
Ligand-induced action of the W2866.48 rotamer toggle switch in the β2-adrenergic receptor†
Anita
Plazinska
 *a,
Wojciech
Plazinski
b,
Rafal
Luchowski
c,
Artur
Wnorowski
a,
Wojciech
Grudzinski
c and
Wieslaw I.
Gruszecki
c
*a,
Wojciech
Plazinski
b,
Rafal
Luchowski
c,
Artur
Wnorowski
a,
Wojciech
Grudzinski
c and
Wieslaw I.
Gruszecki
c
aDepartment of Biopharmacy, Faculty of Pharmacy, Medical University of Lublin, W. Chodzki Str., 4a, 20-093 Lublin, Poland. E-mail: anita.plazinska@umlub.pl
bJ. Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, Niezapominajek Str., 8, 30-239 Cracow, Poland
cDepartment of Biophysics, Institute of Physics, Maria Curie-Sklodowska University, 20-031 Lublin, Poland
First published on 29th November 2017
Abstract
Studies focused on GPCRs, particularly on the β2-adrenergic receptor (β2-AR), have demonstrated the relationship between ligand structure, receptor conformational changes and the corresponding pharmacological outcomes. Herein, we studied the molecular details of the rotameric flip of the W2866.48 sidechain, i.e. a presumed action switch that has not been reported in native β2-AR thus far. It is believed that although both the ‘active’ and ‘inactive’ conformers of β2-AR exhibit similar conformations of this switch, it may still play a substantial role in the ligand-induced activation of the receptor. By using both experimental methods (time-resolved fluorescence spectroscopy) and molecular modeling techniques (enhanced-sampling molecular dynamics), we characterized the conformational rearrangements of W2866.48 in relation to the type of ligand present in the binding cavity and to the conformation of the receptor (‘active’ vs. ‘inactive’ β2-AR). We found that the conformational behaviour of W2866.48 is correlated with the pharmacological character of the ligand present in the binding cavity but not with the instantaneous conformation of the receptor. Namely, agonists promote the W2866.48 conformations that facilitate the increase of the solvation within the inner receptor channel. In contrast, antagonists and inverse agonists act toward the decrease of the solvation in the inner channel. This creates an opportunity for using computational methodologies in determining the pharmacological properties of various ligands. The combination of the time-resolved fluorescence spectroscopy technique with the enhanced-sampling molecular dynamics simulations is shown to be a powerful tool for studying the ligand-induced conformational rearrangements in GPCRs.
1. Introduction
The β2-adrenergic receptor (β2-AR) is one of the most heavily investigated members of G protein coupled receptors (GPCRs), a ubiquitous cell-surface protein superfamily characterized by a seven transmembrane (TM) domain topography and the ability to activate heterotrimeric G proteins and β-arrestins. It is well established that β2-AR is a key regulator of cardiovascular and pulmonary physiology acting through the binding of epinephrine (adrenaline) and norepinephrine (noradrenaline). These chemical stimuli induce a series of conformational changes in the structure of β2-AR that are transferred from the binding site to the intracellular part of the receptor where the interactions with downstream signaling molecules occur. The diversity of signaling options implies that β2-AR and related receptors are not just binary on/off switches, but exist as dynamic entities that can adopt multiple pathway-specific conformations. The presence of ligand may induce or stabilize a unique conformational state that can be distinguished by its activity toward different signaling molecules (e.g. G proteins, arrestins).β2-AR has been structurally well characterized in its active and inactive states but details of its activation are still not fully understood.1–4 (See Fig. 1 for the graphical illustration of the β2-AR structure.) Ligands of β2-AR are seen to bind at a buried site between TM3, TM5, TM6 and TM7 with the residues D1133.32, S2035.42, S2045.43, S2075.46, N2936.55, Y3087.35, N3127.39 and Y3167.43 (superscript numbers correspond to the general numbering scheme of Ballesteros and Weinstein5) defining the topology of the binding site. However, it should be taken into account that the crystal structures show only the most thermodynamically stable β2-AR conformers. The crystallographic studies are not capable of recognizing the dynamic structural features of β2-AR, thus, other biophysical analyses involving NMR, HDX-MS or DEER spectroscopy as well as molecular modeling approaches have been applied to clarify the details of the β2-AR activation.
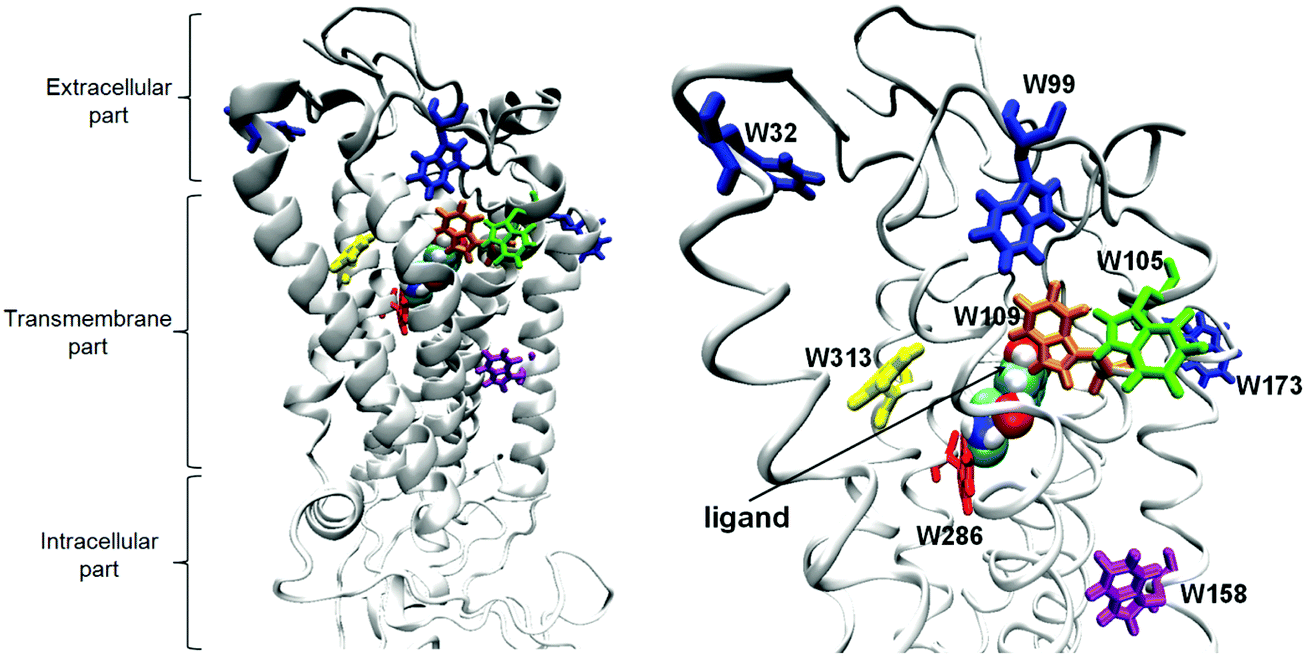 | ||
| Fig. 1 The 3D model of the β2-AR–(R)-epinephrine complex. The eight tryptophans are shown in stick and the epinephrine ligand in ball representations. | ||
The activation of GPCRs depends on the agonist-driven disruption of non-covalent interactions between highly conserved residues that tether together the transmembrane helices in the inactive conformation. The kinetics of achieving the active conformation comprises two fast steps, representing the agonist fitting into a loose binding pocket and conformational changes facilitating its full engagement in the binding pocket, and a slow step representing more substantial movements and deformations of the transmembrane helices.6 The crystal structures of opsin associated with a carboxyl-terminal peptide fragment of its G subunit, transducin,7 and β2-AR co-crystalized with the agonist molecule and the Gs protein indicate the presence of a substantial change in the arrangement of the helical bundle, with prominent movement of TM6.3 The sets of key residues that initiate these discrete rearrangements in the receptor structure ultimately leading to receptor activation are referred to as ‘micro-switches’.8
To achieve even partial activation of β2-AR, an agonist is required to release at least one of the following switches: the ‘ionic lock’ (the D/ERY motif) composed of charged residues that keep the intracellular parts of TM3 and TM6 in the inactive conformation and the ‘rotamer toggle switch’ (the CWxP motif in TM6) created by W2866.48, F2896.51, and F2906.52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9–12 that restrains TM6 twisted in the conformation preventing the activation. The latter is possibly the most common switch among GPCRs. The important role of tryptophan in the CWxP motif as a molecular switch is widely acknowledged by experimental studies concerning mainly the rhodopsin and opsin proteins and indicating the specific conformational rearrangements of this residue.13–17 In the case of rhodopsin, the relocation of conserved residues W6.48 and F6.44 toward P5.50 was observed.18 However, the rotamer change that was originally proposed based on computer simulations was not observed.19 Also, in the case of the A2AR activation, the W6.48 movement and the TM6 rotation were observed. Here, this residue does not undergo a rotamer transition, but rather moves along with the backbone.20 In contrast to rhodopsin and A2AR, in β2-AR, no W286-related movements were observed, only the rotation and movement of the cytoplasmic part of TM6.3
9–12 that restrains TM6 twisted in the conformation preventing the activation. The latter is possibly the most common switch among GPCRs. The important role of tryptophan in the CWxP motif as a molecular switch is widely acknowledged by experimental studies concerning mainly the rhodopsin and opsin proteins and indicating the specific conformational rearrangements of this residue.13–17 In the case of rhodopsin, the relocation of conserved residues W6.48 and F6.44 toward P5.50 was observed.18 However, the rotamer change that was originally proposed based on computer simulations was not observed.19 Also, in the case of the A2AR activation, the W6.48 movement and the TM6 rotation were observed. Here, this residue does not undergo a rotamer transition, but rather moves along with the backbone.20 In contrast to rhodopsin and A2AR, in β2-AR, no W286-related movements were observed, only the rotation and movement of the cytoplasmic part of TM6.3
Although related studies focused on β2-AR confirm the significant role of W2866.48 in the activation process, no experimentally-inferred molecular details are known so far except confirmation that the presence of ligands may influence the preferred orientation of W2866.48.11, 21
The activation of β2-AR relies on the conformational movement of P2886.50 (part of CWxP motif), which provides a kink in TM6. The rotation of this conserved proline depends on its contact with the adjacent aromatic residues: W2866.48, F2896.51, F2906.52, C2856.4711 and F2085.47.12 Agonist ligands can interact with these aromatic residues that further modulate the proline conformation; this results in twisting the cytoplasmic end of TM6 away from TM3. Noteworthily, the rotamer toggle switch is also associated with the hydrogen bond network between the conserved residues (P2886.50, F2896.51, F2906.52, F2085.47 and Y2095.48) and structural water molecules filling the receptor cavity. This network extends from the TM region toward the extracellular surface of the receptor, enabling the structural cross-talk between different segments of β2-AR and ligand approaching the binding site.
The high resolution X-ray structures have displayed surprisingly different pictures of the conformations and contacts exhibited by the discussed micro-switches in the active conformers of different GPCRs. For example, the most highly conserved residue, R3.50 (DRY motif in TM3), is found in rather different conformations and interaction patterns in the active forms of rhodopsin, opsin, β2-AR and A2AR.2,3,7,20,22,23 This is also the case for Y7.53 (TM7) of the NPxxY motif, and W6.48 (TM6) of the CWxP motif, which have not been observed to change the expected rotation.13,14
The theoretical studies performed for other GPCRs, e.g. for A2AR,24 5-HT3,25 and for β2-AR with the point mutation F290S26 indicated the occurrence of conformational rearrangements of tryptophan that creates the ‘toggle-switch’. However, due to the application of standard, unbiased molecular dynamics (MD) simulations, only observation of single events of conformational transitions was possible. For instance, no rotation of the W2866.48 aromatic moiety was reported, which, in theory, could be significant in the context of possible π–π interactions between amino acid residues as well as of hydrogen bonding involving the indole moiety.
Finally, our recent MD simulations27 showed that some of the agonist ligands may exhibit a close contact with W2866.48 when being bound in the binding cavity of β2-AR. This additionally supports the hypotheses of the ligand-induced conformational changes of the W2866.48 sidechain.
It is interesting to note that no reports on the conformational changes of W2866.48 in the case of native β2-AR exist. Considering molecular modeling techniques, this may be due to a large timescale (μs) of the considered process. Unbiased MD simulations of β2-AR interacting with various ligands performed so far (Plazinska et al., unpublished data) revealed that in the ‘active’ structure, the indole sidechain of W2866.48 is still ‘vertical’ with respect to the inner receptor channel. This is in agreement with both the crystal structures of activated β2-AR and the favourable W2866.48 orientation in the ‘inactive’ β2-AR.
In addition, GPCRs generally have wide and open entrances to their binding sites, which are readily accessible to solvent. The conformational changes that occur during activation of GPCRs include reorganizations of inner protein hydrogen bonding networks and contacts with internal waters.3,23,28 The details of how the water molecules enter the interior of the receptor and thereby affect GPCR activation at the atomic level were investigated by using molecular dynamics (MD) simulations.28–31 The simulations indicated that the water molecules can enter into the receptor during activation from two different directions: either from the extracellular or from the intracellular side depending on the receptor type.29 The internal water pathway affects not only the functional state of the GPCRs but also the change of the conformation of the highly conserved Y7.53 residue located in the NPxxY motif.29
There is still a lot of uncertainty about both the molecular details of the β2-AR activation mechanism and the role played by the ligand molecule in this process. In the present work, we are concerned with the influence of the ligand on the behaviour of the crucial molecular switch that governs the initial stages of β2-AR activation, namely W2866.48. The main goals of our present work can be summarized as follows:
1. to qualitatively and quantitatively assess the influence of the presence of ligands in the binding cavity of β2-AR on the conformational behaviour of W2866.48;
2. to investigate the dependence of the conformational motions of W2866.48 on the structural form of the receptor (i.e. ‘active’ vs. ‘inactive’ β2-AR);
3. to confirm whether the conformational behaviour of W2866.48 is associated with the pharmacological character of the ligand (i.e. agonist vs. antagonist vs. inverse agonist) or with the instantaneous receptor conformation;
4. to determine the role played by aromatic amino-acid residues neighbouring W2866.48 (i.e. Y3087.35, F209, F2896.51 and F2906.52) in the conformational rearrangements of W2866.48;
5. to clarify whether the ligand chirality is relevant for the W2866.48 conformational behaviour;
6. to associate the conformational preferences of W2866.48 expressed in geometrical/mechanistic terms with the solvation of the binding cavity.
Let us mention that these goals do not include studying the full activation of the receptor but rather the behaviour of the single W2866.48 residue in the context of the varying molecular environment (i.e. different ligands and receptor conformations). The analogous, more detailed description of the behaviour exhibited by the remaining elements of the toggle switch is postponed to our future work.
Regarding points 1–3, no distinction would be necessary when considering the physical systems and sufficiently large (i.e. of the order of ms) timescales. Then, the presence of the ligand of a given pharmacological type imposes the thermodynamically-stable conformation of the receptor. However, from the perspective of the molecular dynamics simulations, only limited timescales (tens of ns up to μs) are accessible. Therefore, we can distinguish the actual, instantaneous conformation of the receptor (which can be incompatible with the pharmacological type of the bound ligand) and check whether the behaviour of W2866.48 is the inherent feature of exclusively the protein conformation or rather of the type of bound ligand.
Furthermore, the conformational features of W2866.48 can be expressed in terms of its solvation state. This is especially important in the context of the possible role of water in the GPCR activation process and connection of the theoretical results with the experimental measurements.
Our investigations were focused on a group of functionally different ligands that can bind to β2-AR. More precisely, we considered the agonists ((R)-epinephrine, (R)-isoproterenol, (R,R)- and (S,S)-fenoterol), the inverse agonists (S-carazolol, S-timolol) and the antagonists (S-alprenolol, S-metoprolol). The structural formulae of the ligands are given in the ESI† as Fig. S1. Advanced computational methodologies (metadynamics-based, enhanced-sampling MD simulations) have been applied to achieve these goals. In addition, experimental methods (time-resolved fluorescence spectroscopy, TRFS) have been used to verify some of the results obtained with theoretical studies and to provide additional data on the behaviour of the tryptophan residues of β2-AR under different conditions (varying conformation of β2-AR induced by the presence of the agonist ligand).
TRFS is an extremely sensitive technique that has provided a wealth of insight into biological processes. The tryptophan emission typically dominates the fluorescence of a given protein32 and is a highly sensitive probe for changes of the protein secondary and tertiary structures. Moreover, the fluorescence emission from tryptophans is extremely sensitive to changes in the local environment.33 Therefore, the TRFS studies concerning proteins containing tryptophan residues can provide insight into protein conformational changes upon, e.g., ligand binding.14,34–40 Apart from the insight into the structural rearrangements of β2-AR, our work demonstrates the potential of the TRFS technique in connection with the advanced MD simulations for determining the ligand-induced conformational rearrangements that occur in any other, tryptophan-rich GPCR protein.
2. Methods
2.1 Experiment
000 × g for 10 min at 4 °C. The pellet contacting cell membranes was resuspended in buffer composed of 15 mM Tris–HCl, pH 7.4, 120 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, and 5 mM glucose. Protein content of the membrane fraction was assessed using the BCA Protein Assay Kit (Thermo Scientific/Pierce) according to the manufacturer protocol. Stocks containing 1 mg ml−1 protein were stored at −80 °C until further usage.
The spectrometer was equipped with a microchannel plate detector (Hamamatsu) and the monochromator set to 370 nm as an observation wavelength. The spectral bandwidth of the decay measurements was ∼5.4 nm. The intensity decay data were analyzed by FluoFit software using the multi exponential model:
 | (1) |
 | (2) |
The exposure of protein tryptophan residues to water molecules results in a decrease of tryptophan fluorescence lifetime because of the number of non-radiative processes.33 Tryptophan electronic excited states are extremely sensitive to collisions with solvent molecules causing their quenching. Based on this, groups of molecules characterized with the same fluorescence lifetime have been prescribed different accessibility to water. Next, the experimental data have been correlated with calculations regarding the radial distribution of tryptophan sidechains to water (see the next section). Taking into account the Smoluchowski equation describing the efficiency of fluorophore quenching expressed by the diffusion-controlled bimolecular rate constant k:
| k = 4πNRcD, | (3) |
2.2 Simulations
The two β2-AR models (In_β2-AR – representing the inactive state, and the Ac_β2-AR model – representing the active state) with bound ligands were inserted into equilibrated palmitoyl-oleoyl-phosphatidylcholine (POPC) cell membrane model applying the InflateGro procedure (http://www.csb.bit.uni-bonn.de/inflategro.html) and solvated with ∼16300 SPC42 water molecules. We have used the GROMACS-inherent,43 solvate/genbox protocol that fills the protein cavity using the predefined, equilibrated water box and removes the possible steric clashes. The presence of crystalized water molecules was accounted for as well. The sodium ions were added where necessary to neutralize the total charge. Additionally, In_β2-AR and Ac_β2-AR without bound ligands were simulated as well. After solvation, the systems were subjected to the energy minimization protocol (steepest descent algorithm) and a series of constant-volume MD simulations with positional constraints to equilibrate the system and keep the ligand molecules in their initial positions. This stage lasted for a total of 15 ns. Subsequently, 5 ns of unconstrained MD simulation was performed under constant volume conditions, followed by the final, 5 ns-long, unconstrained MD simulations under constant pressure. The receptor structures containing point mutations were prepared from already equilibrated systems by: (i) gradually (5 ns) turning off all the nonbonded interactions between the sidechain of the residue being converted to alanine and the rest of the system; (ii) simultaneous conversion of the nonbonded interactions characteristic of the Cβ carbon atom of the native residue to those of the methyl group; (iii) equilibration of such a system by a further 5 ns of unbiased MD simulation.
In most of the cases, the type of β2-AR structure used to construct the β2-AR–ligand complexes was consistent with the pharmacological role of the ligand, i.e. In_β2-AR corresponded to bound antagonists or inverse agonists whereas Ac_β2-AR corresponded to agonists. The three exceptions were the Ac_β2-AR–carazolol, In_β2-AR–(R)-epinephrine and In_β2-AR–(R,R)-fenoterol systems. The ligand structures used in the study are given in Fig. S1 (ESI†).
The simulations concerned the following nineteen systems: (i) unliganded β2-AR, in its active (Ac_β2-AR) and inactive (In_β2-AR) conformational states; (ii) β2-AR in complex with agonists: Ac_β2-AR–(R)-epinephrine, Ac_β2-AR–(R)-isoproterenol, Ac_β2-AR–(R,R)-fenoterol, Ac_β2-AR–(S,S)-fenoterol, In_β2-AR–(R)-epinephrine, and In_β2-AR–(R,R)-fenoterol; (iii) complexes with the inverse agonists: In_β2-AR–(S)-carazolol, In_β2-AR–(S)-timolol, and Ac_β2-AR–(S)-carazolol; (iv) complexes with the antagonists: In_β2-AR–(S)-alprenolol and In_β2-AR–(S)-metoprolol; (v) complexes of the Ac_β2-AR conformer containing point mutations in the apo form or in complex with various ligands: Ac_β2-ARY308A, Ac_β2-ARF289A–(R)-epinephrine, Ac_β2-ARF290A–(R)-epinephrine, Ac_β2-ARY209A–(R)-epinephrine, Ac_β2-ARY308A–(R)-epinephrine, and Ac_β2-ARY308A–(R,R)-fenoterol. The full list of simulated systems and the corresponding simulation type is provided in Table S1 (ESI†).
The procedure of docking was performed using the binding cavity within the sphere of radius 11 Å, covering the ligands originally co-crystallized with β2-AR and the closest residues, e.g., D113, S203, S204, S207, N293, N312, Y308, C191, D192 and F193.1–4,45 The estimation of the ligand–protein interactions was described by the MVD-implemented scoring function (MolDock Score). The predicted positions of the ligands in the β2-AR cavity were characterized by a simultaneous lowering of the scoring function values; this corresponds to the high values of the ligand binding energy. Further details on the validation procedure, final positions of the docked ligands and the corresponding MolDock Score values are given in the ESI† (Fig. S2, S3 and Table S2).
The PME method56 was applied for treatment of the long-range electrostatic interactions with the 0.9 nm cutoff. The cutoff for the Lennard-Jones interactions was 1.4 nm. These values are required for appropriate POPC bilayer simulation. The equations of motion were integrated using the leapfrog scheme57 with a timestep of 2 fs. During the MD runs, the LINCS algorithm58 was applied to constrain all hydrogen atom-containing bond lengths. The simulations were carried out under periodic boundary conditions based on rectangular computational boxes (initial dimensions of 7.22 × 7.22 × 13.12 nm3). The temperature was maintained close to its reference value (310 K) by applying the V-rescale thermostat59 whereas, for the constant pressure (1 atm, isotropic coordinate scaling), the Parrinello–Rahman barostat was used with a relaxation time of 0.4 ps.60 The center of mass motion was removed every step (separately for the following groups: solvent + ions, protein + ligand, lipid bilayer).
Before the enhanced sampling protocol, standard, unbiased MD simulations were performed for ∼5 ns to fully equilibrate the systems and relax the pressure. During this stage, the position of the ligand in the binding cavity was monitored to ensure it remained stable. The corresponding data (expressed as the distance between the amine group of each of the ligands and the D113 residue) are given in Fig. S4 (ESI†). The calculated 2D free energy maps (FEMs) were associated with the following variables: (i) the χ2 dihedral angle of the W2866.48 residue defined by the Cα–Cβ–Cγ–CD quadruplet of atoms; (ii) the χ1 dihedral angle of the W2866.48 residue defined by the N–Cα–Cβ–Cγ quadruplet of atoms. See Fig. 2 for the notation. During calculations, the well-tempered metadynamics61 protocol was applied, which scales the bias added to the system Hamiltonian. The metadynamics calculations were performed by GROMACS combined with PLUMED 1.3.62 The starting deposition rate was set to 0.6 kJ mol−1 ps−1 with the Gaussian width equal to 0.314 rad and a temperature parameter ΔT (see eqn (2) in ref. 61) equal to 1788 K. The latter parameter regulates the rate of scaling the bias ‘portions’ added to the system as the simulation proceeds. Its current value was chosen by a trial-and-error procedure, ensuring both fast and accurate sampling of 2D free energy maps defined by dihedral angle values. The metadynamics simulations were carried out for a duration of 75 ns for each system and the convergence of the resulting FEMs was checked by the sum_hills tool62 and hand-written bash scripts. If the convergence was not satisfactory, the metadynamics simulations were extended up to 120 ns. See ESI† for the related convergence plots (Fig. S5). The convergence of the FEMs was additionally confirmed during the subsequent local-elevation umbrella sampling calculations, performed for the selected systems. Note that currently no systematic, quantitative error-analysis procedure exists for any MD simulations with bias changing in time (e.g. metadynamics).
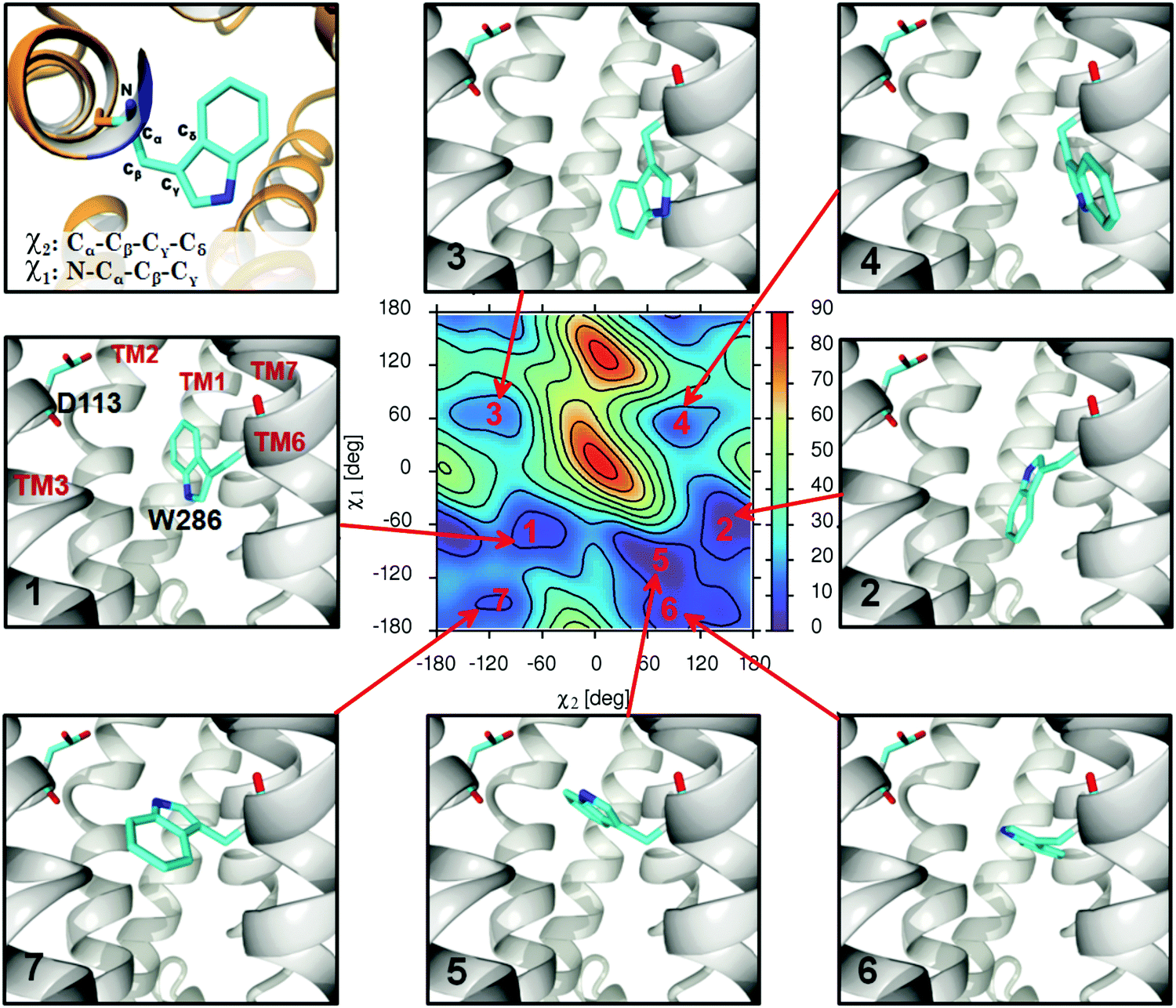 | ||
| Fig. 2 The depiction of the seven main types of the W286 sidechain conformers observed during MD simulations. The free energy map corresponds to the apo-In-β2-AR molecular system and the energy scale is [kJ mol−1]. See the Methods section for definitions of the χ1 and χ2 dihedral angles. | ||
The relative free energies of particular conformational states of W286 were calculated based on the division of the χ1vs. χ2 phase space into three distinct regions (see Fig. S6, ESI† and the discussion in further parts of the paper). The populations and free energies were calculated from the Boltzmann relation; the procedure was repeated every 100 ps to perform the convergence tests.
The radial distribution functions (RDFs) for the distance between water and all the tryptophan residues present in the β2-AR molecule were calculated for the following two systems: (i) the Ac_β2-AR–epinephrine complex and (ii) the unliganded In_β2-AR. This corresponds to the experimentally investigated systems and such simulations were required to facilitate the analysis of the measured data. The aim was to estimate the number of solvent molecules in the vicinity of each of the tryptophans present in the system. The center-of-mass (COM) of tryptophan was defined by using only the heavy atoms of the indole ring. The final data have the form of integrals (within the range 0–1.2 nm) of RDFs which, based on their values, can be translated into the character of the environment surrounding the given residue. Although the upper limit of the integral seems to be arbitrary, qualitatively, the same information can be obtained by changing it within the range 0.8–1.4 nm. Note also that the approximate diameter of the sphere defined by the full rotation of the tryptophan sidechain around the χ2 angle is equal to ∼1.2 nm; thus, the accepted value has a strong physical meaning as the size of the ‘environment’ experienced by the rotating tryptophan sidechain.
The calculated RDFs relied both on the unbiased MD simulations for a duration of 30 ns and on the local-elevation umbrella sampling (LEUS)63 protocol. The LEUS procedure was introduced to account for the conformational mobility of the W2866.48 residue, which is central to the study. LEUS allows calculation of the superposition of RDFs that accounts for all the relevant conformational states of W2866.48. The previously calculated χ1vs. χ2 FEMs with the reversed sign of the free energy values were used as the bias allowing the system to migrate between various free energy minima within a relatively short timescale. The LEUS-based simulations were performed for a duration of 75 ns and the RDFs were calculated every 1 ps. In the subsequent step, all the biased RDFs were reweighted by using the Boltzmann relation, inherent to LEUS,63 and averaged to provide the single, unbiased RDF function accounting for the different conformational states of W2866.48. All the LEUS-related calculations were performed by GROMACS combined with PLUMED 1.3 with transformation of the 2D FEMs into 2D grids of dimension 200 × 200 bins.
In order to show the connection between the conformational states of the W286 residue and its solvation, the biased MD simulations were performed for the apo forms of both Ac_β2-AR and In_β2-AR for a duration of 30 ns. The introduced constraints accounted for maintaining either the OS (χ1 = 60 deg, force constants = 6000 kJ mol−1 rad−2) or the CS conformer of the W286 sidechain (χ1 = −150 deg, force constants = 6000 kJ mol−1 rad−2). See Fig. S6, ESI† for definitions of the conformational states.
3. Results and discussion
3.1 Fluorescence spectroscopy
β2-AR contains eight tryptophan residues throughout the 397 amino-acid sequence of the protein (Fig. 1), thus, the tryptophan fluorescence can be used to probe the conformational changes of the receptor.14,36The TRFS investigation was performed for the unliganded, wild type of β2-AR and the β2-AR–(R)-epinephrine complex. The unliganded β2-AR is expected to correspond to the ‘inactive’ conformation of β2-AR (In_β2-AR), investigated during the computational part of the study. Analogously, the β2-AR-(R)–epinephrine complex corresponds to the ‘active’ conformer of β2-AR (Ac_β2-AR). Fig. 3 presents the fluorescence decays recorded from the β2-AR samples of these two types. The insets show fluorescence lifetime components, determined based on the deconvolution analysis, along with relative amplitudes. The fact that the protein comprises 8 tryptophan residues responsible for fluorescence emission implies that the given tryptophan emits fluorescence with a characteristic lifetime, determined most probably by the polarity of its nearest vicinity. Considering the unliganded β2-AR, the distribution of the amplitudes of fluorescence lifetime suggests that two tryptophans are localized in the most hydrophobic environment (τ1 = 5.76 ns), a further three in a more polar environment (τ2 = 2.21 ns) and also three tryptophans in a highly polar environment (τ3 = 0.25 ns). An attempt to more specifically assign tryptophan residues, localized in the crystallographic structure of β2-AR, to particular fluorescence lifetime components can be based on the local-elevation umbrella sampling (LEUS-MD)-derived probabilities of the localization of water molecules in the vicinity of each of the tryptophans in the In_β2-AR system (see ESI,† Table S3). Based on the probability at a distance of 1.2 nm, one can assign the W313 and W1584.50 residues to the fluorescence lifetime component τ1 = 5.76 ns, W1093.28, W1053.24 and W2866.48 to the fluorescence lifetime component τ2 = 2.21 ns and W173 (the second extracellular loop, ECL2), W99 (ECL1) and W321.31 to the fluorescence lifetime component τ3 = 0.25 ns.
 | ||
| Fig. 3 (lft) Fluorescence intensity decays measured for unliganded β2-AR and (right) β2-AR in complex with (R)-epinephrine. The decays were fitted with a deconvoluted multi-exponential model using the instrument response function (given in red). Insets to figures show fractional amplitudes of the decay analysis; the colour code represents the character of the environment around the given tryptophan residue: polar (violet), intermediate (green) or non-polar (orange). Average lifetimes 〈τ〉 and goodness of fit (χR2) are indicated. | ||
The fluorescence kinetics for the β2-AR–epinephrine system, along with fluorescence lifetime distribution, is presented in Fig. 3. Again, tryptophan residues can be concluded to occupy locations in the protein characterized by different polarities. Interestingly, the binding of β2-AR to epinephrine results in a pronounced redistribution of fluorescence lifetime components, without considerably changing the amplitude averaged fluorescence lifetime (2.35 ns vs. 2.19 ns) in the control; the magnitude of both these values is in agreement with the already existing data.36,64 Note that such a redistribution can be ascribed not only to the presence of ligand in the binding cavity, but also to the agonist-triggered conformational rearrangements of the β2-AR molecule (i.e. receptor activation). The assignment of fluorescence lifetime components to particular residues was based on analogous LEUS-MD simulations concerning the Ac_β2-AR–epinephrine system (see Table S3, ESI†). In this case, the pool of tryptophan residues in a highly hydrophobic environment (τ1 = 6.3 ns) is represented by only one element and the tryptophan pool strongly exposed to interactions with water molecules (τ3 = 0.82 ns) increased to four elements. According to the assignment criteria identical with those of In_β2-AR, W1584.50 emits fluorescence with the characteristic lifetime of τ1 = 6.3 ns, three residues: W3137.40, W173 (ECL2) and W1053.24 emit with the characteristic lifetime τ2 = 2.8 ns and four residues: W321.31, W1093.28, W2866.48 and W99 (ECL1) emit with the characteristic fluorescence lifetime τ3 = 0.8 ns. During the assignment procedure, the weights of the contribution of each particular characteristic lifetime were not imposed by any constraints. The fact that their values are always very close to a multiple of 12.5% (i.e. 1/8, which reflects the total number of tryptophan residues in the system) confirms the correctness of the assignment.
The observation that the binding of epinephrine affects the fluorescence lifetimes assigned to the W2866.48 residue is fully reflected by the results of separate simulations aimed at determining the free energy maps related to the conformational motion of the W2866.48 sidechain. This issue is described and discussed in detail in Section 3.2. Let us mention here that the correct reproduction of the experimental results was achieved only after accounting for all the possible conformers of the W2866.48 sidechain and their relative weights. This was possible due to applying the LEUS procedure and the reweighted RDFs and their integrals (see ESI,† Fig. S7–S9) correspond to the 2D energy maps discussed in the next section. The simpler, unbiased MD simulations may provide incomplete information about the conformational flexibility of the W2866.48 residue, limited to the single minimum of the free energy. This does not change much in the case of unliganded β2-AR but has a large implication for the correct interpretation of the W2866.48 behaviour in the activated β2-AR and emphasizes the necessity of using the enhanced-sampling MD simulations.
The fact that the binding of epinephrine to β2-AR mostly affects the fluorescence lifetimes assigned to the residues W3137.40, W1093.28 and W2866.48 implies localization of the cofactor binding site in this region of the receptor protein. This is not surprising when considering the available crystal structures of the agonist–β2-AR complexes, including the epinephrine itself (PDB: 4LDO). However, the uniform character of the changed lifetimes (i.e. their decrease) suggests the change of the environment of the related tryptophans from a less to a more polar one. As the crystal structures of both the active and inactive conformers of β2-AR are nearly identical with respect to the vicinity of the considered tryptophan residues, this cannot be explained in terms of changes in the secondary protein structure but requires accounting for the presence of solvent and dynamic motion or the molecular system. The decreased τ values characteristic of the β2-AR–epinephrine complex are correlated with the larger number of water molecules in the region of the binding cavity, in comparison to the unliganded receptor. This is in agreement not only with the results of simulations presented further but also with the findings reported elsewhere, stating that the influx of water into the orthosteric site of GPCRs (located close to W3137.40, W1093.28 and W2866.48) is associated with the process of receptor activation.24,29
Finally, let us comment on the behaviour of W173 (ECL2), which, upon the epinephrine-induced activation of β2-AR, increases its characteristic lifetime; this is associated with the transfer from a more to a less polar environment. This residue is exposed outside the receptor channel and located approximately at the interface between the extracellular and transmembrane parts of β2-AR. According to the visual inspection of the MD trajectories, the conformational changes of the β2-AR molecule, accompanying the activation process, facilitate the immersion of W173 deeper into the lipid bilayer and reduce its contact with water. As W173 is located far from the binding cavity and does not exhibit any contact with the ligand molecule, it is not likely that any changes concerning this tryptophan will have an influence on the ligand-induced activation process.
This stage of the study confirms that the agonist induced activation of β2-AR is a complicated process, involving not only residues in the vicinity of the binding cavity but also those located in the extracellular part. More precisely, four (out of eight possible) tryptophan residues changed their environment upon the ‘inactive’ → ‘active’ conformational change. W3137.40, W1093.28 and W2866.48, located closest to the ligand binding site among all of the tryptophans, exhibit an increase of the polarity of their environment, which can be associated with the larger flow of water into the inner receptor channel. Additionally, W1584.50 always occupies the hydrophobic environment, independent of the receptor conformation. This is in line with the recent findings reporting the W158–cholesterol interactions for membrane-embedded β2-AR.65
Finally, let us mention that the choice of the two molecular systems subjected to experimental measurements relied on the results obtained during MD simulations and described in the next section. As will be shown, these two systems exhibit limiting behaviours in the context of conformational states of the W2866.48 residue. Therefore, studying them experimentally was most promising in view of the expected results.
3.2 Analysis of free energy maps (FEMs)
In general, all the calculated FEMs exhibit a similar pattern of free energy minima. Therefore, we introduced the numbering of particular minima common for all systems. The maximum number of seven distinct free energy minima can be observed; however, in some cases not all of them appear. The connection of these free energy minima with the given conformational states of the W2866.48 sidechain is graphically illustrated in Fig. 2.
Minima 1 and 2, which usually correspond to the lowest free energy values (i.e. the most favourable conformations) are associated with similar conformations where aromatic rings of W2866.48 are arranged parallel to TM6. Due to the definition of χ2 and the internal symmetry of the system, related patterns can also be distinguished in the cases of the minima pairs: 3/4 and 6/7. This is because of the rotation of the W2866.48 indole ring around the vector defined by Cβ and Cγ atoms; such a transformation is weakly correlated with the value of the χ1 angle. Therefore, at the fixed χ1 value, two distinct conformers are expected to exist, differing in the phase shift (±180 deg) of the χ2 angle. The only exception is minimum 5, which appears as a distinct conformational state in 6 (out of 19) considered systems, and in the remaining ones, its counterpart is either absent or belongs to a larger basin of free energy shared with minima 1 or 2.
The dihedral angle χ1 better represents those conformational changes of W2866.48 that may be essential for the activation/deactivation processes in comparison to χ2. The latter rather informs about the rotation of the indole ring, which (with one exception of the structure associated with minimum 5 on the FEMs) does not contribute much to channel clearance. Therefore, it is recommended to use χ1 instead of χ2 as the first, one-dimensional approximation. In the majority of the theoretical studies concerning GPCRs, the focus is on the χ1 angle.26 However, in some special cases (i.e. where there exists a significant contribution of the structures characteristic of minimum 5), this may be confusing as similar values of χ1 angle are exhibited by different conformers (assigned here to the GS, OS and CS sets; see the next sub-section for definitions).
Fig. S11 and S12 (ESI†) contain FEMs calculated for various systems containing Ac_β2-AR and In_β2-AR conformers of β2-AR, respectively. All the W2866.48 conformers extracted from the available crystal structures of β2-AR always occupy the same region of the FEMs, namely minimum 1. However, as can be seen in Fig. S11 and S12 (ESI†), the collection of allowed conformations is much wider. The most favourable ones are always those associated with either minimum 1 or 2, i.e. close to the crystallographic structures.
The free energy barriers between particular W2866.48 conformers are relatively large (i.e. of the order of tens of kJ mol−1). Therefore, there is no possibility of proper sampling of all existing energy minima including the metastable states due to a large (μs) timescale of the W286 sidechain reorientation (however, single reorientation events can be observed; see ref. 21 and 26).
The presence of ligand in the binding cavity is not necessary to trigger the conformational rearrangements of the W2866.48 sidechain, as indicated by the complex landscape of FEMs calculated for both apo forms of β2-AR. This is consistent with the experimental observations indicating that β2-AR can undergo the ‘active’–‘inactive’ interconversion even in the absence of agonist.66,67 However, as the conformational behaviour W2866.48 is rather complex and can be expressed in terms of the dynamic equilibrium between particular conformational states, the presence of ligand in the binding cavity can influence this dynamic equilibrium, shifting it in diverse directions. This is discussed in the next paragraphs.
With reference to the experimental data, it is important to associate the particular types of the tryptophan conformers with the solvation state of the given residue. This issue has been addressed for W2866.48 by a series of separate MD simulations with introduced constraints. The results, given in the ESI† (Fig. S10), confirm that the OS conformers facilitate the increase of the solvation of the W2866.48 sidechain in the case of the Ac_β2-AR–epinephrine complex. In contrast, this is not the case of the unliganded In_β2-AR, where only minor changes were observed. Although the results given in Fig. S10 (ESI†) cannot be treated quantitatively due to the introduced constrains, they are qualitatively compatible with both the experimental results and the LEUS-derived data collected in Table S3 and depicted in Fig. S9 (ESI†).
When the ligand type (agonist vs. antagonist/inverse agonist) is compatible with the expected conformation β2-AR (‘active’ vs. ‘inactive’), some trends can be observed in the population of particular conformational states of W2866.48. This trend can be examined by using the assignment scheme (shown in Fig. S6, ESI†) aimed at calculating the relative free energies of the CS, GS, and OS. More precisely, in the case of In_β2-AR and its complexes with antagonists/inverse agonists, the populations of the states can be ordered as follows: GS > CS > OS. The quantitative data are illustrated graphically in Fig. 4. The free energy differences between GS and CS vary between +1.5 and +14 kJ mol−1. In contrast, for complexes including Ac_β2-AR and the agonists as well as for apo-Ac_β2-AR, the related trend is changed into the following: GS > OS > CS. The free energy difference between the first and the second most populated state varies from +5 to +12.5 kJ mol−1. This observation suggests that the occurrence of any of the above trends is correlated with either the particular receptor conformation (Ac_β2-AR vs. In_β2-AR) or with the type of ligand bound to β2-AR (agonist vs. antagonist/inverse agonist). Moreover, the relative energies of the OS in Ac_β2-AR are (on average) significantly lower than those of the OS in In_β2-AR, which suggests that the former system is much more prone to clearance of the inner receptor channel. Finally, it is worth noting that both apo forms of the receptor exhibit a uniform trend in the populations of the OS and CS; namely, in both cases, the CS conformation is preferred over the OS, which implies that β2-AR maintains the inner channel as closed in the absence of any ligand.
 | ||
| Fig. 4 The relative free energies associated with the CS and OS conformational states of W2866.48. The energies are relative with respect to the energy of the most populated state, i.e. the GS, which is assumed to be zero. (A) The values calculated for the In_β2-AR-antagonist/inverse agonist complexes. (B) The values calculated for the systems with a receptor conformation incompatible with ligand type (i.e. Ac_β2-AR-inverse agonist or In_β2-AR-agonist). (C) The values calculated for the Ac_β2-AR-agonist complexes. | ||
In the case of physical systems, the ligand pharmacological type is unequivocally associated with the receptor conformation (i.e. binding of an agonist results in a stable active conformer of β2-AR whereas binding of an antagonist leads to a stable inactive β2-AR conformer). A similar correlation has been assumed in the molecular systems described above. However, such an approach does not resolve the ambiguity in finding the reasons behind the above-mentioned trends in populations of the W2866.48 conformers. They can be associated with either conformation of the receptor (i.e. all liganded Ac_β2-AR exhibit the same trend, diverse in comparison to that of In_β2-AR) or the ligand type (i.e. the trend may change upon binding of different ligands to the same conformer of the receptor). This issue has been addressed by a separate series of simulations. The results are given in Fig. S13 (ESI†) as FEMs and in Fig. 4 as free energy differences between the GS and CS or OS. It appears that both the above-mentioned trends are changed in the case of complexes including the ligand of the pharmacological type not compatible with the receptor conformation, e.g. In_β2-AR–(R)-epinephrine, In_β2-AR–(R,R)-fenoterol and Ac_β2-AR–(S)-carazolol. There, the populations of the states for agonists can be ordered as follows: GS > OS > CS, whereas for the inverse agonist: GS > CS > OS. Thus, the presence of ligand of the pharmacological character incompatible with the receptor conformation induces the changes in the W2866.48 conformation in accordance with the ligand character (i.e. toward CS in the case of the inverse agonist and toward OS in the case of agonists). The change in the above population trends is in agreement with the expectation that the presence of agonist in the binding cavity will facilitate the receptor activation (i.e. will favour the solvation of the inner channel) whereas the presence of antagonist/inverse agonist will cease the process of activation (i.e. will favour the desolvation of the inner channel). This finding also links the pharmacological character of the ligand molecule with its influence on the single molecular switch of β2-AR; in contrast to the full activation process, the behaviour of the latter can easily be predicted on the basis of MD simulations.
The interpretation of FEMs in terms of the three distinct states representing gradually increasing clearance of the inner channel is justified in the context of other observations, indicating that opening the channel is an essential step of the full GPCR activation. Related studies concern both the experimental data obtained for the rhodopsin-like G-protein-coupled receptors11,13,14,68,69 as well as the results of MD simulations of the mutation-containing β2-AR.21,26 However, no analogous results have been reported so far for native and unlabeled β2-AR.
It is worth noting that interconversion between the particular conformers of W2866.48 has the character of dynamic equilibrium, i.e. at a timescale much larger than ∼1 μs, CS, GS and OS coexist in all the considered systems. Furthermore, the timescale that can be ascribed to the height of free energy barriers separating CS, GS and OS is of the order of μs, which may have an important implication in the further steps of the opening of the inner receptor channel. Namely, the time during which the toggle switch remains in the open state may be sufficient for water molecules to further penetrate the channel and contribute to the subsequent stages of the activation process. According to the results of the LEUS-MD and the constrained MD simulations, the orientation of W2866.48 has a significant effect on the average number of water molecules in the binding cavity (see Table S3 and Fig. S11, ESI†). This is additionally confirmed by the results of the experimental studies given in Section 3.1 (see Fig. 3 and the related discussion) and links the results shown in this section (i.e. the populations of the GS, OS, and CS) with the solvation of the binding cavity.
Fig. S14 (ESI†) shows how FEMs are altered upon introducing the four different point mutations: F289A, F290A, Y209A and Y308A, whereas Fig. 5 contains the free energy differences between particular conformational states of W286. The mutated residues either belong to the molecular switch of interest (F2896.51 and F2906.52) or are believed to play a crucial role in the ligand-triggered activation of receptor (Y3087.35).70 Recently, Tandaleet al. observed the rotameric flip of W2866.48 in the system containing β2-AR with the F290S point mutation.26 It has been suggested that the aromatic stacking between F290 and W286 stabilizes the active conformation and the formation of this lock plays an important role in transmembrane domain movements in the activation mechanism for rhodopsin-like receptors (class A GPCRs).72
 | ||
| Fig. 5 The relative free energies associated with the CS and OS conformations of W2866.48 in the ‘active’ conformer of the β2-AR (either unliganded or bound to agonist ligand) containing point mutation. The rest of the details are as in Fig. 4. | ||
The relevance of the considered mutation depends on both ligand type and mutation position. For instance, the Y308A mutation drastically changes the conformation of W2866.48 in the case of the Ac_β2-ARY308A–(R,R)-fenoterol complex and shifts the dynamic equilibrium toward the CS whereas the OS is virtually nonexistent (free energy level above 30 kJ mol−1). Similarly, the larger contribution of the CS is observed in apo-β2-ARY308A in comparison to the native β2-AR. In contrast, the FEM calculated for the β2-ARY308A–(R)-epinephrine complex is more similar in comparison to that corresponding to the native β2-AR. The results obtained for the Ac_β2-ARY308A–(R,R)-fenoterol complex are in agreement with the data indicating the special role of Y3087.35 in both ligand binding and the ligand-induced β2-AR activation.70 Thus, the present results give a molecular interpretation to the experimentally-inferred observation that the specific interactions between the ligand and the Y3087.35 residue of β2-AR stabilize the receptor conformations favouring the receptor–Gs protein coupling and subsequently result in Gs-biased agonism. However, the experimental studies were focused only on fenoterol and did not include epinephrine. Therefore, the diverse results obtained in the latter case suggest that the mechanism of Y3087.35 action may vary depending on the ligand type. Additionally, the different influence of Y3087.35 on the landscape of both considered FEMs can be ascribed to the difference between the dimensions of the epinephrine and fenoterol molecules; the larger fenoterol molecule is able to directly interact with Y3087.35 (by means of hydrogen bonding and π–π interactions), whereas the smaller epinephrine ligand is unable to create such interactions due to the lack of the p-hydroxy substituent.
Among the remaining three point mutations, the Y209A substitution seems to have the smallest effect on the balance between the GS, OS and CS. Thus, in spite of the fact that Y2095.48 is involved in the interactions with the other residues creating the toggle switch, such interactions are not relevant in the context of the mechanism of the channel opening/closing by W2866.48. Interestingly, there exists a shift of the dynamic equilibrium toward positive values of χ2 that results in a deviation of the global energy minimum from that characteristic of the crystal structure of β2-AR. In the case of both the F289A and F290A mutations, additional changes are observed that result in an alteration of the balance between the CS, GS and OS. Introducing the F290A mutation results in a large domination of the CS over the remaining states (this is also the only case when the GS is not the favourable state), whereas the presence of the F289A mutation is correlated with the larger population of the OS in comparison with the native β2-AR–epinephrine complex. Thus, it can be concluded that the elements of the toggle switch may exhibit different directions of action when not considering them independently but not as a whole. There is no experimental data related to the point mutations described above; however, Tandale et al. observed that reorientation of W2866.48 was possible only upon introducing the F290S mutation into the native structure of β2-AR.26 The mentioned reorientation corresponds to the change of the GS into conformations closer to the CS, which is in line with our data. Furthermore, the resulting structure exhibited the rotation of the indole ring that corresponds to the positive values of the χ2 angle, i.e. also in agreement with our results (see Fig. 4 in ref. 26). Thus, we demonstrated that F2906.52 strongly affects the W2866.48 conformation, and its interactions within the rotamer toggle switch stabilize the ‘active’ conformations of W2866.48.
4. Conclusions
Theoretical, molecular dynamics-based studies, accompanied by the time resolved fluorescence spectroscopy experiments, were carried out in order to investigate the conformational behaviour of the W2866.48 residue of β2-AR. This residue and its conformational motions are believed to play a substantial role in the process of receptor activation in spite of the fact that both main conformers of β2-AR (i.e. ‘active’ and ‘inactive’ β2-AR) exhibit roughly the same orientation of the W2866.48 sidechain, which is also the case of the remaining elements of the ‘rotamer toggle switch’. This suggests that the possible role of W2866.48 should be considered in the formation of the dynamic, metastable states that may eventually become essential steps on the path leading to the full activation of β2-AR. In the theoretical part of our study, we focused on how both the conformation of the receptor and the presence of different ligands in its binding cavity influence the conformational behaviour of W2866.48. The results show that W2866.48 exhibits multiple conformational states either in the presence or absence of ligands in the binding cavity. In some cases, the favourable conformation is the same as that exhibited by the crystal structures but in other cases there may exist a substantial contribution of the non-crystallographic conformers. The pharmacological character of the ligand correlates well with the population of the particular conformers of W2866.48. Namely, conformers that increase the solvation in the region of W2866.48 and facilitate the access of water molecules to the binding cavity are characteristic of agonists (epinephrine, fenoterol). On the other hand, conformers that decrease the solvation in the binding cavity are characteristic of antagonists and inverse agonists (metoprolol, carazolol). These findings allow the pharmacological character of the ligand to be connected with its influence on the single molecular switch of the receptor and indicate that the related computational methodology can be used in order to indicate the ligand character. Furthermore, they are compatible with the hypothesis that the opening of the inner receptor channel is an essential step that leads to the full activation of the GPCR receptors. The experimental data are in line with the results of the simulations and show that the presence of the agonist molecule promotes a conformational change in the receptor that leads to an increase in the polarity of the environment around W3137.40, W2866.48 and W1093.28, which are located closest to the binding cavity. However, from the perspective of simulations, accounting for contributions of all the conformers of W2866.48 (including those that are not present in the crystal structures of β2-AR) is necessary to appropriately reflect and interpret the experimental data. We have discussed the role of point mutations in the vicinity of the orthosteric site to conclude that the mutations involving F289A, F290A, Y209A and Y308A may lead to diverse influences on the behaviour of W2866.48, which is additionally dependent on the ligand type. The role of the Y3087.35 residue is confirmed as significant for the ligand-induced β2-AR activation whereas F2906.52 is shown to stabilize the active conformational state of the receptor.Finally, we have demonstrated that time-resolved fluorescence spectroscopy can be an extremely useful tool to provide important information regarding the ligand-induced conformational changes in GPCRs. This especially concerns the dynamic aspects of such processes and other data that can be difficult to extract from crystal structures. However, the supporting role of the enhanced-sampling molecular dynamics simulations is crucial in the context of the appropriate interpretation of the collected data.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge financial support from the National Science Center (grant 2013/09/D/NZ2/02992) and the National Centre for Research and Development (Polish–Norwegian Research Programme, Small Grant Scheme, DZP/POL-NOR/252/2013).References
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS PubMed.
- S. G. Rasmussen, H. J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. Devree, D. M. Rosenbaum, F. S. Thian, T. S. Kobilka, A. Schnapp, I. Konetzki, R. K. Sunahara, S. H. Gellman, A. Pautsch, J. Steyaert, W. I. Weis and B. K. Kobilka, Nature, 2011, 469, 175–180 CrossRef CAS PubMed.
- S. G. Rasmussen, B. T. DeVree, Y. Zou, A. C. Kruse, K. Y. Chung, T. S. Kobilka, F. S. Thian, P. S. Chae, E. Pardon, D. Calinski, J. M. Mathiesen, S. T. Shah, J. A. Lyons, M. Caffrey, S. H. Gellman, J. Steyaert, G. Skiniotis, W. I. Weis, R. K. Sunahara and B. K. Kobilka, Nature, 2011, 477, 549–555 CrossRef CAS PubMed.
- A. M. Ring, A. Manglik, A. C. Kruse, M. D. Enos, W. I. Weis, K. C. Garcia and B. K. Kobilka, Nature, 2013, 502, 575–579 CrossRef CAS PubMed.
- J. A. Ballesteros and H. Weinstein, Methods Neurosci., 1995, 25, 366–428 CAS.
- V. Katritch, K. A. Reynolds, V. Cherezov, M. A. Hanson, C. B. Roth, M. Yeager and R. Abagyan, J. Mol. Recognit., 2009, 22, 307–318 CrossRef CAS PubMed.
- P. Scheerer, J. H. Park, P. W. Hildebrand, Y. J. Kim, N. Krauss, H. W. Choe, K. P. Hofmann and O. P. Ernst, Nature, 2008, 455, 497–502 CrossRef CAS PubMed.
- R. Nygaard, T. M. Frimurer, B. Holst, M. M. Rosenkilde and T. W. Schwartz, Trends Pharmacol. Sci., 2009, 30, 249–259 CrossRef CAS PubMed.
- B. K. Kobilka, Biochim. Biophys. Acta, 2007, 1768, 794–807 CrossRef CAS PubMed.
- T. W. Schwartz and M. M. Rosenkilde, Trends Pharmacol. Sci., 1996, 17, 213–216 CrossRef CAS PubMed.
- L. Shi, G. Liapakis, R. Xu, F. Guarnieri, J. A. Ballesteros and J. A. Javitch, J. Biol. Chem., 2002, 277, 40989–40996 CrossRef CAS PubMed.
- B. Holst, R. Nygaard, L. Valentin-Hansen, A. Bach, M. S. Engelstoft, P. S. Petersen, T. M. Frimurer and T. W. Schwartz, J. Biol. Chem., 2010, 285, 3973–3985 CrossRef CAS PubMed.
- E. Crocker, M. Eilers, S. Ahuja, V. Hornak, A. Hirshfeld, M. Sheves and S. O. Smith, J. Mol. Biol., 2006, 357, 163–172 CrossRef CAS PubMed.
- S. W. Lin and T. P. Sakmar, Biochemistry, 1996, 35, 11149–11159 CrossRef CAS PubMed.
- C. N. Rafferty, C. G. Muellenberg and H. Shichi, Biochemistry, 1980, 19, 2145–2151 CrossRef CAS PubMed.
- T. Okada, O. P. Ernst, K. Palczewski and K. P. Hofmann, Trends Biochem. Sci., 2001, 26, 318–324 CrossRef CAS PubMed.
- O. Fritze, S. Filipek, V. Kuksa, K. Palczewski, K. P. Hofmann and O. P. Ernst, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 2290–2295 CrossRef CAS PubMed.
- G. Lebon, T. Warne, P. C. Edwards, K. Bennett, C. J. Langmead, A. G. Leslie and C. G. Tate, Nature, 2011, 474, 521–525 CrossRef CAS PubMed.
- J. Standfuss, P. C. Edwards, A. D'Antona, M. Fransen, G. Xie, D. D. Oprian and G. F. Schertler, Nature, 2011, 471, 656–660 CrossRef CAS PubMed.
- F. Xu, H. Wu, V. Katritch, G. W. Han, K. A. Jacobson, Z.-G. Gao, V. Cherezov and R. C. Stevens, Science, 2011, 332, 322–327 CrossRef CAS PubMed.
- S. Bhattacharya, S. E. Hall, H. Li and N. Vaidehi, Biophys. J., 2008, 94, 2027–2042 CrossRef CAS PubMed.
- J. H. Park, P. Scheerer, K. P. Hofmann, H. W. Choe and O. P. Ernst, Nature, 2008, 454, 183–187 CrossRef CAS PubMed.
- H. W. Choe, Y. J. Kim, J. H. Park, T. Morizumi, E. F. Pai, N. Krauss, K. P. Hofmann, P. Scheerer and O. P. Ernst, Nature, 2011, 471, 651–655 CrossRef CAS PubMed.
- S. Yuan, Z. Hu, S. Filipek and H. Vogel, Angew. Chem., Int. Ed. Engl., 2015, 54, 556–559 CAS.
- S. Yuan, S. Filipek and H. Vogel, Structure, 2016, 24, 816–825 CrossRef CAS PubMed.
- A. Tandale, M. Joshi and D. Sengupta, Sci. Rep., 2016, 6, 24379 CrossRef CAS PubMed.
- A. Plazinska, W. Plazinski and K. Jozwiak, Eur. Biophys. J., 2015, 44, 149–163 CrossRef CAS PubMed.
- S. Yuan, H. Vogel and S. Filipek, Angew. Chem., 2013, 52, 10112–10115 CrossRef CAS PubMed.
- S. Yuan, S. Filipek, K. Palczewski and H. Vogel, Nat. Commun., 2014, 5, 4733–4743 CrossRef CAS PubMed.
- S. Yuan, U. Ghoshdastider, B. Trzaskowski, D. Latek, A. Debinski, W. Pulawski, R. Wu, V. Gerke and S. Filipek, PLoS One, 2012, 7, e47114 CAS.
- B. Isin, K. Schulten, E. Tajkhorshid and I. Bahar, Biophys. J., 2008, 95, 789–803 CrossRef CAS PubMed.
- J. T. Vivian and P. R. Callis, Biophys. J., 2001, 80, 2093–2109 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2010 Search PubMed.
- J. R. Alcala, E. Gratton and F. G. Prendergast, Biophys. J., 1987, 51, 597–604 CrossRef CAS PubMed.
- J. Fidy, M. Laberge, B. Ullrich, L. Polgar, Z. Szeltner, J. Gallay and M. Vincent, Pure Appl. Chem., 2001, 73, 415–419 CrossRef CAS.
- S. Lin, U. Gether and B. K. Kobilka, Biochemistry, 1996, 35, 14445–14451 CrossRef CAS PubMed.
- Y. Engelborghs, Spectrochim. Acta, Part A, 2001, 57, 2255–2270 CrossRef CAS.
- J. L. Baneres, A. Martin, P. Hullot, J. P. Girard, J. C. Rossi and J. Parello, J. Mol. Biol., 2003, 329, 801–814 CrossRef CAS PubMed.
- Y. Suzuki, E. Moriyoshi, D. Tsuchiya and H. Jingami, J. Biol. Chem., 2004, 279, 35526–35534 CrossRef CAS PubMed.
- K.-H. Ruan, V. Cervantes and J. Wu, Biochemistry, 2009, 48, 3157–3165 CrossRef CAS PubMed.
- M. Zubik, R. Luchowski, M. Puzio, E. Janik, J. Bednarska, W. Grudzinski and W. I. Gruszecki, Biochim. Biophys. Acta, 2013, 1827, 355–364 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, IntermolecularForces, ed. B. Pullman, Reidel Publishing Company, Dordrecht, 1981, pp. 331–342 Search PubMed.
- S. Pronk, S. Pall, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef CAS.
- A. Plazinska, M. Kolinski, I. W. Wainer and K. Jozwiak, J. Mol. Model., 2013, 19, 4919–4930 CrossRef CAS PubMed.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- A. Kukol and J. Chem, J. Chem. Theory Comput., 2009, 5, 615–626 CrossRef CAS PubMed.
- S. Canzar, M. El-Kebir, R. Pool, K. Elbassioni, A. K. Malde, A. E. Mark, D. P. Geerke, L. Stougie and G. W. Klau, J. Comput. Biol., 2013, 20, 188–198 CrossRef CAS PubMed.
- K. B. Koziara, M. Stroet, A. K. Malde and A. E. Mark, J. Comput.-Aided Mol. Des., 2014, 28, 221–233 CrossRef CAS PubMed.
- E. A. Cino, W.-Y. Choy and M. Karttunen, J. Chem. Theory Comput., 2012, 8, 2725–2740 CrossRef CAS PubMed.
- O. F. Lange, D. van der Spoel and B. L. de Groot, Biophys. J., 2010, 99, 647–655 CrossRef CAS PubMed.
- R. B. Best, N.-V. Buchete and G. Hummer, Biophys. J., 2008, 95, L07–L09 CrossRef CAS PubMed.
- W. Plazinski and A. Plazinska, Pure Appl. Chem., 2017, 89, 1283–1294 CrossRef CAS.
- A. Plazinska, W. Plazinski and K. Jozwiak, J. Comput. Chem., 2014, 35, 876–882 CrossRef CAS PubMed.
- A. Plazinska and W. Plazinski, Mol. BioSyst., 2017, 13, 910–920 RSC.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- R. W. Hockney, S. P. Goel and J. Eastwood, J. Comput. Phys., 1974, 14, 148–158 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- H. S. Hansen and P. H. Hünenberger, J. Comput. Chem., 2010, 31, 1–23 CrossRef CAS PubMed.
- P. Ghanouni, Z. Gryczynski, J. J. Steenhuis, T. W. Lee, D. L. Farrens, J. R. Lakowicz and B. K. Kobilka, J. Biol. Chem., 2001, 276, 24433–24436 CrossRef CAS PubMed.
- X. Prasanna, A. Chattopadhyay and D. Sengupta, Biophys. J., 2014, 106, 1290–1300 CrossRef CAS PubMed.
- R. Lamichhane, J. J. Liu, G. Pljevaljcic, K. L. White, E. van der Schans, V. Katritch, R. C. Stevens, K. Wüthrich and D. P. Millar, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14254–14259 CrossRef CAS PubMed.
- W. I. Weis and B. K. Kobilka, Curr. Opin. Struct. Biol., 2008, 18, 734–740 CrossRef CAS PubMed.
- S. Ahuja, V. Hornak, E. C. Yan, N. Syrett, J. A. Goncalves, A. Hirshfeld, M. Ziliox, T. P. Sakmar, M. Sheves, P. J. Reeves, S. O. Smith and M. Eilers, Nat. Struct. Mol. Biol., 2009, 16, 168–175 CAS.
- T. D. Dunham and D. L. Farrens, J. Biol. Chem., 1999, 274, 1683–1690 CrossRef CAS PubMed.
- A. Y. Woo, K. Jozwiak, L. Toll, M. J. Tanga, J. A. Kozocas, L. Jimenez, Y. Huang, Y. Song, A. Plazinska, K. Pajak, R. K. Paul, M. Bernier, I. W. Wainer and R. P. Xiao, J. Biol. Chem., 2014, 289, 19351–19363 CrossRef PubMed.
- K. Jozwiak, L. Toll, L. Jimenez, A. Y. Woo, R. P. Xiao and I. W. Wainer, Biochem. Pharmacol., 2010, 79, 1610–1615 CrossRef CAS PubMed.
- J. Mokrosiński and B. Holst, Methods Enzymol., 2010, 484, 53–73 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp04808d |
| This journal is © the Owner Societies 2018 |