
Carbon disulfide. Just toxic or also bioregulatory and/or therapeutic?
Anthony W.
DeMartino
a,
David F.
Zigler
b,
Jon M.
Fukuto
c and
Peter C.
Ford
 *a
*a
aDepartment of Chemistry and Biochemistry, University of California, Santa Barbara, Santa Barbara, CA 93106-9510, USA. E-mail: ford@chem.ucsb.edu
bDepartment of Chemistry & Biochemistry, California Polytechnic State University, San Luis Obispo, CA 93407, USA
cDepartment of Chemistry, Sonoma State University, Rohnert Park, CA 94928, USA
First published on 27th September 2016
Abstract
The overview presented here has the goal of examining whether carbon disulfide (CS2) may play a role as an endogenously generated bioregulator and/or has therapeutic value. The neuro- and reproductive system toxicity of CS2 has been documented from its long-term use in the viscose rayon industry. CS2 is also used in the production of dithiocarbamates (DTCs), which are potent fungicides and pesticides, thus raising concern that CS2 may be an environmental toxin. However, DTCs also have recognized medicinal use in the treatment of heavy metal poisonings as well as having potency for reducing inflammation. Three known small molecule bioregulators (SMBs) nitric oxide, carbon monoxide, and hydrogen sulfide were initially viewed as environmental toxins. Yet each is now recognized as having intricate, though not fully elucidated, biological functions at concentration regimes far lower than the toxic doses. The literature also implies that the mammalian chemical biology of CS2 has broader implications from inflammatory states to the gut microbiome. On these bases, we suggest that the very nature of CS2 poisoning may be related to interrupting or overwhelming relevant regulatory or signaling process(es), much like other SMBs.
 Anthony W. DeMartino | Anthony (Tony) DeMartino received his BS in biochemistry from Wake Forest University in Winston-Salem, NC where he worked with Prof. Ulrich Bierbach. He is currently finishing his PhD in chemistry with Prof. Peter C. Ford at UC Santa Barbara where he has worked on photochemical NO donors and is currently examining the potential of CS2 as a small molecule bioregulator through the development of effective donors and various collaborative pharmacological studies. |
 David F. Zigler | David F. Zigler is an Assistant Professor in the Department of Chemistry & Biochemistry at California Polytechnic State University in San Luis Obispo, CA. After his graduate work with the late Karen J. Brewer at Virginia Tech, he worked as a postdoc in the Ford lab at UC Santa Barbara to create new compounds with caged CS2. Appointments at the University of North Carolina followed with Malcolm Forbes using EPR to study reactions with photo-generated 1O2 and with John Papanikolas in the UNC-EFRC Center for Solar Fuels to examine photo-initiated electron transfer at semiconductor/electrolyte interfaces. Professor Zigler's current research interests include inorganic phototherapeutics and materials for solar-to-chemical energy conversion. |
 Jon M. Fukuto | Jon M. Fukuto received a BA in chemistry from UC San Diego and PhD in chemistry from UC Berkeley where he worked with Prof. Fritz Jensen. He then did postdoctoral work at UCLA in Pharmacology with Prof. Arthur Cho. After a short stint in industry, he returned to UCLA as faculty in Pharmacology where he resided for over 18 years. At UCLA he worked on the chemical biology of the small molecule signaling agents nitric oxide (NO), nitroxyl (HNO) and hydrogen sulfide (H2S). He is currently a Professor of Chemistry at Sonoma State University where he continues this work. |
 Peter C. Ford | Professor Peter C. Ford joined the UCSB faculty after graduate study with Ken Wiberg at Yale and a postdoctoral fellowship with Henry Taube at Stanford. He has been a Visiting Fellow at the Australian National U, Guest Professor at U Copenhagen, Humboldt Fellow at U Regensberg and U Muenster and Guest Investigator at the US National Cancer Institute. Honors include a Humboldt Senior US Scientist Award, Inter-American Photochemical Society 2008 Award in Photochemistry, ACS 2013 National Award for Distinguished Service to Inorganic Chemistry and RSC 2015 Award in Inorganic Mechanisms. He is an AAAS Fellow and a RSC Fellow. His current research focuses on photochemical delivery of bioactive small molecules to physiological targets and biomass conversion to chemicals and fuels. He has served as research advisor to 65 PhD graduates and a number of postdocs, undergraduates and international visiting scholars. |
1. Introduction
Small molecule bioregulators (SMBs) are physiological signaling agents with interconnected pathways based on their unique chemistry and interactions with specific biological targets.1 This cellular signaling paradigm is a burgeoning field in mammalian chemical biology, and over the past several decades, SMBs central to this network have been identified. Three key SMBs, nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) have been called “gasotransmitters” although they are solubilized at the physiologically pertinent concentrations. A number of specific functions have been elucidated; for example, NO plays a key role in regulating blood pressure via the activation of soluble-guanylyl cyclase (sGC).2 Other known physiological functions are not yet as well understood, and the biological roles of these SMBs and of respective derivatives such as nitroxyl (HNO), peroxynitrite (ONOO−) and persulfides (R–SS−) continue to be the subjects of extensive biomedical research efforts.1–11If one were to envision an unidentified (so far) SMB in mammalian chemical biology, it may be appropriate to consider the shared properties of NO, CO, and H2S. All are relatively nonpolar but are partially soluble in aqueous and lipid systems; thus, they readily diffuse in physiological media via spatial concentration gradients.12 The actions of these SMBs involve chemical reactions with targets such as metal centers or redox active amino acids.7 Furthermore, all were initially considered to be environmental toxins; yet each was found to be formed by endogenous processes. In this context, carbon disulfide (CS2) is a small, nonpolar, readily diffusible molecule long recognized for its toxicity that may be generated endogenously. These similarities led us to contemplate whether CS2 might be an unrecognized SMB and/or have therapeutic potential.13
The purpose of this review is to explore the possibility of such roles for carbon disulfide. We will first outline possible modes of toxicity and then will survey the chemical properties of CS2 in the context of physiologically relevant conditions. Next, we will discuss known mechanisms for CS2 metabolism in order to visualize a framework by which CS2 may be managed endogenously. Finally, some therapeutic targets are suggested.
2. General physical and chemical properties of CS2
2.1. Physical properties and known sources
Pure CS2 is a colorless, pleasant-smelling liquid displaying a high vapor pressure (48.21 kPa at 25 °C), moderate water solubility (2.3 g L−1 at 22 °C), and high lipophilicity.14 It is a linear, nonpolar molecule like its homolog carbon dioxide (CO2) with carbon–sulfur bonds 155.6 pm in length.15 CS2 is slightly more soluble than CO2 in aqueous solutions; the Henry's law constants at 25 °C in pure water are 0.054 M atm−1 and 0.034 M atm−1 respectively.16Although CS2 is a listed environmental pollutant, it is produced naturally by soil-based microorganisms and is formed in vegetation fires and volcanoes. These sources account for 40–50% of atmospheric release, and the rest is anthropogenic.17 CS2 is produced industrially from heating a carbon source such as charcoal or natural gas with sulfur.18 The annual industrial production is ∼75 million kilograms per year (ca. 2010) for use in the rayon and cellophane, oil and gas, agricultural, and metal ore processing industries.18,19 It has been used in production of viscose rayon since the nineteenth century,20 when the deleterious effects on exposed workers in that industry were first described.21
2.2. Toxicity
As noted above, CS2 toxicity was first described in the 1850s21 and is the subject of numerous subsequent studies.22–26 Of particular interest are the neurotoxic effects.27–33 Acute CS2 poisoning causes toxic psychosis and narcotic somnolence. Long-term sub-acute exposures are linked with memory, behavioral and cognitive disturbances, and also concomitant nerve demyelination, cytostructural damage, and progressive paralysis of the extremities.33,34 The mechanism of CS2 neurotoxicity, however, is currently unknown. Epidemiological studies of chronic CS2 exposure are somewhat contradictory, particularly regarding reproductive35–37 and cardiovascular issues.38,39 From a brief overview of the literature, the described toxicity, pathology, and/or metabolism of CS2 indicate a breadth of targeted biochemical effects.2.3. Some key chemical reactions
CS2 is stable to hydration and at biologically relevant pH values is relatively unreactive toward hydrolysis to carbonyl sulfide (COS) in aqueous solutions (eqn (1)). | (1) |
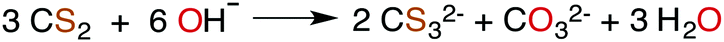 | (2) |
The industrially important reaction of CS2 with primary and secondary alcohols and a metal hydroxide to form xanthates (eqn (3)) was reported as early as 1822.42
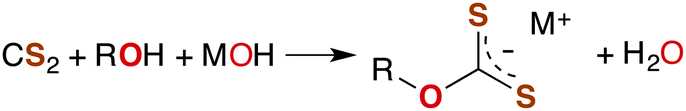 | (3) |
 | (4) |
The analogous reactions of CS2 with primary and secondary amines lead to the formation of dithiocarbamates (DTCs) (eqn (5)). This equilibrium is dependent on the nature of the amine (e.g., sterics and electronics) and on the solution pH. In acidic solutions, dithiocarbamates readily decompose to the constituent amine plus CS2.44–46 At pH 7.4 and 35 °C, primary and most secondary alkyl-DTCs are relatively stable, whereas under mild acidic conditions, secondary alkyl-DTCs revert to the amine and CS2 with rate constants on the order of 10−3 s−1 at pH 5.9.47
 | (5) |
A CS2 reaction that might have particular significance with regard to biological properties is the reversible formation of trithiocarbonates (TTCs, also known as thioxanthates) as illustrated in eqn (6).48–50 We will illustrate possible roles of this reaction in greater detail below.
 | (6) |
2.4. Interactions with metal centers
Carbon disulfide and its adducts (xanthates, DTCs, and TTCs) can serve as ligands in metal complexes. Typically, CS2 coordinates more strongly to metal centers than does either CO2 or COS.51,52 There are a number of examples of η1- and η2-CS2 complexes53–55 as well as polynuclear complexes56 where the CS2 moiety is a bridging ligand (Fig. 1).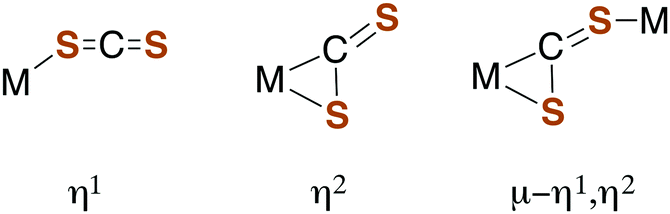 | ||
| Fig. 1 Binding modes of CS2 with metal centers. | ||
Such coordination can activate CS2 toward reaction with nucleophiles. For example, reversible CS2 insertion into a metal–ligand bond is a common reaction (eqn (7)), presumably because of the stronger bonding of the 1,1-dithiolate (–CS2−) functionality.
 | (7) |
 | (8) |
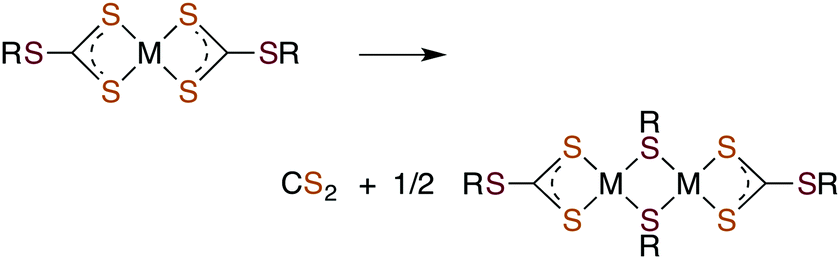
Metal centers also react with various RXCS2− (where X = NR′ or S) ligands to form stable complexes under a variety of conditions. Although generally stable in neutral aqueous solutions, some M(S2CXR) complexes eliminate CS2.64–69 For example, spontaneous release of CS2 from the Ni(II) S-benzyl- and tert-butyl-trithiocarbonate complexes (eqn (9)) occurs with first order rate constants on the order of 10−4 s−1 at 35 °C in lipophilic media.64
 | (9) |
3. Biological chemistry
3.1. Hydrolysis (or not)
Like CO2, carbon disulfide is rather unreactive toward spontaneous hydration and hydrolysis at biological pH values, so a significant proportion of the dissolved compound is present in the solvated form, CS2 (aq). Furthermore, carbonic anhydrase (CA), the enzyme that catalyzes the hydration of CO2, is ineffective in activating the analogous reaction of CS2 (eqn (1)). For example, with excess CS2, the turnover frequency of cytosolic CA II, the most efficient of the fourteen CA isomers,74 is about 9 orders of magnitude slower (∼5 × 10−4 s−1) than the analogous reaction with CO2 (∼106 s−1).75The first step of CS2 hydrolysis, like CO2, is hydration. Using a model for the Zn-active site of CA II, Anders and coworkers computationally probed the formation of dithiocarbonate, the hydrated intermediate of CS2 (Scheme 1) by considering a mechanism analogous to CO2 hydration.76–78 It is argued that dithiocarbonate formation has a slightly higher barrier of activation owing to a decreased electrophilicity of CS2.76 In addition, the intermediate dithiobicarbonate (HCS2O−) is a thermodynamic sink, which further slows hydrolysis.51 The calculations with this active site model suggest that CA does not complete a catalytic cycle with CS2. Rather, CS2 is proposed to inhibit CA. Although CS2 does not appear to interact with CA II,75 xanthates and TTCs are effective inhibitors.79 Nevertheless, there are bacterial-evolved, archaeal CS2 hydrolases that do convert CS2 to CO2 and H2S through intermediate COS (eqn (1) and (10)).80 In spite of the high homology with carbonic anhydrases, these CS2 hydrolases apparently do not use CO2 as a substrate,80 just as CA does not efficiently catalyze CS2 hydrolysis. Notably, COS is readily hydrolyzed by both types.75,77,78
 | (10) |
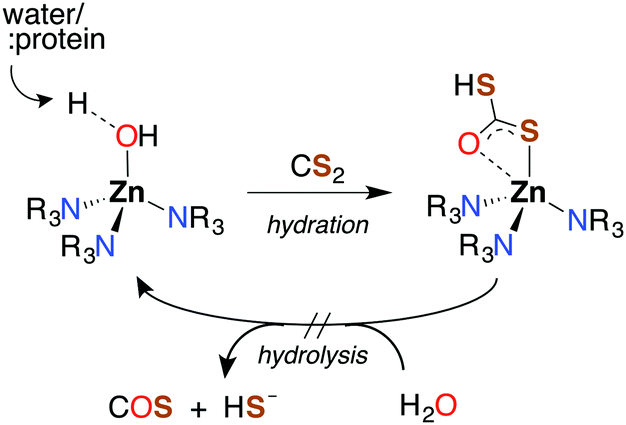 | ||
| Scheme 1 Hydration of CS2 in Zn-based CA model active site. Anders and coworkers calculated a mechanism analogous to that for CO2, with hydration leading to a stable dithiobicarbonate adduct. The subsequent step (hydrolysis) is slow. | ||
The presence of CS2 hydrolases in some microorganisms and clear divergence in evolution imply an evolutionary need for a CS2-specific enzyme. Thiobacillus thioparus and Thiobacillus denitrificans are microbes that use reduced sulfur species (dimethyl sulfide, methyl mercaptan, and H2S) for energy production.81 These may need such hydrolases to function in CS2-rich environments. T. thioparus has been shown to consume CS2.82
3.2. Methane monooxygenase (MMO) and ammonia monooxygenase (AMO)
Carbon disulfide has been shown to be an inhibitor of nitrification in Nitrosomonas and Nitrosococcus bacterial strains83,84 and reported to inhibit methane monooxygenase (MMO).85 From a detoxification viewpoint, MMO is evolutionarily and mechanistically similar to ammonium monooxygenase (AMO),85 and methanotrophs that depend on MMO and live in high sulfur environments (e.g., Methylomicrobium alcaliphilium) have active CS2 hydrolases. The underlying mechanism proposed for CS2 inhibition of nitrification involves reaction at a nucleophilic amine (lysine) or thiol (cysteine) to form a DTC or TTC near the active site of AMO.84,86 The resulting 1,1-dithiolate may then chelate and sequester the active site copper or modify the activity owing to structural or electronic changes. An alternative mechanism would be reversible CS2 insertion into metal–N or –S bonds as discussed above. Thus, one can envision CS2 diffusing to a reactive site and either inserting in a metal–ligand bond motif or binding a proximal amine or thiolate to form a protein bound TTC or DTC as illustrated in pathways i and ii, respectively, in Fig. 2. In either case, the resulting changes in structure, electronics, metal chelation, etc. would impact protein function.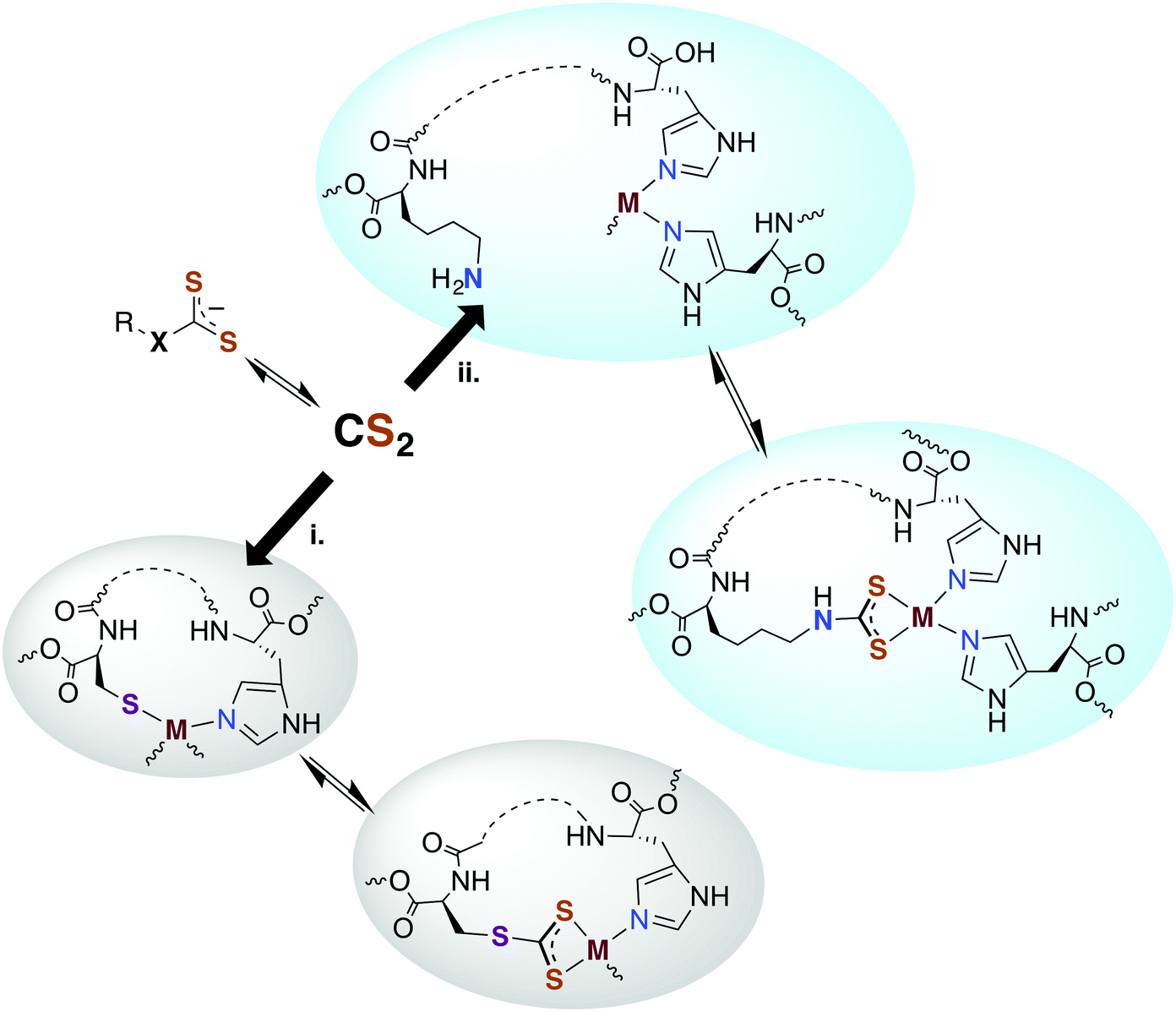 | ||
| Fig. 2 Possible interactions of CS2 with biological metals. Since it is lipophilic, CS2 could readily transport to the active sites of cellular proteins and (i) insert into a metal–cysteine bond to form a trithiocarbonate chelate or (ii) bind a protein amine or thiol proximal to a metal center where the resulting dithiocarbamate or trithiocarbonate can bind. X = NR′ or S. | ||
3.3. Carbon monoxide dehydrogenase (CODH)
CODH interconverts CO to CO2 in anaerobic bacteria and is responsible for the final step of the production of acetyl coenzyme A (acetyl-CoA). Ragsdale and coworkers87 have reported that CS2 is a rapid binding, reversible inhibitor of this Ni/Fe–S cluster protein. Consistent with its interaction with CA, CS2 does not bind or interact with the CO2-binding site. Instead it serves as a competitive inhibitor to the terminal binding site of CO at an iron center of the cluster, the site of acetyl-CoA synthesis. Two proposals were offered regarding CS2 binding at this site: insertion into a ligand–iron bond or coordination as CS2 to the metal. An alternative possibility would be CS2 binding via electrophilic reaction at a protein thiol or amine near the coordination site. Equally likely is that CS2 binds the thiol of acetyl-CoA itself. Since CODH catalyzes the acetylation of the homocysteine thiol moiety of CoA,88 CS2 binding at this thiol would generate a TTC that inhibits CODH via chelation of the iron.3.4. Potential targets in mammalian physiology
In general, CS2 exposure in humans and assorted mammals has been assessed via detection of the metabolites thiazolidine-2-thione-4-carboxylic acid (TTCA), 2-thio-5-thiazolidinones, and occasionally 2-oxothiozolidine-4-carboxylic acid (Fig. 3) in the urine.27,30,32,89–91 TTCA is the product of CS2 reaction with cysteine or cysteine-containing poly/oligopeptides, and its urinary release has been shown to increase linearly with CS2 oral intake by rats.92 Other major metabolites, such as the 2-thio-5-thiazolidinones, likely result from terminal amines in polypeptides and other biological amines acting as nucleophiles at the CS2 carbon to form (initially) DTCs followed by eventual cyclization. These metabolic scenarios are considered below.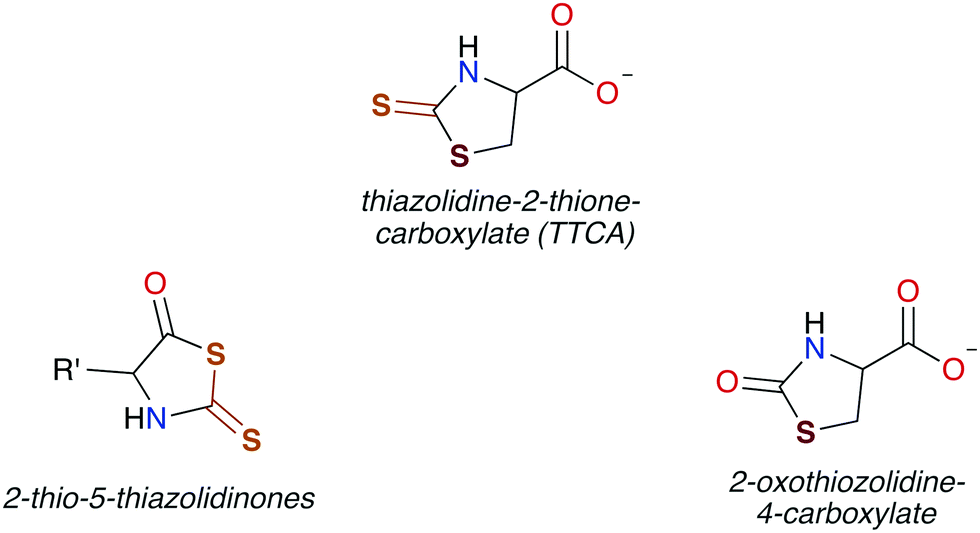 | ||
| Fig. 3 Cyclic metabolites found in the urine of mammals after exposure to CS2. | ||
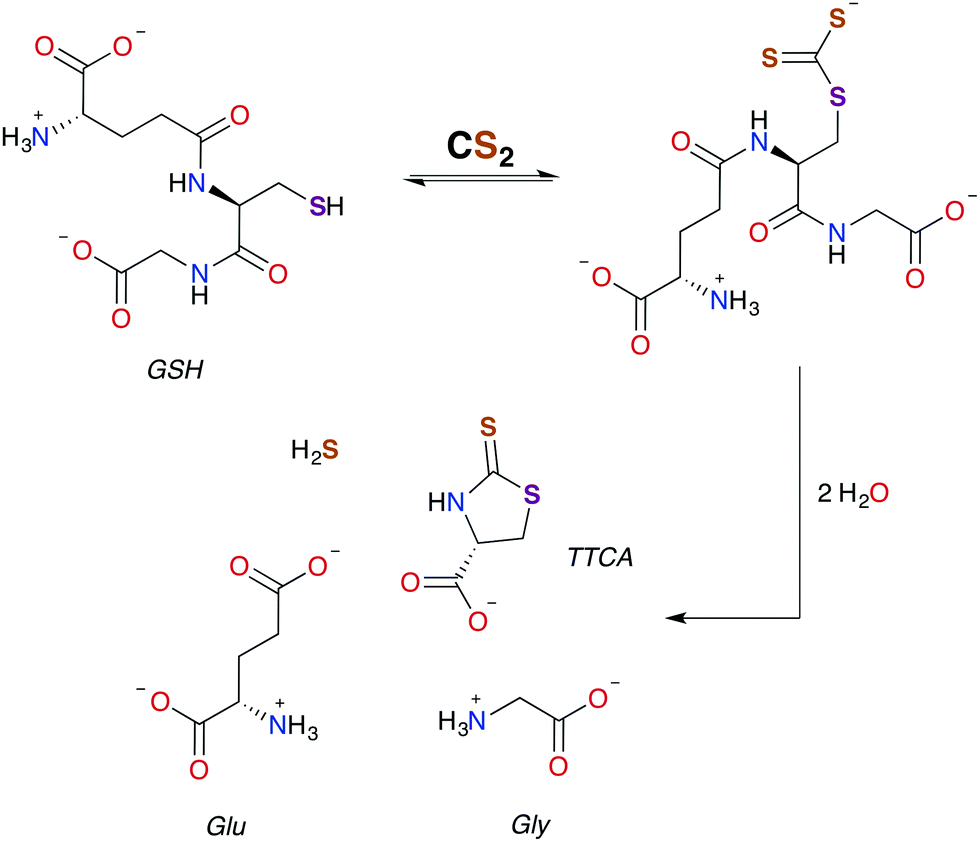 | ||
| Scheme 2 Pathway for the formation of TTCA from the reaction of GSH with CS2.30,49 | ||
As aforementioned, TTCA release through the urinary tract is a signature of external exposure to CS2. This thiazolidine is apparently derived from the reaction of CS2 with the thiolate of cysteine or GSH, in the latter case resulting also in liberation of glycine and L-glutamic acid.89,102,103 However, it is not known whether TTCA formation is enzyme assisted. While reaction with a secondary amine of GSH to give a DTC is a mechanistic possibility, such sites are much less nucleophilic than the thiolate.104 In either case, intramolecular cyclization and liberation of H2S would result in TTCA as roughly outlined in Scheme 2.
The formation of alkyl TTCs in aqueous solution is reversible (eqn (6)),105 although the equilibria are dependent on the pH and on the pKa of the parent thiol. Such equilibria are not well characterized with biological thiols, but these are especially interesting given that TTCs may be a means to sequester or transport CS2 in serum or to provide a pathway for the interactions of CS2 with metalloproteins. In addition, TTC formation as post-translational protein modification at cysteine residues likely leads to important structural and/or functional effects that also may be important signaling pathways, even if such reactions are reversible. Protein microenvironments can markedly affect the acidity of a thiol through interactions with charged amino acid side chains and/or hydrogen bonding.99–101 Such interactions with protein thiols are potential targets and switches for CS2 signaling.
Another signaling mechanism may involve the direct reaction of TTCs with transition metal centers or by reversible CS2 insertion into metal thiolate bonds to form TTC metal complexes (Fig. 2). The latter pathway was demonstrated with a variety of metal–thiolate complexes under ambient conditions57,68,69 including the biologically relevant zinc(II) and copper(I) centers (see above). Thus, one can envision metal thiolate centers such as zinc fingers or iron redox centers as targets for the reversible formation of CS2 adducts. Although the above examples were in non-aqueous media, similar reactivity may be relevant in the cores of particular proteins or other lipophilic areas.
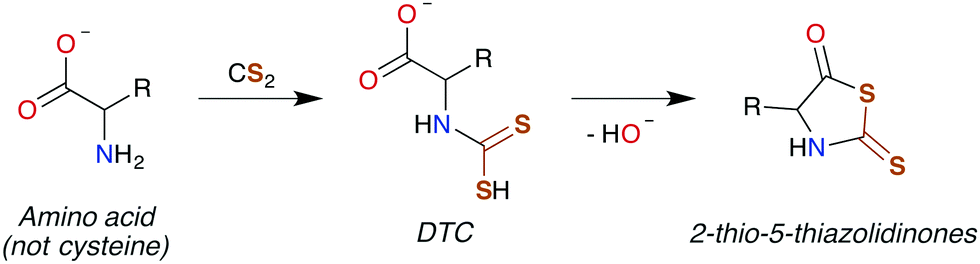 | ||
| Scheme 3 Conversion of an amino acid to a DTC, followed by subsequent cyclization to 2-thio-5-thiazolidinone. | ||
DTCs are commonly used fungicides and pesticides, and thus their toxicity has been reviewed119,120 most recently by Rodrigues-Lima and coworkers.121 Many of the pathologies of DTC toxicity mirror those documented for CS2, including potent central nervous system (CNS) effects under acute exposure as well as links to Parkinson's disease via induction of severe CNS depression.122–124 DTC disulfides also have neuropathies very similar to that of those produced by CS2 exposure,125–129 although a different mechanism of toxicity has been proposed.129
The similar symptoms of DTCs and CS2 toxicity suggest a shared mechanism. Under acidic conditions, N,N-diethyldithiocarbamate (DEDTC) releases CS2, but it is generally assumed that DEDTC is stable at or above neutral pH.130,131 Valentine and coworkers103 investigated possible in vivo CS2 release from disulfiram and DTCs such as DEDTC to establish whether such release induced symptoms attributed to toxicity characteristic of CS2 exposure. Using radiolabeling, they found a marked increase in urinary TTCA attributed to the presence of free CS2 regardless of the compound tested (disulfiram or a DTC). Not surprisingly, oral exposure to DEDTC gave much higher fluxes of TTCA owing to the acidic environment of the dietary tract. Incorporation of the labeled carbon at the thiocarbonyl of TTCA indicates that CS2 is generated from the various DTCs. This result may answer a question raised by Cvek and Dvorak in a review of DTC cellular interactions,132 namely, that it is unclear how polar DTCs might enter a cell through lipid bilayers. The in vivo study by Valentine et al.103 supports the view that DTCs and CS2 are in labile equilibria. This lability under physiological conditions thereby provides a mechanism to transport this functionality through hydrophobic membranes by passive diffusion of CS2.
The reaction between neuronal amines and CS2 has often been hypothesized to be the source of carbon disulfide-induced neuropathological effects.30,106,107 Catecholamine neurotransmitters, including epinephrine (adrenaline), norepinephrine (noradrenaline), and dopamine have amine-bearing side chains sufficiently nucleophilic133 to react with CS2in vivo. This reaction is a likely cause of the significant norepinephrine depletion in the adrenal glands and storage granules of rodents exposed to CS2 vapor (64 ppb).106 Norepinephrine is produced from dopamine via dopamine β-hydroxylase (DBH), a monooxygenase with copper in the active site,107 and it has been suggested106 that DBH is inhibited by DTC formation and chelation of that copper. Experiments in vitro showed a neuronal amine is needed to inhibit DBH in the presence of CS2 and that this effect is reversible.107 Notably, CS2 proved to be more effective inhibitor of DBH than an added glycine-based DTC.107 This suggests that lipophilic CS2 enters catecholamine storage granules where it reacts to form DTCs and inhibits DBH (Fig. 4). Such a process could play a regulatory role by controlling dopamine/norepinephrine levels.134,135
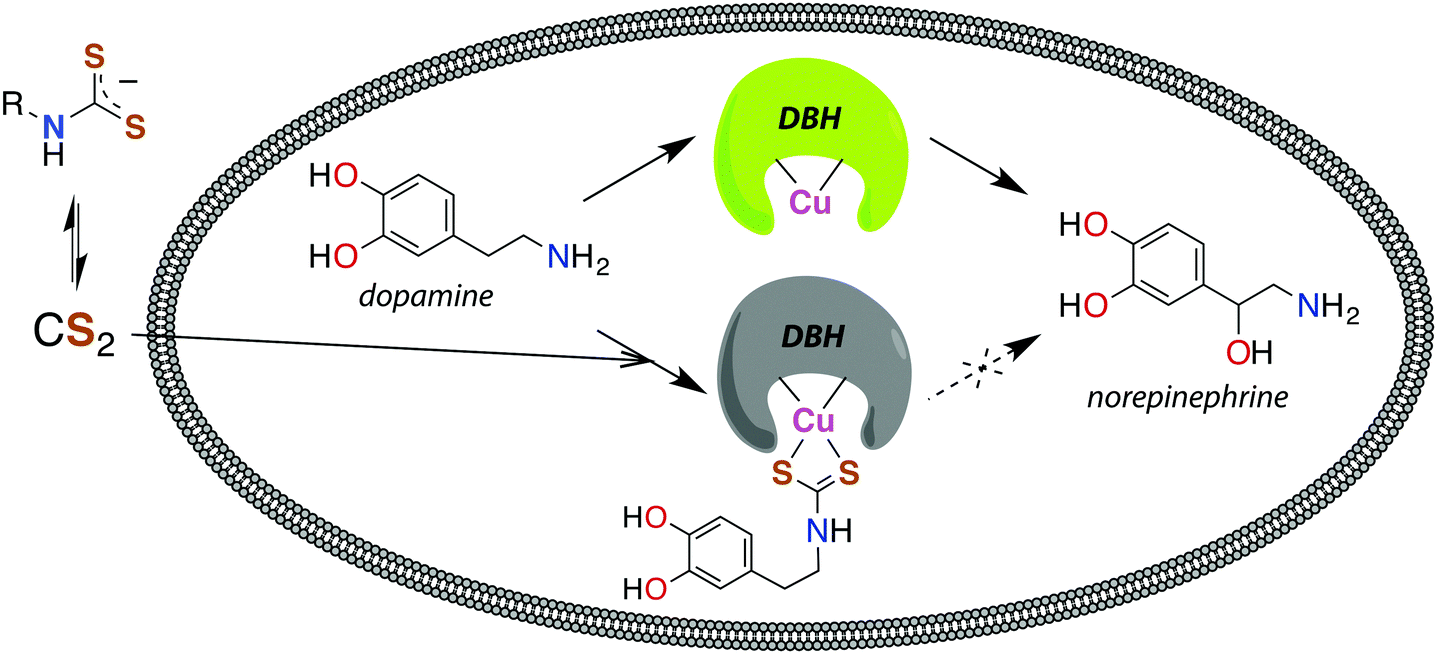 | ||
| Fig. 4 Release of CS2 from DTCs allows for diffusion across membranes, like the storage granules of catecholamines. Dopamine β-hydroxylase (DBH), a copper-centered enzyme, converts dopamine to norepinephrine and is deactivated by CS2via the DTC of catecholamines.107 | ||
Disulfiram (also called “antabuse”), the disulfide of DEDTC, has long been used in alcohol aversion therapy.131 It functions by inhibiting acetaldehyde dehydrogenase (ALDH), thereby greatly enhancing the severity of hangovers due to alcohol consumption. The molecular mechanism of ALDH inhibition is mediated by the metabolic products of disulfiram, the first of which is DEDTC formed by reduction of the disulfide bond (Fig. 5). Metabolites of DEDTC are irreversible inhibitors of ALDH.121 Covalent addition of one of these metabolites to an essential cysteine residue inhibits rodent ALDH in vivo by initiating disulfide bond formation. The current thinking is that oxidation by microsomes forms methyl diethylthiocarbamoyl sulfoxides (Fig. 5) as the active agent.130,136 Direct reactions with DTCs or their disulfides have also been proposed.119,135 Either of these mechanisms provide a route for CS2 to interact with specific proteins having ideally placed cysteines and could be essential in bio-signaling chemistry (Fig. 6).113,136,138
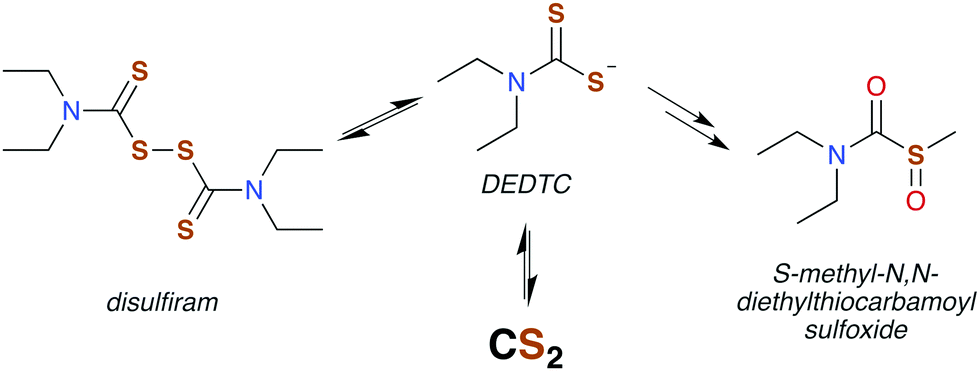 | ||
| Fig. 5 Disulfiram is readily reduced in vivo to form DEDTC, which generates carbon disulfide or is eventually converted to a thiocarbamoyl. All have been implicated in inhibiting ALDH via mixed disulfide formation with protein thiols. | ||
 | ||
| Fig. 6 Possible interactions of CS2-adducts with proteins suggested by the inhibitory action of disulfiram and its metabolites with ALDH. Formation of a mixed disulfide on a critical protein thiol leads to oxidation and intra-protein disulfide formation with a nearby thiol. X = NR′ or S. | ||
Disulfiram is also finding therapeutic use in treating cocaine addiction via an ALDH-independent mechanism.137,139 This may function by inhibiting DBH, the copper-dependent enzyme required for metabolism of dopamine to norepinephrine. These inhibitory effects have been attributed to the copper-chelating activity of DEDTC formed in vivo. Such copper-chelation is also thought to induce proteasome inhibition leading to the proposal that disulfiram could serve in anticancer therapy.140
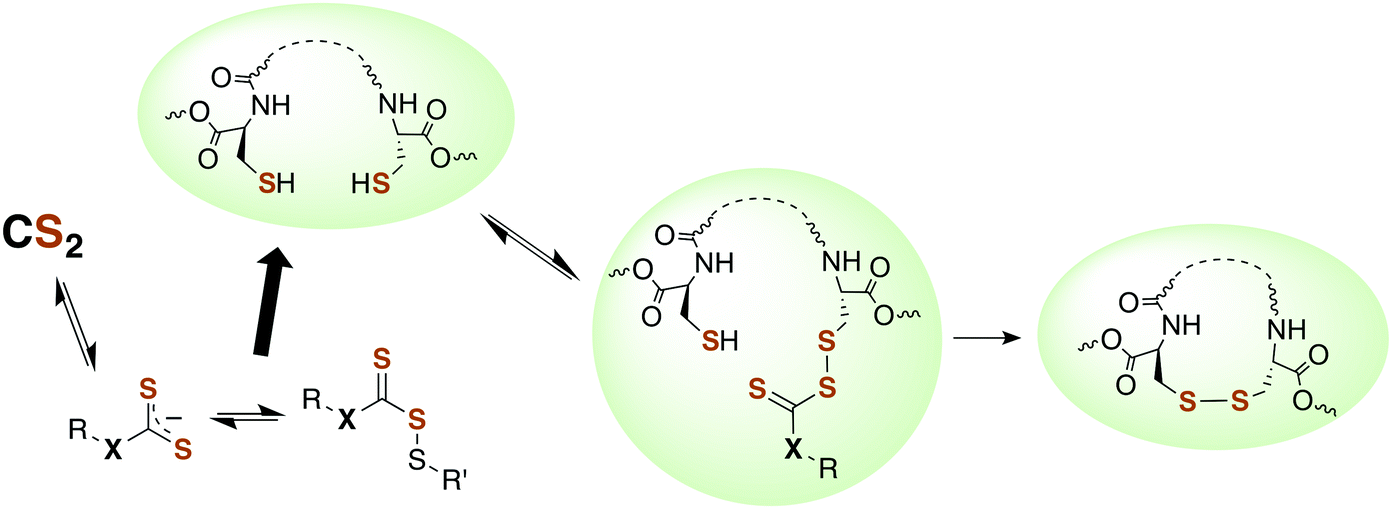
Although there is strong evidence implicating CS2 generation in DTC toxicity,130 it is important to consider concentration regimes. One proposed mechanism for CS2 neuropathy involves the induction of intra- and inter-protein cross-linking. The proteins β-lactoglobulin, porcine neurofilaments, bovine serum albumin, and hemoglobin were each observed to react with CS2 to form thiourea based crosslinks and dimers. This involved predominately lysine ε-amines,129 but also lysine and the N-termini of certain proteins,141,142 as the pKa of such amines can be significantly decreased in protein microenviornments.143 DTC formation on an exposed amine, followed by loss of HS− would give isothiocyanate intermediates that couple with another free amine (Scheme 4).144 However, it should be noted that these in vivo processes were demonstrated in rats exposed to 50 ppm CS2 (5 times the occupational threshold)19 in inhaled air for 2 weeks.
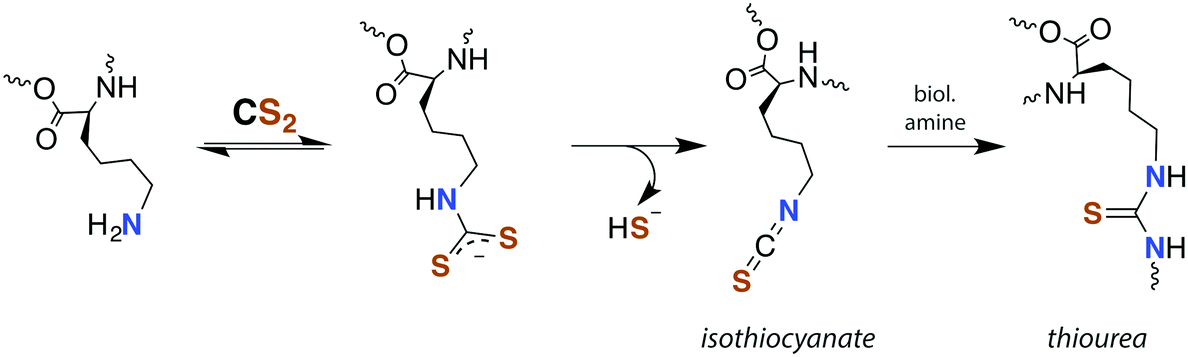 | ||
| Scheme 4 Transformation of CS2 derived lysine dithiocarbamates into dimers responsible for CS2-induced protein cross-linking via thioureas moieties.141 | ||
What stimulates the constitutive expression of NF-κB in cancer cells is not fully understood.155 An extracellular signal triggers a cytosolic-based cascade to release NF-κB from an inhibitor (I-κB). Following translocation to the nucleus, NF-κB tunes the regulation of diverse genes. To activate NF-κB, the I-κB must be phosphorylated by an I-κB kinase (IKK) resulting in its conjugation to ubiquitin (ubiquitination) and subsequent degradation.132,156–159 DTCs can act as thiol antioxidants, which are also known to inhibit NF-κB.152 However, the inhibitory activity of DTCs appears to be isolated from its antioxidant abilities160 and relies on a mechanism that has not been fully elucidated.
A potential target for CS2 is a cysteine (Cys38) in the DNA binding site of NF-κB (Fig. 7, pathway 1).161 Treatment with thiol-reactive compounds such as N-tosyl-L-phenylalanine chloromethyl ketone (TPCK) prevents DNA binding.152 TPCK reacts far more freely at this residue than it does with the cellular GSH, suggesting that the Cys38 thiol is more nucleophilic or has a lower pKa,161 thereby making it a likely target for CS2via the reversible formation of a TTC.150 Such adduct formation could also provide a mechanism for DTC inhibition of NF-κB, given that dithiocarbamates release CS2in vivo (see above).
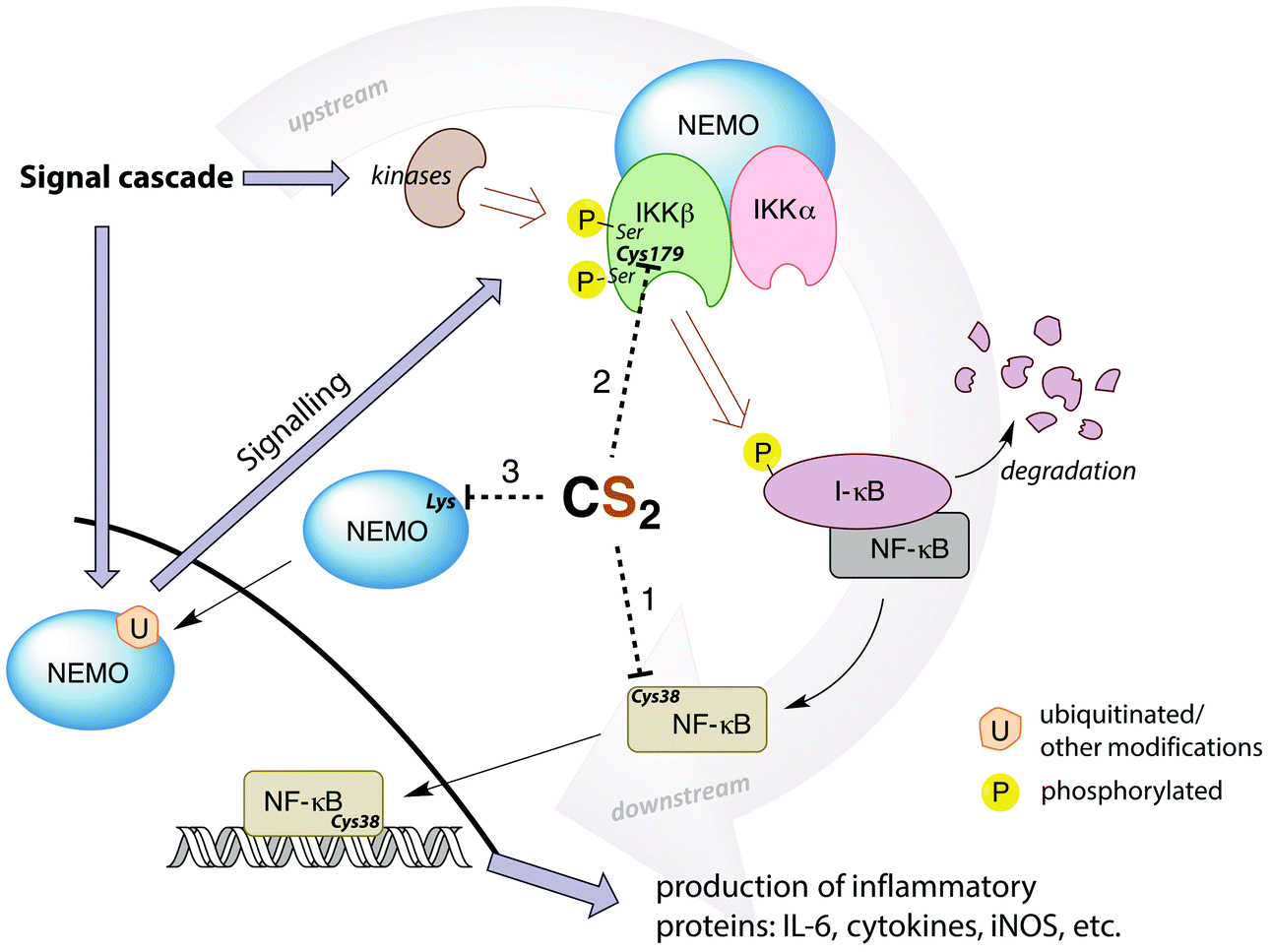 | ||
| Fig. 7 Simplified depiction of NF-κB activation and gene up-regulation to produce pro-inflammatory proteins. The IKK complex (top right) is activated through a signal cascade and in turn activates NF-κB. Possible targets for CS2 inhibition are the Cys38 in the DNA binding region of NF-κB (pathway 1), the Cys179 in IKKβ (pathway 2), or lysines in the zinc finger binding domains of the pre-ubiquitinated scaffold protein NEMO (pathway 3). Cys179 appears to be the most ubiquitous and likely target. (IL-6 = interleukin-6, a proinflammatory cytokine, iNOS = inducible nitric oxide synthase, I-κB = inhibitor of NF-κB, NEMO = NF-κB essential modulator (also known as IKKγ), IKK = I-κB kinase).146,148,152,155,159,161–164 | ||
Thiolate-reactive compounds also inhibit the IKK complex further upstream in the signaling cascade (Fig. 7, pathway 2). The presumed target is again a critical cysteine (Cys179), located between two serine residues (Ser177/181) in the activation loop of the protein β-subunit.164 Ser177/181 are each phosphorylated by another kinase to activate IKK (Fig. 7).165 Replacing Cys179 with alanine prevents phosphorylation, indicating that this cysteine is non-innocent in the activation step.166 Further, Cys179 is implicated as a participant in the catalytic domain of activated IKK164,167 due to significant (albeit reversible) enzyme deactivation occurring with DTC treatment of pre-phosphorylated IKKβ. The fact that DTC inhibition is reversible suggests a mechanism distinct from the disulfide formation step that is responsible for ALDH inhibition. Indeed, CS2 liberated from a dithiocarbamate may form a TTC at Cys179, thereby providing a reversible IKK inhibition pathway.
Another possible upstream target for CS2 is the NF-κB essential modulator (NEMO), which recruits other IKK activators (Fig. 7, pathway 3). NEMO can be activated by ubiquitination at lysine residues including those in a zinc finger binding domain.159,162 This suggests another site for CS2/DTC interference. DTCs have been shown to decrease the expression of NEMO155 in addition to the IKKβ, consistent with this hypothesis.
NF-κB is an alluring therapeutic target due to the prevalence of heightened inflammation in a variety of disease states.70,132,152 If CS2 is indeed an SMB that plays a critical role in the regulation of these important transcription factors, then it stands to reason that CS2 delivery may have therapeutic value, notably in the context of inflammation reduction. Along this line of thought, delivery of CS2 has been proposed in the patent literature for the treatment of inflammatory conditions and was claimed to directly inhibit NF-κB.168
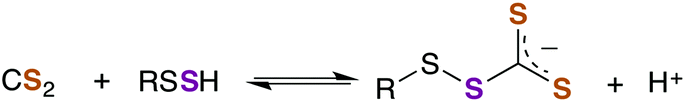 | (11) |
Another potent class of biological nucleophiles is represented by selenoproteins. Like hydropersulfides, selenocysteine is more acidic than cysteine by 2–3 pKa units. Thus, selenocysteines exist predominantly as the anionic selenide at physiological pH. In general, selenols are stronger nucleophiles than are thiols under conditions of equal ionization and even more so under physiological conditions where a higher percentage of the selenide anion would be present compared to thiolate.181 Of course, selenoproteins are far less prevalent compared to thiol proteins. To date, only about 25 human selenoproteins have been identified with functions ranging from thyroid hormone regulation to antioxidant function.182 For comparison, the human genome encodes for over 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cysteines that are widely distributed among proteins.183 The reaction of a selenide with CS2 can be envisioned to be analogous to that shown for the thiol/thiolate with the generation of the selenoxanthate (RSeC(S)S−). Therefore, due to the nucleophilicities of persulfides and selenols, future studies into the chemical biology of CS2 should consider the possible interactions with persulfides and/or selenoproteins.
000 cysteines that are widely distributed among proteins.183 The reaction of a selenide with CS2 can be envisioned to be analogous to that shown for the thiol/thiolate with the generation of the selenoxanthate (RSeC(S)S−). Therefore, due to the nucleophilicities of persulfides and selenols, future studies into the chemical biology of CS2 should consider the possible interactions with persulfides and/or selenoproteins.
3.5. Endogenous production?
A hallmark of the SMBs NO, CO, and H2S is that each is generated endogenously by enzymatic processes.1 While at present there is no direct evidence for mammalian CS2 production by a specific mammalian enzyme, there is ancillary evidence for endogenous production.184,185 For example, humans (controlled for exposure, medication, etc.) exhale CS2. While healthy individuals do not exhale it at levels where there is a significant difference in alveolar gradient,186 elevated CS2 levels on the breath are associated with a variety of disease states.185 For instance, patients with schizophrenia have high positive CS2 gradients, indicating higher CS2 levels in their blood steams.187 Given CS2's interaction with catecholamines resulting in increased dopamine levels,107 and the association of high levels of dopamine with schizophrenia,134,135 an abundance of CS2 may play a role. Exhaled CS2 is also associated with gastric cancers, especially those induced by Helicobacter pylori.184 A significant increase in CS2 is found in human feces after eating, suggesting a role of the gut microbiome in its production.188,189 Moreover, it is found in all samples of healthy donors and missing in many patients with foodborne bacterial infections like Campylobacter jejuni,188 implicating a positive association of CS2 production with a healthy gut microbiome.Exhaled CS2 is also claimed to be a marker for organ rejection after lung transplantation,184,185 although COS has also been claimed as such a marker.190 It is notable that CS2 or COS would be associated with organ transplantation, as it is well-established that CO has protective effects in organ transplantation, reducing probability of rejection.4
What are the sources of exhaled CS2? It is possible that, like other sulfur containing volatiles, it is a by-product of metabolic pathways of methionine.184,185 Alternatively, it has been inferred that it derives from a collection of gut bacteria and micro-organisms of the human microbiome that produces other sulfur-containing small metabolites.187–189 However, one should also consider the possibility of a dedicated, enzymatic source. This is most apparent in certain rodents that exhale CS2 in micromolar concentrations.191 The olfactory system of such rodents appears to have sensory neurons sensitized to CS2 through an isoform of membrane-bound receptor guanylyl cyclase, type D (GC-D).192 This receptor triggers a cyclic guanosine monophosphate (cGMP)-signaling cascade, much like the response of sGC to NO. This CS2-initiated cascade, when combined with an odor, promotes in rodents a preference for the food associated with that odor, an important instinctual survival mechanism.191 Knockouts for the gene encoding the olfactory GC-D resulted in loss of the ability to form these preferences.192,193 This result implies that, at least with these rodents, CS2 has a signaling-inducing target rather than being simply a metabolic byproduct. A form of CA II has also been reported to play a role in such signal transduction,192 even though other CAs generally do not interact strongly with CS2.75 The murine olfactory system is also sensitive to CO2, but by several orders of magnitude less than to CS2. These observations suggest that CS2 is endogenously produced in mammals at concentration regimes appropriate for an SMB.
Notably, TTCA is detected in the urine of individuals not directly exposed to CS2.204 Specifically, consumption of brassica vegetables such as broccoli, cauliflower, and cabbage results in urine excretions of 10 μmol TTCA per liter of urine. This observation suggests that these vegetables may be a natural source of CS2 given the nature of TTCA formation.17 While TTCA is also found in the vegetables themselves, far greater quantities are found when they are boiled.205 Given that TTCA is derived from the reaction of GSH or cysteine with CS2, it is reasonable that heat simply increases the rate of this reaction and thus of TTCA production. Hence, it stands to reason that these food sources may contain trace amounts of CS2 or a precursor thereof. Moreover, brassica vegetables are promoted as healthy and beneficial foods due to their association with lower cancer risk in a multitude of studies.206 The bioactive compounds are the sulfur containing phytochemicals glucosinolates (Fig. 8). They have anti-inflammatory, genetic, and epigenetic properties once metabolized and have been probed for therapeutic potential. Glucosinolates undergo enzymatic conversion to form isothiocyanates and DTCs, which in turn can generate CS2 and TTCA.205,207
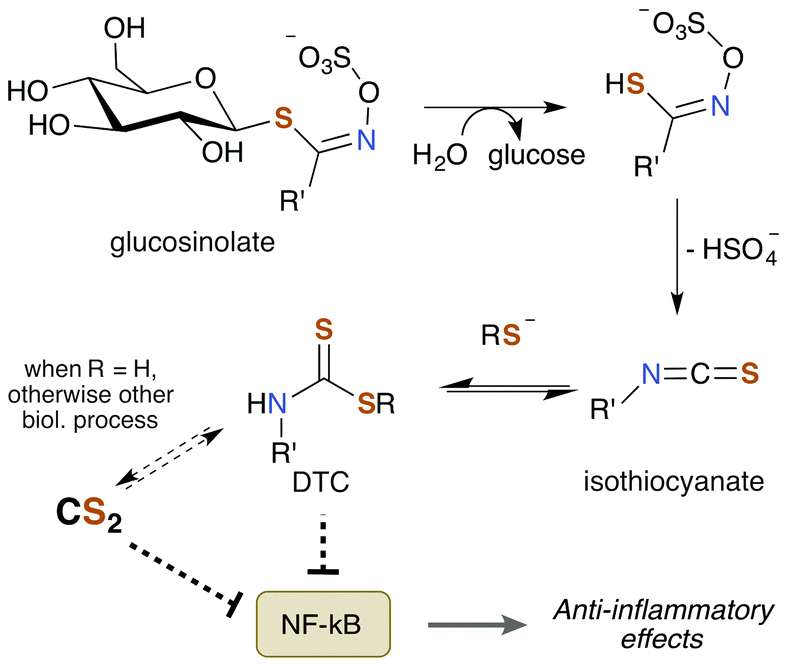 | ||
| Fig. 8 Brassica vegetables contain glucosinolates, which undergo enzymatic conversion to a variety of metabolites, with various effects, including anti-inflammatory action. Since DTCs are generated, these compounds may also yield CS2. | ||
4. Summary and outlook
In this review, we've attempted to draw together literature evidence indicating that carbon disulfide may have roles in mammalian biology beyond its toxicity. The various chemical pathways of CS2 that might contribute to such properties are summarized in Fig. 9. We have posed the questions: does CS2, its precursors, or the resulting metabolites have therapeutic potential? and/or Is CS2 an unrecognized small molecule bioregulator? Like NO, CO, and H2S, carbon disulfide is a small molecule that is nonpolar and diffusible and is also known to be toxic at relatively high concentrations. Furthermore, there is fragmentary evidence that CS2 is produced endogenously, even in humans, although it is possible that this production is at least in part from the closely associated microbiome. The biological chemistry and metabolism of CS2 suggest a commonality in biochemical targets with other SMBs. This includes reactivity with amino acids like cysteine and lysine and the formation of adducts in cytosol, serum, and proteins. Metal centers are also likely targets via the insertion of CS2 into metal ligand bonds to form chelating ligands. These reactions readily occur at ambient temperatures and in aqueous media. Such pathways may initiate interconnected signaling cascades. For examples, reactions of CS2 with biological nucleophiles in some cases release H2S, and the inhibition of NF-κB is critically involved in inducible nitric oxide synthase activation. Furthermore, it is entirely likely that CS2 generated by a specific enzyme, system, etc. would be ultimately consumed and metabolized in a specific manner when functioning normally, e.g., through oxidative desulfurization in the liver.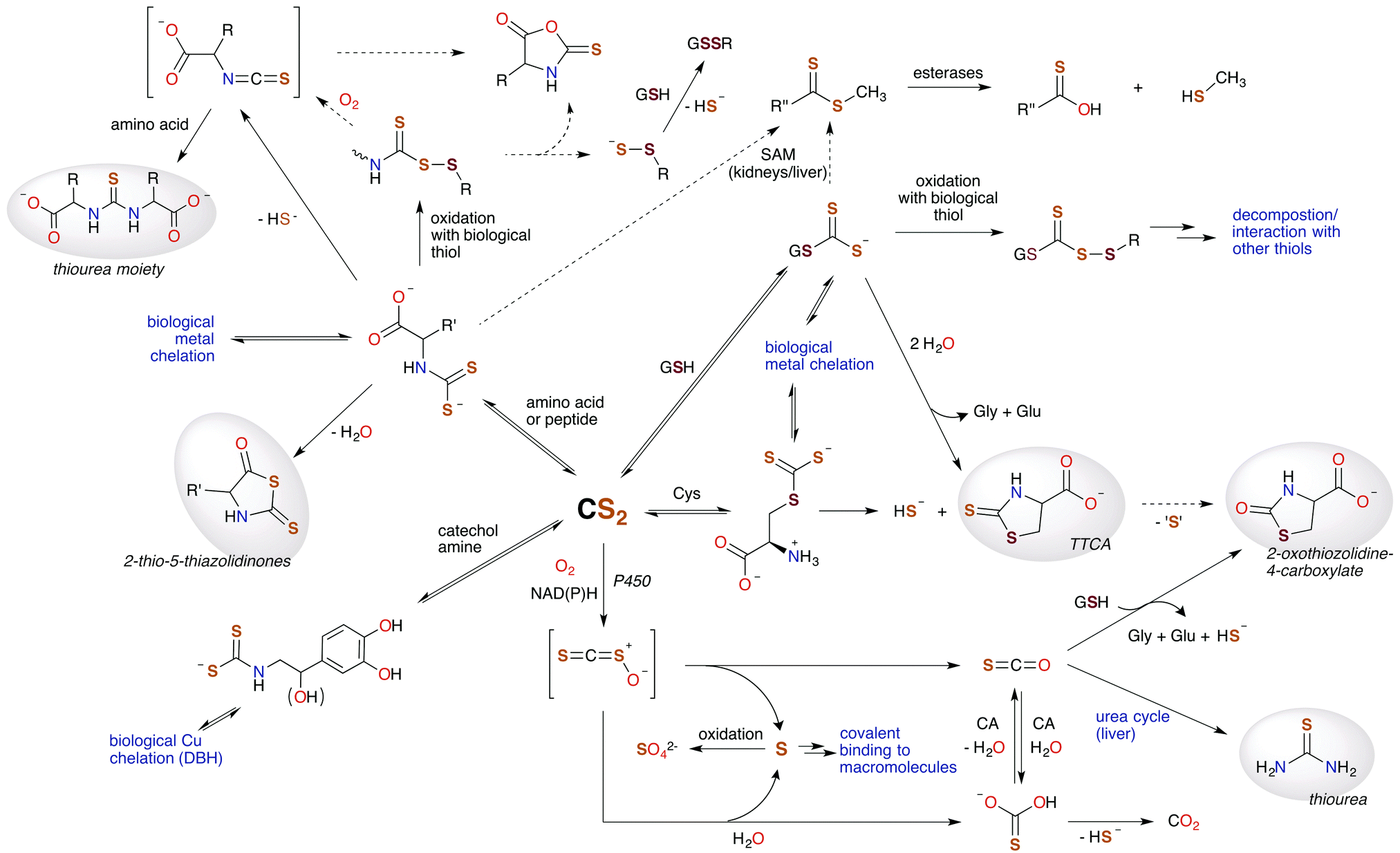 | ||
| Fig. 9 Hypothetical metabolic map for CS2. Species highlighted in grey are known human urinary metabolites of CS2.30,90,171–175 The signaling chemistry is proposed to be through CS2 reactions with biological nucleophiles. Dotted arrows represent possible, albeit minor, pathways. H2S (HS− under physiological conditions) is produced at several sites, suggesting an interplay between CS2 and this SMB. (SAM = S-adenosyl methionine methyltransferases, CA = carbonic anhydrases, P450 = cytochrome P450 monooxygenase, DBH = dopamine β-hydroxylase, TTCA = thiazolidine-2-thione-4-carboxylate.) | ||
Major advances in the investigation of the bioregulatory, therapeutic, etc. roles of SMBs involved the development of strategies for controlled delivery. For example, researchers into the chemical biology of NO have long drawn on the availability of water-soluble salts of various “NONOate” anions, R2NN(O)NO−, that allow for in vitro and in vivo NO generation with well-defined half-lives ranging from minutes to hours.208 Ultimately, analogous donors with predictable kinetics for CS2 release under physiological conditions will need to be utilized to evince CS2 interactions with specific biological targets. In this context, one can envision designing DTC or TTC compounds (or their metal complexes) that, given their reversible equilibria with amines or thiols, would release controlled quantities of CS2 over time to in vitro or in vivo targets. An alternative strategy would be to use photochemical methods to uncage CS2 as is being explored extensively with NO and CO.209 The advantage of the latter strategy would be the ability to control location, timing, and dosage of such release.
The observations discussed herein by no means establish an unambiguous case for the bioregulatory or therapeutic roles of carbon disulfide. But they do point to the exciting potential represented by the chemical biology of CS2 and the relatively unexplored research challenges and opportunities presented by these possibilities.
Author contributions
AWD, DFZ, and PCF each contributed to the preparation and editing of the full manuscript. AWD prepared the initial draft and all the figures and PCF completed the final draft. JMF critically read the manuscript and contributed Section 3.4.5.Financial interest
None of the authors have competing financial interests relevant to the contents of this article.Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| ALDH | Acetaldehyde dehydrogenase |
| AMO | Ammonium monooxygenase |
| CA | Carbonic anhydrase |
| cGMP | Cyclic guanosine monophosphate |
| CNS | Central nervous system |
| CO | Carbon monoxide |
| CO2 | Carbon dioxide |
| CoA | Coenzyme A |
| CODH | Carbon monoxide dehydrogenase |
| COS | Carbonyl sulfide |
| CS2 | Carbon disulfide |
| Cys | Cysteine |
| DBH | Dopamine β-hydroxylase |
| DEDTC | N,N-Diethyldithiocarbamate |
| DTC | Dithiocarbamate |
| GC-D | Guanylyl cyclase, isoform D |
| Glu | L-Glutamic acid |
| Gly | Glycine |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione dimer |
| H2S | Hydrogen sulfide |
| HIV | Human immunodeficiency virus |
| I-κB | Inhibitor NF-κB |
| IKK (α,β) | I-κB kinase (α,β-subunits) |
| IL-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide synthase |
| Lys | L-lysine |
| MMO | Methane monooxygenase |
| NAD(P)H | Reduced nicotinamide adenine dinucleotide (phosphate) |
| NEMO | NF-κB essential modulator (also IKKγ) |
| NF-κB | Nuclear factor κB |
| NO | Nitric oxide |
| P450 | Cytochrome P450 |
| SAM | S-Adenosyl methionine transmethylase |
| Ser | L-Serine |
| sGC | Soluble guanylyl cyclase |
| SMB | Small molecule bioregulator |
| TPCK | N-Tosyl-L-phenylalanine chloromethyl ketone |
| TTC | Trithiocarbonate |
| TTCA | Thiazolidine-2-thione-4-carboxylic acid |
Acknowledgements
We thank Drs Maykon Lima Souza and Mary E. Howe-Grant for their careful reading of the manuscript and suggestions. Studies in the Ford laboratory on SMBs have long been supported by the Chemistry Division of the National Science Foundation.References
- J. M. Fukuto, S. J. Carrington, D. J. Tantillo, J. G. Harrison, L. J. Ignarro, B. A. Freeman, A. Chen and D. A. Wink, Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species, Chem. Res. Toxicol., 2012, 25, 769–793 CrossRef CAS PubMed.
- E. Buys and P. Sips, New insights into the role of soluble guanylate cyclase in blood pressure regulation, Curr. Opin. Nephrol. Hypertens., 2014, 23, 135–142 CrossRef CAS PubMed.
- C. Szabo, Gasotransmitters in cancer: from pathophysiology to experimental therapy, Nat. Rev. Drug Discovery, 2016, 15, 185–203 CrossRef CAS PubMed.
- S. W. Ryter and A. M. K. Choi, Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation, Transl. Res., 2016, 167, 7–34 CrossRef CAS PubMed.
- K. R. Olson, The therapeutic potential of hydrogen sulfide: separating hype from hope, Am. J. Physiol.: Regul., Integr. Comp. Physiol., 2011, 301, R297–R312 CrossRef CAS PubMed.
- I. Andreadou, E. K. Iliodromitis, T. Rassaf, R. Schulz, A. Papapetropoulos and P. Ferdinandy, The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning, Br. J. Pharmacol., 2015, 172, 1587–1606 CrossRef CAS PubMed.
- Nitric Oxide: Biology and Pathobiology, ed. L. J. Ignarro, Elsevier Inc., Burlington MA, 2nd edn, 2010 Search PubMed.
- S. H. Heinemann, T. Hoshi, M. Westerhausen and S. Schiller, Carbon monoxide – physiology, detection and controlled release, Chem. Commun., 2014, 50, 3644–3660 RSC.
- D. A. Wink, L. A. Ridnour, S. P. Hussain and C. C. Harris, The reemergence of nitric oxide and cancer, Nitric Oxide, 2008, 19, 65–67 CrossRef CAS PubMed.
- D. Wu, W. Si, M. Wang, S. Lv, A. Ji and Y. Li, Hydrogen sulfide in cancer: friend or foe?, Nitric Oxide, 2015, 50, 38–45 CrossRef CAS PubMed.
- J. Muntané and B. Bonavida, Special collection: nitric oxide in cancer, Redox Biol., 2015, 6, 505–506 CrossRef PubMed.
- J. R. Lancaster Jr., A Tutorial on the diffusibility and reactivity of free nitric oxide, Nitric Oxide, 1997, 1, 18–30 CrossRef CAS PubMed.
- C. M. Bernt, P. T. Burks, A. W. DeMartino, A. E. Pierri, E. S. Levy, D. F. Zigler and P. C. Ford, Photocatalytic carbon disulfide production via charge transfer quenching of quantum dots, J. Am. Chem. Soc., 2014, 136, 2192–2195 CrossRef CAS PubMed.
- R. W. McKee, Solubility of carbon disulfide vapor in body fluids and tissues, J. Ind. Hyg. Toxicol., 1941, 23, 484–489 CAS.
- G. L. Gutsev and R. J. Bartlett, Electron affinities of CO2, OCS, and CS2, J. Chem. Phys., 1998, 108, 6756–6762 CrossRef CAS.
- R. Sander, Compilation of Henry's Law constants (version 4) for water as solvent, Atmos. Chem. Phys., 2015, 15, 4399–4981 CAS.
- S. F. Watts, The mass budgets of carbonyl sulfide, dimethyl sulfide, carbon disulfide and hydrogen sulfide, Atmos. Environ., 2000, 34, 761–779 CrossRef CAS.
- N. Rojo, G. Gallastegi, A. Barona, L. Gurtubay, G. Ibarra-Berastegi and A. Elías, Biotechnology as an alternative for carbon disulfide treatment in air pollution control, Environ. Rev., 2010, 18, 321–332 CrossRef CAS.
- WHO Concise International Chemical Assessment Document (CICAD) 46: Carbon Disulfide, World Health Organization, United Nations Environment Program, International Labor Organization, 2002.
- H. P. Gelbke, T. Göen, M. Mäurer and S. I. Sulsky, A review of health effects of carbon disulfide in viscose industry and a proposal for an occupational exposure limit, Crit. Rev. Toxicol., 2009, 39, 1–126 CrossRef CAS PubMed.
- A. Delpech, Note sur les accidents que dévelope, chez les ouvriers en caoutchouc, l'inhalation du sulfure de carbone en vapeur, Académie Impériale de Médecine, 1856, vol. 21, p. 350 Search PubMed.
- S. G. Selevan, R. Hornung, J. Fajen and C. Cottrill, Paternal exposure to carbon disulfide and spouse's pregnancy experience, Department of Health and Human Services, Centers for Disease Control, NIOSH, Hazard Evaluations and Field Studies, Cincinnati OH, 1983 Search PubMed.
- A. M. Cirla, P. A. Bertazzi, M. Tomasini, A. Villa, C. Graziano, R. Invernizzi and R. Gilioli, Study of endocrinological functions and sexual behavior in carbon disulfide workers, Med. Lav., 1978, 69, 118–129 CAS.
- G. Wagar, M. Tolonen and E. Helpio, Endocrinological studies of Finnish viscose rayon workers exposed to carbon disulfide, G. Ital. Med. Lav., 1981, 3, 103–105 CAS.
- S. Y. Zhou, Y. X. Liang, Z. Q. Chen and Y. L. Wang, Effects of occupational exposure to low-level carbon disulfide (CS2) on menstruation and pregnancy, Ind. Health, 1988, 26, 203–214 CrossRef CAS PubMed.
- J. Y. Ma, J. J. Ji, Q. Ding, W. D. Liu, S. Q. Wang, N. Wang and G. Y. Chen, The effects of carbon disulfide on male sexual function and semen quality, Toxicol. Ind. Health, 2010, 26, 375–382 CrossRef CAS PubMed.
- D. G. Graham, V. Amarnath, W. M. Valentine, S. J. Pyle and D. C. Anthony, Pathogenetic studies of hexane and carbon disulfide neurotoxicity, Crit. Rev. Toxicol., 1995, 25, 91–112 CrossRef CAS PubMed.
- A. E. Cohen, L. D. Scheel, J. F. Kopp, F. R. Stockell, R. G. Keenan, J. T. Mountain and H. J. Paulus, Biochemical mechanisms in chronic carbon disulfide poisoning, Am. Ind. Hyg. Assoc. J., 1959, 20, 303–323 CrossRef CAS PubMed.
- B. Soucek, Transformations of carbon disulfide in organisms, J. Hyg., Epidemiol., Microbiol., Immunol., 1957, 1, 10–22 CAS.
- R. O. Beauchamp, J. S. Bus, J. A. Popp, C. J. Boreiko, L. Goldberg and M. J. McKenna, A critical review of the literature on carbon disulfide toxicity, Crit. Rev. Toxicol., 1983, 11, 169–278 CrossRef CAS PubMed.
- M. Davidson and M. Feinleib, Carbon disulfide poisoning: A review, Am. Heart J., 1972, 83, 100–114 CrossRef CAS PubMed.
- H. Drexler, T. Göen, J. Angerer, S. Abou-el-ela and G. Lehnert, Carbon disulphide, Int. Arch. Occup. Environ. Health, 1994, 65, 359–365 CrossRef CAS PubMed.
- F. Song, S. Yu, X. Zhau, C. Zhang and K. Xie, Carbon disulfide-induced changes in cytoskeleton protein content of rat cerebral cortex, Neurochem. Res., 2006, 31, 71–79 CrossRef CAS PubMed.
- S. Krstev, B. Perunicic, B. Farkic and R. Banicevic, Neuropsychiatric effects in workers with occupational exposure to carbon disulfide, J. Occup. Health, 2003, 45, 81–87 CrossRef CAS PubMed.
- T. Takebayashi, Y. Nishiwaki, T. Nomiyama, T. Uemura, T. Yamauchi, S. Tanaka, H. Sakurai and K. Omae, Lack of relationship between occupational exposure to carbon disulfide and endocrine dysfunction: a six-year cohort study of the Japanese rayon workers, J. Occup. Health, 2003, 45, 111–118 CrossRef CAS PubMed.
- C. Meyer, Semen quality in workers exposed to carbon disulfide compared to a control group from the same plant, J. Occup. Med., 1981, 23, 435–439 CrossRef CAS.
- A. M. Cirla and C. Graziano, Health impairment in viscose-rayon workers with carbon disulfide risk below 30 mg m−3. An exposed-controls study, G. Ital. Med. Lav., 1981, 3, 69–73 CAS.
- J. Domergue, D. Lison and V. Haufroid, No evidence of cardiovascular toxicity in workers exposed below 5 ppm carbon disulfide, Int. Arch. Occup. Environ. Health, 2016, 89, 835–845 CrossRef CAS PubMed.
- A. Schramm, W. Uter, M. Brandt, T. Goen, M. Kohrmann, T. Baumeister and H. Drexler, Increased intima-media thickness in rayon workers after long-term exposure to carbon disulfide, Int. Arch. Occup. Environ. Health, 2016, 89, 513–519 CrossRef CAS PubMed.
- T. O. Peyton, R. V. Steel and W. R. Mabey, Carbon disulfide, carbonyl sulfide: literature review and environmental assessment, Stanford Res. Inst., 1976 Search PubMed.
- S. R. Rao, Xanthates and Related Compounds, Marcel Dekker, Inc., New York City NY, 1971 Search PubMed.
- W. Zeise, Vorleufige Anzeige einer neuen Klasse von Schwefelverbindungen, J. Chem. Phys., 1822, 35, 175–182 Search PubMed.
- S. R. Rao and C. C. Patel, Kinetics of decomposition of xanthates, J. Mines, Met. Fuels, 1960, 8, 9–14 CAS.
- E. Humeres, N. A. Debacher, M. M. de S. Sierra, J. D. Franco and A. Schutz, Mechanisms of acid of dithiocarbamates. 1. Alkyl dithiocarbamates, J. Org. Chem., 1998, 63, 1598–1603 CrossRef CAS.
- E. Humeres, N. A. Debacher and M. M. de S. Sierra, Mechanisms of acid decomposition of dithiocarbamates. 2. Efficiency of the intramolecular general acid catalysis, J. Org. Chem., 1999, 64, 1807–1813 CrossRef CAS PubMed.
- E. Humeres, B. Sun Lee and N. A. Debacher, Mechanisms of acid decomposition of dithiocarbamates. 5. Piperidyl dithiocarbamate and analogues, J. Org. Chem., 2008, 73, 7189–7196 CrossRef CAS PubMed.
- D. De Filippo, P. Deplano, F. Devillanova, E. F. Trogu and G. Verani, Inductive effect in dithiocarbamate decomposition mechanism, J. Org. Chem., 1973, 38, 560–563 CrossRef CAS.
- W. O. Foye, J. R. Marshall and J. Mickles, Structure of the carbon disulfide adduct of β-mercaptoethylamine, J. Pharm. Sci., 1963, 52, 406–407 CrossRef CAS PubMed.
- T. Weiss, J. Hardt and J. Angerer, Determination of urinary 2-thiazolidinethione-4-carboxylic acid after exposure to alkylene bisdithiocarbamates using gas chromatography–mass spectrometry, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 726, 85–94 CrossRef CAS.
- B. Holmberg, Über Thiocarbonate, J. Prakt. Chem., 1906, 73, 239–248 CrossRef CAS.
- K. K. Pandey, Reactivities of carbonyl sulfide (COS), carbon disulfide (CS2) and carbon dioxide (CO2) with transition metal complexes, Coord. Chem. Rev., 1995, 140, 37–114 CrossRef CAS.
- J. A. Ibers, Centenary Lecture. Reactivities of carbon disulphide, carbon dioxide, and carbonyl sulphide towards some transition-metal systems, Chem. Soc. Rev., 1982, 11, 57–73 RSC.
- M. M. Doeff, Reaction of matrix-isolated iron atoms with carbon disulfide, Inorg. Chem., 1986, 25, 2474–2476 CrossRef CAS.
- H. Werner, Novel Coordination Compounds Formed from CS2 and heteroallenes, Coord. Chem. Rev., 1982, 43, 165–185 CrossRef CAS.
- A. Bheemaraju, J. W. Beattie, R. L. Lord, P. D. Martin and S. Groysman, Carbon disulfide binding at dinuclear and mononuclear nickel complexes ligated by a redox-active ligand: iminopyridine serving as an accumulator of redox equivalents for the activation of heteroallenes, Chem. Commun., 2012, 48, 9595–9597 RSC.
- T. K. Muraoka, A. P. Duarte, P. M. Takahashi, V. M. Nogueira, A. V. G. Netto and A. E. Mauro, Reactivity of Fe(CO)2(η2-CS2)(PPh3)2 towards copper(I) compounds: synthesis of binuclear heterometallic species having CS2 bridging Fe(0) and Cu(I), Eclética Quím., 2007, 32, 7–11 CrossRef CAS.
- K. Tang, X. Jin and Y. Tang, Insertion of CS2 into M–S bonds and its Application in Synthesis of Clusters, Rev. Heteroat. Chem., 1996, 15, 83–114 CAS.
- I. K. Bezougli, A. Bashall, M. McPartlin and D. M. P. Mingos, Insertion of CS2 into the Group 2 metal-alkoxide bonds of [{M(OR)2}n] (M = Mg, Ca, Sr, or Ba; R = Et or Pri), J. Chem. Soc., Dalton Trans., 1998, 2671–2678 RSC.
- K. H. Yih, S. C. Chen, Y. C. Lin, G. H. Lee and Y. Wang, New syntheses of [Et4N][(C5H10NCS2)M(CO)4] (M = Cr, Mo and W); Insertion reaction of carbon disulfide into the metal-nitrogen bond, J. Organomet. Chem., 1995, 494, 149–155 CrossRef CAS.
- N. J. Hartmann, G. Wu and T. W. Hayton, Activation of CS2 by a masked terminal nickel sulfide, J. Chem. Soc., Dalton Trans., 2016 10.1039/C6DT00885B.
- K. B. Ghiassi, D. T. Walters, M. M. Aristov, N. D. Loewen, L. A. Berben, M. Rivera, M. M. Olmstead and A. L. Balch, Formation of a Stable Complex, RuCl2(S2CPPh3)(PPh3)2, containing an unstable zwitterion from the reaction of RuCl2(PPh3)3 with carbon disulfide, Inorg. Chem., 2015, 54, 4565–4573 CrossRef CAS PubMed.
- A. C. Lane, M. V. Vollmer, C. H. Laber, D. Y. Melgarejo, G. M. Chiarella, J. P. Fackler, X. Yang, G. A. Baker and J. R. Walensky, Multinuclear copper(I) and silver(I) amidinate complexes: synthesis, luminescence, and CS2 insertion reactivity, Inorg. Chem., 2014, 53, 11357–11366 CrossRef CAS PubMed.
- B. J. McCormick and R. I. Kaplan, Reaction of carbon disulfide with nickel(II)–amine complexes, Can. J. Chem., 1970, 48, 1876–1880 CrossRef.
- J. M. Andrews, D. Coucouvanis and J. P. Fackler, Thioxanthate complexes. Carbon disulfide eliminations to form bridged mercaptide dimers of nickel(II) and palladium(II), Inorg. Chem., 1972, 11, 493–499 CrossRef CAS.
- A. H. Ewald and E. Sinn, Magnetically anomalous thio complexes of iron(III) and nickel(II), Aust. J. Chem., 1968, 21, 927–938 CrossRef CAS.
- D. Coucouvanis, S. J. Lippard and J. A. Zubieta, Sulfur-bridged dimeric complexes of iron(III), J. Am. Chem. Soc., 1970, 92, 3342–3347 CrossRef CAS.
- D. Coucouvanis, S. J. Lippard and J. A. Zubieta, Dimeric iron(III) complex containing two ethyl mercaptide and two ethyl thioxanthate groups as bridging ligands, J. Am. Chem. Soc., 1969, 91, 761–762 CrossRef CAS.
- H. E. Mabrouk and D. G. Tuck, The direct electrochemical synthesis of zinc and cadmium derivatives of α,ω-alkanedithiols and their reaction with carbon disulphide, Inorg. Chim. Acta, 1988, 145, 237–241 CrossRef CAS.
- A. Avdeef and J. P. Fackler Jr., Bis(triphenylphosphine)ethyltrithiocarbonatocopper(I), J. Coord. Chem., 1975, 4, 211–215 CrossRef CAS.
- S. M. Schmitt, M. Frezza and Q. P. Dou, New applications of old metal-binding drugs in the treatment of human cancer, Front. Biosci., Scholar Ed., 2012, S4, 375–391 CrossRef CAS.
- E. R. T. Tiekink and I. Haiduc, Stereochemical aspects of metal xanthate complexes: molecular structures and supramolecular self-assembly, Progress in Inorganic Chemistry, John Wiley & Sons, Inc., 2005, pp. 127–319 Search PubMed.
- E. R. T. Tiekink and J. Zukerman-Schpector, Stereochemical activity of lone pairs of electrons and supramolecular aggregation patterns based on secondary interactions involving tellurium in its 1,1-dithiolate structures, Coord. Chem. Rev., 2010, 254, 46–76 CrossRef CAS.
- J. N. Verpeaux, M. H. Desbois, A. Madonik, C. Amatore and D. Astruc, Electron-transfer-catalyzed chelation of dithiocarbamate iron complexes [Fe(η5-C5R5)(η1SC(S)NMe2)(CO)2] (R = H, Me) induced by oxidation, Organometallics, 1990, 9, 630–640 CrossRef CAS.
- C. T. Supuran, Carbonic anhydrases: catalytic and inhibition mechanisms, distribution and physiological roles, Carbonic Anhydrase, CRC Press, 2004, 1, pp. 1–23 Search PubMed.
- V. S. Haritos and G. Dojchinov, Carbonic anhydrase metabolism is a key factor in the toxicity of CO2 and COS but not CS2 toward the flour beetle Tribolium castaneum [Coleoptera: Tenebrionidae], Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2005, 140, 139–147 CrossRef PubMed.
- S. Sinnecker, M. Bräuer, W. Koch and E. Anders, CS2 Fixation by carbonic anhydrase model systems: A new substrate in the catalytic cycle, Inorg. Chem., 2001, 40, 1006–1013 CrossRef CAS PubMed.
- S. Schenk, J. Kesselmeier and E. Anders, How does the exchange of one oxygen atom with sulfur affect the catalytic cycle of carbonic anhydrase?, Chem. – Eur. J., 2004, 10, 3091–3105 CrossRef CAS PubMed.
- J. Notni, S. Schenk, G. Protoschill-Krebs, J. Kesselmeier and E. Anders, The missing link in COS metabolism: a model study on the reactivation of carbonic anhydrase from its hydrosulfide analogue, ChemBioChem, 2007, 8, 530–536 CrossRef CAS PubMed.
- F. Carta, A. Akdemir, A. Scozzafava, E. Masini and C. T. Supuran, Xanthates and trithiocarbonates strongly inhibit carbonic anhydrases and show antiglaucoma effects in vivo, J. Med. Chem., 2013, 56, 4691–4700 CrossRef CAS PubMed.
- M. J. Smeulders, T. R. M. Barends, A. Pol, A. Scherer, M. H. Zandvoort, A. Udvarhelyi, A. F. Khadem, A. Menzel, J. Hermans, R. L. Shoeman, H. J. C. T. Wessels, L. P. van den Heuvel, L. Russ, I. Schlichting, M. S. M. Jetten and H. J. M. Op den Camp, Evolution of a new enzyme for carbon disulphide conversion by an acidothermophilic archaeon, Nature, 2011, 478, 412–416 CrossRef CAS PubMed.
- S. Alcántara, I. Estrada, M. S. Vásquez and S. Revah, Carbon disulfide oxidation by a microbial consortium from a trickling filter, Biotechnol. Lett., 1999, 21, 815–819 CrossRef.
- N. A. Smith and D. P. Kelly, Oxidation of carbon disulfide as the sole source of energy for the autotrophic growth of Thiobacillus thioparus strain TK-m, J. Gen. Microbiol., 1988, 134, 3041–3048 CAS.
- R. Warington, IV. On nitrification, J. Chem. Soc., Trans., 1878, 33, 44–51 RSC.
- L. C. Seefeldt, M. E. Rasche and S. A. Ensign, Carbonyl sulfide and carbon dioxide as new substrates, and carbon disulfide as a new inhibitor of nitrogenase, Biochemistry, 1995, 34, 5382–5389 CrossRef CAS PubMed.
- M. J. Smeulders, A. Pol, H. Venselaar, T. R. M. Barends, J. Hermans, M. S. M. Jetten and H. J. M. Op den Camp, Bacterial CS2 hydrolases from Acidithiobacillus thiooxidans strains are homologous to the archaeal catenane CS2 hydrolase, J. Bacteriol., 2013, 195, 4046–4056 CrossRef CAS PubMed.
- M. R. Hyman, C. Y. Kim and D. J. Arp, Inhibition of ammonia monooxygenase in Nitrosomonas europaea by carbon disulfide, J. Bacteriol., 1990, 172, 4775–4782 CAS.
- M. Kumar, W. P. Lu and S. W. Ragsdale, Binding of carbon disulfide to the site of acetyl-CoA synthesis by the nickel-iron-sulfur protein, carbon monoxide dehydrogenase, from Clostridium thermoaceticum, Biochemistry, 1994, 33, 9769–9777 CrossRef CAS PubMed.
- S. W. Ragsdale and H. G. Wood, Acetate biosynthesis by acetogenic bacteria. Evidence that carbon monoxide dehydrogenase is the condensing enzyme that catalyzes the final steps of the synthesis, J. Biol. Chem., 1985, 260, 3970–3977 CAS.
- R. van Doorn, P. T. Henderson, M. Vanhoorne, P. G. Vertin and C. P. M. J. M. Leijdekkers, Determination of thio compounds in urine of workers exposed to carbon disulfide, Arch. Environ. Health Int. J., 1981, 36, 289–297 CrossRef CAS.
- M. Silva, A review of developmental and reproductive toxicity of CS2 and H2S generated by the pesticide sodium tetrathiocarbonate, Birth Defects Res., Part B, 2013, 98, 119–138 CrossRef CAS PubMed.
- M. J. McKenna and V. DiStefano, Carbon disulfide. I. The metabolism of inhaled carbon disulfide in the rat, J. Pharmacol. Exp. Ther., 1977, 202, 245–252 CAS.
- H. Kivistö, E. Elovaara, V. Riihimäki and A. Aitio, Effect of cytochrome P450 isozyme induction and glutathione depletion on the metabolism of CS2 to TTCA in rats, Arch. Toxicol., 1995, 69, 185–190 CrossRef.
- C. V. Smith, D. P. Jones, T. M. Guenthner, L. H. Lash and B. H. Lauterburg, Compartmentation of glutathione: Implications for the study of toxicity and disease, Toxicol. Appl. Pharmacol., 1996, 140, 1–12 CrossRef CAS PubMed.
- D. P. Jones, J. L. Carlson, P. S. Samiec, P. Sternberg Jr., V. C. Mody Jr., R. L. Reed and L. A. S. Brown, Glutathione measurement in human plasma; evaluation of sample collection, storage and derivatization conditions for analysis of dansyl derivatives by HPLC, Clin. Chim. Acta, 1998, 275, 175–184 CrossRef CAS.
- R. Rossi, D. Giustarini, A. Milzani and I. Dalle-Donne, Cysteinylation and homocysteinylation of plasma protein thiols during ageing of healthy human beings, J. Cell. Mol. Med., 2009, 9B, 3131–3140 CrossRef PubMed.
- S. S. Tang and G. G. Chang, Kinetic characterization of the endogenous glutathione transferase activity of octopus lens S-crystallin, J. Biochem., 1996, 119, 1182–1188 CrossRef CAS PubMed.
- N. Suwandaratne, J. Hu, K. Siriwardana, M. Gadogbe and D. Zhang, Evaluation of thiol raman activities and pKa values using internally referenced raman-based pH titration, Anal. Chem., 2016, 88, 3624–3631 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple, Free Radical Biol. Med., 2001, 30, 1191–1212 CrossRef CAS PubMed.
- A. K. Tummanapelli and S. Vasudevan, Ab initio MD simulations of the Brønsted acidity of glutathione in aqueous solutions: predicting pKa shifts of the cysteine residue, J. Phys. Chem. B, 2015, 119, 15353–15358 CrossRef CAS PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, Direct fetermination of thiol pKa by isothermal titration microcalorimetry, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS PubMed.
- Oxidative Stress and Redox Regulation, ed. U. Jakob and D. Reichmann, Springer, Netherlands, 2013 Search PubMed.
- J. S. Bus, The relationship of carbon disulfide metabolism to development of toxicity, Neurotoxicology, 1985, 6, 73–80 CAS.
- D. J. Johnson, D. G. Graham, V. Amarnath, K. Amarnath and W. M. Valentine, The measurement of 2-thiothiazolidine-4-carboxylic acid as an index of the in vivo release of CS2 by dithiocarbamates, Chem. Res. Toxicol., 1996, 9, 910–916 CrossRef CAS PubMed.
- A. Srivastava, S. K. Singh and A. Gupta, Determination of organotrithiocarbonates with o-diacetoxyiodobenzoate and N-chlorosuccinimide in aqueous and non-aqueous media, Analyst, 1990, 115, 421–423 RSC.
- J. E. Drake and J. Yang, Preparation and characterization of trimethylgermanium−sulfur compounds derived from 2-dithiocarbamoyl-3-dithiocarbonylthiopropionate. Crystal Structures of [NH4]3[S2CSCH2CH(NHCS2)CO2] and Me3GeO2C(−CHNHC(S)SCH2−), Inorg. Chem., 1998, 37, 2968–2974 CrossRef CAS.
- L. Magos and J. A. E. Jarvis, Effects of diethyldithiocarbamate and carbon disulphide on brain tyrosine, J. Pharm. Pharmacol., 1970, 22, 936–938 CrossRef CAS PubMed.
- M. J. McKenna and V. DiStefano, Carbon disulfide. II. A proposed mechanism for the action of carbon disulfide on dopamine β-hydroxylase, J. Pharmacol. Exp. Ther., 1977, 202, 253–266 CAS.
- R. J. Rubin, Role of metabolic activation in the renal toxicity of carbon disulfide (CS2), Toxicol. Lett., 1990, 53, 211–213 CrossRef CAS PubMed.
- R. F. M. Herber and H. Poppe, A new method for the estimation of exposure to carbon disulphide, J. Chromatogr. A, 1976, 118, 23–34 CrossRef CAS PubMed.
- R. E. Heikkila, F. S. Cabbat and G. Cohen, In vivo inhibition of superoxide dismutase in mice by diethyldithiocarbamate, J. Biol. Chem., 1976, 251, 2182–2185 CAS.
- M. Wiedau-Pazos, J. J. Goto, S. Rabizadeh, E. B. Gralla, J. A. Roe, M. K. Lee, J. S. Valentine and D. E. Bredesen, Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis, Science, 1996, 271, 515–518 CAS.
- M. Bozdag, F. Carta, D. Vullo, A. Akdemir, S. Isik, C. Lanzi, A. Scozzafava, E. Masini and C. T. Supuran, Synthesis of a new series of dithiocarbamates with effective human carbonic anhydrase inhibitory activity and antiglaucoma action, Bioorg. Med. Chem., 2015, 23, 2368–2376 CrossRef CAS PubMed.
- L. A. Shinobu, S. G. Jones and M. M. Jones, Mobilization of aged cadmium deposits by dithiocarbamates, Arch. Toxicol., 1983, 54, 235–242 CrossRef CAS PubMed.
- F. W. Sunderman, Chelation therapy in nickel poisoning, Ann. Clin. Lab. Sci., 1981, 11, 1–8 CAS.
- E. C. Reisinger, P. Kern, M. Dietrich, M. Ernst, H. D. Flad, P. Bock and German Dtc Study Group, Inhibition of HIV progression by dithiocarb, Lancet, 1990, 335, 679–682 CrossRef CAS.
- E. M. Hersh, G. Brewton, D. Abrams, J. Bartlett, J. Galpin, P. Gill, R. Gorter, M. Gottlieb, J. J. Jonikas, S. Landesman, A. Levine, A. Marcel, E. A. Petersen, M. Whiteside, J. Zahradnik, C. Negron, F. Boultitie, J. Caraux, J. M. Dupuy and R. Salmi, Ditiocarb sodium (diethyldithiocarbamate) therapy in patients with symptomatic HIV infection and AIDS: a randomized, double-blind, placebo-controlled, multicenter study, JAMA, J. Am. Med. Assoc., 1991, 265, 1538–1544 CrossRef CAS PubMed.
- V. Bala, S. Jangir, D. Mandalapu, S. Gupta, Y. S. Chhonker, N. Lal, B. Kushwaha, H. Chandasana, S. Krishna, K. Rawat, J. P. Maikhuri, R. S. Bhatta, M. I. Siddiqi, R. Tripathi, G. Gupta and V. L. Sharma, Dithiocarbamate–thiourea hybrids useful as vaginal microbicides also show reverse transcriptase inhibition: design, synthesis, docking and pharmacokinetic studies, Bioorg. Med. Chem. Lett., 2015, 25, 881–886 CrossRef CAS PubMed.
- H. Mansour, M. Levacher, M. A. Gougerot-Pocidalo, B. Rouveix and J. J. Pocidalo, Diethyldithiocarbamate provides partial protection against pulmonary and lymphoid oxygen toxicity, J. Pharmacol. Exp. Ther., 1986, 236, 476–480 CAS.
- S. Orrenius, C. S. I. Nobel, D. J. van den Dobbelsteen, M. J. Burkitt and A. F. G. Slater, Dithiocarbamates and the redox regulation of cell death, Biochem. Soc. Trans., 1996, 24, 1032–1038 CrossRef CAS PubMed.
- G. Vettorazzi, W. F. Almeida, G. J. Burin, R. B. Jaeger, F. R. Puga, A. F. Rahde, F. G. Reyes and S. Schvartsman, International safety assessment of pesticides: dithiocarbamate pesticides, ETU, and PTU – A review and update, Teratog., Carcinog., Mutagen., 1995, 15, 313–337 CrossRef CAS.
- C. Mathieu, R. Duval, X. Xu, F. Rodrigues-Lima and J. M. Dupret, Effects of pesticide chemicals on the activity of metabolic enzymes: focus on thiocarbamates, Expert Opin. Drug Metab. Toxicol., 2014, 11, 1–14 Search PubMed.
- A. Moretto and C. Colosio, Biochemical and toxicological evidence of neurological effects of pesticides: the example of Parkinson's disease, NeuroToxicology, 2011, 32, 383–391 CrossRef CAS PubMed.
- T. P. Brown, P. C. Rumsby, A. C. Capleton, L. Rushton and L. S. Levy, Pesticides and parkinson's disease-is there a link?, Environ. Health Perspect., 2006, 114, 156–164 CrossRef PubMed.
- Y. Zhou, F. S. Shie, P. Piccardo, T. J. Montine and J. Zhang, Proteasomal inhibition induced by manganese ethylene-bis-dithiocarbamate: relevance to Parkinson's disease, Neuroscience, 2004, 128, 281–291 CrossRef CAS PubMed.
- N. Edington and J. M. Howell, Changes in the nervous system of rabbits following the administration of sodium diethyldithiocarbamate, Nature, 1966, 210, 1060–1062 CrossRef CAS PubMed.
- A. R. Rasul and J. M. Howell, Further observations on the response of the peripheral and central nervous system of the rabbit to sodium diethyldithiocarbamate, Acta Neuropathol., 1973, 24, 161–173 CrossRef CAS PubMed.
- G. B. Frisoni and V. Monda, Disulfiram neuropathy: a review (1971–1988) and report of a case, Alcohol Alcohol., 1989, 24, 429–437 CAS.
- B. Mokri, A. Ohnishi and P. J. Dyck, Disulfiram neuropathy, Neurology, 1981, 31, 730 CrossRef CAS PubMed.
- W. M. Valentine, V. Amarnath, K. Amarnath, F. Rimmele and D. G. Graham, Carbon disulfide mediated protein crosslinking by N,N-diethyldithiocarbamate, Chem. Res. Toxicol., 1995, 8, 96–102 CrossRef CAS PubMed.
- E. G. Tonkin, J. C. L. Erve and W. M. Valentine, Disulfiram produces a non-carbon disulfide-dependent schwannopathy in the rat, J. Neuropathol. Exp. Neurol., 2000, 59, 786–797 CrossRef CAS PubMed.
- D. I. Eneanya, J. R. Bianchine, D. O. Duran and B. D. Andresen, The actions and metabolic fate of disulfiram, Annu. Rev. Pharmacol. Toxicol., 1981, 21, 575–596 CrossRef CAS PubMed.
- B. Cvek and Z. Dvorak, Targeting of nuclear factor-κB and proteasome by dithiocarbamate complexes with metals, Curr. Pharm. Des., 2007, 13, 3155–3167 CrossRef CAS PubMed.
- P. I. Nagy, G. Alagona, C. Ghio and K. Takács-Novák, Theoretical conformational analysis for neurotransmitters in the gas phase and in aqueous solution. Norepinephrine, J. Am. Chem. Soc., 2003, 125, 2770–2785 CrossRef CAS PubMed.
- R. Brisch, A. Saniotis, R. Wolf, H. Bielau, H. G. Bernstein, J. Steiner, B. Bogerts, K. Braun, Z. Jankowski, J. Kumaratilake, M. Henneberg and T. Gos, The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue, Front. Mol. Psychiatry, 2014, 5 DOI:10.3389/fpsyt.2014.00047.
- F. L. O'Connor, The role of serotonin and dopamine in schizophrenia, J. Am. Psychiatr. Nurses Assoc., 1998, 4, S30–S34 CrossRef.
- K. A. Veverka, K. L. Johnson, D. C. Mays, J. J. Lipsky and S. Naylor, Inhibition of aldehyde dehydrogenase by disulfiram and its metabolite methyl diethylthiocarbamoyl-sulfoxide, Biochem. Pharmacol., 1997, 53, 511–518 CrossRef CAS PubMed.
- J. P. Schroeder, D. A. Cooper, J. R. Schank, M. A. Lyle, M. Gaval-Cruz, Y. E. Ogbonmwan, N. Pozdeyev, K. G. Freeman, P. M. Iuvone, G. L. Edwards, P. V. Holmes and D. Weinshenker, Disulfiram attenuates drug-primed reinstatement of cocaine seeking via inhibition of dopamine β-hydroxylase, Neuropsychopharmacology, 2010, 35, 2440–2449 CrossRef CAS PubMed.
- J. J. Lipsky, M. L. Shen and S. Naylor, In vivo inhibition of aldehyde dehydrogenase by disulfiram, Chem.-Biol. Interact., 2001, 130, 93–102 CrossRef PubMed.
- K. S. Barth and R. J. Malcolm, Disulfiram: an old therapeutic with new applications, CNS Neurol. Disord.: Drug Targets, 2010, 9, 5–12 CAS.
- L. Z. Lin and J. Lin, Antabuse (disulfiram) as an affordable and promising anticancer drug, Int. J. Cancer, 2011, 129, 1285–1286 CrossRef CAS PubMed.
- J. C. L. Erve, V. Amarnath, D. G. Graham, R. C. Sills, A. L. Morgan and W. M. Valentine, Carbon disulfide and N,N-diethyldithiocarbamate generate thiourea cross-links on erythrocyte spectrin in vivo, Chem. Res. Toxicol., 1998, 11, 544–549 CrossRef CAS PubMed.
- R. C. Sills, W. M. Valentine, V. Moser, D. G. Graham and D. L. Morgan, Characterization of carbon disulfide neurotoxicity in C57BL6 mice: behavioral, morphologic, and molecular effects, Toxicol. Pathol., 2000, 28, 142–148 CrossRef CAS PubMed.
- D. G. Isom, C. A. Castañeda, B. R. Cannon and E. B. García-Moreno, Large shifts in pKa values of lysine residues buried inside a protein, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 5260–5265 CrossRef CAS PubMed.
- A. P. DeCaprio, D. C. Spink, X. Chen, J. H. Fowke, M. Zhu and S. Bank, Characterization of isothiocyanates, thioureas, and other lysine adduction products in carbon disulfide-treated peptides and protein, Chem. Res. Toxicol., 1992, 5, 496–504 CrossRef CAS PubMed.
- R. Schreck, B. Meier, D. N. Männel, W. Dröge and P. A. Baeuerle, Dithiocarbamates as potent inhibitors of nuclear factor κB activation in intact cells, J. Exp. Med., 1992, 175, 1181–1194 CrossRef CAS PubMed.
- M. Karin and F. R. Greten, NF-κB: linking inflammation and immunity to cancer development and progression, Nat. Rev. Immunol., 2005, 5, 749–759 CrossRef CAS PubMed.
- X. Dolcet, D. Llobet, J. Pallares and X. Matias-Guiu, NF-κB in development and progression of human cancer, Virchows Arch., 2005, 446, 475–482 CrossRef CAS PubMed.
- J. Dutta, Y. Fan, N. Gupta, G. Fan and C. Gelinas, Current insights into the regulation of programmed cell death by NF-κB, Oncogene, 2006, 25, 6800–6816 CrossRef CAS PubMed.
- S. Beinke and S. C. Ley, Functions of NF-kB1 and NF-kB2 in immune cell biology, Biochem. J., 2004, 382, 393–409 CrossRef CAS PubMed.
- M. S. Hayden, A. P. West and S. Ghosh, NF-κB and the immune response, Oncogene, 2006, 25, 6758–6780 CrossRef CAS PubMed.
- D. S. Basseres and A. S. Baldwin, Nuclear factor-κB and inhibitor of κB kinase pathways in oncogenic initiation and progression, Oncogene, 2006, 25, 6817–6830 CrossRef CAS PubMed.
- T. D. Gilmore and M. Herscovitch, Inhibitors of NF-κB signaling: 785 and counting, Oncogene, 2006, 25, 6887–6899 CrossRef CAS PubMed.
- G. Bonizzi and M. Karin, The two NF-κB activation pathways and their role in innate and adaptive immunity, Trends Immunol., 2004, 25, 280–288 CrossRef CAS PubMed.
- H. L. Pahl, Activators and target genes of Rel/Nf-kB transcription factors, Oncogene, 1999, 18, 6853–6866 CrossRef CAS PubMed.
- C. Morais, B. Pat, G. Gobe, D. W. Johnson and H. Healy, Pyrrolidine dithiocarbamate exerts anti-proliferative and pro-apoptotic effects in renal cell carcinoma cell lines, Nephrol., Dial., Transplant., 2006, 21, 3377–3388 CrossRef CAS PubMed.
- S. Miyamoto and I. M. Verma, REL/NF-κB/IκB Story, Adv. Cancer Res., 1995, 66, 255–292 CrossRef CAS PubMed.
- M. J. May and S. Ghosh, Rel/NF-κB and IκB proteins: an overview, Semin. Cancer Biol., 1997, 8, 63–73 CrossRef CAS PubMed.
- P. A. Baeuerle, IκB–NF-κB structures: at the interface of inflammation control, Cell, 1998, 95, 729–731 CrossRef CAS PubMed.
- C. Scheidereit, IκB kinase complexes: gateways to NF-κB activation and transcription, Oncogene, 2006, 25, 6685–6705 CrossRef CAS PubMed.
- M. Hayakawa, H. Miyashita, I. Sakamoto, M. Kitagawa, H. Tanaka, H. Yasuda, M. Karin and K. Kikugawa, Evidence that reactive oxygen species do not mediate NF-κB activation, EMBO J., 2003, 22, 3356–3366 CrossRef CAS PubMed.
- K. H. Ha, M. S. Byun, J. Choi, J. Jeong, K. J. Lee and D. M. Jue, N-Tosyl-l-phenylalanine chloromethyl ketone inhibits NF-κB activation by blocking specific cysteine residues of IκB Kinase β and p65/RelA, Biochemistry, 2009, 48, 7271–7278 CrossRef CAS PubMed.
- H. Sebban, S. Yamaoka and G. Courtois, Posttranslational modifications of NEMO and its partners in NF-κB signaling, Trends Cell Biol., 2006, 16, 569–577 CrossRef CAS PubMed.
- N. L. Reynaert, A. van der Vliet, A. S. Guala, T. McGovern, M. Hristova, C. Pantano, N. H. Heintz, J. Heim, Y. S. Ho, D. E. Matthews, E. F. M. Wouters and Y. M. W. Janssen-Heininger, Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13086–13091 CrossRef CAS PubMed.
- M. S. Byun, J. Choi and D. M. Jue, Cysteine-179 of IκB kinase β plays a critical role in enzyme activation by promoting phosphorylation of activation loop serines, Exp. Mol. Med., 2006, 38, 546–552 CrossRef CAS PubMed.
- S. Liu, Y. R. Misquitta, A. Olland, M. A. Johnson, K. S. Kelleher, R. Kriz, L. L. Lin, M. Stahl and L. Mosyak, Crystal structure of a human IκB kinase β asymmetric dimer, J. Biol. Chem., 2013, 288, 22758–22767 CrossRef CAS PubMed.
- K. I. Jeon, M. S. Byun and D. M. Jue, Gold compound auranofin inhibits IκB kinase (IKK) by modifying Cys-179 of IKKβ subunit, Exp. Mol. Med., 2003, 35, 61–66 CrossRef CAS PubMed.
- K. Heyninck, M. Lahtela-Kakkonen, P. Van der Veken, G. Haegeman and W. Vanden Berghe, Withaferin A inhibits NF-κB activation by targeting cysteine 179 in IKKβ, Biochem. Pharmacol., 2014, 91, 501–509 CrossRef CAS PubMed.
- C. S. Lai, Methods for the controlled delivery of carbon disulfide for the treatment of inflammatory conditions, WO Pat. App., WO1999040907 A1, US, 1999 Search PubMed.
- F. De Matteis, Covalent binding of sulfur to microsomes and loss of cytochrome P-450 during the oxidative desulfuration of several chemicals, Mol. Pharmacol., 1974, 10, 849–854 CAS.
- F. De Matteis and A. A. Seawright, Liver toxicity of carbon disulfide. Possible significance of its metabolism by oxidative desulphuration, Panminerva Med., 1976, 18, 364–366 CAS.
- C. J. Decker, D. R. Doerge and J. R. Cashman, Metabolism of benzimidazoline-2-thiones by rat hepatic microsomes and hog liver flavin-containing monooxygenase, Chem. Res. Toxicol., 1992, 5, 726–733 CrossRef CAS PubMed.
- C. P. Chengelis and R. A. Neal, Hepatic carbonyl sulfide metabolism, Biochem. Biophys. Res. Commun., 1979, 90, 993–999 CrossRef CAS PubMed.
- R. A. Neal, Microsomal metabolism of thiono-sulfur compounds: mechanisms and toxicological significance, Rev. Biochem. Toxicol., 1980, 2, 131–171 CAS.
- J. Halpert and R. A. Neal, Inactivation of rat liver cytochrome P-450 by the suicide substrates parathion and chloramphenicol, Drug Metab. Rev., 1981, 12, 239–259 CrossRef CAS PubMed.
- Q. Li and J. R. Lancaster Jr., Chemical foundations of hydrogen sulfide biology, Nitric Oxide, 2013, 35, 21–34 CrossRef CAS PubMed.
- K. G. Reddie and K. S. Carroll, Expanding the functional diversity of proteins through cysteine oxidation, Curr. Opin. Chem. Biol., 2008, 12, 746–754 CrossRef CAS PubMed.
- T. Ida, T. Sawa, H. Ihara, Y. Tsuchiya, Y. Watanabe, Y. Kumagai, M. Suematsu, H. Motohashi, S. Fujii, T. Matsunaga, M. Yamamoto, K. Ono, N. O. Devarie-Baez, M. Xian, J. M. Fukuto and T. Akaike, Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling, Proc. Natl. Acad. Sci., U. S. A., 2014, 111, 7606–7611 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla and S. H. Snyder, Signaling by gasotransmitters, Sci. Signaling, 2009, 2(68) DOI:10.1126/scisignal.268re2.
- K. Ono, T. Akaike, T. Sawa, Y. Kumagai, D. A. Wink, D. J. Tantillo, A. J. Hobbs, P. Nagy, M. Xian, J. Lin and J. M. Fukuto, The redox chemistry and chemical biology of H2S, hydropersulfides and derived species: implications of their possible biological activity and utility, Free Radical Biol. Med., 2014, 77, 82–94 CrossRef CAS PubMed.
- S. S. Saund, V. Sosa, S. Henriquez, Q. N. N. Nguyen, C. L. Bianco, S. Soeda, R. Millikin, C. White, H. Le, D. J. Tantillo, Y. Kumagai, T. Akaike, J. Lin and J. M. Fukuto, The chemical biology of hydropersulfides (RSSH): chemical stability, reactivity and redox roles, Arch. Biochem. Biophys., 2015, 588, 15–24 CrossRef CAS PubMed.
- R. J. Hondal, S. M. Marino and V. N. Gladyshev, Selenocysteine in thiol/disulfide-like exchange reactions, Antioxid. Redox Signalling, 2013, 18, 1675–1689 CrossRef CAS PubMed.
- J. K. Wrobel, R. Power and M. Toboek, Biological activity of selenium: revisited, IUBMB Life, 2016, 68, 97–105 CrossRef CAS PubMed.
- Y. M. Go, J. D. Chandler and D. P. Jones, The cysteine proteome, Free Radical Biol. Med., 2015, 84, 227–245 CrossRef CAS PubMed.
- B. Buszewski, A. Ulanowska, T. Ligor, M. Jackowski, E. Kłodzińska and J. Szeliga, Identification of volatile organic compounds secreted from cancer tissues and bacterial cultures, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2008, 868, 88–94 CrossRef CAS PubMed.
- W. Miekisch, J. K. Schubert and G. F. Noeldge-Schomburg, Diagnostic potential of breath analysis – focus on volatile organic compounds, Clin. Chim. Acta, 2004, 347, 25–39 CrossRef CAS PubMed.
- M. Phillips, Detection of carbon disulfide in breath and air: a possible new risk factor for coronary artery disease, Int. Arch. Occup. Environ. Health, 1992, 64, 119–123 CrossRef CAS PubMed.
- M. Phillips, M. Sabas and J. Greenberg, Increased pentane and carbon disulfide in the breath of patients with schizophrenia, J. Clin. Pathol., 1993, 46, 861–864 CrossRef CAS PubMed.
- B. Vitali, M. Ndagijimana, F. Cruciani, P. Carnevali, M. Candela, M. E. Guerzoni and P. Brigidi, Impact of a symbiotic food on the gut microbial ecology and metabolic profiles, BMC Microbiol., 2010, 10(4) DOI:10.1186/1471-2180-10-4.
- B. Vitali, M. Ndagijimana, S. Maccaferri, E. Biagi, M. E. Guerzoni and P. Brigidi, An in vitro evaluation of the effect of probiotics and prebiotics on the metabolic profile of human microbiota, Anaerobe, 2012, 18, 386–391 CrossRef CAS PubMed.
- S. Studer, J. Orens, I. Rosas, J. Krishnan, K. Cope, S. Yang, J. Conte, P. Becker and T. Risby, Patterns and significance of exhaled-breath biomarkers in lung transplant recipients with acute allograft rejection, J. Heart Lung Transplant., 2001, 20, 1158–1166 CrossRef CAS PubMed.
- B. G. Galef Jr., J. R. Mason, G. Preti and N. J. Bean, Carbon disulfide: a semiochemical mediating socially-induced diet choice in rats, Physiol. Behav., 1988, 42, 119–124 CrossRef CAS.
- S. D. Munger, T. Leinders-Zufall, L. M. McDougall, R. E. Cockerham, A. Schmid, P. Wandernoth, G. Wennemuth, M. Biel, F. Zufall and K. R. Kelliher, An Olfactory Subsystem that Detects Carbon Disulfide and Mediates Food-Related Social Learning, Curr. Biol., 2010, 20, 1438–1444 CrossRef CAS PubMed.
- H. Arakawa, K. R. Kelliher, F. Zufall and S. D. Munger, The receptor guanylyl cyclase type D (GC-D) ligand uroguanylin promotes the acquisition of food preferences in mice, Chem. Senses, 2013, 38, 391–397 CrossRef CAS PubMed.
- World Health Organization, Carbon disulfide, Air Quality Guidelines, WHO Regional Office for Europe, Copenhagen, Denmark, 2nd edn, 2000, ch. 5.4 Search PubMed.
- T. Göen, A. Schramm, T. Baumeister, W. Uter and H. Drexler, Current and historical individual data about exposure of workers in the rayon industry to carbon disulfide and their validity in calculating the cumulative dose, Int. Arch. Occup. Environ. Health, 2013, 87, 675–683 CrossRef PubMed.
- A. Schramm, W. Uter, M. Brandt, T. Göen, M. Köhrmann, T. Baumeister and H. Drexler, Increased intima-media thickness in rayon workers after long-term exposure to carbon disulfide, Int. Arch. Occup. Environ. Health, 2015, 89, 513–519 CrossRef PubMed.
- A. Eben and F. P. Freudlsperger, 2-Thioxothiazolidine-4-carboxylic acid (TTCA) [Biomonitoring Methods, 1994], The MAK-Collection for Occupational Health and Safety, Wiley-VCH Verlag GmbH & Co. KGaA, 2002 DOI:10.1002/3527600418.
- V. Riihimäki, H. Kivistö, K. Peltonen, E. Helpiö and A. Aitio, Assessment of exposure to carbon disulfide in viscose production workers from urinary 2-thiothiazolidine-4-carboxylic acid determinations, Am. J. Ind. Med., 1992, 22, 85–97 CrossRef.
- L. Campbell, A. H. Jones and H. K. Wilson, Evaluation of occupational exposure to carbon disulphide by blood, exhaled air, and urine analysis, Am. J. Ind. Med., 1985, 8, 143–153 CrossRef CAS PubMed.
- J. Rosier, M. Vanhoorne, R. Grosjean, E. Van De Walle, G. Billemont and C. Van Peteghem, , Preliminary evaluation of urinary 2-thio-thiazolidine-4-carboxylic-acid (TTCA) levels as a test for exposure to carbon disulfide, Int. Arch. Occup. Environ. Health, 1982, 51, 159–167 CrossRef CAS PubMed.
- R. van Doorn, L. P. C. Delbressine, C. M. Leijdekkers, P. G. Vertin and P. T. Henderson, Identification and determination of 2-thiothiazolidine-4-carboxylic acid in urine of workers exposed to carbon disulfide, Arch. Toxicol., 1981, 47, 51–58 CrossRef CAS PubMed.
- J. Roh, C. N. Kim, N. G. Lim, J. H. Chang and Y. B. Cho, Simultaneous analysis of urinary 2-thiothiazolidine-4-carboxylic acid and thiocarbamide as a biological exposure index for carbon disulfide exposure, Yonsei Med. J., 1999, 40, 265–272 CrossRef CAS PubMed.
- L. A. Ridnour, D. D. Thomas, C. Switzer, W. Flores-Santana, J. S. Isenberg, S. Ambs, D. D. Roberts and D. A. Wink, Molecular mechanisms for discrete nitric oxide levels in cancer, Nitric Oxide, 2008, 19, 73–76 CrossRef CAS PubMed.
- P. Simon, T. Nicot and M. Dieudonné, Dietary habits, a non-negligible source of 2-thiothiazolidine-4-carboxylic acid and possible overestimation of carbon disulfide exposure, Int. Arch. Occup. Environ. Health, 1994, 66, 85–90 CrossRef CAS PubMed.
- Y. Kikuchi, T. Uemura, T. Yamuchi, T. Takebayashi, Y. Nishiwaki, K. Yamada, H. Sakurai and K. Omae, Urinary excretion of TTCA after intake of brassica vegetables, J. Occup. Health, 2002, 44, 151–155 CrossRef CAS.
- International Agency for Research on Cancer, Cruciferous vegetables, isothiocyanates and Indoles, IARC Press, Lyon, France, vol. 9, 2004 Search PubMed.
- P. Riso, C. Del Bo' and S. Vendrame, Preventive effects of broccoli bioactives: role on oxidative stress and cancer risk, in Cancer: Oxidative Stress and Dietary Antioxidants, ed. V. Preedy, Elsevier Inc., Academic Press, San Diego, 2014, ch. 11, pp. 115–126 Search PubMed.
- L. K. Keefer, R. W. Nims, K. M. Davies and D. A. Wink, ‘NONOates’ (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms, Methods Enzymol., 1996, 268, 281–293 CAS.
- A. E. Pierri, D. A. Muizzi, A. D. Ostrowski and P. C. Ford, Photo-controlled release of NO and CO with inorganic and organometallic complexes, Struct. Bonding, 2014, 165, 1–45 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |