
Self-assembled uracil complexes containing tautomeric uracils: an IRMPD spectroscopic and computation study of the structures of gaseous uracilnCa2+ (n = 4, 5, or 6) complexes†
Ruodi
Cheng
 ,
Victoria E.
Rose
,
Barry
Power
and
Travis D.
Fridgen
*
,
Victoria E.
Rose
,
Barry
Power
and
Travis D.
Fridgen
*
Department of Chemistry, Memorial University, St. John's, NL A1B 3X7, Canada. E-mail: tfridgen@mun.ca
First published on 29th November 2017
Abstract
The structures of doubly-charged uracil (U) complexes with Ca2+, UnCa2+ (n = 4, 5, 6), were studied by infrared multiple photon dissociation (IRMPD) spectroscopy and computational methods. The ions were produced by electrospray ionization (ESI) and were isolated in the gas phase in a Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR-MS). The recorded IRMPD spectra in both the fingerprint and the C–H/N–H/O–H stretching regions, combined with computed vibrational spectra, reveal that the structures present in the greatest abundance consist of both canonical uracil as well as the lactam (or colloquially “enol”) tautomer of uracil. U4Ca2+ consists of two hydrogen-bonded dimers of uracil, one canonical and one tautomer, with each uracil interacting with Ca2+ through a carbonyl oxygen. The structures most consistent with the vibrational spectrum of U6Ca2+ consist of two hydrogen-bonded uracil trimers, each composed of two canonical and one enolic uracil, with each uracil also interacting with Ca2+ through carbonyl oxygen. U5Ca2+ consists of one of the aforementioned trimers and dimers, each containing one enol tautomerized uracil. The computed structures whose vibrational spectra best agree with the experimental vibrational spectra are also the lowest-energy structures for all three complexes. This study clearly shows that some uracils adopt the normally very high energy enol tautomer in the lowest energy gas phase complexes of uracil with a doubly-charged ion like Ca2+.
1. Introduction
Adenine, guanine, thymine, cytosine, and uracil are nucleobases comprising deoxyribonucleic acid (DNA) and/or ribonucleic acid (RNA). During the process of DNA and RNA replication, metal cations exhibit different effects depending on their concentrations. At high concentration, they can interact with the nucleobases and disrupt normal hydrogen bonding patterns between base pairs. Their presence at high concentrations, therefore, can cause the mismatching of the nucleobases which is considered to be the main reason for mutations as they interfere with the native conformations of the nucleic acid polymers.1,2 However, at lower concentrations, metal cations interact with the phosphate groups of the nucleic acids, stabilizing the double helix of DNA.3 In the 1960s, guanosine 5′-monophosphate was found to form planar G-tetrad complexes that exhibit Hoogsteen paired motifs.4 Further research in the 1980s revealed that these structures could form in DNA, stabilized by monovalent cations.5,6 Studies of G-tetrads are of interest due to their biological relevance as they are found within the guanine-rich telomeric regions near the ends of chromosomes1,5,7 and within the promoter regions of oncogenes.8 Therefore, the G-tetrad has been investigated as a potential target for cancer therapies.9 Whether these structures exist in vivo was controversial; there is evidence that they are formed in living cells, as proteins that recognize the G-tetrad have been found.10,11 More recently, there has been evidence found that telomere end-binding proteins (TEBPs) regulate G-quadruplex DNA formation in vivo.12Cation-bound self-assembled nucleobase complexes have attracted the attention of scientists not only in biological fields, but also materials chemistry. Specific knowledge of self-assembled nucleobase structures bound to metal cations can be applied in the fields of supramolecular chemistry and nanotechnologies where the ability to selectively produce molecular-scale structures is pivotal. It is necessary to better understand how non-covalent interactions such as hydrogen bonds, ion–dipole interactions and π–π interactions affect the structures of complexes containing nucleobases and metal cations. This knowledge can be applied to self-assembly and molecular recognition processes to form supramolecular polymers,13 nanofibres,14 and nano-scale electronics.15 Stable uracil tetramers containing metal cations at their centers, with Hoogsteen-bonding patterns analogous to the guanine quartets, have been found to exist in four-stranded RNA molecules.16
Uracil complexes in the gas phase have been studied by density functional theory (DFT) and electrospray ionization-mass spectrometry (ESI-MS) by Zins et al.17 Lithium, sodium, and calcium cations (Li+, Na+, Ca2+) were found to bond to uracil producing a series of different adducts; notable among these was a decameric complex that appeared to have unusual stability for such a complex structure. The decamer was identified as a magic number cluster, which have a high relative intensity compared to the other ions present. Potassium was not capable of forming the uracil decamer, but the calcium cation led to the formation of particularly large clusters such as 18-mers (U18Ca2+) and even 24-mers (U24Ca2+). These results indicate that both the size and the charge of the metal cation affect uracil aggregation. Theoretical calculations showed that a low energy decamer structure of Na+ and Ca2+ were the same, with two quasiplanar uracil pentamers on two sides with cations in the center. The uracil pentamer is stabilized by ten NH–O hydrogen bonds between uracil molecules. The metal cation interacts with two uracil pentamers by cation dipole interactions. These calculations also showed that the lithium cation may be too small to form sufficient ion–dipole interactions to occupy the space between two uracil pentamers so the Li+ ion forms octameric complexes with the cation existing between two uracil tetramers. Of course, these mass spectral observations did not provide evidence to support the theoretically determined structures.
Further research into the structure of uracil pentamer complex [U5M]+ (M = K+, Na+, Cs+) was conducted using electrospray ionization mass spectrometry (ESI-MS).18 The proposed structure for these pentamers contains two hydrogen bonds between two neighboring uracils forming a cyclic structure with the metal cation in the middle. The uracil pentamer system has a non-planar geometry with K+ in the center while Na+ bonded complex is not exactly a metal-centered structure. The U5Na+ is not as thermodynamically stable as U5K+ because there are fewer interactions with Na+ as a result of its off-center structure. The results also indicated that the suitable size of the metal cation is vital for stabilizing such a complex. Work by Gillis et al.19 investigated the energetics of dissociation of trapped gas phase uracil cluster ions consisting of 4–14 uracil units and a divalent calcium ion center (UnCa2+ where n = 4–14) by blackbody infrared radiative dissociation (BIRD) kinetics and theoretical methods. It was determined that the energy of dissociation for U6Ca2+ is significantly greater than that for the larger clusters (n = 7–14) consistent with collision induced dissociation (CID) studies by Zins et al.20 and indicates that the clusters are composed of an inner solvent shell of six uracil molecules bound directly to the Ca2+, with additional uracils bound in a second solvation shell through hydrogen bonding to the inner shell uracil molecules. Theoretical and experimental results also revealed an odd–even alternation of dissociation threshold energies for the larger clusters, where complexes with an even number of uracil molecules were found to be more stable to dissociation, indicating that the large clusters of uracil, beyond n = 6, may be composed of hydrogen-bonded dimeric units of uracil bound to the inner U6Ca2+ core. In this work, plausible structures were suggested, but only those containing canonical uracil were contemplated, and no spectroscopic evidence for the structures was available.
The present work aims to provide more direct experimental insight into the previously proposed structures of the UnCa2+ (n = 4–6), using infrared multiple photon dissociation (IRMPD) spectroscopy in the 1000–1900 cm−1 and 2800–3800 cm−1 regions. The experimental IRMPD spectra are compared to theoretical infrared (IR) spectra from electronic structure calculations. Different computation methods and basis sets are used to compare the energies of isomeric structures. By comparing theoretical and experimental results, the precise structures of uracil complexes are determined.
2. Methods
2.1 Computational methods
Geometry optimizations and frequency calculations were performed using the Gaussian 09 suite of programs using B3LYP density functional theory and the 6-31+G(d,p) split-valance basis set for all atoms from which the relative 298 K Gibbs energies and enthalpies of various different isomers were compared. The computed IR spectra were scaled by 0.97 and 0.95 in the lower (900–2000 cm−1) and higher (2700–4000 cm−1) energy regions, respectively, to compare with the experimental IRMPD spectra. The single point energies for all lowest energy structures were performed using B3LYP/6-311+G(3df,3pd) for comparison. For all calculations, an empirical dispersion correction was done using Grimme's D3 version with the original D3 damping function, B3LYPD3.212.2 Experimental methods
For the IRMPD spectroscopy, two different instruments were used, one for each of the fingerprint and C–H/N–H/O–H stretching region. For both, a Bruker Apex-Qe 7 Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR-MS) with an Apollo II electrospray ion source was used. Uracil and calcium chloride were purchased from Sigma-Aldrich and used without further purification. The solution producing the uracil tetramer (U4Ca2+) was 1.7 mmol L−1 uracil in 50/50 water and methanol and 5–10 drops of 10 mmol L−1 in CaCl2. U5Ca2+ and U6Ca2+ were prepared to concentrations of 2.5 mmol L−1 uracil in a 50/50 mixture of water and acetonitrile and contained 130 μmol L−1 CaCl2. Solutions were injected using a 1 mL syringe at a flow rate of 112 μL h−1. The temperature of the dry gas was set as 90 °C. Prior to IRMPD, ions were isolated by standard FT-ICR isolation techniques.For IRMPD experiments in the fingerprint region, the FT-ICR-MS was coupled to a free electron laser (FEL) at the Centre Laser Infrarouge d’Orsay.22,23 Irradiation times were between 1 and 2 s and the FEL was scanned at 5 cm−1 intervals. In the C–H/N–H/O–H stretch region isolated ions were irradiated with an optical parametric oscillator/amplifier (OPO/A) in the Laboratory for the Study of Energetics, Structures, and Reactions of Gaseous Ions at Memorial University.24 The ions were irradiated by the laser for 1 s, and the scan step was set as 2 cm−1. The IRMPD efficiencies (intensities) are the negative of the logarithm of the product ion intensities divided by the sum of the total ion intensities.
3. Results and discussion
3.1 Infrared multiple photon dissociation of UnCa2+
Mass spectra following 3420 cm−1 irradiation of the isolated U4Ca2+, U5Ca2+, and U6Ca2+ complexes are displayed in Fig. S10A–C (ESI†), respectively. U4Ca2+ (m/z 244) undergoes charge separation to UH+ (m/z 113) and (U3Ca-H)+ (m/z 375), the latter of which loses uracil to form (U2Ca-H)+ (m/z 263). U5Ca2+ (m/z 300) undergoes loss of uracil (m/z 244) which undergoes the same fragmentation as just described for U4Ca2+.U5Ca2+ also undergoes charge separation forming UH+ and (U4Ca-H)+ (m/z 487) the latter of which undergoes successive losses of uracil forming m/z 375, 263 and 151.
U6Ca2+ (m/z 356) presumably underwent loss of uracil to form m/z 300 exclusively as no (U5Ca-H)+ (m/z 599) was observed. The U5Ca2+ then underwent charge separation and loss of uracil as just described.
All parent and fragment ions were accounted for when determining the IRMPD efficiency to construct the IRMPD spectra which will be discussed below.
3.2 IRMPD spectra for UnCa2+
In Fig. 1, the IRMPD spectra in the fingerprint and C–H/N–H/O–H stretching region for U4Ca2+, U5Ca2+, and U6Ca2+ are presented. Clearly, in both regions, the spectra of each complex are quite similar to one another, so similar structures might be expected. In the high energy region, sharp N–H stretching bands are observed at about 3420 cm−1 and broad hydrogen bonded N–H (or O–H—vide infra) are observed between about 3400 and 2800 cm−1. In the fingerprint region, the most intense bands are observed between about 1575 cm−1 and 1800 cm−1 correspond to strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching for uracil. These bands are quite indicative of the structure of the complex and can be broken down into three regions. The first, indicated by ‘A’ in Fig. 1, is the most red-shifted of the three regions and the band centred at about 1635 cm−1 is the stretching of the CO bound to the metal cation. The second region, B, has absorptions centred around 1730 cm−1 and the third, C, weak shoulders are observed at about 1775 cm−1. Region C is where strong free CO stretching vibrations would be expected and just to the red, region B, CO stretching that has been slightly red shifted by an interaction such as a hydrogen bond is expected.
O stretching for uracil. These bands are quite indicative of the structure of the complex and can be broken down into three regions. The first, indicated by ‘A’ in Fig. 1, is the most red-shifted of the three regions and the band centred at about 1635 cm−1 is the stretching of the CO bound to the metal cation. The second region, B, has absorptions centred around 1730 cm−1 and the third, C, weak shoulders are observed at about 1775 cm−1. Region C is where strong free CO stretching vibrations would be expected and just to the red, region B, CO stretching that has been slightly red shifted by an interaction such as a hydrogen bond is expected.
 | ||
| Fig. 1 Comparison of the experimental IRMPD spectra for UnCa2+ (n = 4, 5, 6) in the fingerprint and the CH/NH/OH stretching regions. | ||
In previous work,17,19,20 only canonical (or diketo) uracil was considered when trying to obtain lowest energy structures of Ca2+/uracil complexes. The lowest energy structure found for U4Ca2+ (Scheme 1A) had each of the uracils bound to Ca2+ through O4 and a hydrogen bond between N3H and O4 of the neighbouring uracil. This structure is called C4a, where C4 indicates four canonical uracils and ‘a’ represents the lowest energy structure. While this structure is consistent with the spectra in the higher energy region—a free N–H group and hydrogen bonding N–H stretching—the four free CO groups would be expected to produce intense features in region C at about 1800 cm−1 in the fingerprint region of the infrared spectrum. The structures of the U4Ca2+, U5Ca2+, and U6Ca2+ complexes will be discussed in more detail below.
 | ||
| Scheme 1 (A) The lowest energy U4Ca2+ composed of all diketo uracils determined in previous works.17,19,20 (B) Dimeric Ca2+/uracil structure composed of one diketo and one N3O4 tautomer comprising another U4Ca2+ isomer. | ||
3.3 U4Ca2+
In some recent studies, it was shown that in the presence of ions uracil25–30 and thymine31 tautomerize preferentially where the N3 hydrogen is instead on O4 (N3O4 tautomer) and the ion interacts with O2 and N3. While the canonical structure for uracil and thymine is significantly lower in energy than any of the enol tautomers, the N3O4 tautomer of thymine bound to a dication can be lower in energy by some 100 kJ mol−1 than the canonical tautomer bound to a dication.31 Therefore, the normally high energy tautomers of uracil, seen in Scheme 2 need to be considered.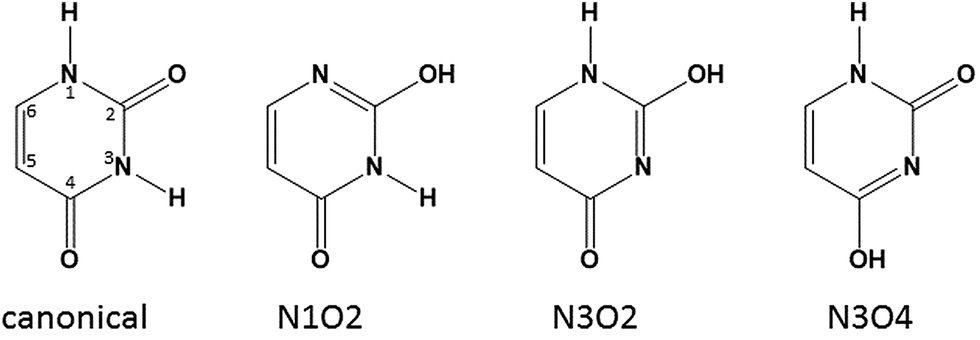 | ||
| Scheme 2 Four tautomers of uracil including the numbering convention for pyrimidine bases such as uracil. | ||
Based on this previous work and armed with the spectroscopy in Fig. 1, a structure for U4Ca2+ composed of two dimers of uracil, each with one canonical and one N3O4 tautomer, both bound to Ca2+ (see Scheme 1B) was constructed. In this structure, canonical uracil is bound to Ca2+ through O4 and the N3O4 tautomer is bound to Ca2+ through O2 providing the strong band in region A. There is a hydrogen bonded N–H group and a hydrogen bonded O–H group, as well as two free N–H groups, consistent with the spectra in the high energy region. There is a hydrogen bonded carbonyl providing the strong band in region B and no free carbonyl group, again consistent with the infrared spectrum. The U4Ca2+ complex based on these dimers is named C2T2a (T for tautomeric uracil) is calculated to be lower in 298 K Gibbs energy by 51.3 kJ mol−1 (see Fig. 2) than the lowest energy structure determined with all canonical uracils.
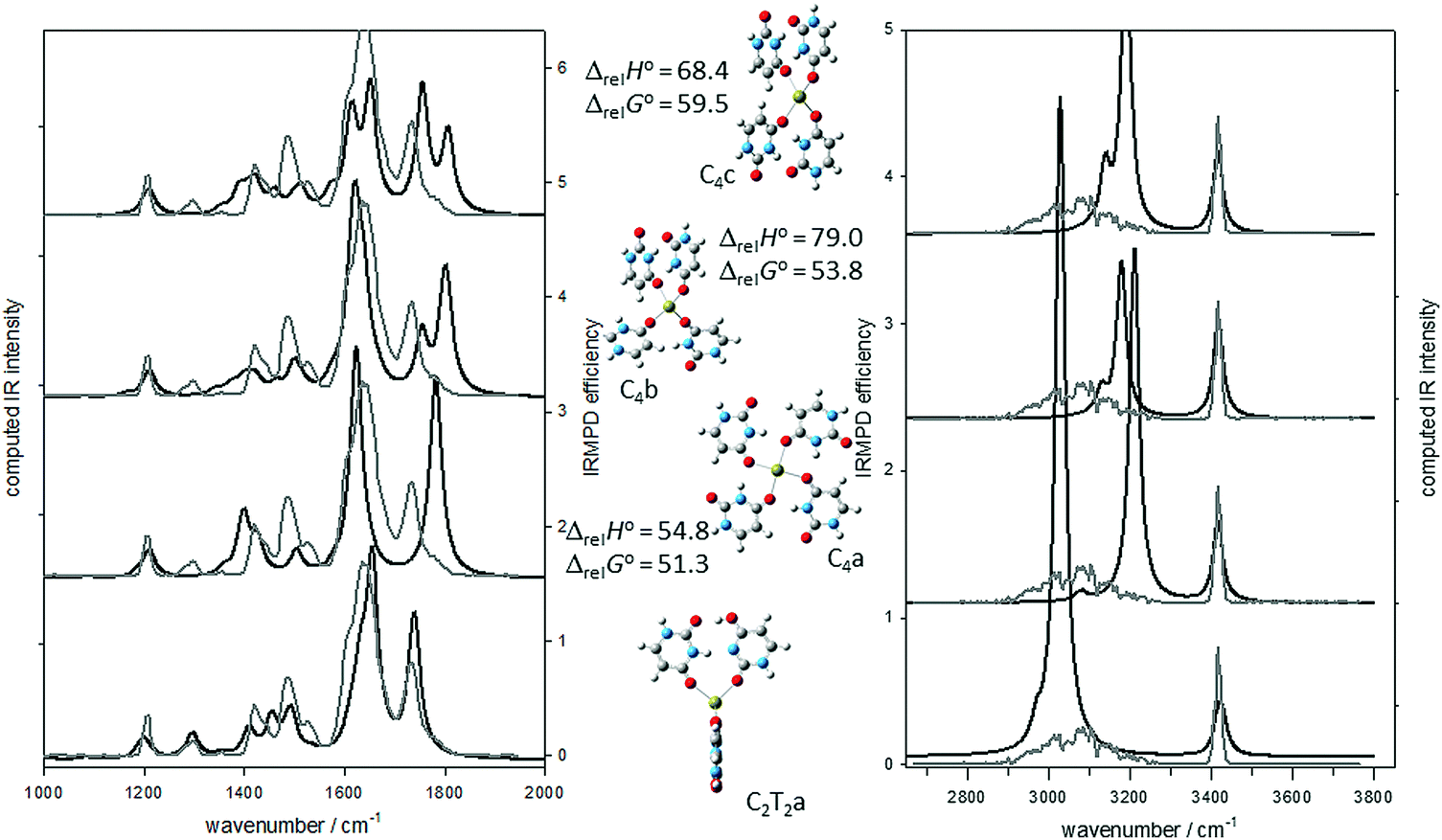 | ||
| Fig. 2 Comparison of the experimental IRMPD spectrum (grey trace) for U4Ca2+ and the B3LYP/6-31+G(d,p) computed IR spectra (black traces) for four different isomers. The 298 K enthalpies and Gibbs energies relative to structure C2T2a are also shown (also computed using B3LYP/6-31+G(d,p)). | ||
In Fig. 2, the experimental IRMPD spectra (grey traces) are compared with the computed IR spectra (black traces) for the structures depicted in the centre of the figure. It is clear that the computed spectrum for the lowest energy structure, C2T2a, compares very well with the entirety of the recorded IRMPD spectrum. It is equally apparent that the computed infrared spectrum for the C4a structure, discussed above, is not consistent with the CO stretching region of the experimental IRMPD spectrum. The obvious disagreement is the absence of a strong free CO stretch in the experimental IRMPD spectrum, which is predicted by the calculations, and the presence of the hydrogen bonded CO stretch in the IRMPD spectrum, which is not predicted by the calculations. Fig. 2 also shows the second and third lowest energy structures from Gillis et al.,19 which are also high in energy compared to C2T2a, and have similar inconsistencies with the IRMPD spectrum mainly in the 1700–1800 cm−1 region.
There are other isomers of U4Ca2+ consisting of two dimers composed of one canonical and one tautomeric uracil. C2T2b is similar to C2T2a except the tautomeric uracil has the N3 hydrogen shifted to O4 and this uracil is bound to Ca2+ through O2, see Fig. S1 (ESI†). This results in a structure that is only 8.0 kJ mol−1 higher in 298 K Gibbs energy than C2T2a. The IRMPD spectrum in the fingerprint region is also consistent with the computed IR spectrum for C2T2b. In the higher energy region, there is a pronounced red-shift computed for the hydrogen bonded O–H stretch, however this is not enough to rule out a contribution to the experimental spectrum from C2T2b. C2T2a/b contains one dimer each as the ones in C2T2a and C2T2b and results in a structure that is computed to be only 3.9 kJ mol−1 higher in 298 K Gibbs energy than the lowest energy C2T2a isomer. Assuming a thermal distribution, it would be expected that C2T2a/b contributes about 20% of the mixture of ions. It is clear that C2T2a/b also reproduces the experimental spectrum quite well and unlike C2T2a predicts the weak band observed at about 1530 cm−1.
C2T2c and C2T2d both have the canonical uracil bound to Ca2+ through O2 and differ from each in the same way that C2T2a and C2T2b differ from one another. C2T2c and C2T2d are more than 20 kJ mol−1 higher in energy than C2T2a and their computed spectra do not compare as well with the experimental IRMPD spectrum. It is concluded from these experiments and calculations that the main contributor to the experimental IRMPD spectrum is the C2T2a structure with a tetrahedral arrangement of two uracil hydrogen bonded dimers with one member of the dimer canonical and one which is a keto–enol tautomer.
Some other higher energy structures for U4Ca2+ are shown in Fig. S7 (ESI†) along with their computed energies and a comparison of the computed and experimental vibrational spectra. These include structures based on four N3O4 or N1O4 tautomeric uracils whose computed vibrational spectra do not reproduce the experimental spectra.
3.4 U6Ca2+
To construct a structure for U6Ca2+, like U4Ca2+, that is consistent with its experimental vibrational spectrum, a third dimer as in Scheme 1B is added to U4Ca2+ to form an octahedral structure. The complex optimizes to the octahedral structure labeled C3T3a in Fig. 3. This isomer, based on the three dimers, “collapses” such that two of the dimers have a B–B stacking interaction. The agreement between the computed IR spectrum for this structure and the IRMPD spectrum is certainly not poor enough to rule the structure out. | ||
| Fig. 3 A comparison of the experimental IRMPD spectrum (grey trace) for U6Ca2+ and the B3LYP/6-31+G(d,p) computed IR spectra (black traces) for five different isomers. The 298 K enthalpies and Gibbs energies relative to structure C4T2a are also shown (also computed using B3LYP/6-31+G(d,p)). | ||
Another way to form the U6Ca2+ is to add canonical uracil to each of the dimers, constructing two trimers with two canonical uracils and one of the tautomers in each, as in Scheme 3, where both canonical uracils are hydrogen bonded to the tautomeric uracil. The added uracil in this trimer (iii in Scheme 3) is also bound to Ca2+ through O4. The addition of this third uracil to each of the dimers in the lowest energy tetramer structure, C2T2a, results in a structure where the two trimers are perpendicular to each other, forming a roughly octahedral complex, and is lower in 298 K Gibbs energy than C4T2f by 36.1 kJ mol−1. Distortion of the complexes from an octahedral structure is due to the inter-trimer uracil hydrogen bonding. The computed infrared spectrum for this lowest-energy structure, C4T2a, can be seen in Fig. 3 and compares very well—better than C4T2f—with the experimental spectrum in the fingerprint region and the higher energy region (see Fig. S2, ESI†). This structure, along with C4T2b, C4T2c, and C4T2e, compared in Fig. S3 (ESI†), maximize the number of hydrogen bonds with two N–H to OC, one O–H to OC, and one N–H to N. However, C2T2a matches the IRMPD spectrum in the fingerprint region much better. C4T2b differs in that one canonical uracil (iii in Scheme 3) is bound through O2 rather than O4 and is 13 kJ mol−1 higher in energy. C4T2c and C4T2e have the two canonical uracils hydrogen bonded together and are higher in energy by 16 and 30 kJ mol−1, respectively.
 | ||
| Scheme 3 Lowest energy trimeric Ca2+/uracil structure composed of two canonical uracils and one N3O4 tautomer comprising the lowest energy U6Ca2+ isomer. | ||
Another structure that has a computed IR spectrum that compares favourably with the IRMPD spectrum is C4T2d (Fig. 3 and Fig. S2, ESI†). This structure, like C4T2c and C4T2e, differs from C4T2a in that the two canonical uracils in each of the trimers is hydrogen bonded together, in the case of C4T2d via an N3–H to O4 hydrogen bond and a C5–H to O2 interaction. While this structure is consistent with the IRMPD spectrum, it is 24.6 kJ mol−1 higher in 298 K Gibbs energy which makes it unlikely to be observed.
As was the case for the tetramers, the previously determined hexameric structures based solely upon canonical uracils,19i.e. C6a and C6b in Fig. 3 and Fig. S2 (ESI†), are higher in energy than C4T2a and their computed IR spectra do not compare well with the IRMPD spectra. The main problem with the computed spectra is that they contain a free CO stretch (region C) and do not predict the hydrogen bonded CO stretch (region B) which is observed experimentally. Some other higher energy structures and their computed spectra are shown in Fig. S9 (ESI†), including those where the two trimers are mixed in that they contain a trimer of C4T2a and one of either C4T2b, c, or e. These ‘mixed’ complexes are intermediate in energy; for example, C4T2a/b is higher in energy than C4T2a, but lower in energy than C4T2b. They are not nearly as good a fit to the IRMPD spectra in comparison to C4T2a (Fig. S9, ESI†).
3.5 U5Ca2+
The IRMPD spectrum of U5Ca2+ is compared to the computed IR spectra of several isomers in Fig. 4 and Fig. S4–S6, S8 (ESI†). Removal of one of the canonical uracils (iii in Scheme 3) from the lowest energy U6Ca2+ structure results in two very similar structures, C3T2a and C3T2b (in Fig. 4 and Fig. S4, respectively, ESI†) which only differ in 298 K Gibbs energy by 0.6 kJ mol−1. They do not differ significantly in structure, simply whether tautomeric or canonical uracil in the remaining dimer is closest to the trimer. The IR spectrum of C3T2a is consistent with the experimental IRMPD spectrum but certainly C3T2b cannot be ruled out spectroscopically or energetically. C3T2c and C3T2d differ from C3T2a and C3T2b in that uracil iii in the trimer is bound through O2 rather than O4 and results in structures that are a little less than 8 kJ mol−1, higher in 298 K Gibbs energy. While neither of these structures can be ruled out spectroscopically, they do not agree as well as the two lower energy structures in the CO stretching region. C3T2e and C3T2f differ in that the trimeric moiety is composed of tautomeric uracil bound to a canonical uracil bound to Ca2+via O2 and the second canonical uracil is bound through O4, but hydrogen bonded to canonical uracil. This results in structures just slightly higher than the previously discussed structure and their computed IR spectra are not as good a match to the experimental vibrational spectrum in the fingerprint region. Other computed IR spectra for higher energy structures are compared in Fig. 4 and the ESI.†
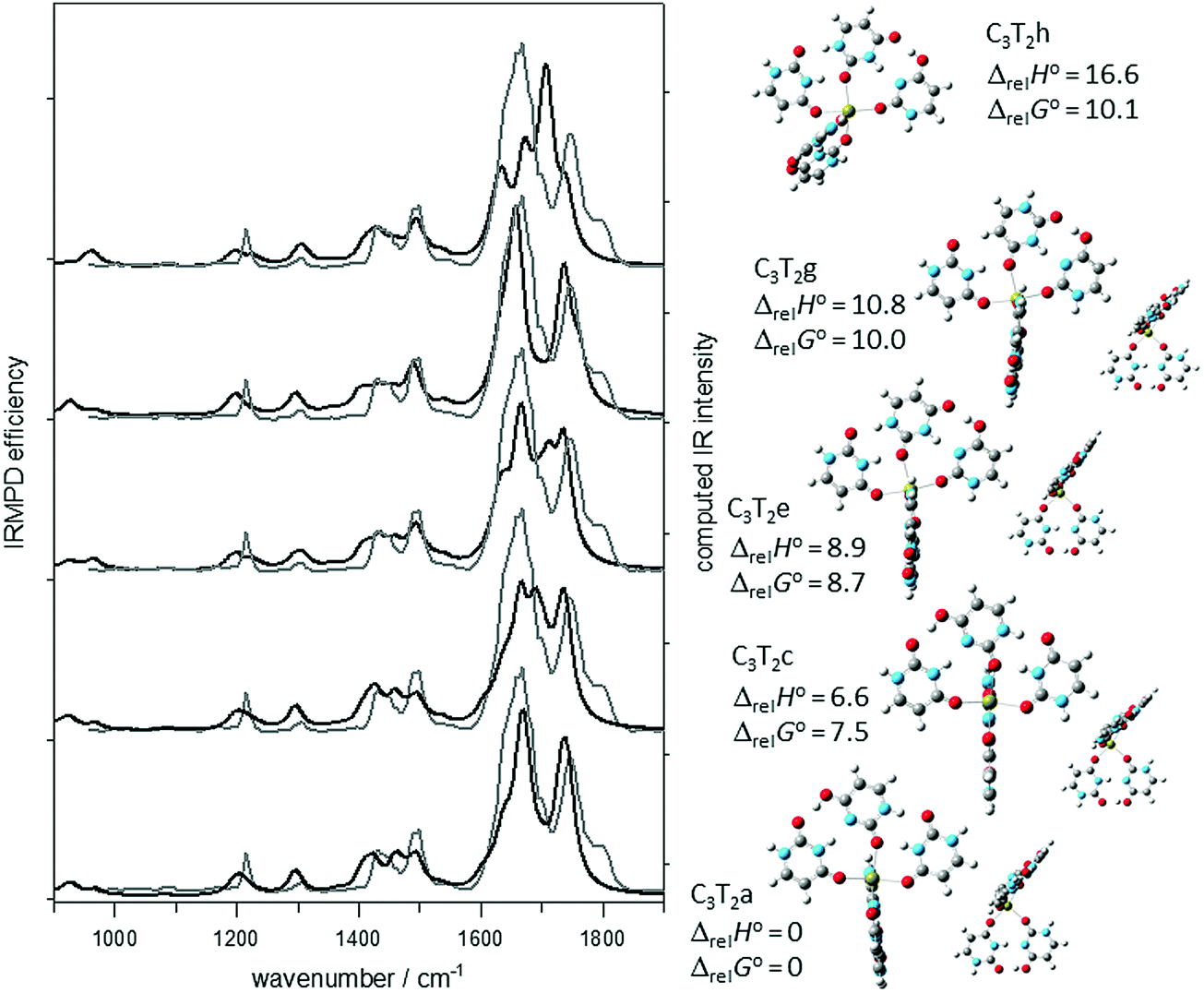 | ||
| Fig. 4 Comparison of the experimental IRMPD spectrum (grey trace) for U5Ca2+ and the B3LYP/6-31+G(d,p) computed IR spectra (black traces) for five different isomers. The 298 K enthalpies and Gibbs energies relative to structure C3T2a are also shown (also computed using B3LYP/6-31+G(d,p)). | ||
In Fig. S6 (ESI†), the computed IR spectra for the three lowest energy C5-based structures that were determined previously19 are compared to the experimental IRMPD spectrum. Besides being between 23 and 33 kJ mol−1 higher in energy than C2T3a, spectroscopically these isomers can be ruled out based on the poor agreement in the CO stretching region as discussed previously for the U4Ca2+ and U6Ca2+ complexes.
3.6 Other observed U5,6Ca2+ structures
In the IRMPD spectra for U5Ca2+ and U6Ca2+, there is a weak but pronounced shoulder observed in region C of Fig. 1, just to the red of 1800 cm−1. The band can only be attributed to a free CO stretch which is not present in any of the lowest energy structures. The only structures found that contain a free CO stretch are those not based on the dimer or trimer containing an N3O4 tautomer. Because the lowest energy structures of pentamer and hexamer clusters lack the free CO stretch, the existence of higher energy clusters is likely due to the presence of this shoulder peak at 1800 cm−1 observed in the experimental spectrum. For example, the lowest energy isomer containing a free CO stretch for U6Ca2+, is C6a. This isomer is also some 33 kJ mol−1 higher in 298 K Gibbs energy than C4T2a, and if present, it must be kinetically trapped, probably during the electrospray process. Tautomerization of the all keto tautomer of uracil to the enol–keto tautomer proceeds with a very high activation energy, some 200 kJ mol−1 for neutral uracil.32 The tautomerization barrier is computed to be lower, but still considerable (70 kJ mol−1, 298 K Gibbs energy barrier) for both the protonated and water-assisted tautomerization33 and an intramolecularly assisted uracil tautomerization in (U2-H)Cu+.28
3.7 Computed energies
The B3LYPD3/6-31+G(d,p) lowest energy structures for U4Ca2+ as well as for the lowest energy C3T1 and C4 structures, were submitted to a single point calculation at the same level of theory but with the larger 6-311+G(3df,3pd) basis. The calculations are compared in Table 1, using the thermal corrections to the 298 K enthalpy and Gibbs energy from the vibrational calculations with the smaller basis. Clearly, there is very little difference between the computed energies using the smaller or larger basis sets. A similar set of calculations were performed on various U5Ca2+ and U6Ca2+ isomers with the same conclusion (see Tables S2 and S3, ESI†).| Structure | B3LYPD3/6-31+G(d,p) | B3LYPD3/6-311+G(3df,3pd) |
|---|---|---|
| C2T2a | 0.0 | 0.0 |
| 0.0 | 0.0 | |
| C2T2b | 7.3 | 6.8 |
| 8.0 | 7.5 | |
| C2T2c | 21.7 | 21.7 |
| 22.4 | 22.3 | |
| C2T2d | 24.6 | 24.3 |
| 25.6 | 25.4 | |
| C3T1a | 35.9 | 35.2 |
| 31.8 | 31.2 | |
| C4a | 54.8 | 58.0 |
| 51.3 | 54.5 | |
4. Conclusions
A combination of IRMPD spectroscopy and computational chemistry was used to show that the structures of three doubly charged uracil/calcium clusters: U4Ca2+, U5Ca2+, and U6Ca2+, all contain two N3O4 enol-tautomerized uracils. The lowest energy structure for the U4Ca2+ complex consists of two uracil dimers, each consisting of one canonical and one tautomeric uracil, with two hydrogen bonds between them. The lowest energy U6Ca2+ complex consists of two uracil trimers, the same dimeric structure as in U4Ca2+ but a second canonical uracil doubly hydrogen bonded to the other side of the tautomeric uracil. The lowest energy U5Ca2+ consists of one dimer and one trimer as in the U4Ca2+ and U6Ca2+, respectively. The computed IR spectra for the lowest energy structures for each of the complexes were consistent with the experimental IRMPD spectra in all cases.For U4Ca2+ a second structure, only a few kJ mol−1 higher than the lowest energy structure, with an IR spectrum also consistent with the IRMPD spectrum also accounts for a small absorption at about 1530 cm−1. A weak shoulder observed in the free CO stretch region for both U4Ca2+ and U4Ca2+ are thought to be due to a small amount of higher energy, kinetically trapped complexes with only canonical uracils, as these are the only ones with predicted absorptions in these regions.
The structures determined for U4Ca2+, U5Ca2+, and U6Ca2+ are vastly different than those previously determined and only composed of canonical uracils.17,19,20 This is easily seen in the CO stretch region where there is the absence of a strong absorption in the IRMPD spectra due to a free CO group which is predicted for canonical uracil based structures and the presence of a hydrogen bonded CO stretch in the IRMPD spectrum which is not predicted for the canonical uracil based structures.
In previous experiments19 the BIRD dissociation kinetics of UnCa2+ were observed where n = 5–14. The thermochemistries of dissociation led to the conclusion that U14Ca2+ was composed of a U6Ca2+ core surrounded by four hydrogen bonded dimers of uracil in the second solvation shell, each hydrogen bonded to the core. This conclusion was based on the even uracil (n = 14, 12, 10, 8) containing complexes being more strongly bound than the odd uracil containing complexes (n = 13, 11, 9, 7), the difference between the successive dissociations, i.e. n = 14 and 13 being about the strength of a hydrogen bond, 10 kJ mol−1. While a detailed computational study of these larger complexes is beyond the scope of this study, it is interesting to contemplate a structure for the U14Ca2+ complex that also explains this even-odd dissociation energy observation. One such structure is proposed in Scheme 4 (only showing half the uracils in the complex) based on a core containing the T4C2a isomer of U6Ca2+. In this structure, each uracil dimer is bound to the complex via three interactions. The uracil labeled ‘a’ in Scheme 4 is bound by three hydrogen bonds, one to the other dimer, and might be the first to be lost, leaving the uracil labeled ‘b’ bound by only two hydrogen bonds and the second to be lost. Uracil ‘c’ is bound by three interactions and would be the third uracil lost followed by ‘d’ which is only bound to the core by two hydrogen bonds.
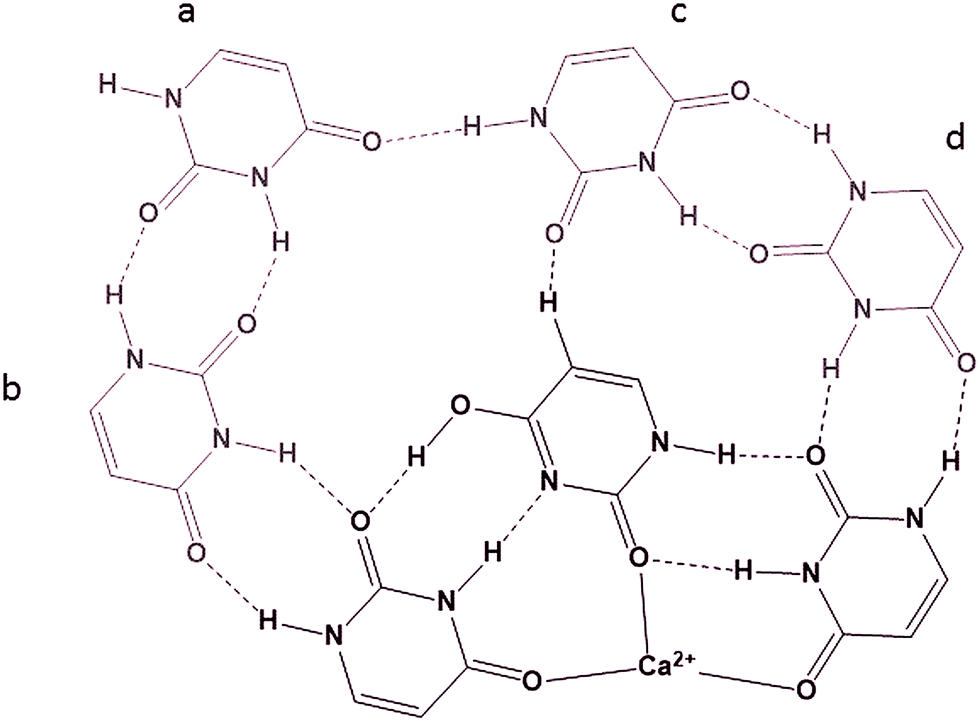 | ||
| Scheme 4 Possible structure of one half of U14Ca2+ with the 6 mer core composed of two trimeric structures. | ||
Conflicts of interest
There are no conflicts to declare.References
- B. Lippert, J. Chem. Soc., Dalton Trans., 1997, 21, 3971–3976 RSC.
- J. E. Reed, A. A. Arnal and S. Neidle, J. Am. Chem. Soc., 2006, 128(18), 5992–5993 CrossRef CAS PubMed.
- G. L. Eichhorn; N. A. Berger; J. J. Butzow; P. Clark; J. Heim; J. Pitha; C. Richardson; J. M. Rifkind; Y. Shin and E. Tarien, in Metal Ions in Biological Systems: Studies of Some Biochemical and Environmental Problems, ed. S. K. Dhar, Springer US, Boston, MA, 1973, pp. 43–66 Search PubMed.
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48(12), 2013–2018 CrossRef CAS.
- W. I. Sundquist and A. Klug, Nature, 1989, 342, 825–829 CrossRef CAS PubMed.
- J. R. Williamson, M. K. Raghuraman and T. R. Cech, Cell, 1989, 59, 871–880 CrossRef CAS PubMed.
- J. R. Williamson, Annu. Rev. Biophys. Biomol. Struct., 1994, 23, 703–730 CrossRef CAS PubMed.
- L. Yuan, T. Tian, Y. Chen, S. Yan, X. Xing, Z. Zhang, Q. Zhai, L. Xu, S. Wang, X. Weng, B. Yuan, Y. Feng and X. Zhou, Sci. Rep., 2013, 3, 01811 CrossRef PubMed.
- L. R. Kelland, Eur. J. Cancer, 2005, 41, 971–979 CrossRef CAS PubMed.
- R. Erlitzki and M. Fry, J. Biol. Chem., 1997, 272(25), 15881–15890 CrossRef CAS PubMed.
- M. P. Horvath and S. C. Schultz, J. Mol. Biol., 2001, 310, 367–377 CrossRef CAS PubMed.
- K. Paeschke, T. Simonsson, J. Postberg, D. Rhodes and H. J. Lipps, Nat. Struct. Mol. Biol., 2005, 12(10), 847–854 CAS.
- S. J. Rowan, P. Suwanmala and S. Sivakova, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3589–3596 CrossRef CAS.
- T. Shimizu, R. Iwaura, M. Masuda and T. Hanada, J. Am. Chem. Soc., 2001, 123(8), 5947–5955 CrossRef CAS PubMed.
- Y. Eichen, E. Braun and U. Sivan, Acta Polym., 1998, 49, 663–670 CrossRef CAS.
- C. Cheong and P. B. Moore, Biochemistry, 1992, 31, 8406–8414 CrossRef CAS PubMed.
- E. Zins, S. Rochut and C. Pepe, J. Mass Spectrom., 2009, 2008(2008), 40–49 CrossRef PubMed.
- B. Qiu, J. Liu, Z. Qin, G. Wang and H. Luo, Chem. Commun., 2009, 2863 RSC.
- E. A. L. Gillis, M. Demireva, K. Nanda, G. Beran, E. R. Williams and T. D. Fridgen, Phys. Chem. Chem. Phys., 2012, 14(10), 3304–3315 RSC.
- E. Zins, S. Rochut and C. Pepe, J. Mass Spectrom., 2009, 2009(2008), 813–820 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 15 CrossRef PubMed.
- J. M. Bakker, T. Besson, D. Scuderi and P. Maı, J. Phys. Chem. A, 2007, 111, 13415–13424 CrossRef CAS PubMed.
- R. Prazeres, F. Glotin, C. Insa, D. A. Jaroszynski and J. M. Ortega, Eur. Phys. J. D, 1998, 3, 87–93 CrossRef CAS.
- M. Azargun, Y. Jami-Alahmadi and T. D. Fridgen, Phys. Chem. Chem. Phys., 2017, 19, 1281–1287 RSC.
- B. Power, V. Haldys, J. Y. Salpin and T. D. Fridgen, Int. J. Mass Spectrom., 2015, 378, 328–335 CrossRef CAS.
- A. A. Power, O. Y. Ali, M. Burt and T. D. Fridgen, Interna, 2012, 330, 233–240 Search PubMed.
- O. Y. Ali, N. M. Randell and T. D. Fridgen, ChemPhysChem, 2012, 13, 1507–1513 CrossRef CAS PubMed.
- O. Y. Ali and T. D. Fridgen, ChemPhysChem, 2012, 13, 588–596 CrossRef CAS PubMed.
- B. Power, S. Rowe and T. D. Fridgen, J. Phys. Chem. B, 2017, 121, 58–65 CrossRef CAS PubMed.
- T. E. Akinyemi, R. R. Wu, Y.-W. Nei, N. A. Cunningham, H. A. Roy, J. D. Steill, G. Berden, J. Oomens and M. T. Rodgers, J. Am. Soc. Mass Spectrom, 2017, 28(11), 2438–2453 CrossRef CAS PubMed.
- M. Kabeláč and P. Hobza, J. Phys. Chem. B, 2006, 110(29), 14515–14523 CrossRef PubMed.
- X. Hu, H. Li, W. Liang and S. Han, J. Phys. Chem. B, 2005, 109(12), 5935–5944 CrossRef CAS PubMed.
- J. M. Bakker, R. K. Sinha, T. Besson, M. Brugnara, P. Tosi, J. Salpin and P. Maitre, J. Phys. Chem. A, 2008, 112, 12393–12400 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp07128k |
| This journal is © the Owner Societies 2018 |