
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The benchmark of 31P NMR parameters in phosphate: a case study on structurally constrained and flexible phosphate†
Jiří
Fukal
a,
Ondřej
Páv
a,
Miloš
Buděšínský
a,
Jakub
Šebera
a and
Vladimír
Sychrovský
*ab
aInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, v.v.i., Flemingovo náměstí 2, 166 10, Praha 6, Czech Republic. E-mail: vladimir.sychrovsky@uochb.cas.cz
bDepartment of Electrotechnology, Electrical Engineering, Czech Technical University, Technická 2, 166 27, Praha 6, Czech Republic
First published on 13th November 2017
Abstract
A benchmark for structural interpretation of the 31P NMR shift and the 2JP,C NMR spin–spin coupling in the phosphate group was obtained by means of theoretical calculations and NMR measurements in diethylphosphate (DEP) and 5,5-dimethyl-2-hydroxy-1,3,2-dioxaphosphinane 2-oxide (cDEP). The NMR parameters were calculated employing the B3LYP, BP86, BPW91, M06-2X, PBE0, KT2, KT3, MP2, and HF methods, and the 6-31+G(d), Iglo-n (n = II, III), cc-pVnZ (n = D, T, Q, 5), aug-cc-pVnZ (n = D, T and Q), and pcS-n and pcJ-n (n = 1, 2, 3, 4) bases, including the solvent effects described with explicit water molecules and/or the implicit Polarizable Continuum Model (PCM). The effect of molecular dynamics (MD) on NMR parameters was MD-calculated using the GAFF force field inclusive of explicit hydration with TIP3P water molecules. Both the optimal geometries and the dynamic behaviors of the DEP and cDEP phosphates differed notably, which allowed a reliable theoretical benchmark of the 31P NMR parameters for highly flexible and structurally constrained phosphate in a one-to-one relationship with the corresponding experiment. The calculated 31P NMR shifts were referenced employing three different NMR reference schemes to highlight the effect of the 31P NMR reference on the accuracy of the calculated 31P NMR shift. The relative Δδ(31P) NMR shift calculated employing the MD/B3LYP/Iglo-III/PCM method differed from the experiment by 0.16 ppm while the NMR shifts referenced to H3PO4 and/or PH3 deviated from the experiment notably more, which illustrated the superior applicability of the relative NMR reference scheme. The 2JP,C coupling in DEP and cDEP calculated employing the MD/B3LYP/Iglo-III(DSO,PSO,SD)/cc-PV5Z(FC)/PCM method inclusive of correction due to explicit hydration differed from the experiment by 0.32 Hz and 0.15 Hz, respectively. The NMR calculations demonstrated that reliable structural interpretation of the 31P NMR parameters in phosphate must involve both the structural and the dynamical components.
1. Introduction
The phosphate group is the key element within the sugar-phosphate backbone of nucleic acids. Although the major stabilizing effects in nucleic acids come from hydrogen bonding and stacking interactions between bases and base-pairs, their 3D framework is given by the conformation of the backbone.1–4 Orientations of the torsion angles adjacent to the backbone phosphate are distinctive for backbone conformational classes in nucleic acids.5 Much of the backbone flexibility originates from rotations around the phosphodiester linkage and that is why interconversions among distinct conformations of the backbone progress typically via the phosphodiester group. Structural resolution of the backbone phosphate is therefore central to all structural studies concerning nucleic acids.To date, the majority of the known structures of nucleic acids have been resolved in crystals by employing X-ray crystallography methods. In liquids, NMR spectroscopy is employed instead. The NMR parameters including the Nuclear Overhauser Effect (NOE), NMR chemical shift of atom A (δ(A)), indirect NMR spin–spin coupling between atoms A and B separated by n bonds (nJA,B), and residual dipolar coupling (RDC) can provide structural constraints for obtaining the 3D structure of DNA and RNA molecules.6 Moreover, probably the only method for the reliable refinement and testing of MD force fields can be currently done with structural data derived from NMR experiments.7 Hence, knowledge of the rules for the structural interpretation of NMR parameters is indispensable. The empirically derived rules can be employed; however, their accuracy need not be guaranteed in contrast to the ab initio calculations of NMR shifts and J-couplings.8 As for the nucleic acids, the NMR measurements and calculations highlighted the base pairing, the orientation of the base with respect to the sugar, the structure of the sugar-phosphate/phosphonate backbone in canonical/modified oligonucleotides aimed at medicinal applications, the oxidative damage of DNA, and the 3D structure of metallo-DNAs including Hg/Ag-mediated base pairs.9–25
The 31P NMR measurements conveniently employ the 100% natural abundance of the isotope. Recent calculations of the 31P NMR shifts in organophosphorus compounds differing by hundreds of ppm illustrated the reliability of the theoretical DFT methods.26 By contrast, the 31P NMR shifts in the nucleic acids differ typically at most by a few ppm which makes their assignment and interpretation truly challenging.27–30 The impact of the accurately interpreted 31P NMR parameters on the quality of NMR-resolved structures of nucleic acids is significant. The theoretical 31P NMR parameters were therefore intensively studied. Already pioneering calculations have unveiled the dependence of the 31P NMR shift on the geometry of phosphate.31,32 Later, by using advanced DFT calculations, Munzarová et al. illustrated that the A-RNA, BI/BII DNA, and Z-DNA backbone conformations are distinguishable with 31P shifts.33,34 Recently, Munzarová et al. calculated the dynamically averaged 31P NMR shifts in RNA and B-DNA by employing the combined MD/DFT calculation method.35,36 The calculations of 31P NMR shift and 2JP,C NMR spin–spin coupling in phosphate illustrated that various conformations of the backbone in nucleic acids can be distinguished with these NMR parameters.37 The effect of solvation due to Mg2+ on the NMR parameters was calculated.38 These NMR calculations demonstrated that the inclusion of the solvent and the dynamics involved affect the magnitude of the NMR parameters in phosphate. In the relation of the theoretical 31P NMR shift to experiments another issue should be considered; the effect of the 31P NMR reference. The δ(31P) NMR shift in nucleic acids is usually measured with respect to phosphoric acid; however, calculation of the δ(31P) may lead to erroneous assignment and misinterpretation. Another 31P NMR reference scheme, the secondary PH3 reference, has nowadays become popular in theoretical 31P NMR calculations.39 General aspects of the 31P NMR referencing were addressed in this work to unambiguously illustrate the performances of the currently available reference schemes and to demonstrate the effect on the structural interpretation of the 31P NMR shift.
As for J-coupling, the NMR parameter provides the so called long-distance structural restraint due to its dependence on the geometries of all the atoms within the NMR spin–spin pathway.40 In nucleic acids, the J-couplings were employed for NMR measurement/detection of the H-bond and for determination of the backbone conformations.41–46 A closer look at the backbone phosphate unveils the lack of suitable atoms measurable by means of NMR, except for the phosphorus and the nearest carbon and hydrogen atoms. The 31P NMR shift and 2JP,C NMR coupling are therefore unique NMR parameters in the phosphate.
Theoretical methods for calculations of the 31P NMR parameters have not been yet benchmarked because of the following reasons and obstacles. The previous 31P NMR calculations aimed at nucleic acids employed simplified models of the phosphate group to include all necessary effects at the level corresponding at least to DFT. The plausibility of the di-methyl phosphate or methyl-ethyl-phosphate that were previously employed to mimic the phosphate group in nucleic acids is therefore unclear. On the other hand, high-accuracy NMR calculations in the nucleic acids are hardly doable even nowadays and the usage of high-precision methods is usually balanced with the adequate size of the molecular model. The DEP and cDEP molecules depicted in Fig. 1 include presumably the smallest possible model of the phosphate group, which is still chemically similar to the phosphate in nucleic acids. As can be anticipated from their chemical structures, the geometries and dynamics of the DEP and cDEP phosphates should allow controllable examination of the effects on 31P NMR parameters. The benchmarked 31P NMR parameters in DEP and cDEP therefore illustrate the accuracy of the calculation methods that are applicable for 31P NMR structural studies of the phosphate.
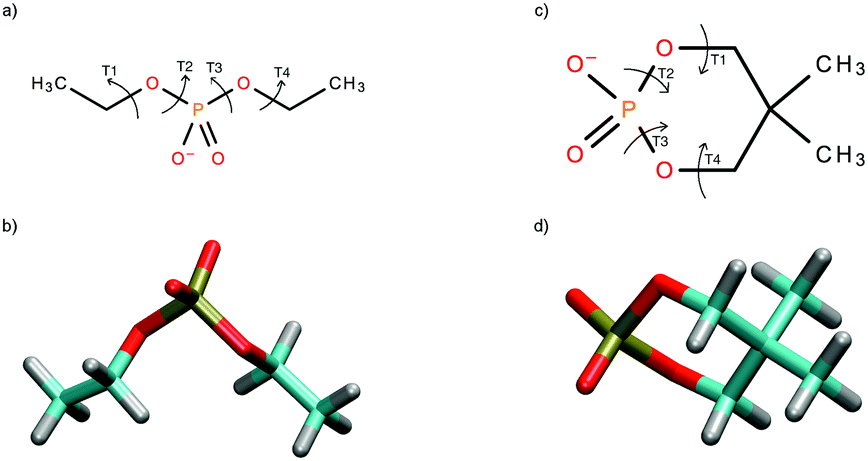 | ||
| Fig. 1 The chemical structure of diethylphosphate (DEP) (a) and 5,5-dimethyl-2-hydroxy-1,3,2-dioxaphosphinane 2-oxide (cDEP) (c) and the geometries of the energy minima calculated for DEP (b) and cDEP (d). The torsion angles T1 = C(H3)–C(H2)–O–P, T2 = C(H2)–O–P–O, T3 = O–P–O–C(H2), and T4 = P–O–C(H2)–C(H3) (the oxygen atoms within C–O–P linkage) were used for describing the geometries of the molecules. | ||
2. Methods
2.1. Experimental
The chemical structures of DEP and cDEP were confirmed by the NMR measurements (Table S1, ESI†).
2.2. The theoretical calculations
The geometry optimizations were carried out with the B3LYP47–50 method, the 6-31+G(d)51–55 atomic basis, and the polarizable continuum implicit solvation model (PCM)56 describing the water solvent. The dependence of the energy on the orientation of the torsion angles adjacent to the phosphorus atom in the phosphate group was calculated with the B3LYP method and the 6-31G(d,p) basis by employing a 30° × 30° geometric grid. The validity of all energy minima was checked with the vibration frequency calculation.The molecular dynamic (MD) calculations employed the GAFF force field57,58 and the ESP atomic charges59,60 calculated using the HF method and the 6-31G(d,p) basis. The 50 Å × 50 Å × 50 Å cubic box in MD calculations included the DEP or cDEP molecule, the TIP3P61 water molecules and one Na+ ion to maintain the electroneutral state of the system. The system was first equilibrated within the two-step procedure including heating and 30 ps of free NPT MD calculation. The production NVT MD calculation included 35 ns under the standard laboratory conditions; the temperature was 300 K and the pressure was 1 atm. The MD snapshot geometries equally separated by 100 ps including water molecules within the first solvation shell were employed. The 350 MD snapshots were geometry optimized keeping the T1, T2, T3, and T4 torsion angles and geometries of the oxygen atoms of the water molecules fixed (Fig. 1). The optimized MD snapshots were used for dynamical averaging of the NMR parameters. The MD snapshots of the NMR reference molecules H3PO4 and PH3 were calculated similarly. The 400 snapshots for H3PO4 and PH3 neglecting explicit water molecules were geometry optimized keeping the O–P–O and H–P–H valence angles fixed. The enhanced MD sampling for the cDEP within 20 ns MD calculation at the temperature 400 K was used to study the interconversions between the conformers of energy minima.
The calculated 31P NMR parameters included the 31P NMR shift of the phosphorus atom and the 2JP,C two-bond J-coupling between the phosphorus and carbon atom. The 31P NMR shift was calculated employing the GIAO approach within the Coupled Perturbed (CP) DFT method.62–64 The 2JP,C was calculated employing the CP DFT method as a sum of the Diamagnetic-spin orbit (DSO), Paramagnetic-spin orbit (PSO), Fermi-contact (FC) and Spin-dipolar (SD) contributions.65,66 The NMR parameters were calculated with the B3LYP method, the Iglo-III basis,67 and the PCM water solvent. Only the FC term of 2JP,C was calculated with the cc-pV5Z basis.68,69 The 31P NMR parameters were also calculated with the KT2,70 KT3,71 BP86,72,73 BPW91,72,74 M06-2X,75 PBE0,76 MP2,77,78 and HF79 methods. The explicit water molecules in NMR calculations were described with the 6-31+G(d) basis. The effect of atomic basis on NMR parameters was calculated with the 6-31+G(d), Iglo-II, Iglo-III and cc-pVnZ (n = D, T, Q and 5), aug-cc-pVnZ (n = D, T and Q), and Jensen's pcS-n and pcJ-n, n = 1, 2, 3, 4 bases.51–55,67–69,80–83 The NMR parameters calculated for the geometry of the global energy minima are called the static NMR parameters in the text. Accordingly, the statistically averaged NMR parameters are called the MD-averaged NMR parameters. The standard mean deviation from the statistical average SM was calculated for the MD-averaged NMR parameters as a sum SM = ∑((Ai − Aav)1/2/N), where Ai is the value of the respective NMR parameter in the i-th snapshot, and Aav = ∑(Ai/N) is the statistical average for N MD snapshots. The 31P NMR shift was calculated using three different NMR references.
The δ(31P) referenced to H3PO4 was calculated as δ(31P) = σ(31P) (in H3PO4) − σ(31P) (in DEP or cDEP) where the σ(31P) was the calculated 31P NMR shielding. The δ(31P)PH3 using the secondary PH3 reference was calculated according to van Wüllen as δ(31P)PH3 = σ(31P) (in PH3, neglecting solvent) − σ(31P) (in DEP or cDEP) − 266.1.39 The relative Δδ(31P) NMR shift was calculated as Δδ(31P) = σ(31P) (in DEP) − σ(31P) (in cDEP) = δ(31P) (in cDEP) − δ(31P) (in DEP), which allowed unbiased comparison of the calculated 31P NMR shift with the experiment neglecting the external NMR reference. The geometry optimizations and NMR calculations were carried out with Gaussian 09.D.01.84 Only the KT2 and KT3 31P NMR shifts were calculated with the Dalton 2016.0 program.85 The MD calculations were carried out with Amber 10.86
3. Results and discussion
3.1. The geometry of the DEP and cDEP phosphates
The DEP and cDEP phosphates are chemically similar due to the equivalent chemical bonding in the vicinity of the phosphorus atom. The magnitudes of the 31P NMR parameters therefore depend on the geometries of the DEP and cDEP phosphates. Two energy-equivalent minima were calculated for both DEP and cDEP because of the molecular symmetry (Table 1). In DEP, the orientations of the T2 and T3 torsion angles were both 72.8° and for the symmetry-associated energy minimum it was −72.8°. We used the −180° to 180° convention rather than 0° to 360° to display clearly the dependences of the energy and NMR parameters on T2 and T3. In cDEP, T2 and T3 were −59.2° and 59.2°, and for the symmetry-associated minimum they were 59.2° and −59.2°, respectively. The optimal geometries of DEP and cDEP thus differed notably.| Parametera | Global energy minimab | Local energy minimab | |||||
|---|---|---|---|---|---|---|---|
| DEP | cDEP | DEP | cDEP | ||||
| a The torsion angles in degrees calculated with the B3LYP method and 6-31G(d,p) basis (Fig. 1). b The energy minima for DEP and cDEP are depicted in Fig. 2b and d, respectively. The parameters for energy-equivalent symmetrical minima are in parentheses. | |||||||
| T1 | 171.4 (−171.4) | 64.4 (−64.6) | 170.3 | −170.3 | 169.4 | −169.4 | −75.4 (75.4) |
| T2 | 72.8 (−72.8) | −59.2 (59.2) | −155.9 | 155.8 | 71.4 | −71.4 | 35.7 (−35.7) |
| T3 | 72.8 (−72.8) | 59.2 (−59.2) | 71.4 | −71.3 | −155.8 | 155.8 | 35.7 (−35.7) |
| T4 | 171.4 (−171.4) | −64.4 (64.4) | 169.4 | −169.4 | 170.2 | −170.3 | −75.4 (75.4) |
The dependences of energy on T2 and T3 calculated for DEP and cDEP unveiled the geometries of the local energy minima (Fig. 2). For DEP, the four symmetrically placed energy-minima were 0.8 kcal mol−1 above the global energy minimum (Fig. 2b). For cDEP, the two energy-minima were 3.8 kcal mol−1 above the global energy minimum (Fig. 2d). Importantly, the potential energy surfaces unveiled the rotational freedom of T2 and T3 in DEP and cDEP. The notable flexibility of the DEP phosphate that can be assumed from the sizable low-energy area within the potential energy surface was actually also MD-calculated (Fig. 2a). The MD snapshots even reflected the curvature of the low-energy profile, which illustrated the reliable MD sampling. By contrast, the flexibility of the cDEP phosphate was notably confined. The T2 and T3 rotational motions were constrained within a small area including the global energy minimum; elsewhere a sharp increase in energy was calculated (Fig. 2c). Interconversions between the two energy-equivalent energy minima of cDEP separated by the energy barrier of ca. 9 kcal mol−1 were not MD-calculated (Fig. 2c). The assumption of blocked interconversions, however, contrasted with the NMR experiment. The 13C NMR shift of the methyl group and the 1H NMR shift of the –CH2– group measured in cDEP clearly demonstrated fast interconversions between the energy minima (Table S1, ESI†).
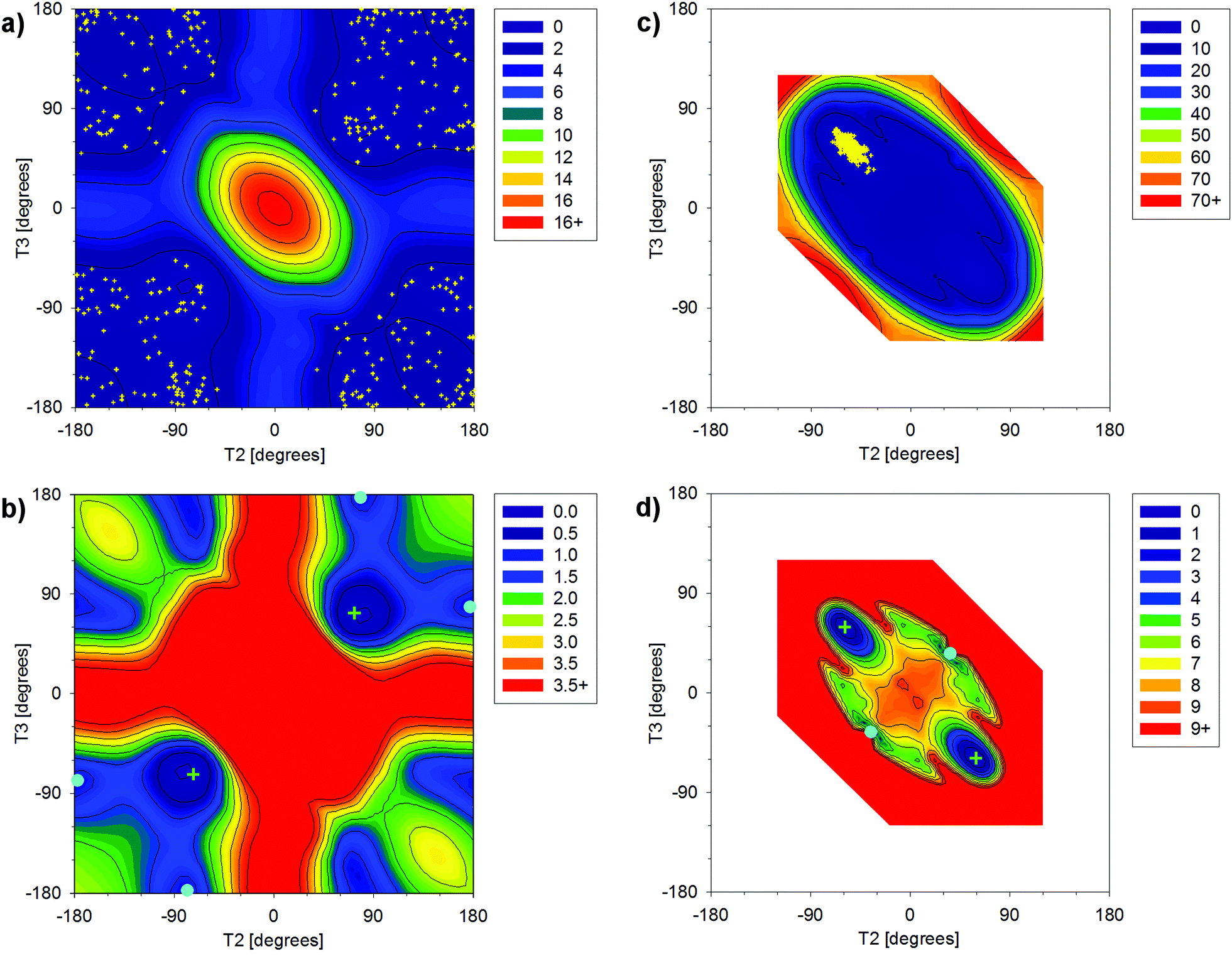 | ||
| Fig. 2 The dependence of energy on T2 and T3 torsion angles relative to the energy of the global energy minimum calculated for DEP (a and b) and cDEP (c and d) with the B3LYP method and the 6-31G(d,p) atomic basis. The relative energies in kcal mol−1 are indicated with colours, as depicted in the right-upper boxes. The global and local energy minima in figures b and d are indicated with green crosses and cyan full cycles, respectively. The MD-snapshot geometries in figures a and c are indicated with yellow dots. The unfilled area for cDEP was not calculated (explanation is given in the main text). | ||
The energy barrier depicted in Fig. 2d thus can be affected due to the neglected involvement of other inner-ring torsional motions. The enhanced MD-sampling of the cDEP geometries calculated at a temperature of 400 K unveiled one accomplished interconversion (Fig. S1 and S2, ESI†). The relative energies of the respective MD snapshots up to 10.7 kcal mol−1 illustrated the feasibility of the interconversions (Fig. S3, ESI†). The flexibility of the cDEP phosphate was notably constrained due to the molecular junction within the ring; however, fast interconversions between the two NMR-equivalent conformers occur.
DEP and cDEP phosphates differed notably as regards their optimal geometries and dynamic behaviour. None of the six DEP energy-minima geometries were similar to any energy-minima geometries of cDEP. The flexibility of the DEP phosphate contrasted with the rigidity of the cDEP phosphate. The effect of the PCM implicit water solvent on the optimized geometries of DEP and cDEP was small. The calculated T2 and T3 torsion angles changed upon PCM hydration by less than 6.0° (Tables 1 and 2). The energy-optimized and MD-calculated geometries of cDEP were practically the same. For DEP, the MD-calculated average geometry was meaningless as the T2 and T3 averaged to ≈180°, whereas these orientations of T2 and T3 were in fact energy-forbidden. The cDEP and DEP phosphates thus can be regarded as geometrical antipodes; the calculated geometries of cDEP were not allowed for DEP and vice versa. The MD snapshot geometries describing the dynamic behaviour of DEP and cDEP were used for MD-averaging of the 31P NMR parameters.
| Parametera | Global energy minimumb | MD-averagec | ||
|---|---|---|---|---|
| DEP | cDEP | DEP | cDEP | |
| a The torsion angles in degrees. b The global energy minimum calculated using the B3LYP method, 6-31+G(d) basis and PCM water solvent. Parameters for the symmetry associated minima are in parentheses. c The statistically averaged geometric parameters in MD snapshots with the SM deviation from the mean in the square brackets. | ||||
| T1 | 177.2 (−177.2) | 60.2 (−60.2) | 179.9 [0.8] | 59.0 [0.3] |
| T2 | 67.1 (−67.1) | −53.2 (53.2) | 179.2 [4.0] | −54.8 [0.4] |
| T3 | 67.1 (−67.1) | 53.2 (−53.2) | 178.4 [3.7] | 55.3 [0.4] |
| T4 | 177.2 (−177.2) | −60.2 (60.2) | 179.9 [1.0] | −60.6 [0.3] |
3.2. Preliminary NMR calculations – the basis set effect
The basis set dependence of NMR parameters was calculated using the 6-31+G(d), Iglo-n (n = II, III), cc-pVnZ (n = D, T, Q and 5), aug-cc-pVnZ (n = D, T and Q), and pcS-n and pcJ-n (n = 1, 2, 3, 4) atomic bases employing the B3LYP method and the PCM water solvent. The geometries of DEP and cDEP were optimized using the B3LYP method, 6-31+G(d) basis and the PCM water solvent if not written otherwise.First, the σ(31P) NMR shielding and 2JP,C coupling in DEP and cDEP were calculated with Iglo-III for the grid point geometries used in the calculations of the potential energy surfaces in Fig. 2 (Fig. S13 and S14, ESI†). The σ(31P) calculated for the global energy minima of DEP and cDEP ranged from 279.48 ppm to 383.37 ppm (Tables S2–S5, ESI†). The relative Δδ(31P) NMR shifts ranged from −9.16 ppm to −0.67 ppm, except for the aug-cc-pVQZ where Δδ(31P) was 5.52 ppm (Table S6, ESI†). The 2JP,C coupling in DEP and cDEP ranged from −8.04 Hz to −5.13 Hz and from −7.13 Hz to −4.47 Hz, respectively.
The convergent-like behaviour of the NMR parameters was calculated for almost all basis sets; however, some NMR parameters obtained with the high-quality bases within particular series differed notably (Fig. 3). The relative Δδ(31P) NMR shift within the pcS-n series converged rapidly, which illustrated the good performance of the Jensen's basis set. The basis set limit for Δδ(31P) in this work with the pcS-4 was −2.14 ppm. The Δδ(31P) within the cc-pVnZ series varied more and the Δδ(31P) with the cc-PV5Z was −2.18 ppm. The Δδ(31P) with Iglo-III was −2.17 ppm. The aug-cc-pVnZ set was not applicable for Δδ(31P) calculation (Fig. 3a). The 2JP,C coupling with the pcJ-n bases was calculated as the FC contribution with the pcJ-n plus the DSO, PSO and SD contributions with Iglo-III. Calculations of the three contributions with high-order pcJ-n bases would be impractical. The 2JP,C in DEP and cDEP with the pcJ-n bases converged to −7.30 Hz and −6.56 Hz, respectively. The 2JP,C in DEP and cDEP with the cc-pVnZ bases decreased to −7.02 Hz and −6.32 Hz, respectively. The Iglo-calculated 2JP,C in DEP and cDEP increased to −7.63 Hz and −6.83 Hz, respectively. The 2JP,C with aug-cc-pVQZ in DEP and cDEP was −6.62 Hz and −5.96 Hz, respectively.
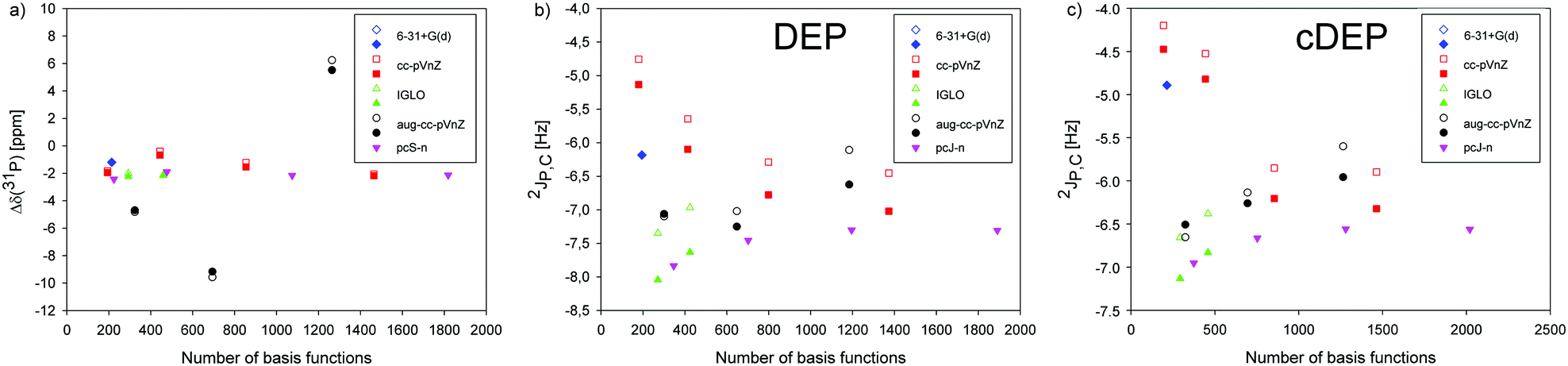 | ||
| Fig. 3 The dependence of relative Δδ(31P) NMR shift and 2JP,C spin–spin coupling on the number of basis functions calculated with the 6-31+G(d), Iglo-II, Iglo-III, cc-pVnZ (n = D, T, Q and 5), aug-cc-pVnZ (n = D, T and Q), and pcS-n and pcJ-n (n = 1, 2, 3, 4) atomic bases for the geometries of DEP and cDEP molecules optimized with the 6-31+G(d) basis (filled symbols) and other atomic bases (open symbols). For the pcJ-n series, only the FC term of 2JP,C was calculated with the bases, and the DSO, PSO, and SD terms were calculated with Iglo III. | ||
The effect of geometry optimization on the NMR parameters was calculated by employing the atomic bases used in NMR calculations. The σ(31P) NMR shielding in DEP and cDEP increased upon geometry reoptimization by up to 11.8 ppm, but for cc-pVDZ and aug-cc-pVDZ σ(31P) decreased (Tables S3 and S5, Fig. S4, ESI†). The effect on the relative Δδ(31P) shift was far smaller as the σ(31P) in DEP and cDEP increased or decreased simultaneously (Fig. 3a). The Δδ(31P) changed due to geometry reoptimization by less than 0.7 ppm and for cc-pV5Z it changed only by 0.1 ppm (Table S6, ESI†). The 2JP,C mostly decreased, except for the aug-cc-pVDZ (Tables S3 and S5, ESI†). The effect of geometry optimization on 2JP,C in DEP and cDEP ranged from −0.2 Hz to −0.7 Hz and from −0.1 Hz to −0.5 Hz, respectively.
The basis set effect on MD-averaged NMR parameters was estimated for the more flexible DEP phosphate using only the first 20 MD snapshots (Fig. S5 and Table S7, ESI†). The σ(31P) decreased upon MD-averaging, by up to 2.5 ppm with cc-pVTZ. The 2JP,C increased, by up to 1.4 Hz with Iglo-II. The 2JP,C calculated with cc-pV5Z increased upon MD-averaging from −7.0 Hz to −6.1 Hz. The effect of molecular dynamics on the NMR parameters should be therefore included.
The preliminary NMR calculations indicated suitable atomic bases. As regards Δδ(31P), the high-quality bases within the conceivable series, pcS-4, cc-pV5Z and Iglo-III gave the same magnitudes of the relative NMR shift (Table S6, ESI†).
Out of the three bases, Iglo-III is well-suited for MD averaging of the 31P NMR shift due to high accuracy for low computational demands. Regarding 2JP,C, the basis set effect was examined for all of the four J-coupling contributions.
The FC contribution was prevalent and depended on atomic bases much more than the other three contributions where the DSO and SD were negligible and the PSO was de facto basis set independent (Tables S2 and S4, ESI†). The FC was determinative for the calculated magnitude of the 2JP,C. To maintain high accuracy of MD-averaged 2JP,C, the FC term was calculated with the cc-pV5Z basis while the remaining terms were calculated with the Iglo-III basis.
3.3. The 31P NMR shift
The δ(31P) NMR shifts measured in DEP and cDEP were referenced to H3PO4. In the theoretical calculations, three NMR references were employed. The δ(31P) referenced to H3PO4 was equivalent to the reference in the experiment. The δ(31P)PH3 was referenced to secondary NMR reference PH3 according to van Wüllen.39 Lastly, the relative Δδ(31P) NMR shift was calculated as the mutual NMR shift of the two phosphates neglecting the external NMR reference. In this way, the effect of the NMR reference on the accuracy of the calculated 31P NMR shift was unveiled.The δ(31P) NMR shift is usually measured in nucleic acids. The accuracy of the calculated δ(31P), however, may be affected when calculation errors for σ(31P) in the phosphate and in H3PO4 are different. The effect can be anticipated due to different chemical bonding of the phosphorus atoms in phosphate and in H3PO4. By contrast, systematic errors in the calculations of σ(31P) for chemically equivalent phosphates should likely cancel out in the calculation of the Δδ(31P) NMR shift. The secondary PH3 NMR reference was introduced to bypass the problematic H3PO4 reference in theoretical calculations.39 However, the calculation of small up-field and down-field 31P NMR shifts such as in the phosphate still remained challenging. In the following, we will show that the choice of NMR reference may become critical for the assignment and interpretation of the 31P NMR shifts.
The δ(31P) measured in DEP and cDEP was 1.34 ppm and −2.68 ppm, respectively. The δ(31P) calculated in DEP and cDEP with the B3LYP method and the pcS-4 basis was 2.14 ppm and 0.00 ppm, respectively. The Iglo-III-calculated δ(31P) in DEP and cDEP was 2.03 ppm and −0.14 ppm, respectively. The cc-PV5Z-calculated δ(31P) in DEP and cDEP was 2.80 ppm and 0.62 ppm, respectively. The calculated δ(31P) shifts depended on high-quality atomic bases rather notably. The δ(31P)PH3 shifts varied similarly; however, their magnitudes deviated from the experiment far more than those of the δ(31P) shifts (Table 3). By contrast, the Δδ(31P) calculated with the three high-quality atomic bases differed by less than 0.1 ppm, which indicated cancellation of the systematic errors in the calculated σ(31P) for DEP and cDEP phosphates. The concurrent similar variations of the σ(31P) in DEP and cDEP due to different atomic bases almost cancelled out, which made the Δδ(31P) numerically stable (Table S6, ESI†). And vice versa, the σ(31P) in H3PO4 and PH3 depended on atomic bases less systematically which explained the notable variations of the δ(31P) and δ(31P)PH3 shifts (Table S8, ESI†). Importantly, the Δδ(31P) calculated with the pcS-4, cc-pV5Z and Iglo-III bases demonstrated that accurate dynamical averaging of the Δδ(31P) shift is achievable at low cost with the Iglo-III. The Δδ(31P) calculated within the static approach involving the global energy minima of DEP and cDEP was −2.18 ppm whereas the measured Δδ(31P) was −4.02 ppm.
| Molecule | Calculations | Experiment | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 6-31+G(d)a | Iglo-IIIa | cc-pV5Za | pcS-4a | 6-31+G(d)bc | 6-31+G(d)b | Iglo-IIIb | Iglo-IIIb,g | ||
| a The NMR calculation for the global energy minimum with different atomic bases. b The statistically averaged NMR parameter and the standard mean deviation from mean SM in parentheses. The SM was obtained as SM for phosphate plus SM for NMR reference. c The calculation including explicit hydration of MD snapshots. d δ(31P) = σ(31P) (H3PO4) − σ(31P) (phosphate). e δ(31P)PH3 = σ(31P) (PH3) − σ(31P) (phosphate) −266.1 ppm (ref. 39). f Δδ (31P) = σ(31P) (DEP) − σ(31P) (cDEP) = δ(31P) (cDEP) − δ(31P) (DEP). All geometries were optimized with the B3LYP method, 6-31+G(d) basis and PCM water, except for the PH3 molecule that was optimized neglecting solvent. g The NMR calculations for original MD snapshots without geometry optimization. N.c. stands for not calculated. | |||||||||
| δ(31P)d | |||||||||
| DEP | 4.72 | 2.03 | 2.80 | 2.14 | 3.19 (0.28) | 3.90 (0.22) | 0.86 (0.23) | −17.35 (0.71) | 1.34 |
| cDEP | 3.52 | −0.14 | 0.62 | 0.00 | 0.71 (0.31) | 1.29 (0.23) | −3.00 (0.23) | −21.74 (0.74) | −2.68 |
| δ(31P)PH3e | |||||||||
| DEP | −42.92 | 6.16 | 6.84 | 7.77 | N.c. | −45.53 (0.91) | 1.96 (1.06) | 10.93 (1.31) | 1.34 |
| cDEP | −44.12 | 3.99 | 4.66 | 5.63 | N.c. | −48.14 (0.92) | −1.90 (1.06) | 7.07 (1.34) | −2.68 |
| Δδ(31P)f | |||||||||
| cDEP wrt DEP | −1.20 | −2.17 | −2.18 | −2.14 | −2.48 (0.39) | −2.61 (0.25) | −3.86 (0.26) | −4.93 (0.65) | −4.02 |
The effect of MD averaging on σ(31P) NMR shielding was noticeable (Fig. S6–S8, ESI†). The σ(31P) in PH3, DEP, cDEP and H3PO4 MD snapshots fluctuated by 95.03 ppm, 22.18 ppm, 23.16 ppm, and 12.67 ppm, respectively. The larger the fluctuation of σ(31P), the larger the dynamic effect on the MD-averaged σ(31P) (Table S8, ESI†). The extent of fluctuations was reflected by statistical SM deviations. The SM for σ(31P) in PH3 was 0.9 ppm, while the SM for the other molecules did not exceed 0.2 ppm. Therefore, the δ(31P)PH3 changed upon MD-averaging more than the δ(31P). The Iglo-III-calculated δ(31P) in DEP and cDEP decreased upon MD-averaging by 1.17 ppm and 2.86 ppm, respectively. The MD-averaged δ(31P) in DEP and cDEP was smaller than those in the experiment by 0.48 ppm and 0.32 ppm, respectively. When considering the error-bars corresponding to the sum of SM deviations for the respective σ(31P), 0.23 ppm, the MD-averaged δ(31P) agreed with the experiment. The MD-averaged δ(31P)PH3 in DEP and cDEP was larger than that in the experiment by 0.62 ppm and 0.78 ppm, respectively. The MD-averaged Δδ(31P) was larger than that in the experiment only by 0.1 ppm. Such accuracy is acceptable for the reliable assignment and interpretation of the 31P NMR shifts in nucleic acids. The effect of explicit water on MD-averaged Δδ(31P) calculated with the 6-31+G(d) basis was only 0.1 ppm (Table 3). The high accuracy and small dependence on both the atomic basis and the model of solvent make the relative NMR referencing superior as compared to the other two NMR reference schemes.
The geometry optimization of MD snapshots was necessary for achieving a high accuracy of MD-averaged 31P NMR shifts. The usage of the original MD snapshots affected the magnitudes of the calculated σ(31P) notably and their large variations were reflected by SM deviations (Table S8, ESI†). Consequently, the δ(31P) and δ(31P)PH3 averaged with non-relaxed MD snapshots deviated from the experiment far more than their static and/or MD-averaged counterparts (Table 3). Strikingly, the Δδ(31P) averaged with non-relaxed snapshots was only 0.9 ppm larger than that of the experiment and the deviation corresponded to the SM deviation. This illustrated again the favourable effect of the relative NMR referencing where the systematic geometrical effect on the NMR parameters due to the original geometry of the MD snapshots almost cancelled out.
The δ(31P) in cDEP decreased upon MD-averaging by 2.86 ppm, whereas the δ(31P) in the more flexible DEP decreased only by 1.17 ppm. This seeming contradiction can be explained by the dynamic effect on σ(31P) (Table S8, ESI†). The σ(31P) in DEP and cDEP decreased upon MD-averaging by 2.13 ppm and 0.44 ppm, respectively. The larger dynamic effect on the local NMR property was thus actually calculated for the more flexible phosphate. The dynamic effect on δ(31P) was due to the effect on the H3PO4 reference where the σ(31P) decreased upon MD-averaging by 3.3 ppm. The Δδ(31P) can be interpreted more straightforwardly in terms of the geometry and dynamics of phosphate. The change of phosphate geometry when going from cDEP to DEP accounted for the “static” Δδ(31P) −2.17 ppm (Table 2). The increased flexibility of the phosphate when going from cDEP to DEP accounted for the “dynamical” Δδ(31P) −1.69 ppm (Fig. 2). The dynamic effect on Δδ(31P) can be highlighted in detail by considering the geometrical dependence of σ(31P) (Fig. S13, ESI†). The σ(31P) in the DEP MD snapshots was mostly below σ(31P) for the energy minimum and σ(31P) decreased upon MD-averaging (Table S8, ESI†). The MD geometries of cDEP were confined within a very narrow elliptic-like area where the main axis was perpendicular with respect to the σ(31P) contour lines (Fig. 2 and Fig. S13, ESI†), which resulted in a notable compensation of the dynamical variations of σ(31P). Consequently, the σ(31P) in cDEP decreased upon MD-averaging only by 0.44 ppm. The structure and dynamics of phosphate were elucidated with the theoretical calculation of the Δδ(31P) NMR shift (Fig. 4).
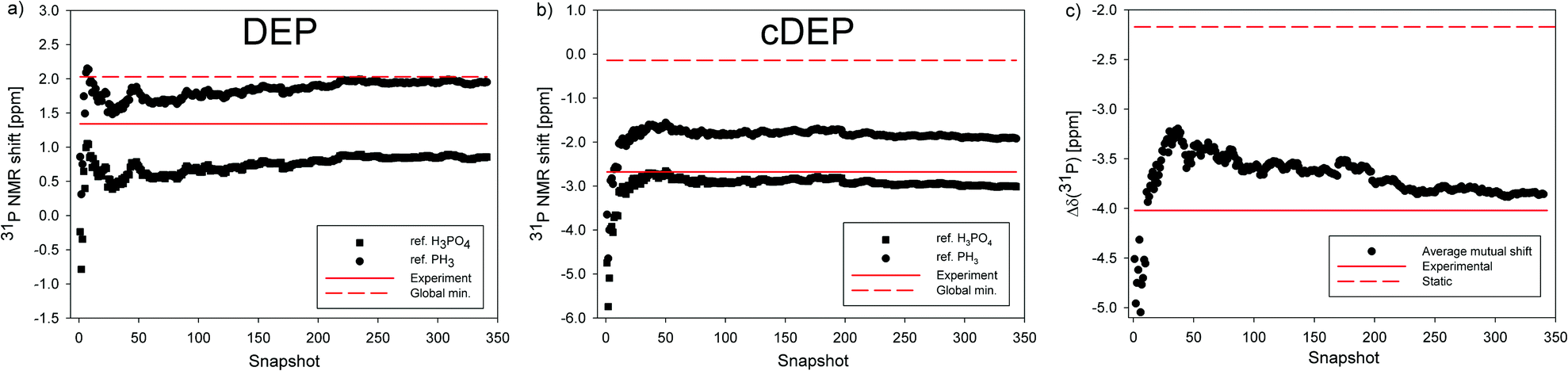 | ||
| Fig. 4 The 31P NMR shift of the phosphate group calculated in DEP and cDEP. (a and b) The cumulative averages of δ(31P) (square) and δ(31P)PH3 (circle) NMR shifts in MD snapshots. (c) The cumulative average of the relative Δδ(31P) NMR shift. The 31P NMR shifts calculated for the global energy minima are indicated with the red-dashed line. The NMR experiment is indicated with the red line. | ||
The plausibility of the B3LYP-calculated 31P NMR shifts was further supported by NMR calculations with other methods inclusive of PCM water hydration and the Iglo-III basis. The geometry optimizations were carried out with the method used in NMR calculation, the 6-31+G(d) basis and the PCM water implicit solvent. The MD-averaged Δδ(31P) calculated with the BP86 method was −4.83 ppm and the static Δδ(31P) was −2.88 ppm. The MD-averaged δ(31P) in DEP and cDEP with BP86 was −1.15 ppm and −5.98 ppm, respectively. The B3LYP method thus provided a better agreement of the calculated Δδ(31P) with the experiment than the BP86 method. The BP86 calculations, however, confirmed the superior performance of the relative NMR reference scheme. Moreover, the dynamic effects on Δδ(31P) −1.95 ppm and −1.69 ppm were calculated with the BP86 and B3LYP method, respectively. Similar magnitudes of the dynamic effect on Δδ(31P) allowed a reliable estimate of MD-averaged Δδ(31P) with other methods by employing the average dynamical correction −1.8 ppm. The static Δδ(31P) with BPW91, M06-2X, PBE0, MP3 and HF was −2.79 ppm, −2.74 ppm, −2.43 ppm, −2.41 ppm and −1.51 ppm, respectively. Assuming the dynamical correction to Δδ(31P), −1.8 ppm, the accuracy of the calculated Δδ(31P) decreased in the order B3LYP, MP2, PBE0, M06-2X, BPW91, HF, and BP86 (Table S9, ESI†).
The Δδ(31P) was furthermore calculated with the KT2 and KT3 DFT methods for geometries optimized with the B3LYP method, 6-31+G(d) basis and PCM water solvent. Excellent performance of the KT2 method has been reported recently for 31P NMR shifts calculated in various organophosphorous compounds.87 The KT2 calculations with the pcS-n bases converged rapidly (Table S10, ESI†). For increasing order of pcS-n, Δδ(31P) was −3.11 ppm, −2.82 ppm, −2.99 ppm and −2.98 ppm. The KT2-calculated Δδ(31P) with Iglo-III was −2.98 ppm, which demonstrated that the performance of Iglo-III is similar to the performance of pcS-4. The Δδ(31P) values with the KT3 method were a little larger (Table S11, ESI†). For increasing order of pcS-n, the Δδ(31P) was −2.65 ppm, −2.40 ppm and −2.59 ppm. The KT3-calculated Δδ(31P) with Iglo-III was −2.58 ppm. Assuming the estimate of the dynamical correction to Δδ(31P) −1.8 ppm, the KT2 and KT3 methods performed less accurately as compared to the B3LYP method.
3.4. The 2JP,C NMR spin–spin coupling
The 2JP,C measured in DEP and cDEP was 5.5 Hz and 5.4 Hz, respectively. The sign of 2JP,C was not measured; however, the negative value of 2JP,C in phosphate was confirmed with the CCSD method.37 The Iglo-III-calculated 2JP,C in geometry-optimized DEP and cDEP was −7.63 Hz and −6.83 Hz, respectively. When Iglo-III was decontracted, the 2JP,C changed only by 0.02 Hz, which illustrated the optimal contraction of the Kutzelnigg's basis (Table 4). When the FC contribution to 2JP,C was calculated with the cc-pV5Z basis, the 2JP,C in DEP and cDEP was −7.01 Hz and −6.31 Hz, respectively. The 2JP,C calculated completely with the cc-pV5Z basis changed only by 0.01 Hz, which demonstrated the negligible basis set effect on the DSO, PSO and SD terms. The FC contribution calculated in DEP and cDEP with cc-pV5Z was larger by ca. 0.3 Hz than the FC with pcJ-4 (Tables S4 and S6, ESI†). The MD-averaging of 2JP,C was calculated with Iglo-III except for the FC term, which was calculated with the cc-pV5Z basis.| Molecule | Calculations | Experiment | ||||||
|---|---|---|---|---|---|---|---|---|
| 6-31+G(d) | 6-31+G(d)c | Iglo-III | Iglo-IIId | Iglo-IIIe | cc-pV5Z | Iglo-IIIf | ||
| a The NMR calculation for the global energy minimum with different atomic bases. b The statistically averaged NMR parameter and the standard mean deviation from the mean SM in the parentheses. c The calculation including explicit hydration of MD snapshots. d The uncontracted basis with added tight polarization functions for the core (the “mixed” keyword in Gaussian 09). e The FC term calculated with the cc-pV5Z basis. f The calculation including correction due to explicit hydration (see the details in main text): 0.13 Hz and 0.34 Hz for DEP and cDEP, respectively. All geometries were optimized with the B3LYP method, 6-31+G(d) basis and PCM water. N.c. stands for not calculated. | ||||||||
| Global energy minimuma | ||||||||
| DEP | −6.18 | −5.88 | −7.63 | −7.65 | −7.01 | −7.02 | −6.71 | 5.5 |
| cDEP | −4.89 | −4.31 | −6.83 | −6.82 | −6.31 | −6.32 | −5.73 | 5.4 |
| MD-averageb | ||||||||
| DEP | −5.34 (0.05) | −5.21 (0.05) | −6.27 (0.08) | N.c. | −5.95 (0.07) | N.c. | −5.82 | 5.5 |
| cDEP | −4.55 (0.03) | −4.21 (0.03) | −6.41 (0.02) | N.c. | −5.89 (0.02) | N.c. | −5.55 | 5.4 |
The 2JP,C in DEP and cDEP increased due to MD-averaging towards the experiment by 1.06 Hz and 0.42 Hz, respectively. The dynamic effect on 2JP,C can be foreseen from the dependence of 2JP,C on the T2 and T3 torsion angles (Fig. S14, ESI†) by considering the MD-calculated geometries (Fig. 2). The 2JP,C in the snapshots fluctuated notably; however, convergence of the MD-averaged 2JP,C with the SM smaller than 0.1 Hz was achieved within 350 snapshots (Fig. S9 and S10). The SM deviation represented ca. 2% of 2JP,C. The SM for σ(31P) represented less than 1% of the σ(31P) magnitude (Table S8, ESI†). The numerical accuracy of the MD-averaged 31P NMR shift was therefore comparable or even better than the accuracy of MD-averaged 2JP,C.
The effect of the explicit water solvent on 2JP,C was calculated with 6-31+G(d) although the basis cannot guarantee the absolute accuracy of 2JP,C (Table 4). The 2JP,C in geometry optimized DEP and cDEP increased due to the explicit hydration towards the experiment by 0.30 Hz and 0.58 Hz, respectively. The MD-averaged 2JP,C in DEP and cDEP increased upon explicit hydration by 0.13 Hz and 0.34 Hz, respectively. Interestingly, a larger effect on 2JP,C in cDEP was calculated throughout the MD simulation, which implies that the two phosphates were hydrated differently (Fig. S11, ESI†). Accessibility of the water solvent to the phosphate was assumed from the overall number of H-bonds between the two non-esterified oxygen atoms of phosphate and the hydrogen atoms of the surrounding water molecules (Fig. S15, ESI†). The average number of H-bonds for DEP and cDEP was 2.7 and 3.2, respectively. The cDEP phosphate was more exposed to solvent due to its locked ring structure while the DEP phosphate was more shielded from water due to the freely moving ethyl ends. Specific hydration of the phosphate therefore affects the magnitude of 2JP,C. The calculated correction of 2JP,C due to explicit hydration was 0.13 Hz and 0.34 Hz in DEP and cDEP, respectively. The MD-averaged 2JP,C in DEP and cDEP including the hydration correction was −5.82 Hz and −5.55 Hz, respectively.
The 2JP,C increased due to geometry reoptimization with the atomic bases used in NMR calculations (Fig. 3, Tables S3 and S5, ESI†). For example, the 2JP,C in DEP and cDEP increased due to cc-PVTZ geometry optimization by 0.45 Hz and 0.29 Hz, respectively. However, the effect on the MD-averaged 2JP,C in cDEP was both increasing and decreasing (Fig. S12, ESI†). Consequently, the MD-averaged 2JP,C decreased due to the cc-PVTZ geometry reoptimization only negligibly, by 0.04 Hz.
The MD-averaged 2JP,C calculated in DEP and cDEP with the BP86 method was −5.73 Hz and −5.48 Hz, respectively. The static value of 2JP,C in DEP and cDEP was −6.60 Hz and −5.87 Hz, respectively. The increase of 2JP,C due to MD-averaging with the BP86 and B3LYP methods was very similar, which allowed a reliable estimate of the dynamic effect on 2JP,C. The average dynamical correction to 2JP,C in DEP and cDEP was 0.97 Hz and 0.41 Hz, respectively. The corrections due to molecular dynamics and explicit hydration allowed reliable estimate of 2JP,C with the BPW91, M06-2X and PBE0 methods based on the static NMR calculations (Table S12, ESI†). The 2JP,C values calculated with the DFT methods ranged from −5.82 Hz to −5.42 Hz for DEP and from −5.89 Hz to −5.14 Hz for cDEP. The DFT methods gave a 2JP,C differing from the experiment by less than 0.4 Hz. The performances of the DFT methods can be compared assuming the accuracy and the mutual difference of 2JP,C in DEP and cDEP. Importantly, the 2JP,C calculated in DEP was smaller than the 2JP,C in cDEP phosphate except for 2JP,C with the M06-2X and PBE0 methods. The B3LYP and BP86 methods provided similarly accurate 2JP,C values in DEP and cDEP in agreement with the measured trend.
The relatively large difference between the static values of 2JP,C in DEP and cDEP phosphates was apparently due to their notably different geometries of energy minima. The two 2JP,C values got close in agreement with the experiment only upon dynamical averaging (Fig. 5). Hence, the very same magnitudes of 2JP,C, which were measured in DEP and cDEP, were due to the particular dynamics of the two phosphates. The dynamic effect on 2JP,C in DEP was twice as large as the effect on 2JP,C in cDEP. The effect of MD-averaging on 2JP,C can be clearly foreseen from the calculated dependence of 2JP,C on the T2 and T3 torsions inclusive of the MD-snapshot geometries (Fig. S14, ESI†). For DEP, the 2JP,C for the energy minimum was notably smaller than the magnitudes of 2JP,C in the MD snapshots. For cDEP, the MD snapshots covered the areas where the 2JP,C values were similar to the 2JP,C values for the energy minimum. Reliable and accurate structural interpretation of the measured 2JP,C thus inevitably must include the dynamic component. Interpretation of the measured 31P NMR parameters employing empirical rules and neglecting the effect of molecular dynamics might be misleading if not contradictory. The measured relative Δδ(31P) NMR shift apparently indicated the structural non-equivalency of the two phosphates that is seemingly in contradiction with similar magnitudes of the 2JP,C couplings although the two NMR parameters provided in fact the same structural information.
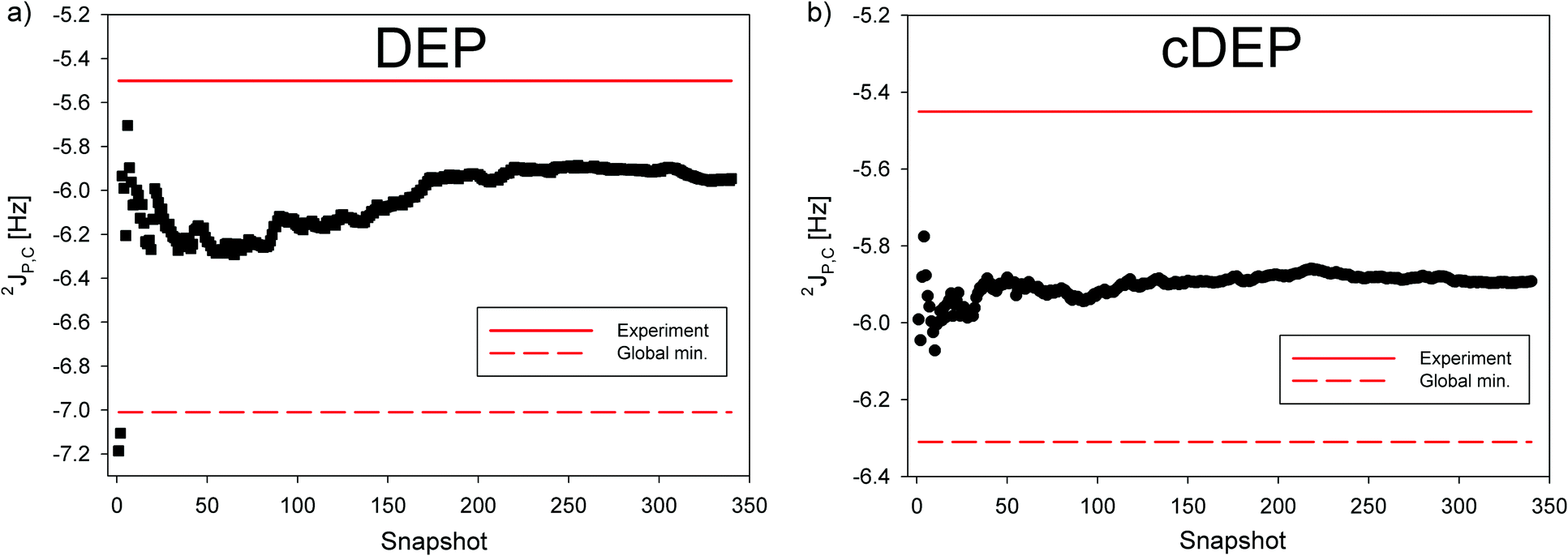 | ||
| Fig. 5 The cumulative average of 2JP,C NMR spin–spin coupling calculated with the B3LYP method in MD snapshots of (a) DEP (black square) and (b) cDEP (black circle). The 2JP,C calculated for the global energy minimum is indicated with the red-dashed line. The measured 2JP,C is indicated with the red line. | ||
4. Conclusions
The benchmark for the 31P NMR shift and the 2JP,C NMR spin–spin coupling in phosphate was obtained by means of theoretical MD/DFT calculations considering the effects of the DFT method, atomic basis, hydration and molecular dynamics in a one-to-one relationship with the NMR measurements in two distinctively different phosphates as regards their optimal geometry and dynamic behaviour. The benchmark for the NMR parameters was aimed at the dynamically flexible DEP phosphate and the dynamically confined cDEP phosphate with notably different optimal geometries.The calculations of the 31P NMR shift demonstrated that the relative referencing of the 31P NMR shift in chemically equivalent phosphates is superior to the NMR reference schemes employing H3PO4 and/or a secondary PH3 reference. The relative MD-averaged Δδ(31P) NMR shift calculated with the B3LYP method, Iglo-III basis and PCM hydration differed from the experiment by 0.16 ppm. The calculation of 2JP,C required a better atomic basis and inclusion of the effect due to explicit hydration that depended on the particular accessibility of the water solvent to DEP and cDEP phosphates. The MD-averaged 2JP,C calculated in DEP and cDEP with the B3LYP method, and Iglo-III(DSO, PSO, SD)/FC(cc-pV5Z) bases inclusive of the hydration effect differed from the experiment by 0.3 Hz and 0.2 Hz, respectively.
The NMR calculations demonstrated that reliable and accurate structural interpretation of the 31P NMR parameters in phosphate inevitably includes both the structural and dynamic components.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Czech Science Foundation GA ČR, by grants with the numbers 16-00270S and 13-26526S. Computational resources were supplied by the Ministry of Education, Youth and Sports of the Czech Republic under the Projects CESNET (Project No. LM2015042) and CERIT-Scientific Cloud (Project No. LM2015085) provided within the program Projects of Large Research, Development and Innovations Infrastructures.References
- B. Schneider, Z. Morávek and H. M. Berman, Nucleic Acids Res., 2004, 32, 1666–1677 CrossRef CAS PubMed.
- D. Svozil, J. Kalina, M. Omelka and B. Schneider, Nucleic Acids Res., 2008, 36, 3690–3706 CrossRef CAS PubMed.
- P. Čech, J. Kukal, J. Černý, B. Schneider and D. Svozil, BMC Bioinf., 2013, 14, 14 CrossRef PubMed.
- J. Černý, P. Božíková and B. Schneider, Nucleic Acids Res., 2016, 44, W284–W287 CrossRef PubMed.
- M. Kowiel, D. Brzezinski and M. Jaskolski, Nucleic Acids Res., 2016, 44, 8479–8489 CrossRef CAS PubMed.
- V. W. S. Schofberger and L. Trantírek, Quantum Chemical Calculations of NMR Parameters, Springer, Dordrecht, Netherlands, 2006, pp. 513–537 Search PubMed.
- A. Cesari, A. Gil-Ley and G. Bussi, J. Chem. Theory Comput., 2016, 12, 6192–6200 CrossRef CAS PubMed.
- T. Helgaker, M. Jaszuński and K. Ruud, Chem. Rev., 1999, 99, 293–352 CrossRef CAS PubMed.
- M. Barfield, A. J. Dingley, J. Feigon and S. Grzesiek, J. Am. Chem. Soc., 2001, 123, 4014–4022 CrossRef CAS PubMed.
- V. Sychrovský, J. Vacek, P. Hobza, L. Žídek, V. Sklenář and D. Cremer, J. Phys. Chem. B, 2002, 106, 10242–10250 CrossRef.
- V. Sychrovský, B. Schneider, P. Hobza, L. Žídek and V. Sklenář, Phys. Chem. Chem. Phys., 2003, 5, 734–739 RSC.
- Z. Vokáčová, F. M. Bickelhaupt, J. Šponer and V. Sychrovský, J. Phys. Chem. A, 2009, 113, 8379–8386 CrossRef PubMed.
- V. Sychrovský, N. Müller, B. Schneider, V. Smrečki, V. Špirko, J. Šponer and L. Trantírek, J. Am. Chem. Soc., 2005, 127, 14663–14667 CrossRef PubMed.
- Z. Vokáčová, L. Trantírek and V. Sychrovský, J. Phys. Chem. A, 2010, 114, 10202–10208 CrossRef PubMed.
- J. M. Fonville, M. Swart, Z. Vokáčová, V. Sychrovský, J. E. Šponer, J. Šponer, C. W. Hilbers, F. M. Bickelhaupt and S. S. Wijmenga, Chem. – Eur. J., 2012, 18, 12372–12387 CrossRef CAS PubMed.
- A. Vavřinská, J. Zelinka, J. Šebera, V. Sychrovský, R. Fiala, R. Boelens, V. Sklenář and L. Trantírek, J. Biomol. NMR, 2016, 64, 53–62 CrossRef PubMed.
- O. Páv, I. Košiová, I. Barvík, R. Pohl, M. Buděšínský and I. Rosenberg, Org. Biomol. Chem., 2011, 9, 6120–6126 Search PubMed.
- R. Liboska, J. Snášel, I. Barvík, M. Buděšínský, R. Pohl, Z. Točík, O. Páv, D. Rejman, P. Novák and I. Rosenberg, Org. Biomol. Chem., 2011, 9, 8261–8267 CAS.
- H. Šípová, T. Špringer, D. Rejman, O. Šimák, M. Petrová, P. Novák, S. Rosenbergová, O. Páv, R. Liboska, I. Barvík, J. Štěpánek, I. Rosenberg and J. Homola, Nucleic Acids Res., 2014, 42, 5378–5389 CrossRef PubMed.
- O. Páv, I. Barvík, R. Liboska, M. Petrová, O. Šimák, S. Rosenbergová, P. Novák, M. Buděšínský and I. Rosenberg, Org. Biomol. Chem., 2017, 15, 701–707 Search PubMed.
- M. Dračínský, M. Šála, B. Klepetářová, J. Šebera, J. Fukal, V. Holečková, Y. Tanaka, R. Nencka and V. Sychrovský, J. Phys. Chem. B, 2016, 120, 915–925 CrossRef PubMed.
- Y. Tanaka, S. Oda, H. Yamaguchi, Y. Kondo, C. Kojima and A. Ono, J. Am. Chem. Soc., 2007, 129, 244–245 CrossRef CAS PubMed.
- Y. Tanaka, J. Kondo, V. Sychrovský, J. Šebera, T. Dairaku, H. Saneyoshi, H. Urata, H. Torigoe and A. Ono, Chem. Commun., 2015, 51, 17343–17360 RSC.
- T. Dairaku, K. Furuita, H. Sato, J. Šebera, K. Nakashima, J. Kondo, D. Yamanaka, Y. Kondo, I. Okamoto, A. Ono, V. Sychrovský, C. Kojima and Y. Tanaka, Chem. – Eur. J., 2016, 22, 13028–13031 CrossRef CAS PubMed.
- T. Dairaku, K. Furuita, H. Sato, J. Šebera, K. Nakashima, A. Ono, V. Sychrovský, C. Kojima and Y. Tanaka, Inorg. Chim. Acta, 2016, 452, 34–42 CrossRef CAS.
- S. K. Latypov, F. M. Polyancev, D. G. Yakhvarov and O. G. Sinyashin, Phys. Chem. Chem. Phys., 2015, 17, 6976–6987 RSC.
- D. Gorenstein, Phosphorus-31 NMR: Principles and Applications, Elsevier, Amsterdam, 1984, p. 604 Search PubMed.
- P. Legault and A. Pardi, J. Magn. Reson., Ser. B, 1994, 103, 82–86 CrossRef CAS PubMed.
- P. Zhang, R. Rycyna and P. B. Moore, Nucleic Acids Res., 1989, 17, 7295–7302 CrossRef CAS PubMed.
- B. Luy and J. P. Marino, J. Am. Chem. Soc., 2001, 123, 11306–11307 CrossRef CAS PubMed.
- D. G. Gorenstein and D. Kar, Biochem. Biophys. Res. Commun., 1975, 65, 1073–1080 CrossRef CAS PubMed.
- C. Giessnerprettre, B. Pullman, F. R. Prado, D. M. Cheng, V. Iuorno and P. O. P. Tso, Biopolymers, 1984, 23, 377–388 CrossRef CAS PubMed.
- J. Přecechtělová, M. L. Munzarová, P. Novák and V. Sklenář, J. Phys. Chem. B, 2007, 111, 2658–2667 CrossRef PubMed.
- J. Přecechtělová, P. Padrta, M. L. Munzarová and V. Sklenář, J. Phys. Chem. B, 2008, 112, 3470–3478 CrossRef PubMed.
- J. Přecechtělová, P. Novák, M. L. Munzarová, M. Kaupp and V. Sklenář, J. Am. Chem. Soc., 2010, 132, 17139–17148 CrossRef PubMed.
- J. Přecechtělová, M. L. Munzarová, J. Vaara, J. Novotný, M. Dračínský and V. Sklenář, J. Chem. Theory Comput., 2013, 9, 1641–1656 CrossRef PubMed.
- L. Benda, Z. S. Vokáčová, M. Straka and V. Sychrovský, J. Phys. Chem. B, 2012, 116, 3823–3833 CrossRef CAS PubMed.
- L. Benda, B. Schneider and V. Sychrovský, J. Phys. Chem. A, 2011, 115, 2385–2395 CrossRef CAS PubMed.
- C. van Wüllen, Phys. Chem. Chem. Phys., 2000, 2, 2137–2144 RSC.
- S. S. Wijmenga and B. N. M. van Buuren, Prog. Nucl. Magn. Reson. Spectrosc., 1998, 32, 287–387 CrossRef CAS.
- S. Grzesiek, F. Cordier, V. Jaravine and M. Barfield, Prog. Nucl. Magn. Reson. Spectrosc., 2004, 45, 275–300 CrossRef CAS.
- A. J. Dingley and S. Grzesiek, J. Am. Chem. Soc., 1998, 120, 8293–8297 CrossRef CAS.
- K. Pervushin, A. Ono, C. Fernández, T. Szyperski, M. Kainosho and K. Wuthrich, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14147–14151 CrossRef CAS.
- Z. Vokáčová, J. Šponer, J. E. Šponer and V. Sychrovský, J. Phys. Chem. B, 2007, 111, 10813–10824 CrossRef PubMed.
- V. Sychrovský, J. Šponer, L. Trantírek and B. Schneider, J. Am. Chem. Soc., 2006, 128, 6823–6828 CrossRef PubMed.
- V. Sychrovský, Z. Vokáčová, J. Šponer, N. Špačková and B. Schneider, J. Phys. Chem. B, 2006, 110, 22894–22902 CrossRef PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499–505 CrossRef CAS.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 15 CrossRef PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2005, 26, 114 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- V. Sychrovský, J. Grafenstein and D. Cremer, J. Chem. Phys., 2000, 113, 3530–3547 CrossRef.
- T. Helgaker, M. Watson and N. C. Handy, J. Chem. Phys., 2000, 113, 9402–9409 CrossRef CAS.
- W. Kutzelnigg, U. Fleischer and M. Schindler, The IGLO-Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities. NMR – Basis Principles and Progress, Springer, Verlag, Berlin/Heidelberg, 1991, vol. 213 Search PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- T. W. Keal and D. J. Tozer, J. Chem. Phys., 2003, 119, 3015–3024 CrossRef CAS.
- T. W. Keal and D. J. Tozer, J. Chem. Phys., 2004, 121, 5654–5660 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef.
- K. Burke, J. P. Perdew and Y. Wang, Recent Progress and New Directions, Plenum, 1998 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 0618–0622 CrossRef.
- M. J. Frisch, M. Headgordon and J. A. Pople, Chem. Phys. Lett., 1990, 166, 281–289 CrossRef CAS.
- C. C. J. Roothaan, Rev. Mod. Phys., 1951, 23, 69–89 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- F. Jensen, J. Chem. Theory Comput., 2008, 4, 719–727 CrossRef CAS PubMed.
- F. Jensen, Theor. Chem. Acc., 2010, 126, 371–382 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekstrom, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernandez, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hattig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenaes, S. Host, I. M. Hoyvik, M. F. Iozzi, B. Jansik, H. J. A. Jensen, D. Jonsson, P. Jorgensen, J. Kauczor, S. Kirpekar, T. Kjrgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnaes, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Salek, C. C. M. Samson, A. S. de Meras, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thogersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Agren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham, III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, Amber 10, University of California, San Francisco, 2012 Search PubMed.
- S. V. Fedorov, Y. Y. Rusakov and L. B. Krivdin, Magn. Reson. Chem., 2014, 52, 699–710 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06969c |
| This journal is © the Owner Societies 2017 |