (f) rate capability and capacity retention of the F-G/Sn@C electrode. (Reprinted from ref. 493, Copyright 2016, with permission from Elsevier.) | ||
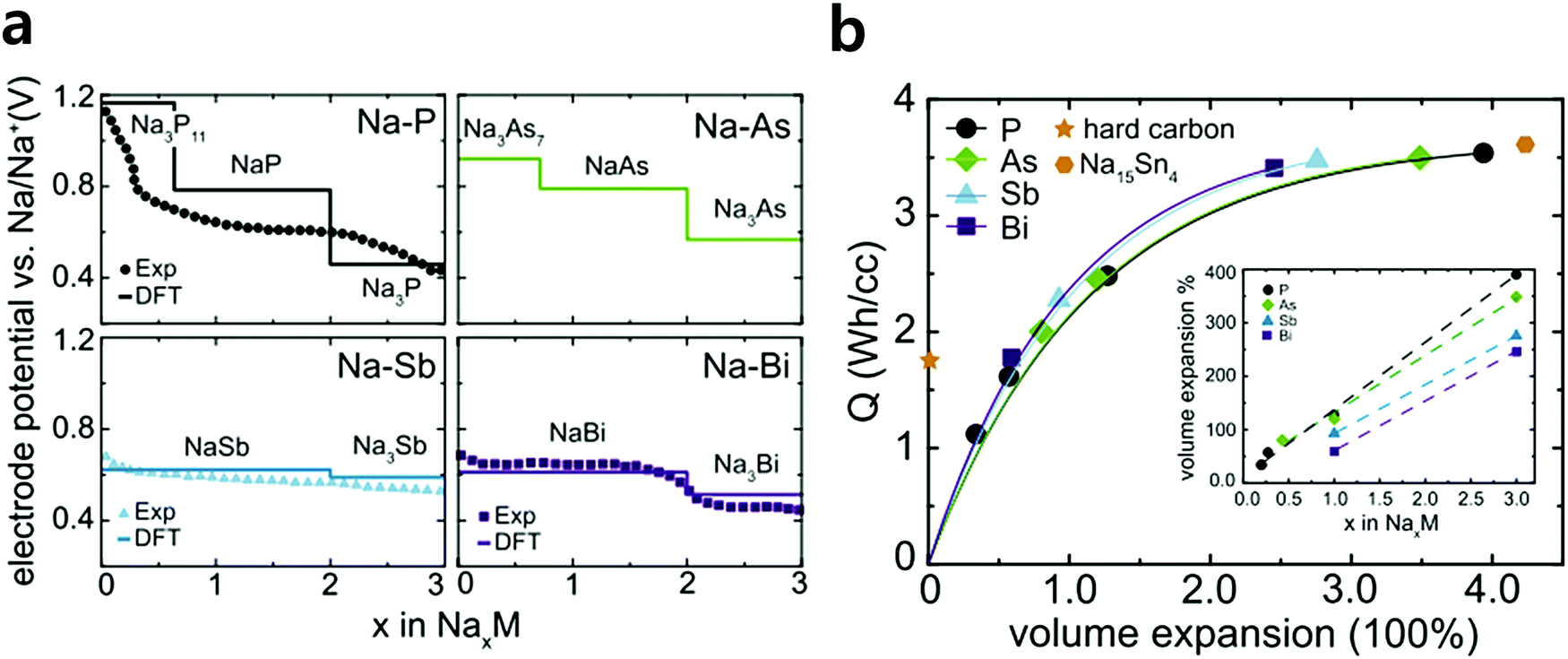 | ||
| Fig. 37 (a) Na–M voltage curves calculated using DFT and (b) the volume changes for group 14 elements in periodic table. (M = P, As, Sb, and Bi.) Experimental values are also given if available. (Reprinted from ref. 245, Copyright 2015, with permission from Elsevier.) | ||
3.3.2.1. Antimony. Antimony delivers a theoretical capacity of 660 mA h g−1 according to formation of Na3Sb (fully sodiation state).496–498 Qian et al. suggested two steps of Na alloying/dealloying reactions as follows: first step: Sb + Na+ + e− ↔ NaSb, second step: NaSb + 2Na+ + 2e− ↔ Na3Sb497 (Fig. 38a). Darwiche et al. more precisely investigated the electrochemical reaction mechanism of Sb with Na and observed the unexpected intermediate amorphous NaxSb phase via an in situ XRD technique498 (Fig. 38b). The crystalline Sb first transforms upon discharge into an intermediate amorphous phase NaxSb; when, all of the Sb is almost completely reacted, this NaxSb amorphous phase starts converting into cubic-hexagonal Na3Sb mixture phases before being stabilized as hexagonal Na3Sb. Upon desodiation, crystalline Na3Sb is transformed into amorphous Sb. (Sodiation: cSb → aNaxSb, aNaxSb → Na3Sbhex/cNa3Sbcub → cNa3Sbhex, desodiation: cNa3Sbhex → aSb, c: cubic, a: amorphous, hex: hexagonal.) They demonstrated that the intermediate amorphous phase may act as a buffer to relieve strain, accounting for the improved cycling in Sb–Na alloying reactions. Through the mechanistic studies, antimony can uptake/release nearly three Na atoms per Sb upon sodiation–desodiation process. However, this multiple electrochemical reaction between tin and Na+ ions leads to drastic volume changes of ∼390% during the Na alloying/dealloying process.499 Therefore, in order to mitigate volumetric changes and to enhance kinetics of alloying reactions, several research efforts are focused on nanostructuring of the active material and employing of the carbon conducting additives.497,498,500–509 He et al. proposed monodisperse antimony nanocrystals with a mean size tunable in the 10–20 nm range as anodes for SIBs.503 Downsizing of the primary Sb to 10–20 nm can result in significantly faster kinetics and more stable operation at higher current densities. In comparison to microcrystalline bulk Sb, nanocrystal Sb exhibited an enhanced rate capability and higher cycling stability in Na cells (Fig. 38c). To provide a conductive pathway and a buffering matrix for effective release of mechanical stress, Wu et al. prepared a uniform nanofiber structure with the Sb nanoparticles embedded homogeneously in the carbon nanofibers.504 The prepared Sb–C electrode delivered a large reversible capacity of 631 mA h g−1 at C/15, a greatly improved rate capability of 337 mA h g−1 at a rate of 5C and a excellent cycling stability for over 400 cycles (Fig. 38d). Recently, Hu et al. proposed an antimony/multilayer graphene hybrid, in which antimony is homogeneously anchored on multilayer graphene.508 A common drawback of carbon composite materials is their structural instability, which originates from the weak interaction between the active component and the carbon matrix. Therefore, to improve the above limitation of carbon composite materials, they proposed the chemically bonded antimony–multilayer graphene (MLG) hybrid nanostructure anode. The MLG strongly couples the Sb nanoparticles via chemical bonding, which can not only increase the electrical conductivity but also allow MLG to serve as a conductive matrix to maintain electrical contact with Sb during the large volume expansion and to help stabilize the SEI layer. This material delivered a high initial charge capacity of 452 mA h g−1 with stable capacity retentions of 90% for 200 cycles and exhibited a superior rate capability of 210 mA h g−1 at 5 A g−1.
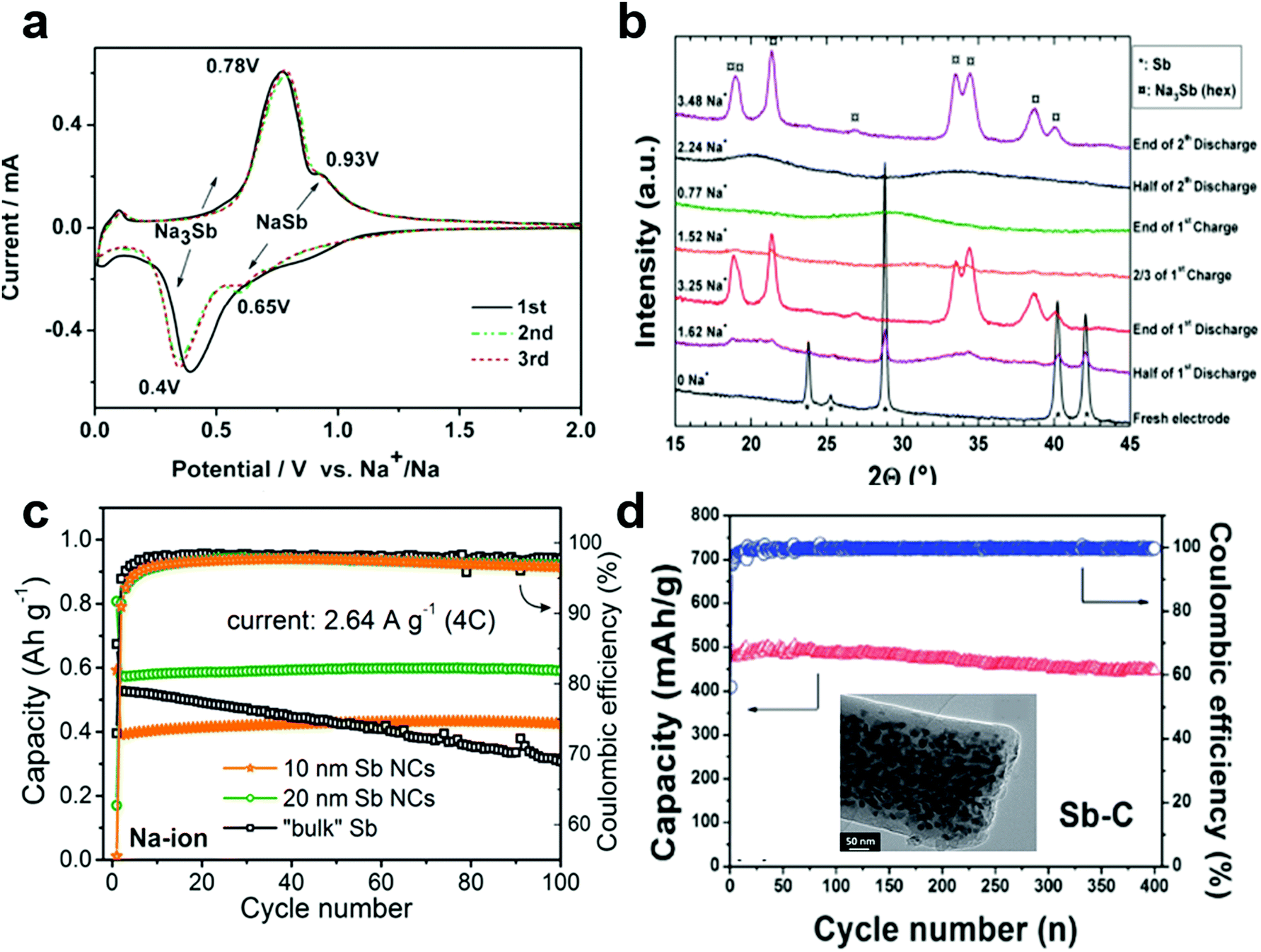 | ||
| Fig. 38 (a) The CV curve of Sb/C nanocomposites. (Reproduced with permission from ref. 497, Copyright 2012 The Royal Society of Chemistry.) (b) Selected operando XRD patterns at various stages of discharge and charge of a Sb/Na cell. (Reproduced with permission from ref. 498, Copyright 2012 American Chemical Society.) (c) Cycling performance of different size Sb NC electrode. (Reproduced with permission from ref. 503, Copyright 2014 American Chemical Society.) (d) Cycling performance of the Sb–C electrode at a cycling rate at 200 mA g−1 and inset image represent that TEM image of the Sb-C nanofibers. (Reproduced with permission from ref. 504, Copyright 2014 The Royal Society of Chemistry.) | ||
3.3.2.2. Phosphorus. Phosphorus electrochemically reacts with sodium to form Na3P at an attractive potential for an anode material, and its light atomic weight can achieve a higher theoretical capacity of 2596 mA h g−1 than any other SIB anodes presently available.510–512 Phosphorous exists in three main allotropes of white phosphorus, red phosphorus, and black phosphorus512,513,518 (Fig. 39a). White phosphorus (white-P) is volatile and unstable; it bursts into flames when it is exposed to the natural atmosphere. Red phosphorus (red-P) is usually amorphous in nature and is widely commercially available. Black phosphorus (black-P) is a crystalline phase, thermodynamically stable below 550 °C, and transforms into red (amorphous) phosphorus at higher temperature.512 For this reason, the amorphous red-P and orthorhombic black-P forms are being widely studied as anodes for SIBs. However, the electrochemical properties of both red-P and black-P are hindered by the enormous volume change (490%) occurring during the electrochemical sodiation/desodiation process.513 Qian et al. reported the improved electrochemical activity of amorphous red-P carbon composites (a-P/C) compared to pure red-P and black-P514 (Fig. 39b). Upon sodiation/desodiation, the pure red P shows a quite large discharge (sodiation) capacity of 897 mA h g−1 but gives only a negligible charge (desodiation) capacity of 15 mA h g−1, indicating the inactivity of this material for a sodium ion insertion reaction because of its insulating electronic nature (electrical conductivity of red-P: below 1 × 10−14 S cm−1). In contrast, a-P/C exhibited greatly enhanced electrochemical performance with initial charge/discharge capacities of 2015 mA h g−1 and 1764 mA h g−1, respectively, and a very high initial Coulombic efficiency of 87%, suggesting that the amorphous structure of phosphorus can effectively buffer the strong volumetric expansion during cycling. Over the same period of time, Kim et al. reported on an amorphous red phosphorus/carbon composite anode, which exhibited an appropriate redox potential of ca. 0.4 V vs. Na/Na+ with a reversible capacity of 1890 mA h g−1 and good rate capability delivering 1540 mA h g−1 at a high current density of 2.86 A g−1
511 (Fig. 39c). They also observed the formation of Na3P at the full sodiated state through ex situ XRD analysis (Fig. 39d). Recently, varieties of amorphous phosphorous with nanoarchitectures and 2D or 3D carbon matrices having high conductivity were applied to achieve a high capacity and a stable cycle life.511,513–519 Song et al. reported a novel phosphorus/graphene nanosheet hybrid through a facile ball milling process513 (Fig. 39e). The graphene stacks are mechanically exfoliated to nanosheets that chemically bond with the surfaces of phosphorus particles. This chemical bonding facilitates robust and intimate contact between phosphorus and graphene nanosheets. Furthermore, the graphene at the particle surfaces can assist to maintain electrical contact and stabilize the solid electrolyte interphase upon the large volume change of phosphorus during cycling. As a result, this composite anode delivers a high reversible capacity of 2077 mA h g−1 with an excellent cycling stability of 1700 mA h g−1 after 60 cycles. On the other hand, black phosphorus is potentially very attractive, as it has a layered structure similar to graphite but a greater interlayer distance.512,520–523 Namely, black phosphors are composed of the two dimensional single layer of phosphorene, which has a large interlayer channel size (3.08 Å), meaning that sodium (1.04 Å) ions can be stored between the phosphorene layers. In addition, orthorhombic black phosphorous with its layered crystal structure is thermodynamically the most stable allotrope.513 The higher bulk conductivity of black phosphorus compared with Red-P is additional advantages for electrochemical activity in Na cells.514,515 Hembram et al. proposed an atomistic mechanism for the sodiation of black phosphorus, based on first principles calculations.521 The layered structure of black phosphorus is maintained up to the composition of Na0.25P, with one-dimensional sodiation (an intercalation process) occurring in the interlayer spaces of the black phosphorus, resulting in sliding of the phosphorene layers because one Na atom tends to bind to four P atoms. At Na levels beyond Na0.25P, the intercalation process changes to an alloying process. Although the sodiation mechanism changes from an intercalation process to an alloying process at critical composition of Na0.25P, the volume expansion of black phosphorus increases linearly with Na concentration. Ramireddy et al. prepared nanocomposites of black (orthorhombic) phosphorus with graphite carbon via ball milling and applied the electrochemical test at different cut-off voltage windows.516 Within the voltage window of 0.01–2 V Na/Na+, the composite anode exhibited a high initial capacity of 1300 mA h g−1, however, the capacity gradually decreased. In contrast, attractive stable cyclic performances over 100 cycles were observed in the voltage windows of 0.33–2.0 V vs. Na/Na+. The post-cycling SEM studies showed that the electrodes gradually disintegrated and delaminated from the current collectors when electrochemical testing was performed within a larger potential window of 0.01–2.0 V vs. Na/Na+. However, this effect was absent for the restricted potential windows of 0.33–2.0 V vs. Na/Na+, leading to stable cyclic performances. To achieve both high capacity and stable cyclability, Sun et al. proposed a nanostructured phosphorene–graphene hybrid with a few phosphorene layers sandwiched between graphene layers512 (Fig. 39f). They also investigated the two-step sodiation mechanism of intercalation and alloying using in situ transmission electron microscopy (TEM) and ex situ X-ray diffraction (XRD) techniques. This nanoarchitecture delivers several advantages: (1) the graphene layers provide an elastic buffer layer to accommodate the anisotropic volumetric expansion during the sodiation process, (2) the phosphorene layers with an increased interlayer distance offer a short diffusion length for sodium ions, and (3) the graphene layers function as an electrical highway. As a result, the phosphorene–graphene hybrid nanostructure exhibited an extremely high specific capacity of 2440 mA h g−1 at 0.05 A g−1 and 83% capacity retention after 100 cycles in the voltage range of 0–1.5 V vs. Na/Na+. Recently, to provide an in-depth understanding of the reaction mechanism and the solid electrolyte interface (SEI) formation process of black phosphorous anodes in Na cells, Dahbi et al. investigated the structural change upon the sodiation/desodiation process via ex situ XRD analysis and electrode/electrolyte interfaces via powerful surface characterization techniques such as HAXPES and TOF-SIMS analyses.522 In the as-prepared electrode, black P exists as a crystalline phase with the orthorhombic lattice. In the full reduction state at 0 V in Na cells, orthorhombic black P changes into trisodium phosphide (Na3P) with a hexagonal lattice. The reduction product is found to be the same as red P. After forming Na3P, the cell is oxidized to 2.0 V, and reformation of the crystalline phase of black P was not found. This observation shows that black P is a metastable polymorph, and amorphous P would be formed as the oxidation product in the Na cell.522,524 They also observed that the VC-added electrolyte especially improves the reversible capacity and achieves a longer cycle life for black P electrodes with NaPF6 in EC/DEC by forming the stable SEI. On the other hand, Xu et al. proposed a black phosphorous–ketjenblack-multiwalled carbon nanotube composite with a high phosphorous loading of 70% as an anode for SIBs.524
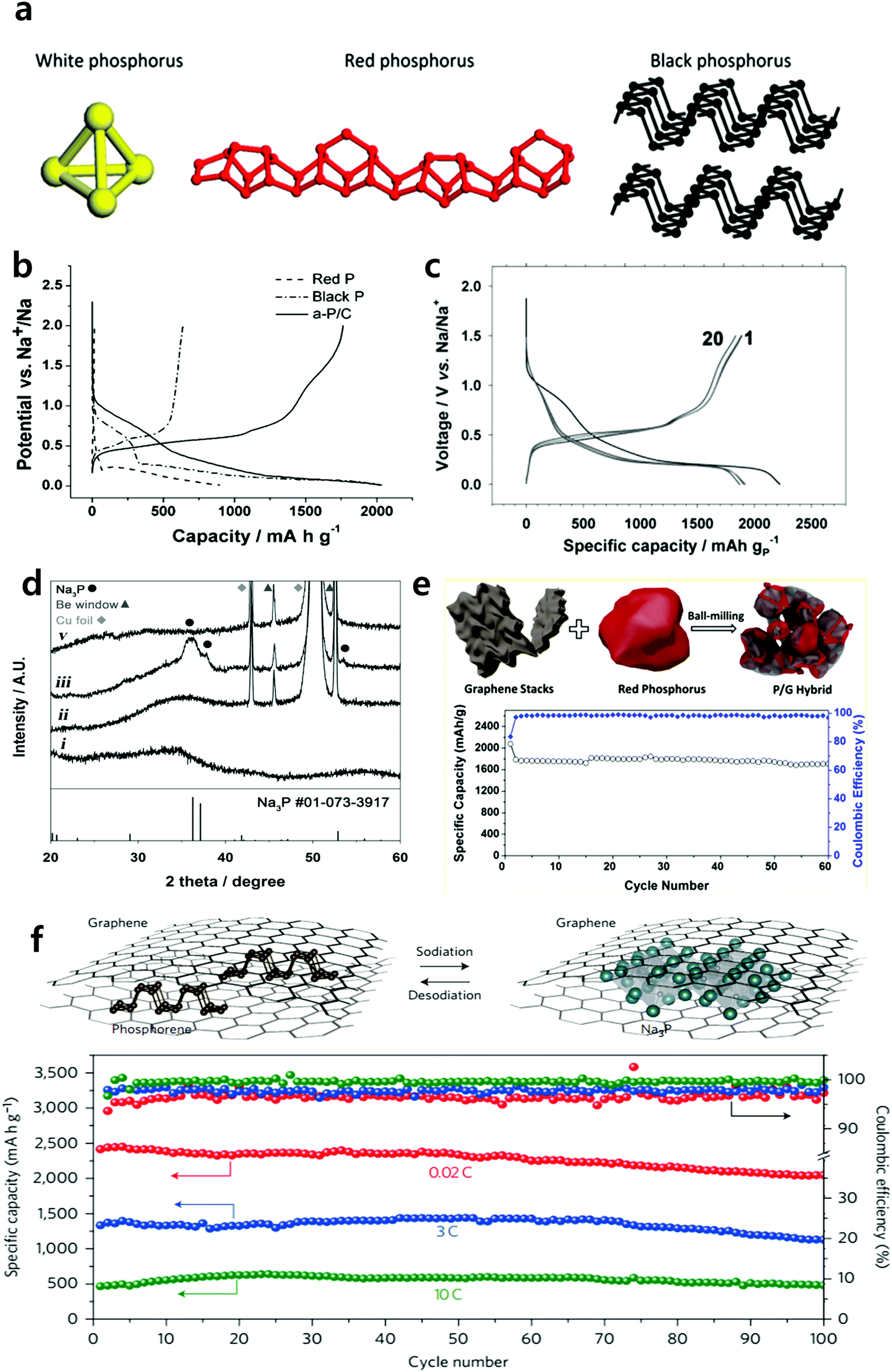 | ||
| Fig. 39 (a) Schematics of white, red, and black phosphorus. (Reproduced with permission from ref. 518, Copyright 2014 American Chemical Society.) (b) Initial charge/discharge curves of three phases of phosphorus: red phosphorus, black phosphorus, and a-P/C nanocomposites. (Reproduced from ref. 514 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) (c) Charge–discharge voltage profile of the red P/C composite electrode. (d) Ex situ XRD patterns of amorphous red P/C composite electrodes. (Reproduced from ref. 511 with permission, Copyright 2013 Wiley-VCH Verlag GmbH & Co. KGaA.) (e) Schematic Illustration of the synthesis of phosphorus/graphene nanosheets (P/G) hybrid and its cycling performance. (Reproduced with permission from ref. 513, Copyright 2014 American Chemical Society.) (f) Structural evolution of the sandwiched phosphorene–graphene structure during sodiation and cycling performance of phosphorene/graphene composites at different current density. (Reproduced by permission from ref. 512, Nature Publishing Group, Copyright 2015.) | ||
3.3.2.3. Bismuth. Bismuth (Bi), belonging to the same group in the periodic table as phosphorus and antimony, has been recently regarded as a potential anode material for LIBs525,526 and SIBs527–531 due to the unique layered crystal structure with a large interlayer spacing. Ellis et al. reported that the sodiation and desodiation mechanisms reversibly follow the Na–Bi equilibrium phase diagram with the formation of NaBi and Na3Bi.527 Bi reacts with Na to form Na3Bi, giving a theoretical capacity of 385 mA h g−1. Later, Sottmann et al. showed that alloying of sodium and bismuth proceeds via two distinct structural mechanisms depending on the crystallite size in the Bi/C anode.529 The transformation of NaBi into c-Na3Bi (c: cubic) requires less disturbance of the crystal structure than NaBi into h-Na3Bi (h: hexagonal) conversion. According to their report, phase fractions of the Na–Bi phases in the charged (2 V) and discharged state (0 V) in the 100th cycle show that c-Na3Bi is favored in the nanocrystalline anode as it forms on the crystallite surfaces. On the other hand, through DFT simulations, Su et al. calculated that Bi could provide facile sites for Na+ ion diffusion and accommodation, based on the intercalation mechanism instead of the alloying process.530 Their ex situ XRD and TEM results consistently showed that bismuth undergoes the Na+ ion intercalation process in Na cells. They also prepared a bismuth–graphene nanocomposite (Bi@graphene) and demonstrated its sodium storage performances in a bismuth crystal structure. The Bi@graphene nanocomposite demonstrated reasonable rate performance ascribed to the unique layered crystal structure of Bi, which has a large interlayer spacing along the c-axis (d(003) = 3.95 Å) to accommodate the Na+ ions.
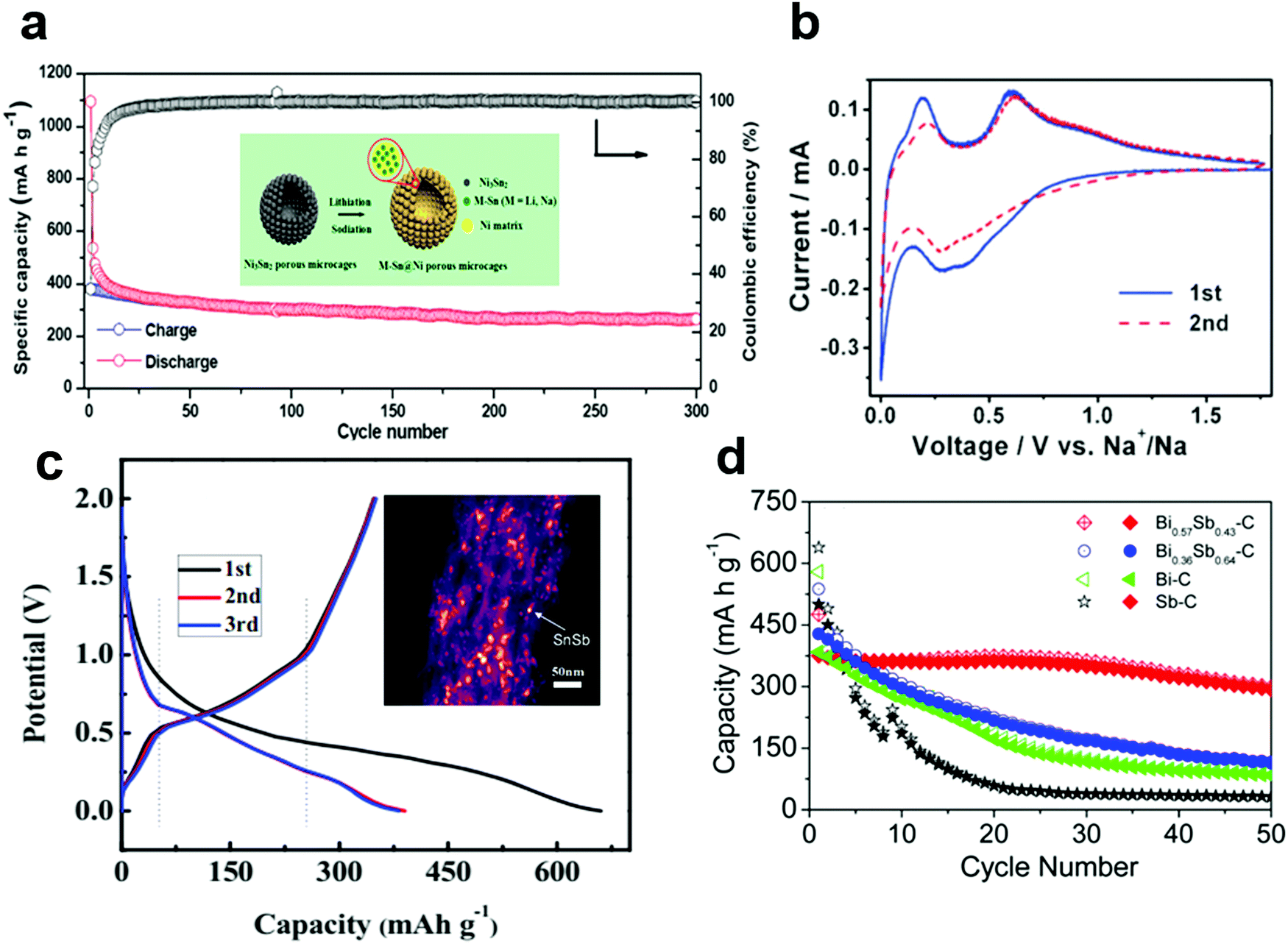 | ||
| Fig. 40 (a) Cycling performances of Ni3Sn2 at a 1C-rate. Inset image represents the schematic representation of the first lithiation and sodiation of porous Ni3Sn2 intermetallic microcages. (Reproduced with permission from ref. 532, Copyright 2014 American Chemical Society.) (b) The initial two CV curves of the SnSb/C nanocomposite electrode from open circuit voltage to 0.0 V vs. Na+/Na at a scan rate of 0.1 mV s−1. (Reproduced with permission from ref. 499, Copyright 2012 The Royal Society of Chemistry.) (c) First galvanostatic charge and discharge profiles of porous CNF–SnSb electrodes. (Reproduced from ref. 539 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) (d) Cycle performance of the Sb–C, Bi–C, Bi0.57–Sb0.43–C and Bi0.36–Sb0.64–C electrode in SIB under galvanostatic conditions with a current density of 100 mA g−1. (Reproduced with permission from ref. 541, Copyright 2014 American Chemical Society.) | ||
As mentioned above, carbon conducting techniques for conversion and alloying materials are very effective for buffering the volume strain and creating the electric conducting pathways. Unfortunately, despite the enormous advancement, the use of such approaches still requires further development for their practical applications. The major drawbacks of carbon conducting techniques such as carbon coating and/or formation of composites for practical applications are as follows. First, a high electrolyte/electrode surface area may lead to more significant side reactions with the electrolyte. For instance, typical disadvantage of carbon coating and/or formation of composites is the irreversible capacity charge loss in the first cycle due to the SEI formation on the carbon surface that needs to be overcome during the battery charge phase. Second, the tap density of carbon coated and/or composite materials is generally decreased compared with the same materials formed from without carbon contents. As a result, the whole capacity of the active material in the electrode is decreased and limited the total energy of the cell. Therefore, to achieve the high energy density and excellent battery performances at the same time, we should well-balance the carbon additive amount with active materials.
3.4. Organic compounds
Organic compounds have so far received less attention as potential anode materials for Li- and/or Na-ion batteries, mainly because of the success of inorganic materials in both research and commercial applications.543 However, new requirements in secondary batteries such as flexibility accompanied by low production costs and environmental friendliness, particularly for portable devices, reach the limit of inorganic electrode materials. In this respect, organic compounds have several advantages, including their chemical diversity, tunable redox property, lightweight, mechanical flexibility and cost-effective aspects, thus offering a wide selection of applications for use in batteries.544,545 Recently, oxocarbon salts with tailored frameworks and metal ions have been introduced as electrodes in rechargeable (Li, Na, K) batteries by Zaho et al.546 Most of all, for SIBs, inorganic compounds display poor cycling stability, mainly due to the larger ion radius of Na+.99,545 In contrast, the redox reaction of organic carbonyl materials is less influenced by the ionic size of alkali ions due to their soft nature.543 Therefore, the major organic carbonyl compounds, including the conjugated carboxylates-, imides- and quinones- and Schiff base-groups, are widely investigated as promising alternative anode materials for SIBs. Despite these advantages, there still remain three major challenges to compete with high performance carbonaceous and/or inorganic compounds: (1) slow reaction kinetics from the extremely low electronic conductivity, (2) particle pulverization induced by a large volume change during the Na+ ion insertion/extraction process and (3) chemical instability in the organic solvent upon cycling.543 Such thresholds can induce the serious capacity fading and loss of active mass upon cycling. In this section, we discuss the Na storage performances of various organic compounds and summarized the present research achievements.Conjugated carboxylates group compounds reveal reversible redox-reactions at low potential (0.2 to 0.5 V vs. Na+/Na) and stable charge/discharge behavior over multiple cycles.543–555 Carboxylate-based organic materials and terephthalate based materials were widely investigated as potential anode materials for SIBs.532–538 Zhao et al. firstly proposed the Na2C8H4O4/KB (ketjen black) composite electrode for SIBs, which has a reversible capacity of 250 mA h g−1, corresponding to a two electron transfer, with excellent cycling performance544 (Fig. 41a). In addition, ALD Al2O3 coating on the surface of the Na2C8H4O4/KB electrode significantly improves the sodium storage performances. Several disodium terephthalate derivatives such as No2–Na2TP, NH2–Na2TP, and Br–Na2TP were introduced by Park et al.545 According to their reports, the substituents on a phenyl ring (amino-, bromo-, and nitro-functional groups) and the regio-isomerism form of dicarboxylates (meta and para positions) can affect the thermodynamic and kinetic properties of disodium terephthalate and its derivatives. The results demonstrated that the Na2TP electrode delivered a reversible capacity of about 295 mA h g−1 during the sodium insertion/extraction process. This reversible Na+ ion (de)insertion of Na2TP can be ascribed to the stabilized conjugated structure between the carbonyl group and the phenyl rings. Br–Na2TP and No2–Na2TP electrodes delivered a high capacity of 300 mA h g−1; however, NH2–Na2TP delivered a relatively low capacity of 200 mA h g−1. Abouimrane et al. first incorporated a disodium terephthalate-based organic anode material into sodium-ion full cells with transition-metal cathode materials547 (Fig. 41b). The Na4C8H2O6 electrode revealed that two reversible electrochemical reactions occurred with two redox couples of Na2C8H2O6/Na4C8H2O6 as the cathodes at 2.3 V and Na4C8H2O6/Na6C8H2O6 as the anode at 0.3 V.236 By using symmetric reactions, Wang et al. fabricated an all-organic SIB using Na4C8H2O6, which delivered a reversible capacity of 180 mA h g−1 with an average operation voltage of 1.8 V. Later, to enhance the fast insertion/extraction of Na-ions at high current densities, Wang et al. suggested an extension of the π-conjugated system by using sodium 4,4′-stilbene-dicarboxylate (SSDC).552 Remarkably, the designed electrodes exhibited a much enhanced high rate performance with reversible capacities of 105 mA h g−1 at a current density of 2 A g−1 and 72 mA h g−1 at a current density as high as 10 A g−1. According to their report, excellent sodium storage performances under high rate charge–discharge conditions can be ascribed to the following two main reasons: (1) improvement of the charge transport and stabilization of the charged and discharged states, and (2) enhancement of the intermolecular interactions and the resulting terrace packing structure; both of these can facilitate the insertion/extraction of Na+ ions. On the other hand, one main reason for the capacity fading of organic compounds is the dissolution of the active compound in polar liquid electrolytes.556 To circumvent unwanted active mass dissolution during cycling, various strategies are introduced such as the application of polymers as active materials or the immobilization of active molecules onto the conductive additives.543,556–558 Recently, Chen et al. proposed the use of PNTCDA, a kind of polyimide.557 The intrinsic stability and insolubility of the polyimide ensure that it is not dissolved in the electrolyte, and thus allows for an excellent cycling stability and a high initial coulomb efficiency of 97.6%. Another promising candidate for organic compounds, biomolecule-based electrodes have also been widely studied. A biomolecule-based organic compounds contained the quinone and carbonyl group.559–563 Recently, Wang et al. proposed renewable-juglone biomolecules with well-defined redox-active quinone carbonyl groups, which exhibit promising electrochemical performance in reversibly transferring sodium ions561 (Fig. 41c). Also, juglone can be immobilized onto reduced graphene oxide (rGO) nanosheets owing to the strong π–π interaction between the aromatic structure and the carbon scaffold. Optical and photoelectron spectra results demonstrated non-covalent immobilization of the redox molecules via π–π interactions on the rGO carbon scaffold, which suppresses the dissolution puzzle of organic materials and enhances both the conductivity and sodium-ion accessibility of the electrode. As a result, juglone/RGO electrode demonstrated a high capacity of 305 mA h g−1 and stable cycle retention after 100 cycles. Armand's group reported polymeric and oligomeric Schiff-based electrodes564,565 (Fig. 41d). Polymeric Schiff bases have the (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–Ar–HCN) repeat unit, which can work as a redox center for sodium storage.551 The reduction reaction proceeds in two steps in the voltage range from 0.005 to 1.6 V versus Na+/Na corresponding to two different processes. Later, the oligomeric Schiff bases are also investigated. López-Herraiz et al. reported for the first time the electrochemical activity of the 10-π-electron end group (–OOC–ϕ–CQN–) (ϕ: refers to the phenyl group) and the central (–NQC–ϕ–CQN–) Hückel units.565 They claimed that the maximum capacities are achieved for oligomers in which H+ ions are replaced by Na+ ions due to the fact that the hydrogen bond of the carboxylic end groups acting as crosslinks is removed, thus helping to accommodate more easily for the inserted Na+ ions.
CH–Ar–HCN) repeat unit, which can work as a redox center for sodium storage.551 The reduction reaction proceeds in two steps in the voltage range from 0.005 to 1.6 V versus Na+/Na corresponding to two different processes. Later, the oligomeric Schiff bases are also investigated. López-Herraiz et al. reported for the first time the electrochemical activity of the 10-π-electron end group (–OOC–ϕ–CQN–) (ϕ: refers to the phenyl group) and the central (–NQC–ϕ–CQN–) Hückel units.565 They claimed that the maximum capacities are achieved for oligomers in which H+ ions are replaced by Na+ ions due to the fact that the hydrogen bond of the carboxylic end groups acting as crosslinks is removed, thus helping to accommodate more easily for the inserted Na+ ions.
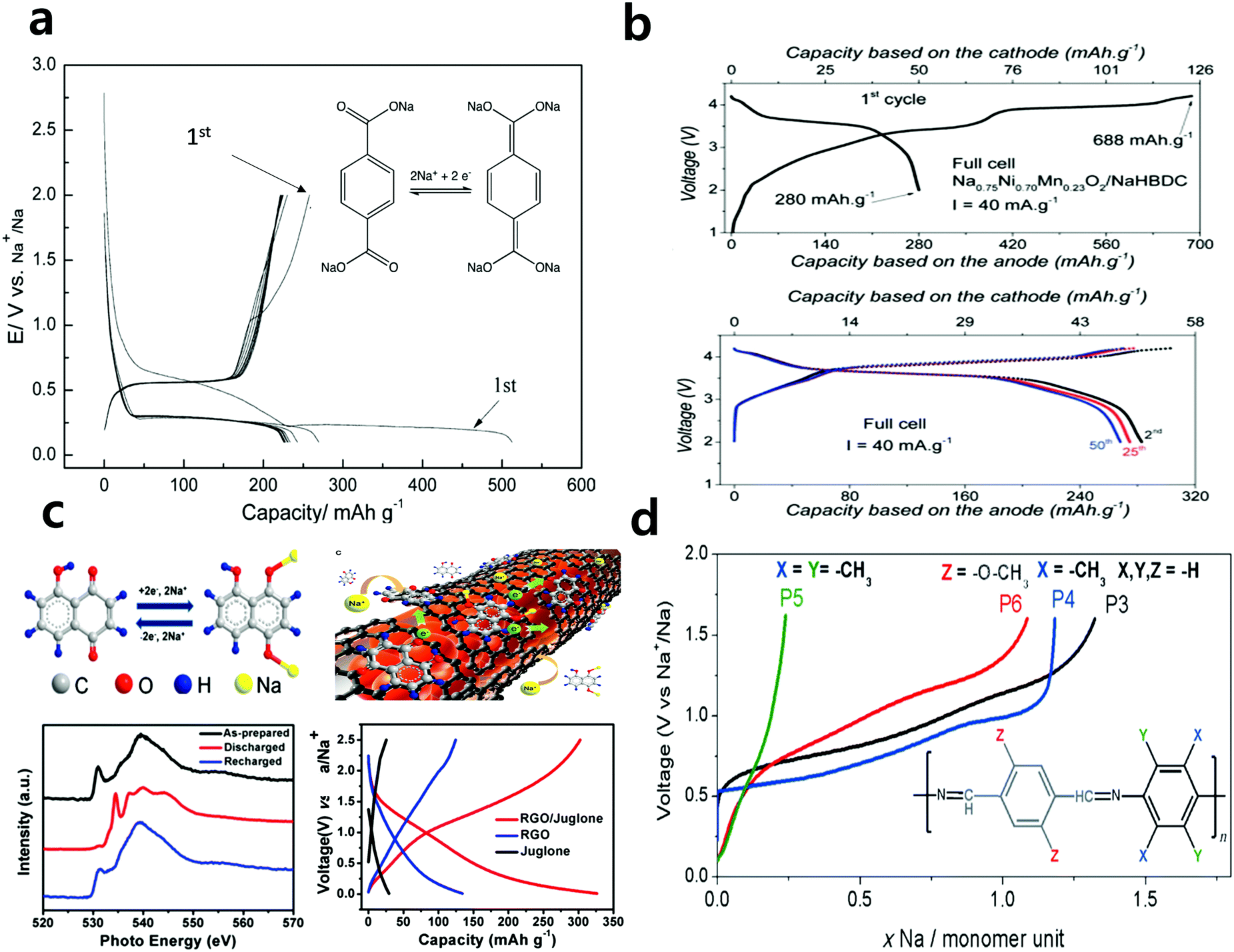 | ||
| Fig. 41 (a) The discharge–charge cycles of the Na2C8H4O4/KB composite electrode. Inset shows the molecular structure of disodium terephthalate (Na2C8H4O4) and the Na insertion/deinsertion mechanism. (Reproduced from ref. 544 with permission, Copyright 2012 Wiley-VCH Verlag GmbH & Co. KGaA.) (b) First charge–discharge curves and cycling performance of Na0.75Mn0.7Ni0.23O2/NaHBDC full cell. (Reproduced from ref. 547 with permission, Copyright 2012 The Royal Society of Chemistry.) (c) Schematic diagram of juglone molecules with RGO nanosheets and the reversible sodium-ion de-/insertion mechanism (top). XANES spectra of Juglone/RGO electrodes and its capacity–voltage profiles at 0.1 A g−1. (Reproduced from ref. 561 with permission, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA.) (d) Voltage versus specific capacity for the first galvanostatic oxidation for the different polymeric Schiff bases. (Reproduced from ref. 564 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
4. Electrolytes, additives, and binders
SIB technology is a very important and promising follow-up to LIB technology. The significance of SIBs is cost effectiveness owing to the geographical distribution of sodium in comparison with lithium. Therefore, in the long term, sodium-ion batteries will replace and/or substitute lithium-ion batteries in mid-large scale battery market.566 However, SIBs have still faced several challenges in terms of developing optimized electrode materials and electrolytes with a suitable capability for stable sodium storage. To date, while a number of efforts have been directed toward the searching for new electrode materials for SIBs, studies dealing with the electrolyte itself are much scarcer.28,567 However, looking back through the history of LIBs, it is clear that a suitable choice of electrolyte and binders is equally as important as the choice of electrode material for making operational SIBs; this is because the electrolyte and binders form a protective layer at both the cathode and anode, the surface layer (SL) and the solid electrolyte interfaces (SEI), respectively.568 Therefore, identifying suitable formulation electrolytes is indispensable to developing high performance SIBs. To accomplish this, we borrow ideas and techniques from those typically used in LIB electrolyte development. In the case of LIBs, various electrolytes, including organic electrolyte solutions, solid- and gel-polymer electrolytes, inorganic solid electrolytes, and ionic liquids have been investigated, and their development is still in progress.99 Organic electrolyte solution based on carbonate-ester polar solvents, where sodium salts are dissolved with complex containing functional additives, are mainly used in the practical development of SIBs due to their large potential window, high ionic conductivity and good temperature performance. On the other hand, a water-based electrolyte has also been proposed as a cost-effective energy storage system, which was successfully commercialized.18,569 In this section, we discuss SIB electrolytes, including salts, solvents, and additives. In addition, we briefly summarized the currently employed binders as well as their effects on the electrochemical performances based on various electrodes and/or sodium ion full cells.4.1. Electrolytes
A general list of properties needed for SIB electrolytes complies with those usually compiled for LIB oriented electrolytes: (1) chemically stable, (2) electrochemically stable, (3) thermally stable, (4) ionically conductive and electronically insulating and (5) low toxicity with low production cost.570,571 These features intrinsically depend on the nature of the salts and the solvents as well as the possible use of additives. For suitable electrolytes, we should consider the major parameter of solvents and salts, which can mainly affect the cell performances. The salt should exhibit: (1) solubility in the solvent, (2) stability vs. reduction as well as oxidation, and (3) chemical stability with the cell component.568 The solvent should: (1) be polar with a high dielectric constant, (2) exhibit low viscosity in order to improve the ionic mobility, (3) remain inert to the charged surfaces of the cathode and the anode during cell operation, and (4) have a wide liquid range (i.e. a low melting point and a high boiling point).568 Over the past few decades, based on such basic properties, the scientific community has made tremendous efforts in finding the best combination of suitable electrolytes; however, not yet currently available for the direct use for practical SIBs. While being of only academic interest, carbonate ester-based electrolyte solution containing sodium salts is considered to be one of the most appropriate electrolyte media for SIB applications. In early reports, in the 1980s, the properties of sodium ion cells are tested using NaI, NaPF6, and NaClO4 salts in organic solvents as the electrolyte.572,573 Then, either NaClO4 or NaPF6 is used as the electrolyte salts in carbonate-ester binary or ternary mixtures, which have become the main stream electrolyte composition used for Na+ chemistry.571Although the sodium perchlorates have some concern of safety and are notoriously difficult to dry, in terms of only academic interest, the commonly used salt is NaClO4 due to a combination of historical and cost reasons.28,570,573 Delmas et al. demonstrated the electrochemical properties of a P2-type NaxCoO2 electrode in NaClO4 in a propylene carbonate solvent.573 In 2000s, D. A. Stevens and J. R. Dahn et al. investigated the reversible insertion of sodium into hard-carbon host structures at room temperature in 1 M NaClO4 in EC:DEC (30:70, v/v) as an electrolyte.247 Alcantara et al. reported the dependency of sodium salts on the electrochemical properties of carbonaceous materials using NaPF6 and NaClO4 salts in EC:DMC 1:1 mixture solvents.575 They suggested that using a sodium NaClO4 based electrolyte for a carbonaceous electrode demonstrated a higher capacity with high Coulombic efficiency. They also reported the dependency of electrolyte solvents on the electrochemical properties of carbonaceous materials using an EC:DMC, DME, and EC:THF solvent containing 1 M NaClO4 salt (Fig. 42a). The THF-solvent and EC:THF mixture have shown to improve the electrochemical performance as compared to only carbonate-based solvents. Komaba et al. investigated electrochemical performance of hard carbon anodes in Na cells with various electrolyte solvents of EC, PC, BC, EC:DMC (50:50, v/v), EC:EMC (50:50, v/v), EC:DEC (50:50, v/v), and PC:VC (98:2, v/v) containing 1 M NaClO4 salt in beaker-type cells.27 The results show that 1 M NaClO4 in PC, EC and EC:DEC solution demonstrated better electrochemical performances than the other mixture solutions. Interestingly, VC is commonly accepted as a suitable additive to modify the interface via thin film forming of the electrodes; however, the addition of VC in a PC solvent was not found to play the same role in hard carbon electrodes. Appropriate additives for SIBs are specifically discussed in the additive part. On the other hand, anodic stability without electrolyte decomposition at high voltage is also a crucial issue for achieving practical SIBs without gas evolution and battery swelling.568 Recently, Oh et al. proposed an ethyl methanesulfonate-based (EMS) electrolyte containing the NaClO4 salt (Fig. 42b). A high anodic stability (up to 5.6 V versus Na/Na+) and ionic conductivity (6.0 × 10−3 S cm−1) can be achieved by replacing the PC with EMS.51
 | ||
| Fig. 42 (a) Voltage/capacity plots corresponding to the first discharge/charge cycles of carbon aerogel microspheres in sodium cells, 1 M NaClO4 dissolved in EC:DMC, DME, THF, and EC:THF as electrolytes. (Reproduced with permission from ref. 575 Copyright 2005 The Electrochemical Society.) (b) Linear sweep voltammetry of a sodium cell using two electrolytes, namely, 1 M NaClO4 in PC + 2 vol% FEC and 1 M NaClO4 in EMS + 2 vol% FEC. (Reproduced with permission from ref. 51, Copyright 2014 American Chemical Society.) (c) DSC heating curves of fully sodiated hard carbon in various electrolyte formulations. (Reproduced with permission from ref. 28, Copyright 2012 The Royal Society of Chemistry.) (d) Comparison of capacity retention of hard carbon electrodes between reagent grade and battery grade NaPF6 PC solution. Photographs of the electrolyte solution use are inserted. (Reproduced with permission from ref. 18, Copyright 2015 The Electrochemical Society.) (e) Initial reduction/oxidation curves for hard-carbon electrodes in 1 M NaClO4 PC solution without and with FEC. Inset shows variation in reversible oxidative capacities for the hard-carbon during successive cycle test. (Reproduced with permission from ref. 577, Copyright 2011 American Chemical Society.) (f) Cycle performance of Sn4P3 and Sn electrodes obtained with or without an FEC additive. (Reproduced from ref. 460 with permission, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA.) (g) Structure of polymer binder: PVDF, PVA, CMC-NA, PAA. (Reproduced with permission from ref. 610, Copyright 2011 American Chemical Society.) (a) Cyclic performance of Na0.11Li3.89Ti5O12 electrodes with different binders. (Reproduced with permission from ref. 616, Copyright 2016 The Electrochemical Society.) | ||
On the other hand, in order to realize practical SIBs, many researchers have explored alternative Na salts with high battery performances as well as good battery safety. Ponrouch et al. systematically investigated the most appropriate electrolyte formulation so as to minimize the interface reactions and enhance both cell performances and safety aspects.28 They evaluated the basic properties of the electrolyte solution such as viscosity, ionic conductivity, and thermal and electrochemical stability in diverse solvent mixtures and Na-based salts having either F-based or perchlorate anions. Especially, safety issues, in which they evaluated the thermal stability using fully reduced hard carbon powders in the selected electrolytes via DSC measurements: (i) NaClO4 in PC, (ii) NaClO4 in EC:PC, (iii) NaClO4 in EC:DEC and (iv) NaPF6 in EC:PC. Among them, NaPF6 in the EC:PC electrolyte exhibited the high thermal stability with a first exothermic peak onset temperature of 156.2 °C and a low heat generation of 717.2 J g−1 (Fig. 42c). The appearance of an exothermic peak at a high temperature of NaPF6 in the EC:PC electrolyte can be ascribed to how a significantly more thermally stable SEI layer is formed on a hard carbon surface after cycling compared to those of the other electrolytes. On the other hand, Komaba et al. observed the stable cycling performances in hard carbon electrodes with the NaPF6-based electrolyte solution in comparison to those of the NaClO4-based electrolyte.27 Bhide et al. carried out a comparative study of the physico-chemical properties of non-aqueous liquid electrolytes based on NaPF6, NaClO4 and NaCF3SO3 salts in the binary mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC).576 Bhide et al. also observed that the NaPF6-based electrolyte demonstrated a higher ionic conductivity as a function of composition and temperature. Additionally, in view of practical Na-ion battery systems, the electrolytes are electrochemically tested with Na0.7CoO2 as a cathode. The results showed that the electrolyte NaPF6–EC:DMC is favorable for the formation of a stable surface film and the reversibility of the Na0.7CoO2 cathode material. Ponrouch et al. also achieved a high rate capability for the Na3V2(PO4)2F3 cathode and the hard carbon/Na3V2(PO4)2F3 full cell in 1 M NaPF6 in the EC:PC:DMC (45:45:10, v/v) electrolyte.574 However, when using the NaPF6 containing electrolyte for the electrode materials, negative effects also occurred, such as low ionic conductivity of the SEI formation associated with the presence of NaF. In addition, Bhide et al. proved the presence of NaF in NaPF6 using X-ray diffraction and the observation of the insoluble fraction in NaPF6/EC:DMC solution by the naked eye when exceeding concentrations of 0.4 M.576 Recently, Kubota et al. discuss the influence of the NaPF6 salts with different purities on the solubility of the organic solvents and the electrochemical properties.18 When they tried to dissolve the reagent grade NaPF6, 1 M NaPF6 was not completely dissolved in PC. However, by selecting the battery grade NaPF6, 1 M NaPF6 in the PC solution was revealed to be colorless and transparent because of the complete dissolution of 1 M NaPF6. As anticipated from the electrolyte coloration, the hard carbon anode exhibited better electrochemical performances in the battery grade NaPF6 containing electrolyte solution (Fig. 42d). The above results convinced that the choice of a well-balanced mixture solvent and high purity Na-salt is very important for identifying suitable electrolyte solutions.567,568,570
Recently, for improving the Na storage performances, ether-based electrolytes also have been reported for SIBs.259,261,262,413,429,453,578–581 Especially in graphite anodes, it is found that by using ether-based electrolytes rather than conventionally used ester-based electrolytes, the co-insertion of solvent and sodium ions can be successfully achieved. Kim et al. and Zhu et al. systematically studied the application and the mechanism of graphite as an anode material for SIBs in some ether-based electrolytes.259,262 Some research groups enabled transition-metal disulfides to achieve high-rate capability and long-term cyclability by optimizing the electrolytes and voltage windows in ether-based electrolytes. With regard to transition-metal chalcogenides, ether-based electrolytes exhibited a higher solvent-salt stability and a lower reaction energy barrier than those of carbonate-based electrolytes.407,413,429,578
4.2. Additives
Another major component, often needed to create a functional electrolyte is the choice of additives. Generally, additives are used in functional electrolyte formulations for stable SEI formation and to enhance its electrochemical stability, and are also required for battery safety issues such as reducing flammability and preventing overcharging processes.99,569,577 Komaba et al. examined and compared the electrochemical performance of Na cells with several additives in 1 M NaClO4 in PC solution, including fluoroethylene carbonate (FEC), transdifluoroethylene carbonate (DFEC), ethylene sulfite (ES), and vinylene carbonate (VC), which are well-known to be efficient electrolyte additives for Li-ion batteries.577 The results demonstrated that FEC is the only efficient electrolyte additive for both the cathode and anode. According to Komaba et al.'s reports, an appropriate amount of FEC additive (2 volume%) is helpful for forming high quality passivation films on the electrode surface, as well as for suppressing the side reactions between the Na metal and propylene carbonate solution containing Na salts (Fig. 42e). Meanwhile, Ponrouch et al. observed a decrease in specific capacity and Coulombic efficiency of the hard carbon electrode when FEC was used in the EC:PC solution containing 1 M NaClO4 salt.249 Unlike the PC only based electrolyte, the EC:PC mixture based electrolyte promoted efficient SEI layer formation on the hard carbon electrode surface without the need of FEC additives. Recently, Dahbi et al. reported the electrochemical sodium insertion for hard carbon in a cyclic alkylene carbonate based (PC, PC/EC) solution containing a NaClO4 or NaPF6 salt with or without a fluoroethylene carbonate (FEC) additive to study electrolyte dependency for sodium-ion batteries.588 According to their report, by adding a FEC additive in both NaPF6 and NaClO4 based electrolytes, the hard carbon electrode exhibited an additional voltage plateau at approximately 0.7 V vs. Na/Na+ during the first reduction due to decomposition of FEC and delivered a higher reversible capacity than without a FEC additive electrolyte. Namely, the irreversible reaction during the initial cycle in both NaPF6 and NaClO4 based electrolytes was effectively suppressed by the FEC additive, which enhanced the reversibility of hard carbon anodes. The passivation layer would be further stabilized and become a thinner surface layer in the presence of FEC because the fluorine compounds dissolved in the electrolyte would efficiently induce the formation of an electronically insulating, ionically conducting, and insoluble surface layer to effectively passivate the electrode surface.On the other hand, the significant influence of the electrolyte additive is clearly observed in alloying and/or conversion reaction based anodes.384,460,539,561,564 Such electrode materials accompany dramatic volume changes, which has led to severe electrode pulverization upon the sodiation–desodiation process. Therefore, a stable passivation film on an electrode surface is required for effectively preventing such drawback. The formation of stable SEI films can stabilize the electrode and improve the migration kinetics of Na by using FEC additives. The main components of FEC-containing electrolytes were proposed as important compounds in effectively protecting surface films. The final SEI composition can be affected by a multitude of different factors, such as the order of the bond breaking events, the relative stability of resulting radical ions, and the thermodynamic feasibility of different decomposition reactions.582 When FEC free electrolytes were used in Na cells, the XPS results suggest that the SEI in SIBs has a higher percentage of inorganic salts, alkyl carbonates, and CO rich compounds, which are unstable upon electrochemical cycling.352,486 Recently, Dahbi et al. reported the possible mechanism for FEC reduction reactions in Na cells.522 In addition, Kumar et al. proposed the reaction pathways for the reduction induced decomposition of FEC–Na+ in one electron reduction states.583 When FEC is added in a Na cell, it is believed that FEC reduces first, then polymerizes through the opening of a five-membered ring, and forms very thin and stable protective films which may contain NaF, Na polycarbonate and polyenes (oxygen-free polymer).584–587 As a result, previous reports have indicated that FEC addition in organic electrolyte solutions leads to a significant improvement of the cyclability. Kim et al. and Li et al. examined and compared electrochemical performances of tin–phosphide electrodes with/without FEC additives460–462 (Fig. 42f). More recently, Dahbi et al. investigated the electrode/electrolyte interface and reported the positive effect of VC additives in black phosphorous electrodes.522 VC added electrolytes especially improve the reversible capacity and achieve a longer cycle life for black P electrodes with NaPF6 in EC/DEC. The outermost surface layer of the SEI in the VC additive is composed of both inorganic and organic compounds. According to their report, the organic compounds that contain fragments (m/z = 55 (C5H7+), 57 (C4H9+), 67 (C5H7+), 69 (C5H9+), 71 (C5H11+), and 83 (C6H11+) in TOF-SIMS result) play an important role as components of SEI layers in the electrode, which results in the improvement of electrochemical performance and suppression of SEI dissolution. On the other hand, VC did not form efficient surface layer films on hard-carbon electrodes with NaClO4 in PC.577
As mentioned above, a solid electrolyte interphase (SEI) is one of the crucial determinants of battery life. The final SEI composition can be affected by a multitude of different factors, such as the order of the bond breaking events, the relative stability of resulting radical ions, and the thermodynamic feasibility of different decomposition reactions.582 Previous studies suggested that prolonged decomposition of electrolytes forming a thick and unstable solid–electrolyte interphase continues to be a major drawback in designing sodium-ion batteries. Recently, Kumar et al. investigated the fundamental mechanisms of solvent decomposition involved in SEI formation in SIBs using the density functional theory (DFT) method.583 According to their calculations, in EC based electrolytes, the high reduction potential and a low barrier for the ring opening of EC is the main cause for the continuous growth of a SEI in SIBs. They also studied the effect of additive molecules on the decomposition pathways of the EC. On the other hand, Dahbi et al. reported the formation mechanism and the main component of surface films on the phosphorus and hard carbon electrode with different electrolyte solutions.522,588 In the NaPF6 based electrolyte, they proposed the decomposition mechanism of NaPF6 in the electrolyte in the following four step. (1) NaPF6 ↔ NaF + PF5, (2) PF5 + nNa+ + ne− → NaxPFy + NaF, (3) NaPF6 + H2O → NaF + POF3 + 2HF and (4) POF3 + nNa+ + ne− → NaxPOFy + NaF. In other words, NaPF6 is reduced to a gaseous product, phosphorus pentafluoride (PF5), according to step 1. F− ions are also released and react with Na+ ions in the electrolyte to form NaF on the electrode surface. They also suggested the possible mechanism for additive (FEC and VC) reduction reactions in Na cells. When FEC is added in a Na cell, it is believed that FEC reduces first, then polymerizes through the opening of a five-membered ring, and forms very thin and stable protective films which may contain NaF, Na polycarbonate and polyenes (oxygen-free polymers). In contrast, VC-based electrolytes are also reduced, resulting in the formation of oxygen-containing species. The electrochemical reduction of VC forms CHCHOCO2− radical anions, which could be stabilized by Na+ cations in Na cells. They speculate that such organic compounds that contain fragments play an important role as components of the SEI in the electrode. Such results clearly show that the additive molecules protect solvent molecules from reductive decomposition but also they can promote alternate pathways for decomposition, leading to qualitatively different and potentially stable SEI products.584–587
4.3. Ionic liquids, polymer electrolytes, all-solid electrolytes
The study of ionic liquids (ILs) and polymer electrolytes containing a fraction of sodium salts is still at a very early stage.99,568 Ionic liquids generally have a negligibly low volatility, non-flammability, and high thermal and electrochemical stability. The use of an ionic liquid electrolyte solution has been introduced by several research groups.589–592 However, unsolved drawbacks such as high viscosities at room temperature and expensive production cost still remain for practical utilization as an electrolyte in SIBs. Polymer electrolytes are mainly considered as solvents because of their thermal, and chemical stabilities, as well as their flexibility for cell manufacturing.568 In addition, all-solid-state batteries with inorganic solid electrolytes and electrodes are promising power sources for a wide range of applications because of their safety, long-cycle lives and versatile geometries.18,99,568,593–597 By West et al. a Na/NaClO4-PEO/V2O5 cell was proposed in 1985 to avoid the use of a Na metal anode.593 Later, Ma et al. reported on a SIB cell built using Na0.7CoO2 as the cathode, Na15Pb4 as the anode and NaTf(PEO)8 as the solid polymer electrolyte.72 A phosphate-based all-solid-state symmetrical cell, Na3V2(PO4)/Na3Zr2Si2PO12/Na3V2(PO4), has been successfully fabricated, but the reported electrode performances are for under 80 °C conditions.596 Recently, Hayashi et al. realized an all-solid-state rechargeable sodium cell based on a Na-Sn/Na3PS4 glass-ceramic/TiS2 cell configuration at room temperature. Although the capacity was limited to approximately 40% of the theoretical capacity of TiS2, the all-solid-state cell has the potential to realize good charge–discharge reversibility at ambient and moderate temperatures.597In summary, the main electrolyte for practical use in SIBs is as follows. First, NaPF6 has been largely used as electrolyte salts in carbonate-ester binary or ternary mixtures, which have become the main electrolyte composition used for SIBs. Second, propylene carbonate (PC) is a key component in both binary and ternary solvent mixtures, which has intrinsic advantages derived from its high dielectric constant and wide operating electrochemical and temperature windows.568 Third, the use of FEC as an additive plays the important role of stable passivation film formation on the electrode surface, which effectively prevents electrode destruction as a result of the sodiation–desodiation process.577 Finally, the choice of high purity solvents and Na-salt is necessary for identifying a suitable electrolyte solution.18,28,568,574
4.4. Binders
Another important key component to improving the electrode performance is the choice of a binder used for powdery active materials. Especially, in anode materials, Na storage performances are dramatically affected by selecting the appropriate binders. Thus, investigation of desirable binders is also necessary to stabilize the electrode surface and suppress electrode distortion during the sodiation–desodiation process. Commonly, most electrodes of commercial and/or scientific studies are made using poly(vinylidene fluoride) (PVDF) as a binder to form an electrode layer from a powdery electrode material on a metal current collector due to its good chemical and electrochemical stabilities.99 However, some problems remain in terms of making electrode slurries, such as the relatively high production cost and the necessary use of a volatile and toxic organic solvent (N-methyl pyrrolidone).99,598,599,610 Recently, in both LIBs and SIBs, alternative water-soluble binders have been introduced such as sodium carboxymethyl cellulose (Na-CMC), poly(acrylic acid) (PAA) and sodium alginate (Na-Alg)600–612 (Fig. 42g). Generally, such binders were introduced for alloying reaction materials undergoing a large volume change during lithiation–delithiation or sodiation–desodiation to enhance cycle performance due to the thermally cross-linked three-dimensional interconnection of those binders. A Na-CMC binder is an environmentally friendly and inexpensive material that is derived from cellulose as natural polymers. During the electrochemical reaction process, the Na-CMC binder could play an important role in improving the solid electrolyte interface (SEI) passive layer on the electrode surface, which reduces the irreversible capacity and somehow leads to a better cycle life.601–603 Na-Alg is a high-modulus natural polysaccharide extracted from brown algae, which yields a remarkably stable battery anode.604–606 In particular, Na-Alg is much more polar than the Na-CMC polymer chains, which can ensure a better interfacial interaction between the polymer binder and particles, as well as stronger adhesion between the electrode layer and the Cu substrate. Comparing the chemical character between PVDF and PAA, it is a general assumption that the carboxylic groups of the PAA binder play an important role in achieving better uniformity and stronger binding ability in the composite electrode; this is due to their amorphous and cross-linking nature via the hydrogen bonds between carboxylic groups.608–611,613 A PAA binder allows for the formation of a stable deformable SEI layer on the elastic binder-coated electrode surface, in which the elasticity of the polymer matrix may prevent cracking in the SEI when the volume changes.609,610 The polymeric cross-linkage network can regulate the mechanical/chemical stress that results from large volume expansion in the composite electrode.517,610 In addition, the decomposition reaction of an electrolyte was reduced compared to the PVDF binder due to the stable SEI layer formation via a PAA binder.458,613,614 In 2014, Dahbi et al. compared and examined the binder and electrolyte additive effects in a hard carbon electrode using Na-CMC and PVDF binders.601 Decomposition of the electrolyte on hard-carbon during the initial cycle was sufficiently suppressed by the CMC binder. Moreover, Dahbi et al. noted that uniform coverage with CMC was proposed to be advantageous in stabilizing the electrochemical performance, leading to excellent capacity retention with a higher efficiency compared to the PVDF binder.601,615 To overcome the capacity fading resulting from volume expansion of insertion materials, Sun et al. used the Na-CMC binder in their Li4Ti5O12 electrode.337 Recently, Zhao et al. introduced the Na-doped Li4Ti5O12 electrode using the Na-CMC and Na-alginate binder instead of the PVDF binder. These binders exhibited a slightly higher Coulombic efficiency than that of the PVDF binder. After 100 cycles, the Na-CMC and Na-alginate binders exhibited an excellent cyclability compared to that of the PVDF binder616 (Fig. 42h). When applied to alloy and conversion reaction anode materials in SIBs, their electrochemical performance is dramatically changed depending on binders. Kim et al. achieved excellent cycling performances of red phosphorous carbon composite anodes by using the PAA binder. Upon the sodiation–desodiation process, the PAA binder effectively suppresses a large volume change of phosphorous electrodes compared to that of the PVDF binder.5115. Sodium-ion full cells
Along with the development of LIBs, SIB technologies have been rapidly developing. The gravimetric energy density of SIBs as full cells operable at room temperature has become competitive in the state-of-the-art LIBs with graphite and layered oxides.99 However, current research concentrated mostly on sodium-ion half cells (employing Na metal). Therefore, development of practical sodium ion full cells (without Na metal) remains critical challenges. Recently, in order to realize the practical SIBs, many research groups focus on developing rational full cell design. Furthermore, battery companies such as Aquion Energy (USA), FARADION (UK) and Sumitomo Electric Industries Ltd (Japan) are constantly striving to develop full cell designs, thus resulting in the remarkable improvement of practical SIBs.Historically, sodium-ion full cell studies were conducted earlier than the commercialization of LIBs.71,617–619 In 1988, sodium-ion full cells were proposed based on sodium-lead alloy composite anodes and the P2-type NaxCoO2 cathode, which demonstrated excellent cyclability for over 300 cycles.71 Later, a full cell with a P2-type Na0.6CoO2 cathode and a petroleum coke anode was also proposed with the solid polymer electrolyte P(EO)8NaCF3SO3, at a high operation temperature of 100 °C251 (Fig. 43a). However, these full cell systems did not attract much attention compared to LIBs due to their average discharge potential being lower than 3 V. On the other hand, the performances of sodium-ion full cells have dramatically improved since hard carbon anodes were introduced by Dahn et al.247 As mentioned in the anode section in Section 3.1, hard carbon anodes usually demonstrated a reasonable capacity of 300 mA h g−1 with a low operating potential (nearly ∼0 V), which is beneficial for increasing the average discharge potential and energy density of fabricated full cells with the appropriate cathode. In the case of cathode materials, various types of phosphate- and pyrophosphate-based polyanion materials, which exhibit high operating potentials, are employed for high energy density sodium ion full cells.237 In 2003, Barker et al. reported the preliminary performance data of sodium-ion full cells based on hard carbon/NaVPO4F chemistry181 (Fig. 43b). The average discharge voltage for this cell was demonstrated to be around 3.7 V, which is comparable with commercially available lithium-ion cells based on LiCoO2 or LiMn2O4 cathode materials. However, this full cell consisting of hard carbon/NaVPO4F exhibited a relatively low discharge capacity of 82 mA h g−1. Therefore, to increase both the operating potential and discharge capacity, various full cell configurations have been investigated.27,181,206,574,620–629 Applying the Na3V2(PO4)2F3 cathode and hard carbon anode for fabricating sodium-ion full cells, this full cell exhibited a high capacity of 110 mA h g−1 with an operation voltage of 3.65 V, as well as excellent capacity retention upon cycling with a satisfactory Coulombic efficiency (>98.5%) and very good power performance in the EC:PC:DMC (45:45:10, v/v) electrolyte solution574 (Fig. 43c). Later, Nose et al. assembled a new type of Na-ion full cell by combining Na4Co3(PO4)2P2O7 as a cathode and hard carbon as an anode; this battery demonstrated a 4.0 V-class high operating voltage with long-term operation.206 Layered structured cathodes such as P2-type and O3-type materials are also considered for sodium ion full cells with hard carbon anodes. Previous work has shown that P2-type cathode materials (NaxMO2; x ≤ 0.7, M = transition metal) result in high rechargeable capacities.46 However, a lower initial sodium content in the crystal structure of P2-type layered cathode led to an abnormal Coulombic efficiency above ∼100% in the 1st cycle.100 Therefore, the intrinsic properties hinder practical full cell fabrication. Meanwhile, the practical benefits of O3-type (NaxMO2; x ≈ 1.0, M = transition metal) cathodes are that they are able to fabricate sodium-ion full cells similar to commercial LIBs. Komaba et al. fabricated the full cell combined with a hard carbon anode and O3-tpye Na[Ni1/2Mn1/2]O2 cathode, which demonstrated acceptable battery performance with approximately 3 V for the operating voltage.27 Later, Kim et al. proposed O3-type layered Na[Ni1/3Fe1/3Mn1/3]O2 cathode materials and fabricated a high capacity full cell with hard carbon, which exhibited a high discharge capacity of 100 mA h g−1 and stable cycle retention after 150 cycles with a high operating potential above 3 V50 (Fig. 43d). On the other hand, the sodium ion-full cells used in some works utilize a hard carbon anode that was presodiated in order to reduce the irreversible capacity during the first cycle. Moreover, such a presodiated system needs a different electrode balancing to prevent sodium plating.624 This technique is not intended for commercial applications, but rather for scientific queries, to test the long-term electrochemical performance of such materials using full cells; this is because metallic sodium deposition on the Na metal anode does not warrant a long-term cycling test.87 Hwang et al. fabricated sodium ion full cells with a compositional graded O3-type Na[Ni0.60Co0.05Mn0.35]O2 cathode and presodiated hard carbon anodes, which demonstrated a reversible discharge capacity of 257 mA h g−1 (Na0.68C6) with non-irreversible capacity (Fig. 43e). This full cell exhibited an excellent rate capability of 132.6 mA h g−1 at 1.5 A g−1 and stable cycle retentions at various temperatures (−20, 0, 30 and 55 °C). Especially, it demonstrated a superior cycle retention of ∼80% under extensive cycling conditions for over 300 cycles with an average operation voltage of 2.84 V on discharge at 30 °C. More recently, Keller et al. proposed sodium-ion full cells based on a presodiated hard carbon anode and a mixed layered oxide cathode of P2/P3/O2-Na0.76[Mn0.5Ni0.3Fe0.1Mg0.1]O2.624 Presodiation of hard carbon was carried out until 135 mA h g−1 to prevent excess sodium in the full cell. The developed sodium-ion full cells demonstrated high specific energies (200–240 W h per kg of cathode and anode active materials), high average discharge voltages (3.3 V), high energy efficiencies and an extraordinary long-term cycling stability of 80% capacity retention after 700 cycles.
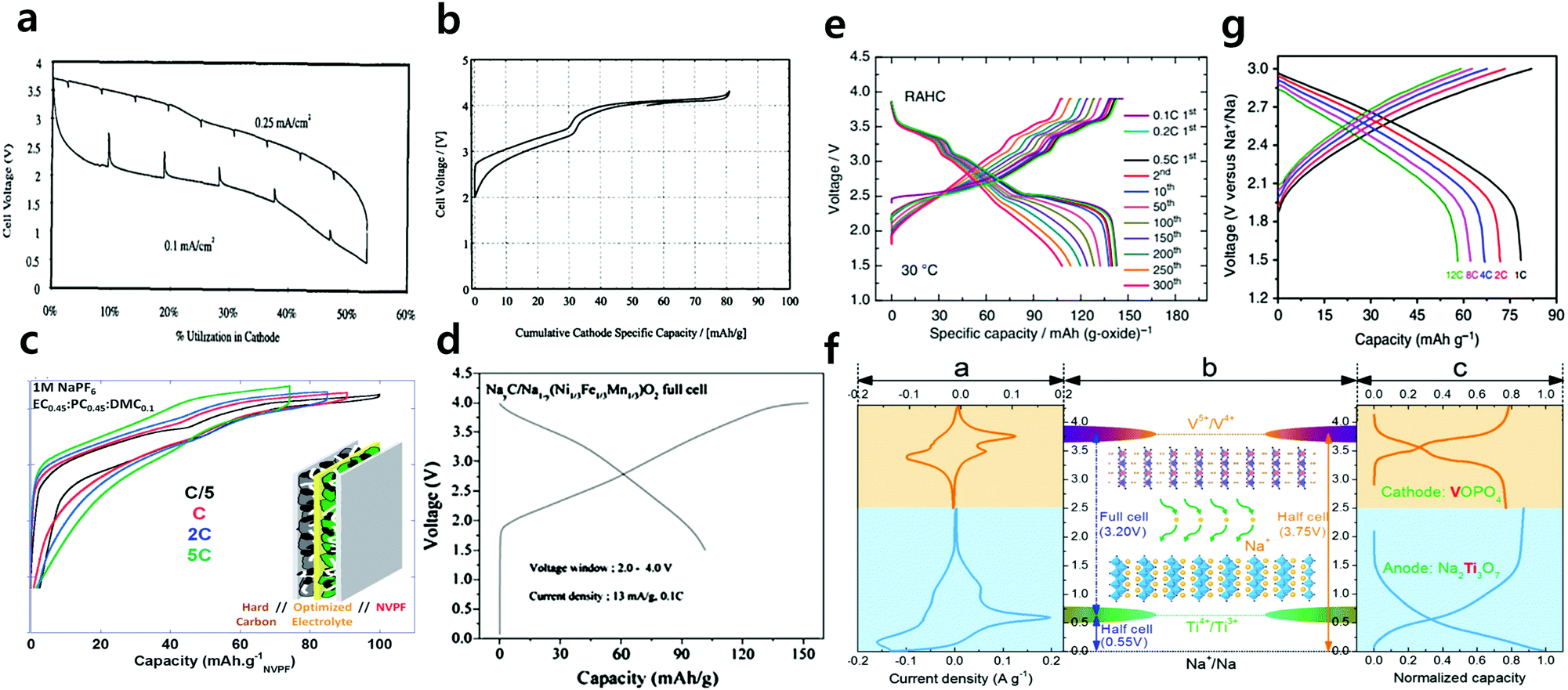 | ||
| Fig. 43 (a) First cycle of an Na0.6CoO2/P(EO)8NaCF3SO3/(ground) petroleum coke cell. (Reproduced with permission from ref. 251, Copyright 1993 The Electrochemical Society.) (b) EVS voltage profile for a typical hard carbon//NaVPO4F full cell cycled between 2.00 and 4.30 V in 1 M solution of NaClO4 in EC/DMC (2:1 weight). (Reproduced with permission from ref. 181, Copyright 2003 The Electrochemical Society.) (c) Voltage versus capacity profiles for NVPF//HC full Na-ion cells cycled in 1 M NaPF6 in EC0.45:PC0.45:DMC0.1. (Reproduced with permission from ref. 574, Copyright 2013 The Royal Society of Chemistry.) (d) First cycle of a full Na-ion cell NayC/Na1–y(Ni1/3Fe1/3Mn1/3)O2. (Reprinted from ref. 50, Copyright 2012, with permission from Elsevier.) (e) Long-term cycle test of C/RAHC full cells in the voltage range of 1.5–3.9 V. (Reproduced by permission from ref. 87, Nature Publishing Group, Copyright 2015.) (f) Schematic of Na2Ti3O7//VOPO4 full cells with electrochemical characterization. (Reproduced with permission from ref. 625, Copyright 2016 The Royal Society of Chemistry.) (g) Discharge profiles of the Na0.6[Cr0.6Ti0.4]O2/Na0.6[Cr0.6Ti0.4]O2 sodium-ion full cell at various rates. (Reproduced by permission from ref. 626, Nature Publishing Group, Copyright 2015.) | ||
However, such full cells based on hard carbon anodes suffer from an inferior rate capability, and the voltage plateau related to most capacities is too close to the sodium plating voltage, causing a safety concern. Li et al. designed a full sodium-ion battery based on nanostructured Na2Ti3O7 and VOPO4 materials as the anodes and cathodes625 (Fig. 43f). In order to reduce the polarization and irreversibility effect of the electrode materials in the first discharge process (both for the anode and cathode sides), pre-desodiated Na2Ti3O7 and pre-sodiated VOPO4 were prepared in advance. This full cell shows outstanding rate capability and excellent cycling stability in the wide temperature range of −20 to 55 °C. Recently, sodium-ion full cells were proposed using the same materials as both cathodes and anodes, namely symmetric full cells.626–629 The greatest advantage of using the same materials for both cathodes and anodes is that they significantly decrease the material's processing cost. Wang et al. proposed the P2-Na0.6[Cr0.6Ti0.4]O2 cation-disordered electrode for high-rate symmetric rechargeable SIBs626 (Fig. 43g). This symmetric full cell exhibited an average operating voltage plateau at ∼2.53 V, as well as an extraordinary rate capability of 75% retention at 12C-rate and superior cycling performance. The Na3V2(PO4)3 materials also exhibit both cathode and anode potentials (3.4 V with a V4+/V3+ redox reaction and 1.6 V with a V3+/V2+ redox reaction), so a novel symmetric full cell system could be designed by employing Na3V2(PO4)3 as a bipolar electrode material.628
Such present results regarding sodium-ion full cells based on several platforms of cathodes and nonmetallic anodes have brought forth great advances in practical issues. Nevertheless, to obtain practical SIBs with high safety, outstanding rate capability, and cycling stability, further investigation of rationally designed full cells, including capacity balancing between the cathode and the anode, the voltage range, and stable electrolyte solution, is still necessary. Further studies of functional additives and binders to effectively control the solid electrolyte interface formation of cathodes and anodes are essential parameters for high safety SIBs. In addition, the total production cost of active materials of the electrode and the cost of battery components should be considered. In summary, a strategy for battery design in realizing practical SIBs is needed to find a good balance between an increase in battery performance with high safety and a decrease in the total cost of the batteries.
6. Summary
Even though SIBs were studied around the same time as LIBs, they were also abandoned at one point, particularly after the commercialization of LIBs by Sony in the early 1990s. However, the growth of technology and the exigency for large-scale applications such as ESSs have opened the door for SIBs to be utilized again. Lithium is not uniformly scattered on the Earth's crust, and given the demand that it is continually increasing, its reserves (specifically in lithium-constrained areas) will be exhausted. This will lead to an escalating price for lithium, and thereby the use of LIBs for ESS applications will not be cost-effective. With the recognition that SIBs have recently received, the hurdles for making them as applicable as LIBs cannot be taken lightly.Electrodes with open frameworks are more desirable in sodium systems, as the larger-sized Na+ (in comparison to Li+) can be introduced into the framework. However, given how the immense structural changes that occur when introducing Na+ cannot be avoided, disruption of structure's durability while trying to maintain its original state is bound to happen.630 Moreover, sodiated transition metal materials are particularly hygroscopic, even upon brief exposure to air. Thus, the preparation of sodiated cathode materials and batteries requires meticulous handling and moisture-free conditions. The hydration of materials will deteriorate the electrochemical performance of the electrode due to the formation of intrinsically insulating NaOH. One recent study on cathode materials for SIBs has focused on this hygroscopic problem. Specifically, the Sumitomo Chemical Co., Ltd research group has successfully synthesized O3-type Ca-doped NaFe0.4Ni0.3Mn0.3O2, and they managed to suppress its tendency to absorb moisture through Ca doping. Although it is reasonable to suspect that the kinetics of Na+ ion diffusion might be a problem due to the larger ionic size relative to that of Li+, Ceder et al. reported that Na+ extraction/re-insertion from/into Na-containing materials is experimentally and computationally faster compared to Li systems due to the low Lewis acidity of Na+. By contrast, the insertion of Na+ into non Na-containing compounds with a smaller bottleneck size may be slower compared to the insertion of Li+. Furthermore, β-Al2O3; the fast Na+ ionic conductor, was discovered almost 50 years ago, which is even earlier than Li+.
Studies on cathode materials are considerably broad, including oxides, polyanions, NASICON (Na Super Ionic Conductor) types and organic compounds. Through studies on the O3-type sodium-containing layered transition metal oxides Na1−xFeO2, it has been found that this material is electrochemically active based on Fe3+/Fe4+ redox couples, which is in contrast to O3-type lithium containing the same layered transition metal oxides in a Li system. Moreover, the irreversible capacity that results from the migration of iron ions in Na1−xFeO2 can be suppressed with the help of Co or Mn occupation in transition metal sites as a substitute to Fe.39,47,50 However, the problem of insufficient cycle stability has yet to be solved. In the case of polyanion compounds, they exhibit better thermal stabilities compared to those of oxides, owing to the presence of the P–O covalent bonds in the crystal structure. They have an operating voltage comparable to that of the Li system. In addition, as opposed to the earlier mentioned information regarding the kinetic diffusion of Na+ ions, a few studies on NaFePO4 have reported that the migration energy of Na+ ions is 0.05 eV higher than that of Li+, which is actually a slower kinetic diffusion than reported here.173 Hong et al.631 and Goodenough et al.632 initially proposed NASICON type compounds for use in Na+ ion solid electrolytes due to the high Na+ ionic conductivity associated with their three-dimensional open framework. However, these compounds can be used as electrode materials with the incorporation of transition metals into their structure. Na3V2(PO4)3, for example, showed improved capacity at both higher and lower voltage regions after the modification of its surface with carbons derived from sugar. Their earlier work showed unsatisfactory results when tested as cathodes in a full symmetrical cell. Prussian blue and its analogues have also attracted attention due to their high energy, power density and electrochemical properties. However, unlike polyanions, their poor thermal stabilities remain a hindrance. Types of cathode materials, their redox couple, capacity, and operation voltage are summarized in Table 1.
| Cathode materials for sodium-ion batteries | |||||||
|---|---|---|---|---|---|---|---|
| Structure | Compositions | Redox couples | Voltages (V) | Theoretical capacity (mA h g−1) | Experimental capacity (mA h g−1) | Ref. | |
| Two-dimensional layer transition metal oxide | Na1−xFeO2 | NaFeO2 | Fe3+/4+ | 1.3–4 | 241 | 125 | 49 |
| Na[Fe0.5Mn0.5]O2 |
Fe3+/4+
Mn3+/4+ |
1.5–4.2 | 242 | 100–110 | 46 | ||
| Na[Fe0.5Co0.5]O2 |
Fe3+/4+
Co3+/4+ |
2.5–4.0 | 240 | 160 | 47 | ||
| Na[Fe0.3Ni0.7]O2 |
Fe3+/4+
Ni3+/4+ |
2.0–3.6 | 237 | 135 | 39 | ||
| Na[Ni1/3Fe1/3Mn1/3]O2 |
Ni2+/4+
Fe3+/4+ Co4+ |
2–4 | 239 | 120 | 50 | ||
| Na[Ni0.25Fe0.5Co0.25]O2 |
Ni2+/4+
Fe3+/4+ Co4+ |
2.1–3.9 | 239 | 140 | 51 | ||
| Na[Ni0.25Fe0.5Mn0.25]O2 |
Ni2+/4+
Fe3+/4+ |
2.1–3.9 | 240 | 140 | 52 | ||
| Na[Li0.05(Ni0.25Fe0.5Mn0.25)0.95]O2 |
Ni2+/4+
Fe3+/4+ |
1.7–4.4 | 246 | 180 | 52 | ||
| Na1−x[Fe1−yMny]O2 | Na2/3[Fe0.5Mn0.5]O2 |
Fe3+/4+
Mn3+/4+ |
1.5–4.3 | 260 | 190 | 54 | |
| Na0.62[Fe0.5Mn0.5]O2 |
Fe3+/4+
Mn3+/4+ |
1.5–4.3 | 263 | 185 | 55 | ||
| Na0.7[Fe0.5Co0.5]O2 |
Fe3+/4+
Co3+/4+ |
2.5–4.0 | 256 | 170 | 47 | ||
| Na1−xCoO2 | NaCoO2 | Co3+/4+ | 2.0–4.0 | 239 | 170 | 33 | |
| Na0.55CoO2 | Co3+/4+ | 2.7–3.5 | 263 | 140 | 62 | ||
| Na2/3[Co2/3Mn1/3]O2 |
Co2+/3+
Mn3+/4+ |
1.5–4.3 | 258 | 180 | 73 and 74 | ||
| Na5/8Ca1/24CoO2 | Co3+/4+ | 2.0–4.0 | 255 | 124 | 79 | ||
| Na1−xNiO2 | NaNiO2 | Ni3+/4+ | 2.0–4.5 | 235 | 145 | 81 | |
| 1.25–3.75 | 235 | 121 | 81 | ||||
| Na[Ni0.5Mn0.5]O2 | Ni2+/4+ | 2.0–4.5 | 239 | 185 | 83 | ||
| 2.2–3.8 | 239 | 125 | 83 | ||||
| 2.0–4.0 | 239 | 124 | 84 | ||||
| NaFe0.2Ni0.4Mn0.4O2 |
Fe3+/4+
Ni3+/4+ |
2.0–4.0 | 240 | 130 | 84 | ||
| Na[Ni0.5Ti0.5]O2 | Ni2+/4+ | 1.5–4.7 | 247 | 121 | 88 | ||
| NaxMnO2 | NaMnO2 | Mn3+/4+ | 2–3.8 | 243 | 197 | 90 | |
| Na0.7MnO2 | Mn3+/4+ | 2–3.8 | 260 | 150 | 98 | ||
| Mn3+/4+ | 1.5–4.4 | 260 | 216 | 97 | |||
| Mn3+/4+ | 1.5–4.4 | 260 | 198 | 97 | |||
| Na2/3[Ni1/3Mn2/3]O2 | Ni2+/4+ | 2.0–4.5 | 258 | 162 | 101, 107 and 113 | ||
| Na2/3[Ni1/3−xMgxMn2/3]O2 | Ni2+/4+ | 2.0–4.5 | Around 258 | 120–145 | 107 | ||
| Na2/3[Ni1/3−xAlxMn2/3]O2 | Ni2+/4+ | 2.0–4.5 | Around 258 | 147 | 113 | ||
| Na2/3[Ni1/3−xFexMn2/3]O2 |
Fe3+/4+
Ni2+/4+ |
2.0–4.5 | Around 258 | 145 | 113 | ||
| Na0.7[Mn0.65Ni0.15Fe0.2]O2 |
Fe3+/4+
Ni2+/4+ |
1.5–4.3 | 258 | 208 | 117 | ||
| Na2/3[Ni1/3−xCoxMn2/3]O2 | Ni2+/4+ | 2.0–4.5 | Around 258 | 144 | 113 | ||
| Na2/3[Ni1/3−xZnxMn2/3]O2 | Ni2+/4+ | 2.2–4.3 | Around 258 | 140 | 111 and 112 | ||
| Na0.7[Mn0.6Ni0.3Co0.1]O2 | Ni2+/4+ | 1.5–4.3 | 256 | 185 | 114 | ||
| Na2/3[Ni1/3Mn1/6Ti1/6]O2 | Ni2+/4+ | 2.5–4.5 | 319 | 127 | 118 and 119 | ||
| Na2/3[Mg0.28Mn0.72]O2 | Mn3+/4+ | 1.5–4.4 | 286 | 220 | 120 | ||
| Na5/6[Li1/4Mn3/4]O2 | Mn3+/4+ | 1.5–4.4 | 267 | 180 | 121 | ||
| Na0.85[Li0.17Ni0.21Mn0.64]O2 |
Ni2+/4+
Mn3+/4+ |
1.5–4.4 | 266 | 200 | 7 | ||
| Na1−xTiO2 | NaTiO2 | Ti3+/4+ | 2.0–3.5 | 260 | 75 | 124 and 125 | |
| Na1−xCrO2 | NaCrO2 | Cr3+/4+ | 2–3.6 | 250 | 110 | 126 | |
| Na1−xVO2 | NaVO2 | V4+/3+ | 1.2–2.4 | 253 | 120 | 134 | |
| Na0.7VO2 | V4+/3+ | 1.2–2.6 | 270 | 105 | 136 | ||
| Na2RuO3 | Na2RuO3 | Ru4+/5+ | 1.5–4.0 | 137 | 147 | 137 | |
| Two-or three-dimensional layer transition metal oxide and fluorides | Manganese oxide | α-MnO2 [hollandite] | Mn3+/4+ | 1.0–4.3 | 308 | 280 | 146 and 147 |
| β-MnO2[hollandite] | Mn3+/4+ | 1.0–4.3 | 308 | 300 | 146 | ||
| Vanadium oxide | α-V2O5 | V5+/4+ | 1.5–3.8 | 294 | 250 | 151 | |
| VO2(B) | V4+/3+ | 1.0–3.8 | 323 | 189 | 153 | ||
| Metal fluorides | FeF3 | Fe3+/2+ | 1.5–4.0 | 712 | 150 | 153–156 and 178 | |
| NaFeF3 | Fe3+/2+ | 1.5–4.0 | 198 | 128 | 178 | ||
| NaNiF3 | Ni3+/2+ | 1.5–4.0 | 193 | Below 40 | 155 | ||
| NaMnF3 | Mn3+/2+ | 1.5–4.0 | 198 | Below 40 | 155 | ||
| Three-dimensional polyanion compounds | Phosphates and fluorophosphates | Amorphous NaFePO4 | Fe3+/2+ | Around 2.4 | 154 | 150 | 156 |
| Olivine NaFePO4 | Fe3+/2+ | Around 2.8 | 154 | 120 | 163–173 | ||
| Na2FePO4F | Fe3+/2+ | 2.0–3.8 | 124 | 100 | 178 | ||
| Na2[Fe0.5Mn0.5]PO4F |
Mn2+/3+
Fe2+/3+ |
2.0–4.5 | 124 | 110 | 180 | ||
| NaV0.96Cr0.04PO4F | V5+/3+ | 3.0–4.5 | 142 | 80 | 184 and 185 | ||
| Na3V2(PO4)2F | V4+/5+ | 3.0–4.2 | 156 | 87 | 186 | ||
| Na1.5VPO4.8F0.7 | V3.8+/5+ | 2.0–4.5 | 134 | 130 | 187 | ||
| Na3V2(PO4)3F3 | V3+/4+ | 2.0–4.2 | 128.2 | 120 | 188–190 | ||
| Pyrophosphates | (MoO2)2P2O7 | Mo6+ | 1.0–3.5 | 250 | 190 | 192 | |
| Na2FeP2O7 | Fe2+/3+ | 2.0–3.8 | 97 | 88 | 196 | ||
| Na3.32Fe2.34(P2O7)2 [Na1.66Fe1.17P2O7] | Fe2+/3+ | 1.7–4.0 | 117.6 | 90 | 201 | ||
| β-Na2MnP2O7 | Mn3+/2+ | 2.0–4.45 | 97.5 | 80 | 202 | ||
| Na2(VO)P2O7 | V5+/4+ | 2.0–4.2 | 93.4 | 80 | 203 | ||
| Mixed phosphates | Na4Co3(PO4)2P2O7 | Co2+/3+ | 3.0–4.7 | 170 | 95 | 206 | |
| Na4Co2.4Mn0.3Ni0.3(PO4)2P2O7 |
Co2+/3+
Mn3+/4+ Ni2+/3+ |
3.0–4.8 | 170 | 80 | 207 | ||
| Na7V4(P2O7)PO4 | V3+/4+ | 2.0–4.2 | 92.8 | 90 | 208 | ||
| NASICON type phosphates | Na3V2(PO4)3 | V4+/3+ | 2.7–3.8 | 117.6 | 93 | 209 | |
| Sulfates, fluorosulfates, and carbonophosphates | Na3MnPO4CO3 |
Mn2+/3+
Mn3+/4+ |
2.0–4.5 | 250 | 125 | 221 | |
| Cyanides and organic compounds | KMnFe[CN]6 |
Mn3+
Fe3+/2+ |
2.0–4.0 | 81 | 70 | 228 | |
| KFeFe[CN]6 | Fe3+/2+ | 2.0–4.0 | 81 | 90 | 228 | ||
| KCoFe[CN]6 |
Co3+
Fe3+/2+ |
2.0–4.0 | 81 | 55 | 228 | ||
| KNiFe[CN]6 |
Ni2+
Fe3+/2+ |
2.0–4.0 | 80 | 50 | 228 | ||
| KCuFe[CN]6 |
Cu2+
Fe3+/2+ |
2.0–4.0 | 79 | 55 | 228 | ||
| KZnFe[CN]6 |
Zn2+
Fe3+/2+ |
2.0–4.0 | 79 | 32 | 228 | ||
| Na1.4MnFe[CN]6 |
Mn3+/2+
Fe3+/2+ |
2.0–4.2 | 82 | 131 | 229 | ||
| Na1.72MnFe[CN]6 |
Mn3+/2+
Fe3+/2+ |
2.0–4.2 | 81 | 120 | 229 | ||
| Na4Fe[CN]6 | Fe3+/2+ | 2.0–3.8 | 89 | 87 | 231 | ||
| Na4C8H2O6 | 1.6–2.8 | 187 | 180 | 236 | |||
| Na2C6O6 | 1.5–2.9 | 237 | 180 | 237 | |||
| Aniline–nitroaniline copolymer | 2.0–4.0 | 180 | 238 | ||||
Given that the electrochemical insertion of Na+ into graphite, which is a commonly used anode material in LIBs, has not been considered suitable for Na-ion batteries, an alternative system for large-scale energy storage is needed because of the thermodynamic instability of binary Na-intercalated GICs.258,259 Therefore, the search for appropriate anodes with proper Na voltage storage, large reversible capacity and high structural stability is necessary in order for SIBs to be successfully developed. Sodium metal is not recommended for use in an anodes due to its high reactivity and formation of dendrite. Many non-graphitic materials such as carbon black, pitch-based carbon-fibers have been demonstrated to insert Na+. Hard carbons, i.e., non-graphitic but graphene containing, carbonaceous materials, are considered as the “the first-generation” anode of choice for SIBs.22,23,241 The low BET surface area of hard carbon appears to be a pivotal factor in achieving good reversible cycling of SIBs. However, the low operational potential of carbonaceous anodes raises severe safety issues for practical applications. Hence, transition metal oxide compounds, especially titanium-based oxides, have been widely studied.300 The most common form of titanium oxide compounds, titanium dioxide (TiO2), has been tested in Na systems by downsizing the size to the nanometer-scale. It was found that the pseudocapacitance mechanism was responsible for the charge storage. The pseudocapacitive effect on typical insertion electrodes is believed to be a potential solution to overcoming the capacity limitation for Na insertion anodes. Conversion reactions of metal oxides can also be useful for anodes in SIBs.243 These conversion reactions, which depend on the transition metals, occasionally combined with the insertion–extraction or alloy-dealloying reactions. The reaction concept was first introduced with spinel NiCo2O4, which delivered a reversible capacity of ∼200 mA h g−1 after the initial discharge of ∼600 mA h g−1. Previous research has shown that the conversion reaction that forms sodium oxide is the primary reaction because spinel NiCo2O4 cannot accommodate for the large size of Na+ into its vacant sites.365 Various metal sulfides such as cobalt sulfides (CoS, CoS2), molybdenum sulfides (Mo2S, MoS2), iron sulfides (FeS, FeS2), and tin sulfides (SnS, SnS2) have been extensively investigated as high capacity anode materials for NIBs. The weaker M–S bond in metal sulfides compared to the corresponding M–O bond in metal oxides is kinetically favorable for conversion reactions with Na ions.410 Hence, the smaller volume changes and better reversibility of Na2S compared to that of Na2O during the sodiation–desodiation process result in better mechanical stabilities and higher initial Coulombic efficiency, respectively.
Apart from electrode materials, suitable electrolytes, additives and binders are equally important for the development of NIBs. With appropriate electrolyte formulation, the interface reaction can be minimized and both cell performances and safety aspects can be strengthened. As reported by Bhide et al., NaPF6 in EC:DMC is favorable for the formation of a stable surface film, as well as the reversibility of the Na0.7CoO2 cathode.576 NaPF6-based electrolyte solution demonstrated higher ionic conductivity, while in a report by Komaba et al., NaPF6-based electrolytes with hard carbon electrodes exhibited more stable cycling performance than that of hard carbon electrodes with NaClO4-based electrolytes.27 According to studies reported by Palacin's group, they have discovered that NaPF6 in the EC:PC:DMC solvent mixture is the best electrolyte for the hard carbon anode.574 On the other hand, Ponrouch et al. reported that NaPF6 in EC:PC exhibited a low generation of heat and a high thermal stability, which can be attributed to the more thermally stable SEI layer that is formed on the HC after cycling.28 As organic liquid electrolytes are less safe than the other electrolytes, using an aqueous electrolyte is a good approach to prevent safety hazards, and additionally they are of a relatively low cost. Developing aqueous Na-ion batteries is meaningful and practical because of the huge abundance of Na resources (NaCl, Na2SO4, NaNO3, etc.). Using poly(vinylidene fluoride) (PVDF) as a binder is very common due to its good chemical and electrochemical stabilities. However, with the use of a volatile and toxic organic solvent (N-methyl pyrrolidone) and relatively high production cost when using PVDF to make the slurry, water-soluble binders such as sodium carboxymethyl cellulose (Na-CMC), poly(acrylic acid) (PAA) and sodium alginate (NaAlg) have been introduced. Na-CMC, an environmentally friendly and inexpensive material, could play an important role in improving the solid electrolyte interface (SEI) passive layer, which reduces the irreversible capacity and somehow leads to a better cycle life. Na-Alg, which is more polarized than CMC, can ensure better interfacial interaction between the polymer binder and the particles, as well as create stronger adhesion between the electrode layer and the Cu substrate. Use of the PAA binder allows for the formation of a stable SEI layer on the elastic binder-coated electrode surface, in which the elasticity of a polymer matrix may prevent cracking at the SEI upon volume changes.
Na-ion based electrochemical systems are alternatives to Li-ion systems. The study of electrode materials, carbon additives, binders, electrolyte salts, solvents, and current collectors is important and must be established in order to realize the practicality of SIBs in large-scale applications such as ESS. We believe that improving the understanding and more findings of materials can accelerate the development of SIBs which are compatible to commercial level LIBs.
7. Perspectives
LIBs have realized their adoption from portable devices to vehicle applications because of their high operation voltage and energy density, so that battery performances including price and safety are very important concern in technological evolution of electronic devices in future. In addition, demands on batteries for zero-emission EVs and energy storage are expected to increase exponentially from the year of 2020, and the secondary battery market will expand almost twice of the current level, 112 billion US dollars.633 Albeit the expandability of LIBs, the geological issue, in which lithium resources are localized in mainly South America, is thought of as a serious situation to be resolved. Use of cobalt or other rare metals as electrode materials is another concern on the price of batteries for future mass production for the purpose of mounting LIBs to EVs and energy storage applications. Indeed, the battery pack price should reach 20–30% of the current value to spread LIBs for those applications from 2020. This is controversial for the use of present high capacity lithiated Ni-rich cathode materials because each metal price except manganese is expected to further increase as more of the present cathode materials are consumed in future. Depletion of these resources, particularly cobalt, should be considered at the same time.Academic and industrial societies are recently raising the alarm about this situation and speaking up about the necessity of alternative rechargeable batteries that can substantially substitute present LIBs. Indeed, sodium-ion batteries (SIBs) were developed together with LIBs in 1980s; however, the inferior battery performance of SIBs relative to LIBs was the main reason for the fade of SIBs, which led to the birth of commercialized LIBs by Sony in 1990s. However, the recent upsurge of EVs and energy storage using LIBs as power sources could cause a shortage of LIBs so a great deal of attention has been paid again to development of SIBs, in which sodium is inexpensive and evenly distributed. Since 2010 with the slogan of beyond lithium, research trends in SIBs have been intensively focused on electrode materials rather than battery systems in order to distinguish the feasibility of commercialization. There are some merits of SIBs versus LIBs as follows; (i) possibility of high performance electrode materials because of the abundance of sodium transition metal compounds relative to lithium compounds, (ii) fast diffusion of sodium ions in the solid phase, which indicates high rate performance of Na cells, (iii) lowering of the manufacturing cost due to the use of Al foil instead of Cu current collectors for anodes and the transfer of the production line of LIBs to SIBs, which does not need new production facility, (iv) superior safety properties of the cathode even at a highly charged state, and (v) less risk for sodium supply. By contrast, there are some demerits of SIBs; (i) low operation approximately 0.3 V versus Li, (ii) a large ionic radius of Na+ (1.02 Å) relative to Li+ (0.76 Å), which induces a simultaneous structure change during Na+ insertion and extraction that may cause gradual capacity fade, and (iii) high reactivity of Na metal which necessitates basic solution for formation of metallic sodium deposition onto anodes that can seriously threaten safety of Na cells.
As mentioned in Fig. 1 and Table 1 development of cathode materials has been progressing towards layer and polyanion structure materials. In particular, know-hows accumulated by the development of LIBs are leading to significant progress in layered structure materials. O3 type layer compounds are very fascinating because they have the same crystal structure as commercial LiCoO2 and Ni-rich derivatives of LIBs, but their capacities are usually limited below 130 mA h g−1 due to simultaneous structural changes induced from the presence of large Na+ ions in the crystal structure.83,634 Different from the O3 type materials, P2 type cathode materials are of interest due to their high capacity approaching approximately 200 mA h g−1.46 Despite the high capacities, the serious flaw of the P2 materials is the lack of extractable sodium content in the compounds, namely, Na0.7MO2 (M: transition metals), of which the first discharge capacity is similar to that of O3 type compounds. Nonetheless, many works have been directed toward stabilization of cycling performances. Appropriate sacrificing agents can show oxidative decomposition at the first charge, which can be used to solve the irreversible capacity at the first cycle. This enables the resulting Coulombic efficiency approaching close to 100%. Provided that the abnormal behavior is resolved commercialization of high capacity SIBs can be readily advanced, because many kinds of dopants are known to be effective to retain the capacity for long term cycling. For this purpose, pioneering works were conducted using Na3N60,635,636 and Na3P637 as the sacrificing agents to minimize the abnormally high Coulombic efficiency of over 130% at the first cycle and to compensate the sodium deficiency. Although such works could improve the first irreversible capacity of P2-type layer cathodes, the resulting Coulombic efficiency at the first cycle is still not sufficient to fabricate the practical SIBs in comparison with O3-type layer cathodes. Therefore, to compete with LIBs, academic community should revisit O3-type layer cathode materials such as Co-free Na[Ni0.5Mn0.5]O283 and carbon-coated NaCrO2131 which show high Coulombic efficiency at initial cycle and excellent cyclability. The know-hows accumulated from LIBs were further used to synthesize O3-type full concentration gradient (FCG) cathode materials that demonstrate high capacity and good cycling performances.87,160,566 Full cells adopting hard carbon anodes confirmed cycling stability of the materials for a long time. Spoke-like assembly of nanorods greatly improved the strength of the particles, and this enabled unprecedentedly good cycling stability and rate performances. Although the chemistries are very interesting for P2 type layer cathodes for investigation of the structural change during Na+ insertion and extraction, elaboration is required in order to stabilize the Coulombic efficiency at the first cycle. Polyanion materials are attractive because of high operation voltage and cycling stability, whereas low capacities below 100 mA h g−1 and more seriously moisture uptake in air are the critical issues to be resolved for those compounds. These suggest suitability of O3 type cathode materials in practical uses for future EVs and energy storage applications.
Anode materials are activated by insertion, conversion, and alloying reactions as summarized in Fig. 1. Due to the low operation voltage and extraordinary cycling stability, hard carbon proposed by Dahn et al.22 in 2000 is regarded as the better candidate than any other electrode material for anode materials. Since the first LIBs adopted hard carbon as the anode materials, hard carbon can be the first commercial anode materials even in SIBs as well. Most of all, most urgent is the sodium metal deposition onto the surface of hard carbon at low voltage, in which safety issues should be considered because of the high reactivity of sodium metal. In addition, capacity below 300 mA h g−1, which is lower than graphite anodes for LIBs, is not advantageous to improve the energy density of SIBs. Thus, conversion and alloying reaction based materials are intensively being studied to find alternative anode materials that deliver high capacity and do not have sodium metal deposition on discharge. It is evident that these conversion and alloying materials have superiority in capacity, while volume expansion of electrodes in the sodiate state arising from the large ionic size of Na+ is the intrinsic problem for these electrodes. Low Coulombic efficiency at the first cycle is another critical issue to be studied further. Provided that the above-mentioned issues are cleared, phosphorous materials that deliver over 2000 mA h g−1 are expected to be some of the candidate materials for anodes to improve energy density of SIBs. So far, various anode materials are under investigation, it is believed that hard carbon is thought to be the best candidate in practical applications due to its low operation voltage, cycling stability, and high Coulombic efficiency at the first cycle. Reviewing many kinds of cathode and anode materials in the present review, in summary, there is no doubt about the practical use of the O3 type cathode and hard carbon anode assembly for successful full cell configuration.
At present, leading companies are promoting several prototypes of SIBs for commercialization. Sumitomo Chemical Co. Ltd demonstrated a pouch type full cell using the O3-type NaNi0.3Fe0.4Mn0.3O2 cathode in 2013.638 Recently, CNRS and RS2E have launched the first commercial cylindrical 18650 SIBs, which guaranty 2000 cycles with an energy density of 90 W h kg−1.639 In 2015, FRADION developed prototype SIBs and successfully mounted SIBs into E-bikes.640 Sharp Laboratory also promotes SIBs that adopt O3 type or Prussian white cathodes, hard carbon anodes, and conventional carbonate-based electrolytes for high energy density SIBs.641,642 Recent a report by Choi et al. and Doron et al.643 proposed the importance of minimization of energy consumption ($ W h−1) rather than lowering of material costs to successful spread of SIBs towards energy storage applications. Also, SIBs are faced with intrinsic low energy density relative to LIBs, and this further lowers volumetric energy density in the limited space in battery pack. One option proposed is to improve the tap density of cathode materials. Our recent work on the radially aligned hierarchical columnar (RAHC) structure87 and spoke-like nanorod assembly (SNA) in a spherical secondary particle566 can be a good example to have high capacity and, more importantly, to maximize the electrode density achieved from its unique feature of intrinsic robustness under high compression pressure in the electrode pressing process. To date, specific energy density of SIBs was only estimated based on the weight of active materials by calculation with the assumption of 300 mA h g−1 for hard carbon as a negative electrode material for full cells. Such an estimated specific energy density of SIBs with some of the cathode materials reaches and/or exceeds 300 W h kg−1.18,643 Unfortunately, in consideration of not only the active materials but all components that compose SIBs, the actual value of energy density in the first commercial cylindrical 18650 SIBs is dramatically lowered below 100 W h kg−1. Therefore, it is very important to keep in our mind how we can improve the energy density of a SIB system when developing high capacity cathode and anode materials and stable electrolytes including additives that operate in a high voltage region, in particular, to maximize the energy density. Furthermore, more systematic studies on surface modification of active materials to minimize the side reactions with electrolytes, binders, current collectors, and the other components should be intensively progressed so as to advance the SIBs in practical use towards energy storage applications.
Conflict of interest
The authors declare that there is no conflict of interest for publishing this review in Chemical Society Reviews.Acknowledgements
This work was supported by the Global Frontier R&D Program (2013M3A6B1078875) of the Center for Hybrid Interface Materials (HIM) funded by the Ministry of Science, ICT, & Future Planning and by a Human Resources Development program (No. 20154010200840) of a Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korean government Ministry of Trade, Industry, and Energy.References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2360 CAS.
- Y. Nishi, J. Power Sources, 2001, 100, 101–106 CrossRef CAS.
- J.-M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- C.-X. Zhu and H. Li, Energy Environ. Sci., 2011, 4, 2614–2624 Search PubMed.
- E. de la Llave, V. Borgel, K. J. Park, J.-Y. Hwang, Y.-K. Sun, P. Hartmann, F. F. Chesneau and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 1867–1875 CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- T. B. Reddy and D. Linden, Linden's Handbook of Batteries, McGraw-Hill, 2010 Search PubMed.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, J. Nanotechnol., 2015, 6, 1016–1055 CAS.
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenough, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- A. S. Nagelberg and W. L. Worrell, J. Solid State Chem., 1979, 29, 345–354 CrossRef CAS.
- J. P. Parant, R. Olazcuag, M. Devalett, C. Fouassie and P. Hagenmul, J. Solid State Chem., 1971, 3, 1–5 CrossRef CAS.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS.
- J. J. Braconnier, C. Delmas, C. Fouassier and P. Hagenmuller, Mater. Res. Bull., 1980, 15, 1797–1804 CrossRef CAS.
- M. S. Whittingham, Prog. Solid State Chem., 1978, 12, 41–99 CrossRef CAS.
- L. W. Shacklette, J. E. Toth and R. L. Elsenbaumer, Conjugated polymer as substrate for the plating of alkali metal in a nonaqueous secondary battery, Allied Corp., USA, 1985 Search PubMed.
- T. Shishikura and M. Takeuchi, Secondary batteries, ShowaDenko K. K. Hitachi, Ltd, Japan, 1987 Search PubMed.
- K. Kubota and S. Komaba, J. Electrochem. Soc., 2015, 162, A2538–A2550 CrossRef CAS.
- D. P. Divincenzo and E. J. Mele, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 2538–2553 CrossRef CAS.
- R. Alcántara, J. M. Jiménez-Mateos, P. Lavela and J. L. Tirado, Electrochem. Commun., 2001, 3, 639–642 CrossRef.
- P. Thomas, J. Ghanbaja and D. Billaud, Electrochim. Acta, 1999, 45, 423–430 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 4428–4431 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- L. Joncourt, M. Mermoux, P. H. Touzain, L. Bonnetain, D. Dumas and B. Allard, J. Phys. Chem. Solids, 1996, 57, 877–882 CrossRef CAS.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A515–A519 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- A. Ponrouch, E. Marchante, M. Courty, J. M. Tarascon and M. R. Palacin, Energy Environ. Sci., 2012, 5, 8572–8583 CAS.
- X. Wu, Y. Cao, X. Ai, J. Qian and H. Yang, Electrochem. Commun., 2013, 31, 145–148 CrossRef CAS.
- J. Y. Luo, W. J. Cui, P. He and Y. Y. Xia, Nat. Chem., 2010, 2, 760–765 CrossRef CAS PubMed.
- Y. Wang, J. Yi and Y. Xia, Adv. Energy Mater., 2012, 2, 830–840 CrossRef CAS.
- E. Monyoncho and R. Bissessur, Mater. Res. Bull., 2013, 48, 2678–2688 CrossRef CAS.
- C. Delmas, J.-J. Braconnier, C. Fouassier and P. Hagenmuller, Solid State Ionics, 1981, 3/4, 165–169 CrossRef.
- R. Famery, P. Bassoul and F. Queyroux, J. Solid State Chem., 1985, 57, 178–190 CrossRef CAS.
- R. Famery, P. Bassoul and F. Queyroux, J. Solid State Chem., 1986, 61, 293–300 CrossRef CAS.
- S. Kikkawa, H. Ohkura and M. Koizumi, Mater. Chem. Phys., 1987, 18, 375–380 CrossRef CAS.
- B. Fuchs and S. Kemmler-Sack, Solid State Ionics, 1994, 68, 279–285 CrossRef CAS.
- T. Shirane, R. Kanno, Y. Kawamoto, Y. Takeda, M. Takano, T. Kamiyama and F. Izumi, Solid State Ionics, 1995, 79, 279–285 CrossRef.
- Y. Takeda, K. Nakahara, M. Nishijima, N. Imanishi, O. Yamamoto and M. Takano, Mater. Res. Bull., 1994, 29, 659–666 CrossRef CAS.
- V. L. McLaren, A. R. West, M. Tabuchi, A. Nakashima, H. Takahara, H. Kobayashi, H. Sakaebe, H. Kageyama, A. Hirano and Y. Takeda, J. Electrochem. Soc., 2004, 151, A672–A681 CrossRef CAS.
- T. Matsumura, N. Sonoyama and R. Kanno, Solid State Ionics, 2003, 161, 31–39 CrossRef CAS.
- S. Miyazaki, S. Kikkawa and M. Koizumi, Synth. Met., 1983, 6, 211–217 CrossRef CAS.
- S. Kikkawa, S. Miyazaki and M. Koizumi, J. Power Sources, 1984, 14, 231–234 CrossRef.
- N. Yabuuchi, H. Yoshida and S. Komaba, Electrochemistry, 2012, 80, 716–719 CrossRef CAS.
- J. Zhao, L. Zhao, N. Dimov, S. Okada and T. Nishida, J. Electrochem. Soc., 2013, 160, A3077–A3081 CrossRef CAS.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Mishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef CAS PubMed.
- H. Yoshida, N. Yabuuchi and S. Komaba, Electrochem. Commun., 2013, 34, 60–63 CrossRef CAS.
- X. Wang, G. Liu, T. Iwao, M. Okubo and A. Yamada, J. Phys. Chem. C, 2014, 118, 2970 CAS.
- Y. Nanba, T. Iwao, B. M. D. Boisse, W. Zhao, E. Hosono, D. Asakura, H. Niwa, H. Kiuchi, J. Miyawaki, Y. Harada, M. Okubo and A. Yamada, Chem. Mater., 2016, 28, 1058–1065 CrossRef CAS.
- D. Kim, E. Lee, M. Slater, W. Lu, S. Rood and C. S. Johnson, Electrochem. Commun., 2012, 18, 66–69 CrossRef CAS.
- S.-M. Oh, S.-T. Myung, C. S. Yoon, J. Lu, J. Hassoun, B. Scrosati, K. Amine and Y.-K. Sun, Nano Lett., 2014, 14, 1620–1626 CrossRef CAS PubMed.
- S.-M. Oh, S.-T. Myung, J.-Y. Hwang, B. Scrosati, K. Amine and Y.-K. Sun, Chem. Mater., 2014, 26, 6165–6171 CrossRef CAS.
- S. Komaba, T. Nakayama, A. Ogata, T. Shimizu, C. Takei, S. Takada, A. Hokura and I. Nakai, ECS Trans., 2009, 16, 43–55 CAS.
- G. Singh, J. Miguel Lopez del Amo, M. Calceran, S. Perez-Villar and T. Rojo, J. Mater. Chem. A, 2015, 3, 6954–6961 CAS.
- B. Mortemard de Boisse, D. Carlier, M. Guignard, L. Bourgeois and C. Delmas, Inorg. Chem., 2014, 53, 11197–11205 CrossRef CAS PubMed.
- E. Talaie, V. Duffort, H. L. Smith, B. Fultz and L. F. Nazar, Energy Environ. Sci., 2015, 8, 2512–2523 CAS.
- J. S. Thorne, R. A. Dunlap and M. N. Obrovac, J. Electrochem. Soc., 2013, 160, A361–A367 CrossRef CAS.
- Y. H. Jung, A. S. Christiansen, R. E. Johnsen, P. Norby and D. K. Kim, Adv. Funct. Mater., 2015, 25, 3227–3237 CrossRef CAS.
- B. Mortemard de Boisse, D. Carlier, M. Guignard and C. Delmas, J. Electrochem. Soc., 2013, 160, A569–A574 CrossRef CAS.
- G. Singh, B. Acebedo, M. C. Cabanas, D. Shanmukarag, M. Armand and T. Rojo, Electrochem. Commun., 2013, 37, 61–63 CrossRef CAS.
- V. Duffort, E. Talaie, R. Black and L. F. Nazar, Chem. Mater., 2015, 27, 2515–2524 CrossRef CAS.
- R. Berthelot, D. Carlier and C. Delmas, Nat. Mater., 2011, 10, 74–80 CrossRef CAS PubMed.
- K. Takeda, H. Sakurai, E. Takayama-Muromachi, F. Izumi, R. A. Dilanian and T. Sasaki, Nature, 2003, 422, 53–55 CrossRef PubMed.
- Q. Huang, M. L. Foo, J. W. Lynn, H. W. Zandbergren, G. Lawes, Y. Wang, B. H. Toby, A. P. Ramirez, N. P. Ong and R. J. Cava, J. Phys.: Condens. Matter, 2004, 16, 5803–5814 CrossRef CAS.
- M. Lee, L. Viciu, L. Li, Y. Wang, M. L. Foo, S. Watauchi, R. A. Pascal Jr., R. J. Cava and P. Ong, Nat. Mater., 2006, 5, 537–540 CrossRef CAS PubMed.
- M. Roger, D. J. P. Morris, D. A. Tennant, M. J. Gutmann, J. P. Goff, J.-U. Hoffmann, R. Feyerherm, E. Dudzik, D. Prabhakaran, A. T. Boothroyd, N. Shannon, B. Lake and P. P. Deen, Nature, 2007, 445, 631–634 CrossRef CAS PubMed.
- Y. Hinuma, Y. S. Meng and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 224111 CrossRef.
- T. A. Platova, I. R. Mukhamedshin, H. Alloul, A. V. Dooglav and G. Collin, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 224106 CrossRef.
- J. Molenda, C. Delmas, P. Dordor and A. Stoklosa, Solid State Ionics, 1984, 12, 473–477 CrossRef CAS.
- F. C. Chou, E. T. Abel, J. H. Cho and Y. S. Lee, J. Phys. Chem. Solids, 2005, 66, 155–160 CrossRef CAS.
- L. W. Shacklette, T. R. Jow and L. Townsend, J. Electrochem. Soc., 1988, 135, 2669–2674 CrossRef CAS.
- Y. Ma, M. M. Doeff, J. Steven, J. De and C. Lutgard, J. Electrochem. Soc., 1993, 140, 2726–2733 CrossRef CAS.
- F. Tournadre, L. Croguennec, I. Saadoune, D. Carlier, Y. Shao-Horn, P. Willmann and C. Delmas, J. Solid State Chem., 2004, 177, 2790–2802 CrossRef CAS.
- F. Tournadre, L. Croguennec, P. Willmann and C. Delmas, J. Solid State Chem., 2004, 177, 2803–2809 CrossRef CAS.
- J.-H. Cheng, C.-J. Pan, J.-F. Lee, J.-M. Chen, M. Guignard, C. Delmas, D. Carlier and B.-J. Hwang, Chem. Mater., 2014, 26, 1219–1225 CrossRef CAS.
- D. Carlier, J. H. Cheng, R. Berthelot, M. Guignard, M. Yoncheva, R. Stoyanova, B. J. Hwang and C. Delmas, Dalton Trans., 2011, 40, 9306–9312 RSC.
- P. Yang, C. Zhang, M. Li, X. Yang, C. Wang, X. Bie, Y. Wei, G. Chen and F. Du, ChemPhysChem, 2015, 16, 3408–3412 CrossRef CAS PubMed.
- X. Chen, X. Zhou, M. Hu, J. Liang, D. Wu, J. Wei and Z. Zhou, J. Mater. Chem. A, 2015, 3, 20708–20714 CAS.
- M. Matsui, F. Mizukoshi and N. Imanishi, J. Power Sources, 2015, 280, 205–209 CrossRef CAS.
- X. Xia and J. R. Dahn, J. Electrochem. Soc., 2012, 159, A647–A650 CrossRef CAS.
- P. Vassilaras, X. Ma, X. Li and G. Ceder, J. Electrochem. Soc., 2013, 160, A207–A211 CrossRef CAS.
- M. H. Han, E. Gonzalo, G. Singh and T. Rojo, Energy Environ. Sci., 2015, 8, 81–102 CAS.
- S. Komaba, N. Yabuuchi, T. Nakayama, A. Ogata, T. Ishikawa and I. Nakai, Inorg. Chem., 2012, 51, 6211–6220 CrossRef CAS PubMed.
- D. D. Yuan, Y. X. Wang, Y. L. Cao, X. P. Ai and H. X. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 8585–8591 CAS.
- J.-Y. Hwang, S.-T. Myung, D. Aurbach and Y.-K. Sun, J. Power Sources, 2016, 324, 106–112 CrossRef CAS.
- H.-J. Oh, K.-J. Noh, B.-R. Kim, W. Kang, S.-C. Jung and S.-J. Kim, Jpn. J. Appl. Phys., 2013, 52, 1AC15 1-4 CrossRef.
- J.-Y. Hwang, S.-M. Oh, S.-T. Myung, K. Y. Chung, I. Belharouak and Y.-K. Sun, Nat. Commun., 2014, 6, 6856 Search PubMed.
- H. Yu, S. Guo, Y. Zhu, M. Ishida and H. Zhou, Chem. Commun., 2014, 50, 457–459 RSC.
- J. P. Parant, R. Olazcuaga, M. Devalette, C. Fouassier and P. Hagenmuller, J. Solid State Chem., 1971, 3, 1–11 CrossRef CAS.
- X. Ma, H. Chen and G. Ceder, J. Electrochem. Soc., 2011, 158, A1307–A1312 CrossRef CAS.
- A. Mendiboure, C. Delmas and P. Hagenmuller, J. Solid State Chem., 1985, 57, 323–331 CrossRef CAS.
- X. Li, X. Ma, D. Su, L. Liu, R. Shisnell, S. P. Ong, H. Chen, A. Toumar, J.-C. Idrobo, Y. Lei, J. Bai, F. Wang, J. W. Lynn, Y. S. Lee and G. Ceder, Nat. Mater., 2014, 13, 586–592 CrossRef CAS PubMed.
- O. I. Velikokhantnyi, C.-C. Chang and P. N. Kumta, J. Electrochem. Soc., 2003, 150, A1262–A1266 CrossRef.
- R. Hoppe, G. Brachtel and M. Jansen, Z. Anorg. Allg. Chem., 1975, 417, 1 CrossRef CAS.
- R. Stoyanova, D. Carlier, M. Sendova-Vassileva, M. Yoncheva, E. Zhecheva, D. Nihtianova and C. Delmas, J. Solid State Chem., 2010, 183, 1372–1379 CrossRef CAS.
- M. Yoncheva, R. Stoyanova, E. Zhecheva, E. Kuzmanova, M. Sendova-Vassileva, D. Nihtianova, D. Carlier, M. Guignard and C. Delmas, J. Mater. Chem., 2012, 22, 23418–23427 RSC.
- S. Kumakura, Y. Tahara, K. Kubota, K. Chihara and S. Komaba, Angew. Chem., Int. Ed., 2016, 55, 1–5 CrossRef PubMed.
- A. Caballero, L. Hernan, J. Morales, L. Sanchez, J. Santos Pena and M. A. G. Aranda, J. Mater. Chem., 2012, 12, 1142–1147 RSC.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- N. Yabuuchi and S. Komaba, Sci. Technol. Adv. Mater., 2014, 15, 043501 CrossRef PubMed.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A1225–A1229 CrossRef CAS.
- K. Hemalatha, M. Jayakumar, P. Bera and A. S. Prakash, J. Mater. Chem. A, 2015, 3, 20908–20912 CAS.
- D. H. Lee, J. Xu and Y. S. Meng, Phys. Chem. Chem. Phys., 2013, 15, 3304–3312 RSC.
- A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2000, 2, 301–304 Search PubMed.
- J. Xu, D. H. Lee, R. J. Clement, X. Yu, M. Leskes, A. J. Pell, G. Pintacuda, X.-Q. Yang, C. P. Grey and Y. S. Meng, Chem. Mater., 2014, 26, 1260–1269 CrossRef CAS.
- P.-F. Wang, Y. Yu, Y.-X. Yin, Y.-S. Wang, L.-J. Wan, L. Gu and Y.-G. Guo, Angew. Chem., Int. Ed., 2016, 128, 7571–7575 CrossRef.
- G. Singh, N. Tapia-Ruiz, J. Miguel Lopez del Amo, U. Maitra, J. W. Somerville, A. R. Armstrong, J. Martinez de Ilarduya, T. Rojo and P. G. Bruce, Chem. Mater., 2016, 28, 5087–5094 CrossRef.
- D. Buchholz, C. Vaalma, L. G. Chagas and S. Passerini, J. Power Sources, 2015, 282, 581–585 CrossRef CAS.
- J. Billuad, G. Singh, A. R. Armstrong, E. Gonzalo, V. Roddatis, M. Armand, T. Rojo and P. G. Bruce, Energy Environ. Sci., 2014, 7, 1387–1391 Search PubMed.
- R. J. Clement, J. Billuad, A. R. Armstrong, G. Singh, T. Rojo, P. G. Bruce and C. P. Grey, Energy Environ. Sci., 2016, 9, 3240–3251 CAS.
- X. Wu, G.-L. Xu, G. Zhong, Z. Going, M. J. McDonald, S. Zheng, R. Fu, Z. Chen, K. Amine and Y. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 22227–22237 CAS.
- X. Wu, J. Guo, D. Wang, G. Zhong, M. J. McDonald and Y. Yang, J. Power Sources, 2015, 281, 18–26 CrossRef CAS.
- W. Zhao, H. Kirie, A. Tanaka, M. Unno, S. Yamamoto and H. Noguchi, Mater. Lett., 2014, 135, 131–134 CrossRef CAS.
- J. Yoshida, E. Guerin, M. Arnault, C. Constantin, B. Mortemard de Boisse, D. Calier, M. Guignard and C. Delmas, J. Electrochem. Soc., 2014, 161, A1987–A1991 CrossRef CAS.
- R. Kataoka, T. Mukai, A. Yoshizawa, K. Inoue, T. Kiyobayashi and T. Sakai, J. Electrochem. Soc., 2015, 162, A553–A558 CrossRef CAS.
- Z.-Y. Li, J. Zhang, R. Gao, H. Zhang, Z. Hu and X. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 15439–15448 CAS.
- D. Yuan, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Electrochim. Acta, 2014, 116, 300–305 CrossRef CAS.
- H. Yoshida, N. Yabuuchi, K. Kubota, I. Ikeuchi, A. Garsuch, M. Schulz-Dobric and S. Komaba, Chem. Commun., 2014, 50, 3677–3680 RSC.
- W. Zao, A. Tanaka, K. Momosaki, S. Yamamoto, F. Zhang, Q. Guo and H. Noguchi, Electrochim. Acta, 2015, 170, 171–181 CrossRef.
- N. Yabuuchi, R. Hara, K. Kubota, J. Paulsen, S. Kumakura and S. Komaba, J. Mater. Chem. A, 2014, 2, 16851–16855 CAS.
- N. Yabuuchi, R. Hara, M. Kajiyama, K. Kubota, T. Ishigaki, A. Hoshikawa and S. Komaba, Adv. Energy Mater., 2014, 4, 1301453 CrossRef.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- Y. Liu, X. Fang, A. Zhang, C. Shen, Q. Lium, H. A. Enaya and C. Zhou, Nano Energy, 2016, 27, 27–34 CrossRef CAS.
- P. Hagenmuller, A. Le Cerf and M. Onillon, C. R. Acad. Sci., 1962, 255–928 Search PubMed.
- A. Maazaz, C. Delmas and P. Hagenmuller, J. Inclusion Phenom., 1983, 1, 45–51 CrossRef CAS.
- J. J. Broconnier, C. Delmas and P. Hagenmuller, Mater. Res. Bull., 1982, 17, 993–1000 CrossRef.
- S. Miyazaki, S. Kikkawa and M. Koizumi, Rev. Chim. Miner., 2981, 19, 301 Search PubMed.
- S. Komaba, C. Takei, T. Nakayama, A. Ogata and N. Yabuuchi, Electrochem. Commun., 2010, 12, 355–358 CrossRef CAS.
- X. Xia and J. R. Dahn, Electrochem. Solid-State Lett., 2012, 15, A1–A4 CrossRef CAS.
- J. J. Ding, Y. Zhou, Q. Sun and Z. Fu, Electrochem. Commun., 2012, 22, 85–88 CrossRef CAS.
- C.-Y. Yu, J.-S. Park, H.-G. Jung, K.-Y. Chung, D. Aurbach, Y.-K. Sun and S.-T. Myung, Energy Environ. Sci., 2015, 8, 2019–2026 CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta and S. Ninazawa, J. Power Sources, 2013, 237, 52–57 CrossRef CAS.
- M. Onoda, J. Phys.: Condens. Matter, 2008, 20, 145205 CrossRef.
- T. McQueen, P. Stephenes, Q. Huang, T. E. Klimczuk, F. Ronning and R. Cava, Phys. Rev. Lett., 2008, 101, 166402 CrossRef CAS PubMed.
- D. Hamani, M. Ati, J. M. Tarascon and P. Rozier, Electrochem. Commun., 2011, 13, 938–941 CrossRef CAS.
- M. Guignard, C. Didier, J. Darriet, P. Bordet, E. Elkaim and C. Delmas, Nat. Mater., 2013, 12, 74–80 CrossRef CAS PubMed.
- M. Tamaru, X. Wang, M. Okubo and A. Yamada, Electrochem. Commun., 2013, 33, 23–26 CrossRef CAS.
- S. Guo, Y. Sun, J. Yi, K. Zhu, P. Liu, Y. Zhu, G. Zhu, M. Chen, M. Ishida and H. Zhou, NPG Asia Mater., 2016, 8, e266 CrossRef CAS.
- M. M. Doeff, M. Y. Peng, Y. Ma and L. C. De Jonghe, J. Electrochem. Soc., 1994, 141, L145–L147 CrossRef CAS.
- M. M. Doeff, T. J. Richardson and L. Kepley, J. Electrochem. Soc., 1996, 143, 2507–2516 CrossRef CAS.
- F. Sauvage, L. Faffont, J. M. Tarascon and E. Baudrin, Inorg. Chem., 2007, 46, 3289–3294 CrossRef CAS PubMed.
- Y. Cao, L. Xiao, W. Wang, D. Choi, Z. Nie, J. Yu, L. V. Saraf, Z. Yang and J. Liu, Adv. Mater., 2011, 23, 3155–3160 CrossRef CAS PubMed.
- H. Kim, D. J. Kim, D.-H. Seo, M. S. Yeom, K. Kang, D. K. Kim and Y. Jung, Chem. Mater., 2012, 24, 1205–1211 CrossRef CAS.
- J. F. Whitacre, A. Tevar and S. Sharma, Electrochem. Commun., 2010, 12, 463–466 CrossRef CAS.
- Z. Li, D. Young, K. Xiang, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2013, 3, 290–294 CrossRef CAS.
- D. Su, H.-J. Ahn and G. Wang, J. Mater. Chem. A, 2013, 1, 4845–4850 CAS.
- D. A. Tompsett and M. S. Islam, Chem. Mater., 2013, 25, 2515–2526 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657–660 CrossRef CAS.
- J. M. Tarascon, D. Guyomard, B. Wilkens, W. R. McKinnon and P. Barboux, Solid State Ionics, 1992, 57, 113–120 CrossRef CAS.
- N. Yabuuchi, M. Yano, S. Kuze and S. Komaba, Electrochim. Acta, 2012, 82, 296–301 CrossRef CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Skaarup, Solid State Ionics, 1988, 28–30, 1129–1131 Search PubMed.
- S. Tepavcevic, H. Xiong, V. R. Stamenkovic, X. Zuo, M. Balasubramanian, V. B. Prakapenka, C. S. Johnson and T. Rajh, ACS Nano, 2012, 6, 530–538 CrossRef CAS PubMed.
- N. B. Mahadi, J.-S. Park, J.-H. Park, K. Y. Chung, S. Y. Yi, Y.-K. Sun and S.-T. Myung, J. Power Sources, 2016, 326, 522–532 CrossRef CAS.
- H. Arai, S. Okada, Y. Sakurai and J.-I. Yamaki, J. Power Sources, 1997, 68, 716–719 CrossRef CAS.
- I. D. Gocheva, M. Nishijima, T. Doi, S. Okada, J.-I. Yamaki and T. Nishida, J. Power Sources, 2009, 187, 247–252 CrossRef CAS.
- M. Nishijima, I. D. Gocheva, S. Okada, T. Doi, J.-I. Yamaki and T. Nishida, J. Power Sources, 2009, 190, 558–562 CrossRef CAS.
- Y. Yamada, T. Doi, I. Tanaka, S. Okada and J.-I. Yamaki, J. Power Sources, 2011, 196, 4837–4841 CrossRef CAS.
- S.-T. Myung, A. Ogata, K.-S. Lee, S. Komaba, Y.-K. Sun and H. Yashiro, J. Electrochem. Soc., 2008, 155, A374–A383 CrossRef CAS.
- S.-T. Myung, K.-S. Lee, C. S. Yoon, Y.-K. Sun, K. Amine and H. Yashiro, J. Phys. Chem. C, 2010, 114, 4710–4718 CAS.
- Y.-K. Sun, Z. Chen, H.-J. Noh, D.-J. Lee, H.-G. Jung, Y. Ren, S. Wang, C. S. Yoon, S.-T. Myung and K. Amine, Nat. Mater., 2012, 11, 942–947 CrossRef CAS PubMed.
- C.-H. Jo, D.-H. Cho, J.-W. Lee, H. Yashiro and S.-T. Myung, J. Power Sources, 2016, 282, 511–519 CrossRef.
- J. H. Jo, C.-H. Jo, H. Yashiro and S.-T. Myung, J. Power Sources, 2016, 313, 1–8 CrossRef CAS.
- M. Avdeev, Z. Mohamed, C. D. Ling, J. Lu, M. Tamaru and A. Yamada, Inorg. Chem., 2013, 52, 8685–8693 CrossRef CAS PubMed.
- P. Moreau, D. Guyomard, J. Gaubicher and F. Boucher, Chem. Mater., 2010, 22, 4126–4128 CrossRef CAS.
- C. Li, X. Miao, W. Chu, P. Wu and D. G. Tong, J. Mater. Chem. A, 2015, 3, 8265–8271 CAS.
- A. Whiteside, C. A. Fisher, S. C. Park and M. S. Islam, Phys. Chem. Chem. Phys., 2014, 16, 21788–21794 RSC.
- K. Zaghib, J. Trottier, P. Hovington, F. Brochu, A. Guerfi, A. Mauger and C. M. Julien, J. Power Sources, 2011, 196, 9612–9617 CrossRef CAS.
- K. T. Lee, T. N. Ramesh, F. Nan, G. Botton and L. F. Nazar, Chem. Mater., 2011, 23, 3593–3600 CrossRef CAS.
- S.-M. Oh, S.-T. Myung, J. Hassoun, B. Scrosati and Y.-K. Sun, Electrochem. Commun., 2012, 22, 149–152 CrossRef CAS.
- M. Casas-Cabanas, V. V. Roddatis, D. Saurel, P. Kubiak, J. Carretero-Gonzalez, V. Palomares, Pl. Serras and T. Rojo, J. Mater. Chem., 2012, 22, 17421–17423 RSC.
- W. Tang, X. Song, Y. Du, C. Peng, M. Lin, S. Xi, B. Tian, J. Zheng, Y. Wu, F. Pan and K. P. Loh, J. Mater. Chem. A, 2016, 4, 4822–4892 Search PubMed.
- M. Nakayama, S. Yamada, R. Jalem and T. Kasuga, Solid State Ionics, 2016, 286, 40–44 CrossRef CAS.
- G. Ali, J.-H. Lee, D. Susanto, S.-W. Choi, B. W. Cho, K.-W. Nam and K. Y. Chung, ACS Appl. Mater. Interfaces, 2016, 8, 15422–15429 CAS.
- J. N. Reimer and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef.
- Y. Zhu, Y. Xu, Y. Liu, C. Luo and C. Wang, Nanoscale, 2013, 5, 780–787 RSC.
- B. L. Ellis, W. R. M. Makahnouk, Y. Makimura, K. Toghill and L. F. Nazar, Nat. Mater., 2007, 6, 749–753 CrossRef CAS PubMed.
- N. Rechan, J.-N. Chotard, L. Dupont, K. Djellab, M. Armand and J.-M. Tarascon, J. Electrochem. Soc., 2009, 156, A993–A999 CrossRef.
- Y. Kawabe, N. Yabuuchi, M. Kajiyama, N. Fukuhara, T. Inamasu, R. Okuyama, I. Nakai and S. Komaba, Electrochem. Commun., 2011, 13, 1225–1228 CrossRef CAS.
- R. Triphthi, S. M. Wood, M. S. Islam and L. F. Nazar, Energy Environ. Sci., 2013, 6, 2257–2264 Search PubMed.
- Y. Kawabe, N. Yabuuchi, M. Kajiyama, N. Fujuhara, T. Inamasu, R. Okuyama, I. Nakai and S. Komaba, Electrochemistry, 2011, 80, 80–84 CrossRef.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, 6, Al–A4 Search PubMed.
- J.-M. Le Meins, M.-P. Crosnier-Lopez, A. Hemon-Ribaud and G. Courbion, J. Solid State Chem., 1999, 148, 260–277 CrossRef CAS.
- J. Zhao, J. He, X. Ding, J. Zhou, Y. Ma, S. Wu and R. Huang, J. Power Sources, 2010, 198, 6854–6859 CrossRef.
- H. Zhuo, X. Yang, A. Tang, Z. Liu, S. Gamboa and P. J. Sebastian, J. Power Sources, 2006, 160, 698–703 CrossRef CAS.
- Z.-M. Liu, X.-Y. Wang, Y. Wang, A. P. Tang, S.-Y. Yang and L.-F. He, Trans. Nonferrous Met. Soc. China, 2008, 18, 346–350 CrossRef.
- F. Sauvage, E. Quarez, J.-M. Tarascon and E. Baudrin, Solid State Sci., 2006, 8, 1215–1221 CrossRef CAS.
- Y.-U. Park, D.-H. Seo, H.-S. Kwon, B. Kim, J. Kim, H. Kim, I. Kim, H.-I. Yoo and K. Kang, J. Am. Chem. Soc., 2013, 135, 13870–13878 CrossRef CAS PubMed.
- R. A. Shakoor, D.-H. Seo, H. Kim, Y.-U. Park, J. Kim, S.-W. Kim, H. Gwon, S. Lee and K. Kang, J. Mater. Chem., 2012, 22, 20535–20541 RSC.
- P. Serras, V. Palomares, A. Goni, I. Gil de Muro, P. Kubiak, L. Lezama and T. Rojo, J. Mater. Chem., 2012, 22, 22301–22308 RSC.
- K. Chihara, A. Kitajou, I. D. Gocheva, S. Okada and J.-I. Yamaki, J. Power Sources, 2013, 227, 80–85 CrossRef CAS.
- M. Bianchini, N. Brisset, F. Fauth, F. Weill, E. Elkaim, E. Uard, C. Masquelier and L. Croguennec, Chem. Mater., 2014, 26, 4238–4247 CrossRef CAS.
- Y. Uebou, S. Okada and J.-I. Yamaki, J. Power Sources, 2003, 115, 119–124 CrossRef CAS.
- L. Adam, N. Guesdon and B. Raveau, J. Solid State Chem., 2008, 181, 3110–3115 CrossRef CAS.
- S. Nishimura, M. Nakamura, R. Natsui and A. Yamada, J. Am. Chem. Soc., 2010, 132, 13596–13597 CrossRef CAS PubMed.
- H. Zhou, S. Upreti, N. A. Chernova, G. Hautier, G. Ceder and M. S. Whittingham, Chem. Mater., 2011, 23, 293–300 CrossRef CAS.
- T. Honma, T. Togashi, N. Ito and T. Komatsu, J. Ceram. Soc. Jpn., 2012, 120, 344–346 CrossRef CAS.
- P. Barpanda, T. Ye, S. Nishimura, S.-C. Chung, Y. Yamada, M. Okubo, H. Zhou and A. Yamada, Electrochem. Commun., 2012, 24, 116–119 CrossRef CAS.
- H. Kim, R. A. Shakoor, C. Park, S. Y. Lim, J.-S. Kim, Y. N. Jo, W. Cho, K. Miyasaka, R. Kahraman, Y. Jung and J. W. Choi, Adv. Funct. Mater., 2013, 23, 1147–1155 CrossRef CAS.
- J. M. Clark, P. Barpanda, A. Yamada and M. S. Islam, J. Mater. Chem. A, 2014, 2, 11807–11812 CAS.
- C.-Y. Chen, K. Matsumoto, T. Nohira and R. Hagiwara, J. Power Sources, 2014, 246, 783–787 CrossRef CAS.
- K. H. Ha, S. H. Woo, D. Mok, N.-S. Choi, N.-S. Choi, Y. Park, S. M. Oh, Y. Kim, J. Lee, L. F. Nazar and K. T. Lee, Adv. Energy Mater., 2013, 3, 770–776 CrossRef CAS.
- P. Barpanda, T. Ye, M. Avdeev, S.-C. Chung and A. Yamada, J. Mater. Chem. A, 2013, 1, 4194–4197 CAS.
- P. Barpanda, G. Liu, M. Avdeev and A. Yamada, ChemElectroChem, 2014, 1, 1488–1491 CrossRef CAS.
- F. Sanz, C. Parada, J. M. Rojo and C. Ruiz-Valero, Chem. Mater., 2001, 13, 1334–1340 CrossRef CAS.
- H. Kim, I. Park, D.-H. Seo, S. Lee, S.-W. Kim, W. J. Kwon, Y.-U. Park, C. S. Kim, S. Jeon and K. Kang, J. Am. Chem. Soc., 2012, 134, 10369–10372 CrossRef CAS PubMed.
- M. Nose, H. Nakayama, K. Nobuhara, H. Yamaguchi, S. Nakanishi and H. Iba, J. Power Sources, 2013, 234, 175–179 CrossRef CAS.
- M. Nose, S. Shiotani, H. Nakayama, K. Nobuhara, S. Nakanishi and H. Iba, Electrochem. Commun., 2013, 34, 266–269 CrossRef CAS.
- S. Y. Lim, H. Kim, J. Chung, J. H. Lee, B. G. Kim, J.-J. Choi, K. Y. Chung, W. Cho, S.-J. Kim, W. A. Goddard III, Y. Jung and J.-W. Choi, Proc. Natl. Aacd. Sci. U. S. A., 2014, 111, 599–604 CrossRef CAS PubMed.
- Z. Jian, L. Zhao, H. Pan, Y.-S. Hu, H. Li, W. Chen and L. Chen, Electrochem. Commun., 2012, 14, 86–89 CrossRef CAS.
- J. Gopalakrishnan and K. K. Rangan, Chem. Mater., 1992, 4, 745–747 CrossRef CAS.
- C. Masquelier, in Lithium Batteries Science and Technology, ed. G.-A. Nazri and G. Pistoia, Kluwer Academic, Boston, 2004, ch. 15, p. 445 Search PubMed.
- L. S. Plashnitsa, E. Kobayashi, Y. Noguchi, S. Okada and J.-I. Yamaki, J. Electrochem. Soc., 2010, 157, A536–A543 CrossRef CAS.
- Z. Jian, W. Han, X. Lu, H. Yang, Y.-S. Hu, J. Zhou, Z. Zhou, J. Li, W. Chen, D. Chen and L. Chen, Adv. Energy Mater., 2013, 3, 156–160 CrossRef CAS.
- K. Saravanan, C. W. Mason, A. Rudola, K. H. Wong and P. Balaya, Adv. Energy Mater., 2013, 3, 444–450 CrossRef CAS.
- Z. Jian, C. Yuan, W. Han, X. Lu, L. Gu, X. Xi, Y.-S. Hu, H. Li, W. Chen, D. Chen, Y. Ikuhara and L. Chen, Adv. Funct. Mater., 2014, 24, 4265–4272 CrossRef CAS.
- N. Recham, J. N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J.-M. Tarascon, Nat. Mater., 2010, 9, 68–74 CrossRef CAS PubMed.
- P. Barpanda, J.-N. Chotard, N. Rcham, C. Delacourt, M. Ati, L. Dupont, M. Armand and J.-M. Tarascon, Inorg. Chem., 2010, 49, 7401–7413 CrossRef CAS PubMed.
- R. Tripathi, G. R. Gardiner, M. S. Islam and L. F. Nazar, Chem. Mater., 2011, 23, 2278–2284 CrossRef CAS.
- P. Barpanda, G. Oyama, S. Nishimura, S.-C. Chung and A. Yamada, Nat. Commun., 2014, 5, 4358 CAS.
- H. Chen, G. Hautier and G. Ceder, J. Am. Chem. Soc., 2012, 134, 19619–19627 CrossRef CAS PubMed.
- H. Chen, Q. Hao, O. Zivkovic, G. Hautier, L.-S. Du, Y. Tang, Y.-Y. Hu, X. Ma, C. P. Grey and G. Ceder, Chem. Mater., 2013, 25, 2777–2786 CrossRef CAS.
- G. Hautier, A. Jain, H. L. Chen, C. Moore, S. P. Ong and G. Ceder, J. Mater. Chem., 2001, 21, 17147–17153 RSC.
- A. Eftekhari, J. Power Sources, 2004, 126, 221–228 CrossRef CAS.
- N. Imanishi, T. Morikawa, J. Kondo, Y. Takeda, O. Yamamoto, N. Kinugasa and T. Yamagishi, J. Power Sources, 1999, 79, 215–219 CrossRef CAS.
- C. D. Wessells, M. T. McDowell, S. V. Peddada, M. Pasta, R. A. Huggins and Y. Cui, ACS Nano, 2012, 6, 1688–1694 CrossRef CAS PubMed.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2012, 159, A98–A103 CrossRef CAS.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef PubMed.
- Y. H. Lu, L. Wang, J. G. Cheng and J. B. Goodenough, Chem. Commun., 2012, 48, 6544–6546 RSC.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, Angew. Chem., Int. Ed., 2013, 52, 1964–1967 CrossRef CAS PubMed.
- X. Wu, W. Deng, J. Qian, Y. Cao, X. Ai and H. Yang, J. Mater. Chem. A, 2013, 1, 10130–10134 CAS.
- J. Qian, M. Zhou, Y. Cao, Z. Ai and H. Yang, Adv. Energy Mater., 2012, 2, 410–414 CrossRef CAS.
- Z. P. Song and H. S. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- Y. L. Liang, Z. L. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS PubMed.
- H. Y. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J. M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS PubMed.
- K. Chihara, N. Chujo, A. Kitajou and S. Okada, Electrochim. Acta, 2013, 110, 240–246 CrossRef CAS.
- R. Zhao, L. Zhu, Y. Cao, X. Ai and H. X. Yang, Electrochem. Commun., 2012, 21, 36–38 CrossRef CAS.
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS PubMed.
- Y. Kim, K. H. Ha, S. M. Oh and K. T. Lee, Chemistry, 2014, 20, 11980–11992 CrossRef CAS PubMed.
- M.-S. Balogun, Y. Luo, W. Qiu, P. Liu and Y. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- Y. Mei, Y. Huang and X. Hu, J. Mater. Chem. A, 2016, 4, 12001–12013 CAS.
- F. Klein, B. Jache, A. Bhide and P. Adelhelm, Phys. Chem. Chem. Phys., 2013, 15, 15876–15887 RSC.
- V. L. Chevrier and G. Ceder, J. Electrochem. Soc., 2011, 158, A1011–A1014 CrossRef CAS.
- M. Mortazavi, Q. Ye, N. Birbilis and N. V. Medhekar, J. Power Sources, 2015, 285, 29–36 CrossRef CAS.
- S. P. Ong, V. L. Chevrier, G. Hautier, A. Jain, C. Moore, S. Kim, X. Ma and G. Ceder, Energy Environ. Sci., 2011, 4, 3680–3688 CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 1271–1273 CrossRef CAS.
- P.-C. Tsai, S.-C. Chung, S.-K. Lin and A. Yamada, J. Mater. Chem. A, 2015, 3, 9763–9768 CAS.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- C. Bommier, T. W. Surta, M. Dolgos and X. Ji, Nano Lett., 2015, 15, 5888–5892 CrossRef CAS PubMed.
- M. M. Doeff, Y. P. Ma, S. J. Visco and L. C. Dejonghe, J. Electrochem. Soc., 1993, 140, L169–L170 CrossRef CAS.
- D. Aurbach, M. D. Levi, E. Levi, H. Teller, B. Markovsky and G. Salitra, J. Electrochem. Soc., 1988, 145, 3024–3034 CrossRef.
- Y. F. Reynier, R. Yazami and B. Fultz, J. Electrochem. Soc., 2004, 151, A422–A426 CrossRef CAS.
- P. Ge and M. Fouletier, Solid State Ionics, 1988, 1172–1175 CrossRef.
- R. Raccichini, A. Varzi, S. Passerini and B. Scrosati, Nat. Mater., 2015, 14, 271–279 CrossRef CAS PubMed.
- Z. Wang, S. M. Selbach and T. Grande, RSC Adv., 2014, 4, 3973–3983 RSC.
- K. Nobuhara, H. Nakayama, M. Nose, S. Nakanishi and H. Iba, J. Power Sources, 2013, 243, 585–587 CrossRef CAS.
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed.
- Z. Zhu, F. Cheng, Z. Hu and J. Chen, J. Power Sources, 2015, 293, 626–634 CrossRef CAS.
- B. Jache, J. O. Binder, T. Abe and P. Adelgelm, Phys. Chem. Chem. Phys., 2016, 18, 14299–14316 RSC.
- H. Kim, J. Hong, G. Yoon, H. Kim, K.-Y. Park, M.-S. Park, W.-S. Yoon and K. Kang, Energy Environ. Sci., 2015, 8, 2963–2969 CAS.
- H. Kim, J. Hong, Y.-U. Park, J. Kim, I. Hwang and K. Kang, Adv. Funct. Mater., 2015, 25, 534–541 CrossRef CAS.
- Y. Wen, K. He, Y. Zhu, F. Han, Y. Xu, I. Matsuda, Y. Ishii, J. Cumings and C. Wang, Nat. Commun., 2014, 5, 4033 CAS.
- Y.-J. Kang, S. C. Jung, J. W. Choi and Y.-K. Han, Chem. Mater., 2015, 27, 5402–5406 CrossRef CAS.
- K. Gotoh, T. Ishikawa, S. Shimadzu, N. Yabuuchi, S. Komaba, K. Takeda, A. Goto, K. Deguchi, S. Ohki, K. Hashi, T. Shimizu and H. Ishida, J. Power Sources, 2013, 225, 137–140 CrossRef CAS.
- S. Wenzel, T. Hara, J. Janek and P. Adelhelm, Energy Environ. Sci., 2011, 4, 3342–3345 CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1195 CrossRef CAS PubMed.
- J. Liu and X. W. Liu, Adv. Mater., 2012, 24, 4097–4111 CrossRef CAS PubMed.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480–3498 CrossRef CAS PubMed.
- J. Zhu, R. Duanm, S. Zhang, N. Jiang, Y. Zhang and J. Zhu, SpringerPlus, 2014, 3, 585 CrossRef PubMed.
- G. Kucinskis, G. Bajars and J. Kleperis, J. Power Sources, 2013, 240, 66–79 CrossRef CAS.
- S. Wu, R. Xu, M. Lu, R. Ge, J. Iocozzia, C. Han, B. Jiang and Z. Lin, Adv. Energy Mater., 2015, 5, 1500400 CrossRef.
- X.-F. Luo, C.-H. Yang, Y.-Y. Peng, N.-W. Pu, M.-D. Ger, C.-T. Hsieh and J.-K. Chang, J. Mater. Chem. A, 2015, 3, 10320–10326 CAS.
- Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Carbon, 2013, 57, 202–208 CrossRef CAS.
- J. Ding, H. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. Xu, B. Zahiri, X. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed.
- D. Datta, J. Li and V. B. Shenoy, ACS Appl. Mater. Interfaces, 2014, 6, 1788–1795 CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 CAS.
- H. Song, N. Li, H. Cui and C. Wang, Nano Energy, 2014, 4, 81–87 CrossRef CAS.
- Z. Wang, L. Qie, L. Yuan, W. Zhang, X. Hu and Y. Huang, Carbon, 2013, 55, 328–334 CrossRef CAS.
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed.
- B. Ruan, J. Wang, D. Shi, Y. Xu, S. Chou, H. Liu and J. Wang, J. Mater. Chem. A, 2015, 3, 19011–19017 CAS.
- S. Wang, L. Xia, L. Yu, L. Zhang, H. Wang and X. W. D. Lou, Adv. Energy Mater., 2016, 6, 1502217 CrossRef.
- H. G. Wang, Z. Wu, F. L. Meng, D. L. Ma, X. L. Huang, L. M. Wang and X. B. Zhang, ChemSusChem, 2013, 6, 56–60 CrossRef PubMed.
- C. Ling and F. Mizuno, Phys. Chem. Chem. Phys., 2014, 16, 10419–10424 RSC.
- L. Fu, K. Tang, K. Song, P. A. van Aken, Y. Yu and J. Maier, Nanoscale, 2014, 6, 1384–1389 RSC.
- F. Yang, Z. Zhang, K. Du, X. Zhao, W. Chen, Y. Lai and J. Li, Carbon, 2015, 91, 88–95 CrossRef CAS.
- G. Ma, K. Huang, Q. Zhuang and Z. Ju, Mater. Lett., 2016, 174, 221–225 CrossRef CAS.
- W. Li, M. Zhou, H. Li, K. Wang, S. Cheng and K. Jiang, Energy Environ. Sci., 2015, 8, 2916–2921 CAS.
- L. Qie, W. Chen, X. Xiong, C. Hu, F. Zou, P. Hu and Y. Huang, Adv. Sci., 2015, 2, 1500195 Search PubMed.
- J. Ye, J. Zang, Z. Tian, M. Zheng and Q. Dong, J. Mater. Chem. A, 2016, 4, 13223–13227 CAS.
- Y. Li, Z. Wang, L. Li, S. Peng, L. Zhang, M. Srinivasan and S. Ramakrishna, Carbon, 2016, 99, 556–563 CrossRef CAS.
- F. Shen, W. Luo, J. Dai, Y. Yao, M. Zhu, E. Hitz, Y. Tang, Y. Chen, V. L. Sprenkle, X. Li and L. Hu, Adv. Energy Mater., 2016, 6, 1600377 CrossRef.
- E. M. Lotfabad, J. Ding, K. Cui, A. Kohandehghan, W. P. Kalisvaart, M. Hazelton and D. Mitlin, ACS Nano, 2014, 8, 7115–7129 CrossRef CAS PubMed.
- H. Li, F. Shen, W. Luo, J. Dai, X. Han, Y. Chen, Y. Yao, H. Zhu, K. Fu, E. Hitz and L. Hu, ACS Appl. Mater. Interfaces, 2016, 8, 2204–2210 CAS.
- L. Wu, D. Buchholz, C. Vaalma, G. A. Giffin and S. Passerini, ChemElectroChem, 2015, 3, 292–298 CrossRef.
- T. Yang, T. Qian, M. Wang, X. Shen, N. Xu, Z. Sun and C. Yan, Adv. Mater., 2016, 28, 539–545 CrossRef CAS PubMed.
- K.-L. Hong, L. Qie, R. Zeng, Z.-Q. Yi, W. Zhang, D. Wang, W. Yin, C. Wu, Q.-J. Fan, W.-X. Zhang and Y.-H. Huang, J. Mater. Chem. A, 2014, 2, 12733–12738 CAS.
- H. Zheng, J. Li, X. Song, G. Liu and V. S. Battaglia, Electrochim. Acta, 2012, 71, 258–265 CrossRef CAS.
- V. Aravindan, Y.-S. Lee, R. Yazami and S. Madhavi, Mater. Today, 2015, 18, 345–351 CrossRef CAS.
- S. Guo, J. Yi, Y. Sun and H. Zhou, Energy Environ. Sci., 2016, 9, 2978–3006 CAS.
- F. Legrain, O. Malyi and S. Manzhos, J. Power Sources, 2015, 278, 197–202 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Chem. Mater., 2015, 27, 6022–6029 CrossRef CAS.
- S. Lunell, A. Stashans, L. Ojamae, H. Lindstrom and A. Hagfeldt, J. Am. Chem. Soc., 1997, 119, 7374–7380 CrossRef CAS.
- H. Xiong, M. D. Slater, M. Balasubramanian, C. S. Johnson and T. Rajh, J. Phys. Chem. Lett., 2011, 2, 2560–2565 CrossRef CAS.
- M. Stromme Mattsson, M. Veszelei, G. A. Niklasson, C.-G. Granqvist, A. Stashan and S. Lunell, in Cation Diffusion in Electrochromic Fluorinated Ti Dioxide. In Electrochromic Materials and Their Applications III, ed. K. C. Ho, C. B. Greenberg and D. M. MacArthur, The Electrochemical Society, Pennington, NJ, 1997 Search PubMed.
- Y. Xu, E. M. Lotfabad, H. Wang, B. Farbod, Z. Xu, A. Kohandehghan and D. Mitlin, Chem. Commun., 2013, 49, 8973–8975 RSC.
- L. Wu, D. Buchholz, D. Bresser, L. Gomes Chagas and S. Passerini, J. Power Sources, 2014, 251, 379–385 CrossRef CAS.
- J. R. González, R. Alcántara, F. Nacimiento, G. F. Ortiz and J. L. Tirado, CrystEngComm, 2014, 16, 4602–4609 RSC.
- K. T. Kim, G. Ali, K. Y. Chung, C. S. Yoon, H. Yashiro, Y.-K. Sun, J. Lu, K. Amine and S. T. Myung, Nano Lett., 2014, 14, 416–422 CrossRef CAS PubMed.
- L. Wu, D. Bresser, D. Buchholz, G. A. Giffin, C. R. Castro, A. Ochel and S. Passerini, Adv. Energy Mater., 2015, 5, 1401142 CrossRef.
- Y. Yeo, J. W. Jung, K. Park and I. D. Kim, Sci. Rep., 2015, 5, 13862 CrossRef PubMed.
- J.-Y. Hwang, S.-T. Myung, J.-H. Lee, A. Abouimrane, I. Belharouak and Y.-K. Sun, Nano Energy, 2015, 16, 218–226 CrossRef CAS.
- B. Wang, F. Zhao, G. Du, S. Porter, Y. Liu, P. Zhang, Z. Cheng, H. K. Liu and Z. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 16009–16015 CAS.
- Y. Xu, M. Zhou, L. Wen, C. Wang, H. Zhao, Y. Mi, L. Liang, Q. Fu, M. Wu and Y. Lei, Chem. Mater., 2015, 27, 4274–4280 CrossRef CAS.
- N. O. Gopal, H.-H. Lo and S.-C. Ke, J. Am. Chem. Soc., 2008, 130, 2760–2761 CrossRef CAS PubMed.
- Y. Yang, X. Ji, M. Jing, H. Hou, Y. Zhu, L. Fang, X. Yang, Q. Chen and C. E. Banks, J. Mater. Chem. A, 2015, 3, 5648–5655 CAS.
- S. K. Das, B. Jache, H. Lahon, C. L. Bender, J. Janek and P. Adelhelm, Chem. Commun., 2016, 52, 1428–1431 RSC.
- S.-M. Oh, J.-Y. Hwang, C. S. Yoon, J. Lu, K. Amine, I. Belharouak and Y.-K. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 11295–11301 CAS.
- X. Yang, C. Wang, Y. Yang, Y. Zhang, X. Jia, J. Chen and X. Ji, J. Mater. Chem. A, 2015, 3, 8800–8807 CAS.
- Z. Hong, K. Zhou, Z. Huang and M. Wei, Sci. Rep., 2015, 5, 11960 CrossRef PubMed.
- Y. Ge, H. Jiang, J. Zhu, Y. Lu, C. Chen, Y. Hu, Y. Qiu and X. Zhang, Electrochim. Acta, 2015, 157, 142–148 CrossRef CAS.
- M. N. Tahir, B. Oschmann, D. Buchholz, X. Dou, I. Lieberwirth, M. Panthofer, W. Tremel, R. Zentel and S. Passerini, Adv. Energy Mater., 2016, 6, 1501489 CrossRef PubMed.
- J. Ni, S. Fu, C. Wu, J. Maier, Y. Yu and L. Li, Adv. Mater., 2016, 28, 2259–2265 CrossRef CAS PubMed.
- Z. Hong, J. Hong, C. Xie, Z. Huang and M. Wei, Electrochim. Acta, 2016, 202, 203–208 CrossRef CAS.
- Z. Hong, K. Zhou, J. Zhang, Z. Huang and M. Wei, J. Mater. Chem. A, 2015, 3, 17412–17416 CAS.
- H. Usui, S. Yoshioka, K. Wasada, M. Shimizu and H. Sakaguchi, ACS Appl. Mater. Interfaces, 2015, 7, 6567–6573 CAS.
- Y. Zhang, X. Pu, Y. Yang, Y. Zhu, H. Hou, M. Jing, X. Yang, J. Chen and X. Ji, Phys. Chem. Chem. Phys., 2015, 17, 15764–15770 RSC.
- Y. Zhang, C. W. Foster, C. E. Banks, L. Shao, H. Hou, G. Zou, J. Chen, Z. Huang and X. Ji, Adv. Mater., 2016, 28, 9391–9399 CrossRef CAS PubMed.
- J. P. Huang, D. D. Yuan, H. Z. Zhang, Y. L. Cao, G. R. Li, H. X. Yang and X. P. Gao, RSC Adv., 2013, 3, 12593–12597 RSC.
- J. Lee, J. K. Lee, K. Y. Chung, H.-G. Jung, H. Kim, J. Mun and W. Choi, Electrochim. Acta, 2016, 200, 21–28 CrossRef CAS.
- C. Chen, Y. Wen, X. Hu, X. Ji, M. Yan, L. Mai, P. Hu, B. Shan and Y. Huang, Nat. Commun., 2015, 6, 6929 CrossRef CAS PubMed.
- L. Wu, D. Bresser, D. Buchholz and S. Passerini, J. Electrochem. Soc., 2015, 162, A3052–A3058 CrossRef CAS.
- M. Søndergaard, K. J. Dalgaard, E. D. Bøjesen, K. Wonsyld, S. Dahl and B. B. Iversen, J. Mater. Chem. A, 2015, 3, 18667–18674 Search PubMed.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454–456 CrossRef CAS.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345–1351 CAS.
- L. Zhao, H.-L. Pan, Y.-S. Hu, H. Li and L.-Q. Chen, Chin. Phys. B, 2012, 21, 028201 CrossRef.
- Y. Sun, L. Zhao, H. Pan, X. Lu, L. Gu, Y. S. Hu, H. Li, M. Armand, Y. Ikuhara, L. Chen and X. Huang, Nat. Commun., 2013, 4, 1870 CrossRef PubMed.
- M. Kitta, K. Kuratani, M. Tabuchi, R. Kataoka, T. Kiyobayashi and M. Kohyama, Electrochemistry, 2015, 83, 989–992 CrossRef CAS.
- K.-T. Kim, C.-Y. Yu, C. S. Yoon, S.-J. Kim, Y.-K. Sun and S.-T. Myung, Nano Energy, 2015, 12, 725–734 CrossRef CAS.
- X. Yu, H. Pan, W. Wan, C. Ma, J. Bai, Q. Meng, S. N. Ehrlich, Y. S. Hu and X. Q. Yang, Nano Lett., 2013, 13, 4721–4727 CrossRef CAS PubMed.
- G. Hasegawa, K. Kanamori, T. Kiyomura, H. Kurata, K. Nakanishi and T. Abe, Adv. Energy Mater., 2015, 5, 1400730 CrossRef.
- P. Yu, C. Li and X. Guo, J. Phys. Chem. C, 2014, 118, 10616–10624 CAS.
- C. Chen, H. Xu, T. Zhou, Z. Guo, L. Chen, M. Yan, L. Mai, P. Hu, S. Cheng, Y. Huang and J. Xie, Adv. Energy Mater., 2016, 6, 1600322 CrossRef.
- Q. Zhou, L. Liu, J. Tan, Z. Yan, Z. Huang and X. Wang, J. Power Sources, 2015, 283, 243–250 CrossRef CAS.
- L. Y. Yang, H. Z. Li, J. Liu, S. S. Tang, Y. K. Lu, S. T. Li, J. Min, N. Yan and M. Lei, J. Mater. Chem. A, 2015, 3, 24446–24452 CAS.
- X. Feng, H. Zou, H. Xiang, X. Guo, T. Zhou, Y. Wu, W. Xu, P. Yan, C. Wang, J. G. Zhang and Y. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 16718–16726 CAS.
- P. Senguttuvan, G. Rousse, V. Seznec, J.-M. Tarascon and M. R. Palacín, Chem. Mater., 2011, 23, 4109–4111 CrossRef CAS.
- J. Xu, C. Ma, M. Balasubramanian and Y. S. Meng, Chem. Commun., 2014, 50, 12564–12567 RSC.
- A. Rudola, K. Saravanan, C. W. Mason and P. Balaya, J. Mater. Chem. A, 2013, 1, 2653–2662 CAS.
- A. Rudola, N. Sharma and P. Balaya, Electrochem. Commun., 2015, 61, 10–13 CrossRef CAS.
- H. Pan, X. Lu, X. Yu, Y.-S. Hu, H. Li, X.-Q. Yang and L. Chen, Adv. Energy Mater., 2013, 3, 1186–1194 CrossRef CAS.
- M. A. Muñoz-Márquez, M. Zarrabeitia, E. Castillo-Martinez, A. Eguia-Barrio, T. Rojo and M. Casas-Cabanas, ACS Appl. Mater. Interfaces, 2015, 7, 7801–7808 Search PubMed.
- W. Zou, J. Li, Q. Deng, J. Xue, X. Dai, A. Zhou and J. Li, Solid State Ionics, 2014, 262, 192–196 CrossRef CAS.
- N. D. Trinh, O. Crosnier, S. B. Schougaard and T. Broussre, ECS Trans., 2011, 35, 91–98 CAS.
- A. Rudola, K. Saravanan, S. Devaraj, H. Gong and P. Balaya, Chem. Commun., 2013, 49, 7451–7453 RSC.
- K. Shen and M. Wagemaker, Inorg. Chem., 2014, 53, 8250–8256 CrossRef CAS PubMed.
- M. Shirpour, J. Cabana and M. Doeff, Energy Environ. Sci., 2013, 6, 2538–2547 CAS.
- S. H. Woo, Y. Park, W. Y. Choi, N.-S. Choi, S. Nam, B. Park and K. T. Lee, J. Electrochem. Soc., 2012, 159, A2016–2023 CrossRef CAS.
- P. J. P. Naeyaert, M. Avdeev, N. Sharma, H. B. Yahia and C. D. Ling, Chem. Mater., 2014, 26, 7067–7072 CrossRef CAS.
- A. Maazaz, C. Delmas and P. Hagenmuller, J. Inclusion Phenom. Macrocyclic Chem., 1983, 1, 45–51 CrossRef CAS.
- D. Wu, X. Li, B. Xu, N. Twu, L. Liu and G. Ceder, Energy Environ. Sci., 2015, 8, 195–202 CAS.
- Y. Wang, X. Yu, S. Xu, J. Bai, R. Xiao, Y. S. Hu, H. Li, X. Q. Yang, L. Chen and X. Huang, Nat. Commun., 2013, 4, 2365 Search PubMed.
- A. D. Wadsley and W. G. Mumme, Acta Crystallogr., 1968, 24, 392–396 CrossRef CAS.
- H. Li, H. Fei, X. Liu, J. Yang and M. Wei, Chem. Commun., 2015, 51, 9298–9300 RSC.
- R. Alcantara, M. Jaraba, P. Lavela and J. L. Tirado, Chem. Mater., 2002, 14, 2847–2848 CrossRef CAS.
- S. Komaba, T. Mikumo, N. Yabuuchi, A. Ogata, H. Yoshida and Y. Yamada, J. Electrochem. Soc., 2010, 157, A60–A65 CrossRef CAS.
- S. Hariharan, K. Saravanan, V. Ramar and P. Balaya, Phys. Chem. Chem. Phys., 2013, 15, 2945–2953 RSC.
- D. Y. Park and S. T. Myung, ACS Appl. Mater. Interfaces, 2014, 6, 11749–11757 CAS.
- S. Liu, Y. Wang, Y. Dong, Z. Zhao, Z. Wang and J. Qiu, ChemElectroChem, 2016, 3, 38–44 CrossRef CAS.
- B. Koo, S. Chattopadhyay, T. Shibata, V. B. Prakapenka, C. S. Johnson, T. Rajh and E. V. Shevchenko, Chem. Mater., 2013, 25, 245–252 CrossRef CAS.
- P. R. Kumar, Y. H. Jung, K. K. Bharathi, C. H. Lim and D. K. Kim, Electrochim. Acta, 2014, 146, 503–510 CrossRef CAS.
- J. Ming, H. Ming, W. Yang, W.-J. Kwak, J.-B. Park, J. Zheng and Y.-K. Sun, RSC Adv., 2015, 5, 8793–8800 RSC.
- Z. Jian, B. Zhao, P. Liu, F. Li, M. Zheng, M. Chen, Y. Shi and H. Zhou, Chem. Commun., 2014, 50, 1215–1217 RSC.
- B. Huang, K. Tai, M. Zhang, Y. Xiao and S. J. Dillon, Electrochim. Acta, 2014, 118, 143–149 CrossRef CAS.
- X. Liu, T. Chen, H. Chu, L. Niu, Z. Sun, L. Pan and C. Q. Sun, Electrochim. Acta, 2015, 166, 12–16 CrossRef CAS.
- M. Valvo, F. Lindgren, U. Lafont, F. Björefors and K. Edström, J. Power Sources, 2014, 245, 967–978 CrossRef CAS.
- B. Philippe, M. Valvo, F. Lindgren, H. Rensmo and K. Edström, Chem. Mater., 2014, 26, 5028–5041 CrossRef CAS.
- M. M. Rahman, A. M. Glushenkov, T. Ramireddy and Y. Chen, Chem. Commun., 2014, 50, 5057–5060 RSC.
- M. M. Rahman, I. Sultana, Z. Chen, M. Srikanth, L. H. Li, X. J. Dai and Y. Chen, Nanoscale, 2015, 7, 13088–13095 RSC.
- Q. Deng, L. Wang and J. Li, J. Mater. Sci., 2015, 50, 4142–4148 CrossRef CAS.
- Z. Jian, P. Liu, F. Li, M. Chen and H. Zhou, J. Mater. Chem. A, 2014, 2, 13805–13809 CAS.
- K. C. Klavetter, S. Garcia, N. Dahal, J. L. Snider, J. Pedro de Souza, T. H. Cell, M. A. Cassara, A. Heller, S. M. Humphrey and C. B. Mullins, J. Mater. Chem. A, 2014, 2, 14209–14221 CAS.
- M. Gu, A. Kushima, Y. Shao, J. G. Zhang, J. Liu, N. D. Browning, J. Li and C. Wang, Nano Lett., 2013, 13, 5203–5211 CrossRef CAS PubMed.
- Y.-X. Wang, Y.-G. Lim, M.-S. Park, S.-L. Chou, J. H. Kim, H.-K. Liu, S.-X. Dou and Y.-J. Kim, J. Mater. Chem. A, 2014, 2, 529–534 CAS.
- M. Dirican, Y. Lu, Y. Ge, O. Yildiz and X. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 18387–18396 CAS.
- Y. C. Lu, C. Ma, J. Alvarado, T. Kidera, N. Dimov, Y. S. Meng and S. Okada, J. Power Sources, 2015, 284, 287–295 CrossRef CAS.
- D. Su, X. Xie and G. Wang, Chemistry, 2014, 20, 3192–3197 CrossRef CAS PubMed.
- X. Xie, S. Chen, B. Sun, C. Wang and G. Wang, ChemSusChem, 2015, 8, 2948–2955 CrossRef CAS PubMed.
- R. S. Kalubarme, J. Y. Lee and C. J. Park, ACS Appl. Mater. Interfaces, 2015, 7, 17226–17237 CAS.
- X. Xie, D. Su, J. Zhang, S. Chen, A. K. Mondal and G. Wang, Nanoscale, 2015, 7, 3164–3172 RSC.
- L. Wang, K. Zhang, Z. Hu, W. Duan, F. Cheng and J. Chen, Nano Res., 2013, 7, 199–208 CrossRef.
- S. Yuan, X. L. Huang, D. L. Ma, H. G. Wang, F. Z. Meng and X. B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- X. Zhang, W. Qin, D. Li, D. Yan, B. Hu, Z. Sun and L. Pan, Chem. Commun., 2015, 51, 16413–16416 RSC.
- H. Liu, F. Cao, H. Zheng, H. Sheng, L. Li, S. Wu, C. Liu and J. Wang, Chem. Commun., 2015, 51, 10443–10446 RSC.
- Y. Lu, N. Zhang, Q. Zhao, J. Liang and J. Chen, Nanoscale, 2015, 7, 2770–2776 RSC.
- Z. Zhang, J. Feng, L. Ci, Y. Tian and S. Xiong, Mater. Technol., 2016, 31, 497–500 CrossRef CAS.
- L.-L. Feng, G.-D. Li, Y. Liu, Y. Wu, H. Chen, Y. Wang, Y.-C. Zou, D. Wang and X. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 980–988 CAS.
- Y. Jiang, M. Hu, D. Zhang, T. Yuan, W. Sun, B. Xu and M. Yan, Nano Energy, 2014, 5, 60–66 CrossRef CAS.
- F. Zou, Y. M. Chen, K. Liu, Z. Yu, W. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377–386 CrossRef CAS PubMed.
- L. L. Feng, G. D. Li, Y. Liu, Y. Wu, H. Chen, Y. Wang, Y. C. Zou, D. Wang and X. Zou, ACS Appl. Mater. Interfaces, 2015, 7, 980–988 CAS.
- Q. Wang, L. Jiao, H. Du, W. Peng, Y. Han, D. Song, Y. Si, Y. Wang and H. Yuan, J. Chem. Mater., 2011, 21, 327–329 RSC.
- D. He, D. Wu, J. Gao, X. Wu, X. Zeng and W. Ding, J. Power Sources, 2015, 294, 643–649 CrossRef CAS.
- S.-J. Bao, C. M. Li, C.-X. Guo and Y. Qiao, J. Power Sources, 2008, 180, 676–681 CrossRef CAS.
- Q. Wang, L. Jiao, H. Du, W. Peng, Y. Han, D. Song, Y. Si, Y. Wang and H. Yuan, J. Mater. Chem., 2011, 21, 327–329 RSC.
- J. Liu, C. Wu, D. Xiao, P. Kopold, L. Gu, P. A. van Aken, J. Maier and Y. Yu, Small, 2016, 12, 2354–2364 CrossRef CAS PubMed.
- D. Yin, G. Huang, F. Zhang, Y. Qin, Z. Na, Y. Wu and L. Wang, Chemistry, 2016, 22, 1467–1474 CrossRef CAS PubMed.
- Z. Shadike, M. H. Cao, F. Ding, L. Sang and Z. W. Fu, Chem. Commun., 2015, 51, 10486–10489 RSC.
- Q. Zhou, L. Liu, G. Guo, Z. Yan, J. Tan, Z. Huang, X. Chen and X. Wang, RSC Adv., 2015, 5, 71644–71651 RSC.
- S. Peng, X. Han, L. Li, Z. Zhu, F. Cheng, M. Srinivansan, S. Adams and S. Ramakrishna, Small, 2016, 12, 1359–1368 CrossRef CAS PubMed.
- Q. Zhou, L. Liu, Z. Huang, L. Yi, X. Wang and G. Cao, J. Mater. Chem. A, 2016, 4, 5505–5516 CAS.
- J. Park, J.-S. Kim, J.-W. Park, T.-H. Nam, K.-W. Kim, J.-H. Ahn, G. Wang and H.-J. Ahn, Electrochim. Acta, 2013, 92, 427–432 CrossRef CAS.
- L. David, R. Bhandavt and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
- Y. X. Wang, K. H. Seng, S. L. Chou, J. Z. Wang, Z. Guo, D. Wexler, H. K. Liu and S. X. Dou, Chem. Commun., 2014, 50, 10730–10733 RSC.
- W. H. Ryu, J. W. Jung, K. Park, S. J. Kim and I. D. Kim, Nanoscale, 2014, 6, 10975–10981 RSC.
- Y. X. Wang, S. L. Chou, D. Wexler, H. K. Liu and S. X. Dou, Chemistry, 2014, 20, 9607–9612 CrossRef CAS PubMed.
- S. Zhang, X. Yu, H. Yu, Y. Chen, P. Gao, C. Li and C. Zhu, ACS Appl. Mater. Interfaces, 2014, 6, 21880–21885 CAS.
- D. Su, S. Dou and G. Wang, Adv. Energy Mater., 2015, 5, 1401205 CrossRef.
- X. Xie, Z. Ao, D. Su, J. Zhang and G. Wang, Adv. Funct. Mater., 2015, 25, 1393–1403 CrossRef CAS.
- S. Kalluri, K. H. Seng, Z. Guo, A. Du, K. Konstantinov, H. K. Liu and S. X. Dou, Sci. Rep., 2015, 5, 11989 CrossRef CAS PubMed.
- S. H. Choi, Y. N. Ko, J.-K. Lee and Y. C. Kang, Adv. Funct. Mater., 2015, 25, 1780–1788 CrossRef CAS.
- Y. Lu, Q. Zhao, N. Zhang, K. Lei, F. Li and J. Chen, Adv. Funct. Mater., 2016, 26, 911–918 CrossRef CAS.
- T. B. Kim, W. H. Jung, H. S. Ryu, K. W. Kim, J. H. Ahn, K. K. Cho, G. B. Cho, T. H. Nam, I. S. Ahn and H. J. Ahn, J. Alloys Compd., 2008, 449, 304–307 CrossRef CAS.
- T. B. Kim, J. W. Choi, H. S. Ryu, G. B. Cho, K. W. Kim, J. H. Ahn, K. K. Cho and H. J. Ahn, J. Power Sources, 2007, 174, 1275–1278 CrossRef CAS.
- A. Kitajou, J. Yamaguchi, S. Hara and S. Okada, J. Power Sources, 2014, 247, 391–395 CrossRef CAS.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS PubMed.
- Y. Zhu, L. Suo, T. Gao, X. Fan, F. Han and C. Wang, Electrochem. Commun., 2015, 54, 18–22 CrossRef CAS.
- S. Y. Lee and Y. C. Kang, Chemistry, 2016, 22, 2769–2774 CrossRef CAS PubMed.
- Z. Hu, Z. Zhu, F. Cheng, K. Zhang, J. Wang, C. Chen and J. Chen, Energy Environ. Sci., 2015, 8, 1309–1316 CAS.
- M. Walter, T. Zund and M. V. Kovalenko, Nanoscale, 2015, 7, 9158–9163 RSC.
- X. Wei, W. Li, J. A. Shi, L. Gu and Y. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27804–27809 CAS.
- Y. X. Wang, J. Yang, S. L. Chou, H. K. Liu, W. X. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 8689 CrossRef CAS PubMed.
- L. Wu, H. Lu, L. Xiao, J. Qian, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2014, 2, 16424–16428 CAS.
- L. Wu, H. Lu, L. Xiao, X. Ai, H. Yang and Y. Cao, J. Power Sources, 2015, 293, 784–789 CrossRef CAS.
- Y. C. Lu, C. Ma, J. Alvarado, N. Dimov, Y. S. Meng and S. Okada, J. Mater. Chem. A, 2015, 3, 16971–16977 CAS.
- C. Zhu, P. Kopold, W. Li, P. A. van Aken, J. Maier and Y. Yu, Adv. Sci., 2015, 2, 1500200 CrossRef PubMed.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2013, 1, 7181 CAS.
- X. Xie, D. Su, S. Chen, J. Zhang, S. Dou and G. Wang, Chem. – Asian J., 2014, 9, 1611–1617 CrossRef CAS PubMed.
- B. Qu, C. Ma, G. Ji, C. Xu, J. Xu, Y. S. Meng, T. Wang and J. Y. Lee, Adv. Mater., 2014, 26, 3854–3859 CrossRef CAS PubMed.
- Y. Zhang, P. Zhu, L. Huang, J. Xie, S. Zhang, G. Cao and X. Zhao, Adv. Funct. Mater., 2015, 25, 481–489 CrossRef CAS.
- J. Wnag, C. Luo, J. Mao, Y. Zhu, X. Fan, T. Gao, A. C. Mignerey and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11476–11481 Search PubMed.
- Y. Liu, H. Kang, L. Jiao, C. Chen, K. Cao, Y. Wang and H. Yuan, Nanoscale, 2015, 7, 1325–1332 RSC.
- P. V. Prikhodchenko, D. Y. W. Yu, S. K. Batabyal, V. Uvarov, J. Gun, S. Sladkevich, A. A. Mikhaylov, A. G. Medvedev and O. Lev, J. Mater. Chem. A, 2014, 2, 8431–8437 CAS.
- J. Wang, C. Luo, J. Mao, Y. Zhu, X. Fan, T. Gao, A. C. Mignerey and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 11476–11481 CAS.
- Q. Pan, J. Xie, T. Zhu, G. Cao, X. Zhao and S. Zhang, Inorg. Chem., 2014, 53, 3511–3518 CrossRef CAS PubMed.
- J.-S. Kim, D.-Y. Kim, G.-B. Cho, T.-H. Nam, K.-W. Kim, H.-S. Ryu, J.-H. Ahn and H.-J. Ahn, J. Power Sources, 2009, 189, 864–868 CrossRef CAS.
- X. Xu, S. Ji, M. Gu and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20957–20964 CAS.
- Q. Pan, J. Xie, T. Zhu, G. Cao, X. Zhao and S. Zhang, Inorg. Chem., 2014, 53, 3511–3518 CrossRef CAS PubMed.
- T. Wang, P. Hu, C. Zhang, H. Du, Z. Zhang, X. Wang, S. Chen, J. Xiong and G. Cui, ACS Appl. Mater. Interfaces, 2016, 8, 7811–7817 CAS.
- H.-S. Ryu, J.-E. Kim, J.-S. Park, J.-W. Park, K.-W. Kim, J.-H. Ahn, T.-H. Nam, G. Wang and H.-J. Ahn, J. Electrochem. Soc., 2013, 160, A338–A343 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Chem. Commun., 2014, 50, 4192–4195 RSC.
- Y. Liu, N. Zhang, H. Kang, M. Shang, L. Jiao and J. Chen, Chemistry, 2015, 21, 11878–11884 CrossRef CAS PubMed.
- D. Su, K. Kretschmer and G. Wang, Adv. Energy Mater., 2016, 6, 1501785 CrossRef.
- W. Qin, D. Li, X. Zhang, D. Yan, B. Hu and L. Pan, Electrochim. Acta, 2016, 191, 435–443 CrossRef CAS.
- J. Fullenwarth, A. Darwiche, A. Soares, B. Donnadieu and L. Monconduit, J. Mater. Chem. A, 2014, 2, 2050–2059 CAS.
- S. O. Kim and A. Manthiram, Chem. Commun., 2016, 52, 4337–4340 RSC.
- M. Fan, Y. Chen, Y. Xie, T. Yang, X. Shen, N. Xu, H. Yu and C. Yan, Adv. Funct. Mater., 2016, 26, 5019–5027 CrossRef CAS.
- W. J. Li, S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, Chem. Commun., 2015, 51, 3682–3685 RSC.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS PubMed.
- Y. Kim, Y. Kim, A. Choi, S. Woo, D. Mok, N. S. Choi, Y. S. Jung, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2014, 26, 4139–4144 CrossRef CAS PubMed.
- W. Li, S. L. Chou, J. Z. Wang, J. H. Kim, H. K. Liu and S. X. Dou, Adv. Mater., 2014, 26, 4037–4042 CrossRef CAS PubMed.
- X. Fan, J. Mao, Y. Zhu, C. Luo, L. Suo, T. Gao, F. Han, S.-C. Liou and C. Wang, Adv. Energy Mater., 2015, 5, 1500174 CrossRef.
- J. Liu, P. Kopold, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Energy. Environ. Sci., 2015, 8, 3531–3538 CAS.
- W.-J. Li, Q.-R. Yang, S.-L. Chou, J.-Z. Wang and H.-K. Liu, J. Power Sources, 2015, 294, 627–632 CrossRef CAS.
- J. Mao, X. Fan, C. Luo and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7147–7155 CAS.
- W. Zhang, M. Dahbi, S. Amagasa, Y. Yamada and S. Komaba, Electrochem. Commun., 2016, 69, 11–14 CrossRef CAS.
- F. Luo, B. Liu, J. Zheng, G. Chum, K. Zhong, H. Li, X. Huang and L. Chen, J. Electrochem. Soc., 2015, 16, A2509–A2528 CrossRef.
- J. H. Lee, C. S. Yoon, J.-Y. Hwang, S.-J. Kim, F. Maglia, P. Lamp, S.-T. Myung and Y.-K. Sun, Energy Environ. Sci., 2016, 9, 2152–2158 CAS.
- H. Morito, T. Yamada, T. Ikeda and H. Yamane, J. Alloys Compd., 2009, 480, 723–726 CrossRef CAS.
- S. C. Jung, D. S. Jung, J. W. Choi and Y. K. Han, J. Phys. Chem. Lett., 2014, 5, 1283–1288 CrossRef CAS PubMed.
- O. I. Malyi, T. L. Tan and S. Manzhos, Appl. Phys. Express, 2013, 6, 027301 CrossRef.
- O. Malyi, V. V. Kulish, T. L. TA464:C464an and S. Manzhos, Nano Energy, 2013, 2, 1149–1157 CrossRef CAS.
- F. Legrain, O. I. Malyi and S. Manzhos, Comput. Mater. Sci., 2014, 94, 214–217 CrossRef CAS.
- V. V. Kulish, O. I. Malyi, M. F. Ng, Z. Chen, S. Manzhos and P. Wu, Phys. Chem. Chem. Phys., 2014, 16, 4260–4267 RSC.
- Y. Xu, E. Swaans, S. Basak, H. W. Zandbergen, D. M. Borsa and F. M. Mulder, Adv. Energy Mater., 2016, 6, 1501436 CrossRef.
- L. Zhang, X. Hu, C. Chen, H. Guo, X. Liu, G. Xu, H. Zhong, S. Chung, P. Wu, J. Meng, Y. Huang, S. Dou and H. Liu, Adv. Mater., 2017, 29, 1604708 CrossRef PubMed.
- J. Sangster and A. D. Pelton, J. Phase Equilib., 1997, 18, 295 CrossRef CAS.
- C. Yue, Y. Yu, S. Sun, X. He, B. Chen, W. Lin, B. Xu, M. Zheng, S. Wu, J. Li, J. Kang and L. Lin, Adv. Funct. Mater., 2015, 25, 1386–1392 CrossRef CAS.
- P. R. Abel, Y.-M. Lin, T. de Souza, C.-Y. Chou, A. Gupta, J. B. Goodenough, G. S. Hwang, A. Heller and C. B. Mullins, J. Phys. Chem. C, 2013, 117, 18885–18890 CAS.
- L. Baggetto, J. K. Keum, J. F. Browning and G. M. Veith, Electrochem. Commun., 2013, 34, 41–44 CrossRef CAS.
- A. Kohandehghan, K. Cui, M. Kupsta, J. Ding, E. Memarzadeh Lotfabad, W. P. Kalisvaart and D. Mitlin, Nano Lett., 2014, 14, 5873–5882 CrossRef CAS PubMed.
- S. Komaba, Y. Matsuura, T. Ishikawa, N. Yabuuchi, W. Murata and S. Kuze, Electrochem. Commun., 2012, 21, 65–68 CrossRef CAS.
- L. D. Ellis, T. D. Hatchard and M. N. Obrovac, J. Electrochem. Soc., 2012, 159, A1801–A1805 CrossRef CAS.
- J. W. Wang, X. H. Liu, S. X. Mao and J. Y. Huang, Nano Lett., 2012, 12, 5897–5902 CrossRef CAS PubMed.
- J. Wang, C. Eng, Y. C. Chen-Wiegart and J. Wang, Nat. Commun., 2015, 6, 7496 CrossRef CAS PubMed.
- L. Baggetto, P. Ganesh, R. P. Meisner, R. R. Unocic, J.-C. Jumas, C. A. Bridges and G. M. Veith, J. Power Sources, 2013, 234, 48–59 CrossRef CAS.
- Y. Xu, Y. Zhu, Y. Liu and C. Wang, Adv. Energy Mater., 2013, 3, 128–133 CrossRef CAS.
- H. Zhu, Z. Jia, Y. Chen, N. Weadock, J. Wan, O. Vaaland, X. Han, T. Li and L. Hu, Nano Lett., 2013, 13, 3093–3100 CrossRef CAS PubMed.
- D. Bresser, F. Mueller, D. Buchholz, E. Paillard and S. Passerini, Electrochim. Acta, 2014, 128, 163–171 CrossRef CAS.
- X. Xie, K. Kretschmer, J. Zhang, B. Sun, D. Su and G. Wang, Nano Energy, 2015, 13, 208–217 CrossRef CAS.
- Y. Liu, N. Zhang, L. Jiao and J. Chen, Adv. Mater., 2015, 27, 6702–6707 CrossRef CAS PubMed.
- Y. Liu, N. Zhang, L. Jiao, Z. Tao and J. Chen, Adv. Funct. Mater., 2015, 25, 214–220 CrossRef CAS.
- B. Luo, T. Qiu, D. Ye, L. Wang and L. Zhi, Nano Energy, 2016, 22, 232–240 CrossRef CAS.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- C. Kim, K.-Y. Lee, I. Kim, J. Park, G. Cho, K.-W. Kim, J.-H. Ahn and H.-J. Ahn, J. Power Sources, 2016, 317, 153–158 CrossRef CAS.
- J. Sangster and A. D. Pelton, J. Phase Equilib., 1993, 14, 291 Search PubMed.
- J. Qian, Y. Chen, L. Wu, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2012, 48, 7070–7072 RSC.
- A. Darwiche, C. Marino, M. T. Sougrati, B. Fraisse, L. Stievano and L. Monconduit, J. Am. Chem. Soc., 2012, 134, 20805–20811 CrossRef CAS PubMed.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- Y. Zhu, X. Han, Y. Xu, Y. Liu, S. Zheng, K. Xu, L. Hu and C. Wang, ACS Nano, 2013, 7, 6378–6386 CrossRef CAS PubMed.
- X. Zhou, Z. Dai, J. Bao and Y.-G. Guo, J. Mater. Chem. A, 2013, 1, 13727–13731 CAS.
- H. Hou, M. Jing, Y. Yang, Y. Zhang, W. Song, X. Yang, J. Chen, Q. Chen and X. Ji, J. Power Sources, 2015, 284, 227–235 CrossRef CAS.
- M. He, K. Kravchyk, M. Walter and M. V. Kovalenko, Nano Lett., 2014, 14, 1255–1262 CrossRef CAS PubMed.
- L. Wu, X. Hu, J. Qian, F. Pei, F. Wu, R. Mao, X. Ai, H. Yang and Y. Cao, Energy Environ. Sci., 2014, 7, 323–328 CAS.
- H. Hou, Y. Yang, Y. Zhu, M. Jing, C. Pan, L. Fang, W. Song, X. Yang and X. Ji, Electrochim. Acta, 2014, 146, 328–334 CrossRef CAS.
- C. Nithya and S. Gopukumar, J. Mater. Chem. A, 2014, 2, 10516–10525 CAS.
- X. Zhou, Y. Zhong, M. Yang, M. Hu, J. Wei and Z. Zhou, Chem. Commun., 2014, 50, 12888–12891 RSC.
- L. Hu, X. Zhu, Y. Du, Y. Li, X. Zhou and J. Bao, Chem. Mater., 2015, 27, 8138–8145 CrossRef CAS.
- N. Zhang, Y. Liu and Y. Lu, Nano Res., 2015, 8(10), 3384–3393 CrossRef CAS.
- J. M. Sangster, J. Phase Equilib. Diffus., 2009, 31, 62–67 CrossRef.
- Y. Kim, Y. Park, A. Choi, N. S. Choi, J. Kim, J. Lee, J. H. Ryu, S. M. Oh and K. T. Lee, Adv. Mater., 2013, 25, 3045–3049 CrossRef CAS PubMed.
- J. Sun, H. W. Lee, M. Pasta, H. Yuan, G. Zheng, Y. Sun, Y. Li and Y. Cui, Nat. Nanotechnol., 2015, 10, 980–985 CrossRef CAS PubMed.
- J. Song, Z. Yu, M. L. Gordin, S. Hu, R. Yi, D. Tang, T. Walter, M. Regula, D. Choi, X. Li, A. Manivannan and D. Wang, Nano Lett., 2014, 14, 6329–6335 CrossRef CAS PubMed.
- J. Qian, X. Wu, Y. Cao, X. Ai and H. Yang, Angew. Chem., Int. Ed., 2013, 52, 4633–4636 CrossRef CAS PubMed.
- W. J. Li, S. L. Chou, J. Z. Wang, H. K. Liu and S. X. Dou, Nano Lett., 2013, 13, 5480–5484 CrossRef CAS PubMed.
- T. Ramireddy, T. Xing, M. M. Rahman, Y. Chen, Q. Dutercq, D. Gunzelmann and A. M. Glushenkov, J. Mater. Chem. A, 2015, 3, 5572–5584 CAS.
- J. Song, Z. Yu, M. L. Gordin, X. Li, H. Peng and D. Wang, ACS Nano, 2015, 9, 11933–11941 CrossRef CAS PubMed.
- J. Sun, G. Zheng, H. W. Lee, N. Liu, H. Wang, H. Yao, W. Yang and Y. Cui, Nano Lett., 2014, 14, 4573–4580 CrossRef CAS PubMed.
- C. Zhang, X. Wang, Q. Liang, X. Liu, Q. Weng, J. Liu, Y. Yang, Z. Dai, K. Ding, Y. Bando, J. Tang and D. Golberg, Nano Lett., 2016, 16, 2054–2060 CrossRef CAS PubMed.
- A. Morita, Appl. Phys., 1986, 39, 227–242 Search PubMed.
- K. P. S. S. Hembram, H. Jung, B. C. Yeo, S. J. Pai, S. Kim, K.-R. Lee and S. S. Han, J. Phys. Chem. C, 2015, 119, 15041–15046 CAS.
- M. Dahbi, N. Yabuuchi, M. Fukunishi, K. Kubota, K. Chihara, K. Tokiwa, X. Yu, H. Ushiyama, K. Yamashita, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Chem. Mater., 2016, 28, 1625–1635 CrossRef CAS.
- L. Kou, C. Chen and S. C. Smith, J. Phys. Chem. Lett., 2015, 6, 2794–2805 CrossRef CAS PubMed.
- G. L. Xu, Z. Chen, G. M. Zhong, Y. Liu, Y. Yang, T. Ma, Y. Ren, X. Zuo, X. H. Wu, X. Zhang and K. Amine, Nano Lett., 2016, 16, 3955–3965 CrossRef CAS PubMed.
- N. Nitta and G. Yushin, Part. Part. Syst. Charact., 2014, 31, 317–336 CrossRef CAS.
- C.-M. Park, S. Yoon, S.-I. Lee and H.-J. Sohn, J. Power Sources, 2009, 186, 206–210 CrossRef CAS.
- L. D. Ellis, B. N. Wilkes, T. D. Hatachard and M. N. Obrovac, J. Electrochem. Soc., 2014, 161, A416–A421 CrossRef CAS.
- F. Yang, F. Yu, Z. Zhang, K. Zhang, Y. Lai and J. Li, Chemistry, 2016, 22, 2333–2338 CrossRef CAS PubMed.
- J. Sottmann, M. Herrmann, P. Vajeeston, Y. Hu, A. Ruud, C. Drathen, H. Emerich, H. Fjellvåg and D. S. Wragg, Chem. Mater., 2016, 28, 2750–2756 CrossRef CAS.
- D. Su, S. Dou and G. Wang, Nano Energy, 2015, 12, 88–95 CrossRef CAS.
- S. Liu, J. Feng, X. Bian, J. Liu and H. Xu, J. Mater. Chem. A, 2016, 4, 10098–10104 CAS.
- J. Liu, Y. Wen, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 6387–6392 CrossRef CAS PubMed.
- L. Baggetto, K. J. Carroll, H.-Y. Hah, C. E. Johnson, D. R. Mullins, R. R. Unocic, J. A. Johnson, Y. S. Meng and G. M. Veith, J. Phys. Chem. C, 2014, 118, 7856–7864 CAS.
- L. Baggetto, H. Y. Hah, C. E. Johnson, C. A. Bridges, J. A. Johnson and G. M. Veith, Phys. Chem. Chem. Phys., 2014, 16, 9538–9545 RSC.
- Y. M. Lin, P. R. Abel, A. Gupta, J. B. Goodenough, A. Heller and C. B. Mullins, ACS Appl. Mater. Interfaces, 2013, 5, 8273–8277 CAS.
- E. D. Jackson, S. Green and A. L. Prieto, ACS Appl. Mater. Interfaces, 2015, 7, 7447–7450 CAS.
- L. Baggetto, E. Allcorn, R. R. Unocic, A. Manthiram and G. M. Veith, J. Mater. Chem. A, 2013, 1, 11163–11169 CAS.
- I. T. Kim, S.-O. Kim and A. Manthiram, J. Power Sources, 2014, 269, 848–854 CrossRef CAS.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J. G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- L. Ji, W. Zhou, V. Chabot, A. Yu and X. Xiao, ACS Appl. Mater. Interfaces, 2015, 7, 24895–24901 CAS.
- Y. Zhao and A. Manthiram, Chem. Mater., 2015, 27, 3096–3101 CrossRef CAS.
- J. Hassoun, S. Panero, P. Simon, P. L. Taberna and B. Scrosati, Adv. Mater., 2007, 19, 1632–1635 CrossRef CAS.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- L. Zhao, J. Zhao, Y.-S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef CAS.
- Y. Park, D. S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- Q. Zhao, J. Wang, Y. Lu, Y. Li, G. Liang and J. Chen, Angew. Chem., Int. Ed., 2014, 55(38), 12528–12532 Search PubMed.
- A. Abouimrane, W. Weng, H. Eltayeb, Y. Cui, J. Niklas, O. Poluektov and K. Amine, Energy Environ. Sci., 2012, 5, 9632–9638 CAS.
- X. Wu, S. Jin, Z. Zhang, L. Jiang, L. Mu, Y.-S. Hu, H. Li, X. Chen, M. Armand, L. Chen and X. Huang, Sci. Adv., 2015, 1, e1500330 Search PubMed.
- W. Deng, X. Liang, X. Wu, J. Qian, Y. Cao, X. Ai, J. Feng and H. Yang, Sci. Rep., 2013, 3, 2671 CrossRef PubMed.
- W. Deng, J. Qian, Y. Cao, X. Ai and H. Yang, Small, 2016, 12, 583–587 CrossRef CAS PubMed.
- Y. Wang, K. Kretschmer, J. Zhang, A. K. Mondal, X. Guo and G. Wang, RSC Adv., 2016, 6, 57098–57102 RSC.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130 CrossRef CAS PubMed.
- A. Choi, Y. K. Kim, T. K. Kim, M.-S. Kwon, K. T. Lee and H. R. Moon, J. Mater. Chem. A, 2014, 2, 14986–14993 CAS.
- R. R. Zhao, Y. L. Cao, X. P. Ai and H. X. Yang, J. Electroanal. Chem., 2013, 688, 93–97 CrossRef CAS.
- H.-G. Wang, S. Yuan, Z. Si and X.-b. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 CAS.
- S. Renault, V. A. Mihali, K. Edström and D. Brandell, Electrochem. Commun., 2014, 45, 52–55 CrossRef CAS.
- L. Chen, W. Li, Y. Wang, C. Wang and Y. Xia, RSC Adv., 2014, 4, 25369 RSC.
- Z. Li, J. Zhou, R. Xu, S. Liu, Y. Wang, P. Li, W. Wu and M. Wu, Chem. Eng. J., 2016, 287, 516–522 CrossRef CAS.
- C. Luo, Y. Zhu, Y. Xu, Y. Liu, T. Gao, J. Wang and C. Wang, J. Power Sources, 2014, 250, 372–378 CrossRef CAS.
- H. Zhu, J. Yin, X. Zhao, C. Wang and X. Yang, Chem. Commun., 2015, 51, 14708–14711 RSC.
- H. Wang, P. Hu, J. Yang, G. Gong, L. Guo and X. Chen, Adv. Mater., 2015, 27, 2348–2354 CrossRef CAS PubMed.
- C. Luo, J. Wang, X. Fan, Y. Zhu, F. Han, L. Suo and C. Wang, Nano Energy, 2015, 13, 537–545 CrossRef CAS.
- Z. Zhu, H. Li, J. Liang, Z. Tao and J. Chen, Chem. Commun., 2015, 51, 1446–1448 RSC.
- E. Castillo-Martinez, J. Carretero-Gonzalez and M. Armand, Angew. Chem., Int. Ed., 2014, 53, 5341–5345 CrossRef CAS PubMed.
- M. López-Herraiz, E. Castillo-Martínez, J. Carretero-González, J. Carrasco, T. Rojo and M. Armand, Energy Environ. Sci., 2015, 8, 3233–3241 Search PubMed.
- J.-Y. Hwang, S.-T. Myung, C. S. Yoon, S.-S. Kim, D. Aurbach and Y.-K. Sun, Adv. Funct. Mater., 2016, 26, 8083–8093 CrossRef CAS.
- K. Vignarooban, R. Kushagra, A. Elango, P. Badami, B. E. Mellander, X. Xu, T. G. Tucker, C. Nam and A. M. Kannan, Int. J. Hydrogen Energy, 2016, 41, 2829–2846 CrossRef CAS.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, J. Mater. Chem. A, 2015, 3, 22–42 CAS.
- H. Kim, J. Hong, K. Y. Park, H. Kim, S. W. Kim and K. Kang, Chem. Rev., 2014, 114, 11788–11827 CrossRef CAS PubMed.
- D. Aurbach, Y. Talyosef, B. Markovsky, E. Markevich, E. Zinigrad, L. Asraf, J. S. Gnanaraj and H.-J. Kim, Electrochim. Acta, 2004, 50, 247–254 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- G. H. Newman and L. P. Klelmann, J. Electrochem. Soc., 1980, 127, 2097–2099 CrossRef CAS.
- J.-J. Braconnierm, C. Delmas, C. Fouassier and P. Hagenmuller, Mater. Res. Bull., 1980, 15, 1797–1804 CrossRef.
- A. Ponrouch, R. Dedryvère, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacín, Energy Environ. Sci., 2013, 6, 2361–2369 CAS.
- R. Alcántara, P. Lavela, G. F. Ortiz and J. L. Tirado, Electrochem. Solid-State Lett., 2005, 8, A222–A225 CrossRef.
- A. Bhide, J. Hofmann, A. K. Durr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- S. Komaba, T. Ishikawa, N. Yabuuchi, W. Murata, A. Ito and Y. Ohsawa, ACS Appl. Mater. Interfaces, 2011, 3, 4165–4168 CAS.
- T. Kajita and T. Itoh, Phys. Chem. Chem. Phys., 2017, 19, 1003–1009 RSC.
- K. Zhang, M. Park, L. Zhou, G.-H. Lee, J. Shin, Z. Hu, S.-L. Chou, J. Chen and Y.-M. Kang, Angew. Chem., Int. Ed., 2016, 55, 12822–12826 CrossRef CAS PubMed.
- K. Zhang, M. Park, L. Zhou, G.-H. Lee, W. Li and Y.-M. Kang, Adv. Funct. Mater., 2016, 26, 6728–6735 CrossRef CAS.
- K. Zhang, Z. Hu, X. Liu, Z. Tao and J. Chen, Adv. Mater., 2015, 27, 3305–3309 CrossRef CAS PubMed.
- I. A. Shkrob, Y. Zhu, T. W. Marin and D. Abraham, J. Phys. Chem. C, 2013, 117, 19255–19269 CAS.
- H. Kumar, E. Detsi, D. P. Abraham and V. B. Shenoy, Chem. Mater., 2016, 28, 8930–8941 CrossRef CAS.
- V. Etacheri, O. Haik, Y. Goffer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2011, 28, 965–976 CrossRef PubMed.
- X. Chen, X. Li, D. Mei, J. Feng, M. Y. Hu, J. Hu, M. Engelhard, J. Zheng, W. Xu, J. Xiao, J. Liu and J.-G. Zhang, ChemSusChem, 2014, 7, 549–554 CrossRef CAS PubMed.
- L. Ji, M. Gu, Y. Shao, X. Li, M. H. Engelhard, B. W. Arey, W. Wang, Z. Nie, J. Xiao, C. Wang, J.-G. Zhang and J. Liu, Adv. Mater., 2014, 26, 2901–2908 CrossRef CAS PubMed.
- E. Markevich, K. Fridman, R. Sharabi, R. Elazari, G. Salitra, H. E. Gottlieb, G. Gershinsky, A. Garsuch, G. Semrau, M. A. Schmidt and D. Aurbach, J. Electrochem. Soc., 2013, 160, A1824–A1833 CrossRef CAS.
- M. Dahbi, T. Nakano, N. Yabuuchhi, S. Fujimura, K. Chihara, K. Kobota, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2016, 3, 1856–1867 CrossRef CAS.
- M. Egashira, T. Tanaka, N. Yoshimoto and M. Morita, Electrochemistry, 2012, 80, 755–758 CrossRef CAS.
- A. Fukunaga, T. Nohira, Y. Kozawa, R. Hagiwara, S. Sakai, K. Nitta and S. Inazawa, J. Power Sources, 2012, 209, 52–56 CrossRef CAS.
- D. Monti, E. Jónsson, M. R. Palacín and P. Johansson, J. Power Sources, 2014, 245, 630–636 CrossRef CAS.
- S. A. Mohd Noor, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2013, 114, 766–771 CrossRef CAS.
- K. West, B. Zachau-Christiansen, T. Jacobsen and S. Atlung, J. Electrochem. Soc., 1985, 132, 3061–3062 CrossRef CAS.
- H. Gao, B. Guo, J. Song, K. Park and J. B. Goodenough, Adv. Energy Mater., 2015, 5, 1402235 CrossRef.
- Y. L. Ni'mah, M.-Y. Cheng, J. H. Cheng, J. Rick and B.-J. Hwang, J. Power Sources, 2015, 278, 375–381 CrossRef.
- Y. Noguchi, E. Kobayashi, L. S. Plashnitsa, S. Okada and J.-I. Yamaki, Electrochim. Acta, 2013, 101, 59–65 CrossRef CAS.
- A. Hayashi, K. Noi, A. Sakuda and M. Tatsumisago, Nat. Commun., 2012, 3, 856 CrossRef PubMed.
- S. F. Lux, F. Schappacher, A. Balducci, S. Passerini and M. Winter, J. Electrochem. Soc., 2010, 157, A320–A325 CrossRef CAS.
- Z. P. Cai, Y. Liang, W. S. Li, L. D. Xing and Y. H. Liao, J. Power Sources, 2009, 189, 547–551 CrossRef CAS.
- J. Ming, H. Ming, W. J. Kwak, C. Shin, J. Zheng and Y.-K. Sun, Chem. Commun., 2014, 50, 13307–13310 RSC.
- M. Dahbi, T. Nakano, N. Yabuuchi, T. Ishikawa, K. Kubota, M. Fukunishi, S. Shibahara, J.-Y. Son, Y.-T. Cui, H. Oji and S. Komaba, Electrochem. Commun., 2014, 44, 66–69 CrossRef CAS.
- L. Qiu, Z. Shao, W. Wang, F. Wang, D. Wang, Z. Zhou, P. Xiang and C. Xu, RSC Adv., 2014, 4, 2485–24862 Search PubMed.
- B. Koo, H. Kim, Y. Cho, K. T. Lee, N. S. Choi and J. Cho, Angew. Chem., Int. Ed., 2012, 51, 8762–8767 CrossRef CAS PubMed.
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed.
- X. Zhao, S. A. Vail, Y. Lu, J. Song, W. Pan, D. R. Evans and J. J. Lee, ACS Appl. Mater. Interfaces, 2016, 8, 13871–13878 CAS.
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef.
- K. Ui, S. Kikuchi, F. Mikami, Y. Kadoma and N. Kumagai, J. Power Sources, 2007, 173, 518–521 CrossRef CAS.
- V. Palomares, M. Casas-Cabanas, E. Castillo-Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- A. Magasinski, B. Zdyrko, I. Kovalenko, B. Hertzberg, R. Burtovyy, C. F. Huebner, T. F. Fuller, I. Luzinov and G. Yushin, ACS Appl. Mater. Interfaces, 2010, 2, 3004–3010 CAS.
- S. Komaba, K. Shimomura, N. Yabuuchi, T. Ozeki, H. Yui and K. Konno, J. Phys. Chem. C, 2011, 115, 13487–13495 CAS.
- S. L. Chou, Y. Pan, J. Z. Wang, H. K. Liu and S. X. Dou, Phys. Chem. Chem. Phys., 2014, 16, 20347–20359 RSC.
- Q. Fan, W. Zhang, J. Duan, K. Hong, L. Xue and Y. Huang, Electrochim. Acta, 2015, 174, 970–977 CrossRef CAS.
- D. Ma, Z. Cao and A. Hu, Nano-Micro Lett., 2014, 6, 347–358 CrossRef.
- D. Mazouzi, Z. Karkar, C. Reale Hernandez, P. Jimenez Manero, D. Guyomard, L. Roué and B. Lestriez, J. Power Sources, 2015, 280, 533–549 CrossRef CAS.
- W. Zhang, M. Dahbi and S. Komaba, Curr. Opin. Chem. Eng., 2016, 13, 36–44 CrossRef.
- F. Zhao, P. Xue, H. Ge, L. Li and B. Wang, J. Electrochem. Soc., 2016, 163, A690–A695 CrossRef CAS.
- L. W. Shacklette, J. E. Toth and R. L. Elsenbaumer, Conjugated polymer as substrate for the plating of alkali metal in a nonaqueous secondary battery, Allied Corp., USA, 1985 Search PubMed.
- T. Shishikura and M. Takeuchi, Secondary batteries, Showa Denko K. K. Hitachi, Ltd, Japan, 1987 Search PubMed.
- K. Kubota, N. Yabuuchi, H. Yoshida and M. Dahbi, MRS Bull., 2014, 39, 416–422 CrossRef CAS.
- R. Dugas, B. Zhang, P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A867–A874 CrossRef CAS.
- Y. Li, S. Xu, X. Wu, J. Yu, Y. Wang, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2015, 3, 71–77 CAS.
- S. Xu, Y. Wang, L. Ben, Y. Lyu, N. Song, Z. Yang, Y. Li, L. Mu, H.-T. Yang, L. Gu, Y.-S. Hu, H. Li, Z.-H. Cheng, L. Chen and X. Huang, Adv. Energy Mater., 2015, 1501156 CrossRef.
- H. Wang, Y. Xiao, C. Sun, C. Lai and X. Ai, RSC Adv., 2015, 5, 106519–106522 RSC.
- M. Keller, C. Vaalma, D. Buchholz and S. Passerini, ChemElectroChem, 2016, 3, 1124–1132 CrossRef CAS.
- H. Li, L. Peng, Y. Zhu, D. Chen, X. Zhang and G. Yu, Energy Environ. Sci., 2016, 9, 3399–3405 CAS.
- Y. Wang, R. Xiao, Y. S. Hu, M. Avdeev and L. Chen, Nat. Commun., 2015, 6, 6954 CrossRef CAS PubMed.
- S. Guo, H. Yu, P. Liu, Y. Ren, T. Zhang, M. Chen, M. Ishida and H. Zhou, Energy Environ. Sci., 2015, 8, 1237–1244 CAS.
- Y. Zhang, H. Zhao and Y. Du, J. Mater. Chem. A, 2016, 4, 7155–7159 CAS.
- H. Gao and J. B. Goodenough, Angew. Chem., Int. Ed., 2016, 55, 12768–12772 CrossRef CAS PubMed.
- J.-Y. Hwang, C. S. Yoon, I. Belharouak and Y.-K. Sun, J. Mater. Chem. A, 2016, 4, 17952–17959 CAS.
- H. Y.-P. Hong, Mater. Res. Bull., 1976, 11, 173–182 CrossRef CAS.
- J. B. Goodenough, H. Y.-P. Hong and J. A. Kafalas, Mater. Res. Bull., 1976, 11, 203–220 CrossRef CAS.
- http://www.businesswire.com/news/home/20160129005028/en/Global-Rechargeable-Battery-Market-Reach-Close-USD .
- M. Sathiya, K. Hemalatha, K. Ramesha, J.-M. Tarascon and A. S. Prakash, Chem. Mater., 2012, 24, 1846–1853 CrossRef CAS.
- G. Singh, B. Acebedo, M. C. Cabanas, D. Shanmukaraj, M. Armand and T. Rojo, Electrochem. Commun., 2013, 37, 61–63 CrossRef CAS.
- J. M. D. Ilarduya, L. Otaegui, J. M. L. D. Amo, M. Armand and G. Singh, J. Power Sources, 2017, 337, 197–203 CrossRef.
- B. Zhang, R. Dugas, G. Rousse, P. Rozier, A. M. Abakumov and J.-M. Tarascon, Nat. Commun., 2016, 7, 10308 CrossRef CAS PubMed.
- Development of a Sodium Ion Secondary Battery, Sumitomo Chemical Co. Ltd, http://https://www.sumitomo-chem.cO.jp/english/rd/report/theses/docs/2013E_3.pdf.
- A battery Revolution in Motion, CNRS News, http://https://news.cnrs.fr/articles/a-battery-revolution-in-motion.
- Faradion Electric Bike: Prototype Powered By Sodium-Ion Batteries, http://www.greencarreports.com/news/1098434_faradion-electric-bike-prototype-powered-by-sodium-ion-batteries.
- L. Wangm, J. Song, R. Qiao, L. A. Wray, M. A. Hossain, Y.-D. Chuang, W. Yang, Y. Lu, D. Evans, J.-J. Lee, S. Vail, X. Zhao, M. Nishijima, S. Kakimoto and J. B. Goodenough, J. Am. Chem. Soc., 2015, 137, 2548–2554 CrossRef PubMed.
- Noble High Energy Density Sodium Layered Oxide Cathode Materials: from Material to cells, http://https://ecs.confex.com/ecs/230/webprogram/Paper88422.html.
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |