
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-induced radical polarization and liquid-state dynamic nuclear polarization using fullerene nitroxide derivatives†
Guoquan
Liu‡
a,
Shu-Hao
Liou
 a,
Nikolay
Enkin
a,
Igor
Tkach
a and
Marina
Bennati
*ab
a,
Nikolay
Enkin
a,
Igor
Tkach
a and
Marina
Bennati
*ab
aRG EPR Spectroscopy, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany. E-mail: marina.bennati@mpibpc.mpg.de
bDepartment of Chemistry, University of Göttingen, Tammanstr. 2, 37077 Göttingen, Germany
First published on 10th November 2017
Abstract
We report on radical polarization and optically-driven liquid DNP using nitroxide radicals functionalized by photoexcitable fullerene derivatives. Pulse laser excitation of the fullerene moiety leads to transient nitroxide radical polarization that is one order of magnitude larger than that at the Boltzmann equilibrium. The life time of the radical polarization increases with the size of the fullerene derivative and is correlated with the electronic spin-lattice relaxation time T1e. Overhauser NMR signal enhancements of toluene solvent protons were observed under steady-state illumination, which replaced microwave irradiation.
Introduction
Dynamic nuclear polarization (DNP) is a rapidly developing method to enhance the sensitivity of nuclear-magnetic resonance (NMR) techniques.1 The method transfers spin polarization from electrons to coupled nuclear spins by microwave (MW) irradiation. To achieve this, polarizer molecules – usually nitroxide radicals – are mixed in solution with the target molecules, from which the NMR spectrum is recorded. For electron spins in thermal equilibrium prior to MW pumping, NMR signal enhancements up to 3 orders of magnitude have been reported,2 with a theoretical limit determined by the gyromagnetic ratios γ of electrons and nuclear spins (for 1H: |γe|/γH = 658). DNP in liquids is based on electron–nuclear cross relaxation, the so-called Overhauser effect, and the NMR signal enhancement ε is well described by the following equation:3–5| ε = 1 − seff·f·ξ·|γe|/γI | (1) |
Recently, we have reported a series of functionalized nitroxides composed of a fullerene (C60) moiety, a pyrrolidine linkage and a TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) radical (Scheme 1A) as new potential polarizers for liquid DNP.18 In addition, side chains were introduced into the fullerene cage to increase their rotational and diffusional correlation time as well as solubility in water.19 The compounds revealed an improved saturation factor for MW-driven DNP while the coupling factor in toluene was found to be comparable to that of the common polarizer TEMPONE (4-oxo-2,2,6,6-tetramethylpiperidine 1-oxyl).20 Besides, the photo-excited state properties of fullerene–nitroxide (FN) have been extensively investigated by Corvaja and coworkers.17,21–23Scheme 1B illustrates a simplified electronic-state diagram of photo-excited FN.22 The C60 and nitroxide moieties of FN can be considered as two separate electronic entities with electronic excitation localized on the fullerene. The ground state is a doublet (S = 1/2) and denoted as DS0, where the subscript indicates the electronic excitation on C60. Immediately after photo-excitation of C60 to its first excited singlet state S1, the total electronic state is DS1. Intersystem crossing into the 3C60 and exchange coupling of this S = 1 state with the radical S = 1/2 state lead to an excited DT1 doublet and a QT1 quartet state. The exchange interaction was determined to be positive and places DT1 at a higher energy with respect to QT1.22
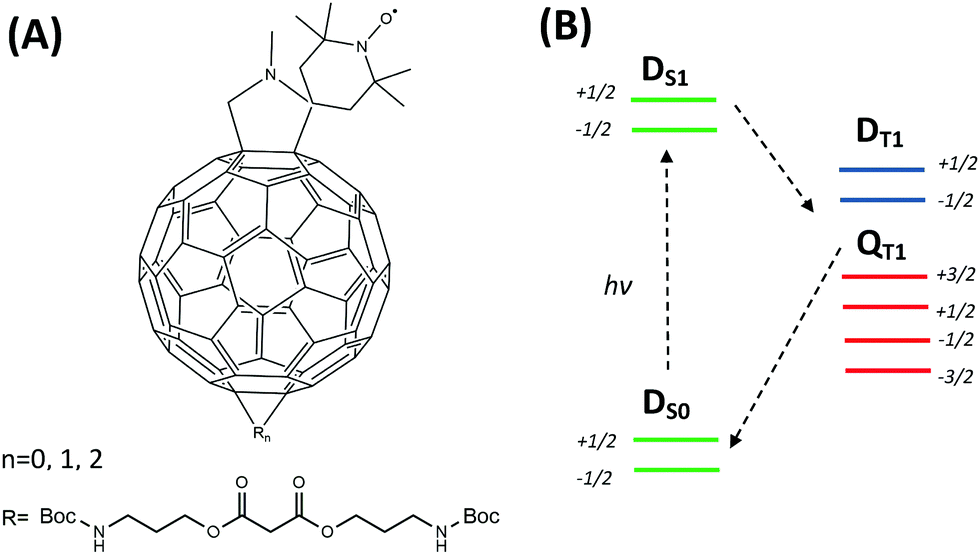 | ||
| Scheme 1 (A) Scheme of the FN (n = 0) structure and its derivatives FN-1a and FN-2a (n = 1, 2) with different numbers of adducts (R). (B) Simplified energy level scheme and transitions of FN after laser excitation according to Corvaja & coworkers.22 | ||
In this paper, we report continuous wave (CW) transient EPR, pulsed EPR and DNP using FN and its derivatives (FN-1a and FN-2a) under light illumination. We show that increasing the size of the polarizing agent has a systematic effect on spin dynamics. Furthermore, we report our first observation of solution DNP by pumping fullerene nitroxide derivatives with CW laser excitation. The results provide a frame to optimize the performance of polarizers for ODNP and pave the way for more systematic investigations.
Experimental methods
Sample preparation
The synthesis of fullerene–nitroxide (FN) derivatives was described in our previous report.18 To prepare the sample for EPR and DNP studies, a desired amount of a particular FN derivative was dissolved in toluene (99.9%, ACS grade, Merck KGaA, Germany) and loaded to either a 1.6 mm O.D. (1.1 mm I.D., volume 10 μl) or a 4 mm O.D. (3 mm I.D., volume 20 μl) quartz tube. Oxygen was removed by five freeze–pump–thaw cycles. The final pressure in the tube was approximately 1 × 10−5 mbar. The tube was then sealed. During sealing the sample was kept frozen in liquid nitrogen. The sample concentration was determined by double-integration of the X-band CW EPR spectrum and by comparing the integrated intensity with a series of samples with known spin concentration.CW transient and pulse EPR at the X-band with laser excitation
CW transient EPR was conducted using a Bruker ElexSys E500 CW/transient EPR spectrometer equipped with a Bruker FlexLine dielectric microwave resonator (EN 4118X-MD4). The resonator holds a rectangular (10 × 3 mm) window, containing a wire grid (0.1 mm wire width, 0.5 mm spacing between wires), which allows for optical illumination of the samples. A LeCroy oscilloscope (WaveJet 354, 500 MHz) and a Philips pulse generator (PM 5786) were used to synchronize the EPR signal detection with laser pulse excitation. Nitrogen gas flow was purged into the cavity during measurements.Pulsed EPR experiments were performed using a commercial Bruker ElexSys E580 spectrometer equipped with a 1 KW TWT MW amplifier (Applied Systems Engineering) and an EN4118X-MD4 Bruker resonator. For FID detection the resonator was overcoupled and the length of a π/2 microwave pulse was approximately 20 ns.
Photoexcitation was performed using a Nd:YAG pulsed laser (Surelite™ SLI-20 Continuum, 20 Hz repetition rate). The FWHM of a laser pulse is specified to be around 6 ns. After a 124 μs Q-switch delay, the average laser power at 532 nm is around 80 mW (≈4 mJ per pulse), which was measured using a thermopile power meter (LabMax™-TO, Coherent).
X band (0.34 T) DNP and 14 MHz 1H-NMR signal detection
Samples were inserted into a Bruker X-band ENDOR resonator (EN 4118X-MD4) and the magnetic field was supplied using a Bruker EleXsys E500 spectrometer. 1H-NMR signals were measured using a Bruker AVANCE III spectrometer. A matching circuit was connected to the ENDOR resonator to tune the NMR circuit at a 1H resonance around 14.6 MHz. Nitrogen gas was purged into the resonator to prevent sample overheating. A 445 nm laser (Laserworld BLUE-4500/445) was used as a CW laser illumination source. A mechanical shutter (SH05 and controller SC10, Thorlab) was controlled using a delay generator to set a suitable illumination time. The Bruker AVANCE console was employed to trigger the delay generator, the timing of the radio frequency (RF) pulse and NMR-FID detection. The length of the RF pulse was set to 3 μs. Signal intensities were defined as the magnitudes calculated from the real and imaginary signal components. The delay time between each scan was set to about 2 minutes to avoid sample overheating. The uncertainty of the enhancement is about 10% and was determined by measuring on three different samples with the same concentration.Results and discussion
CW-transient and pulse EPR of photoexcited FNs
To examine the capability of the FNs to act as a polarizer for liquid DNP, we first examined the polarization of nitroxide radicals in their photoexcited states. Fig. 1 illustrates representative spin polarized CW-transient EPR spectra (X-band/0.34 Tesla) of FN in solution at room temperature as compared to its derivative FN-2a after pulse irradiation with a 532 nm Nd:YAG laser. Consistent with ref. 17 and 21, for FN we observe signatures of a polarized nitroxide radical S = 1/2 corresponding to the DS0 state and of the photo-excited QT1 state.17,21,23,24 These two components can be distinguished according to their g-factors and nitroxide hyperfine splittings and are also observed here in FN-1a and FN-2a (Fig. 1 and Fig. S1, ESI†), albeit with different time evolutions of polarization profiles. The g-factor and hyperfine splitting of QT1, gQT and AQT, are gQT = 1/3gR + 2/3gT and AQT = 1/3AR, where gR = 2.006 and AR = 15.35 G are the isotropic g-factor and the hyperfine splitting of the nitroxide radical, and gT = 2.0014 is the isotropic g-factor of the C60 triplet state.25,26 The solution-state EPR resonance of 3C60 is displayed in Fig. S2 (ESI†) for comparison.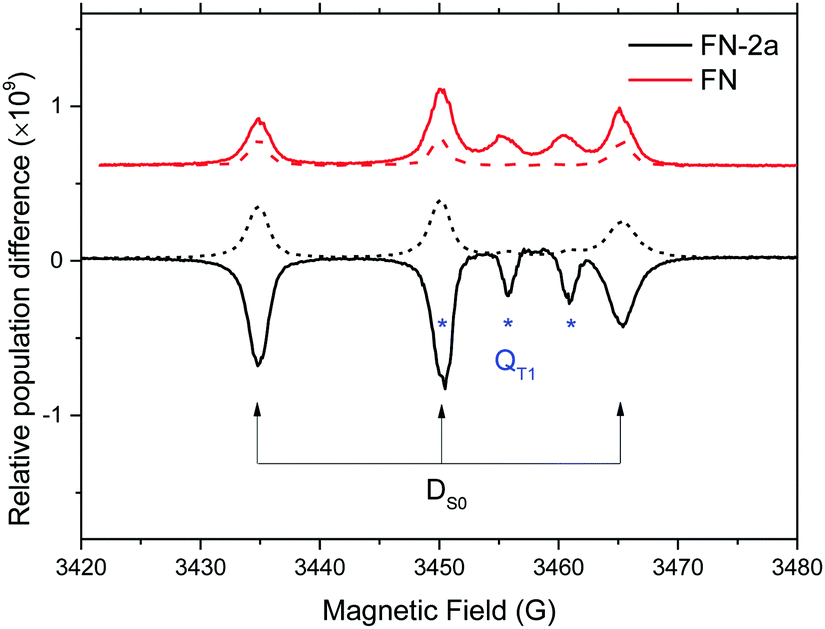 | ||
| Fig. 1 X-band (9.8 GHz) CW transient EPR spectra of 1.5 mM FN (red) and FN-2a (black) in toluene recorded 2.5 μs (solid trace) and 4.1 μs (dash trace) after pulse laser excitation. Transitions assigned to the DS0 and QT1 states are marked. | ||
The time evolution of the CW transient EPR signals of DS0 and QT1 at selected magnetic field positions is displayed in Fig. 2A. As the concentration of the nitroxide radicals remains constant during the experiment, it is clear from Fig. 2A that light illumination drives the ground state DS0 nitroxide polarization (P ∝ 〈SZ〉) of the FNs away from thermal equilibrium. However, to our knowledge, all studies performed using CW transient EPR spectroscopy did not report a direct determination of this transient polarization. The reason for this is that this method detects only changes in signal intensities and is blind toward time-independent signals.27 To obtain more quantitative information, we repeated the experiment as shown in Fig. 2A using pulsed EPR, which allows for a direct comparison of a polarized signal with a reference Boltzmann signal (Fig. 2B). Moreover, the analysis of transient pulsed EPR signals is more straightforward as detection is accomplished in the absence of MW irradiation. Fig. 2B displays the FID-detected time evolution of the nitroxide low-field hyperfine line in the DS0 state as compared to the initial Boltzmann signal (t < 0 and transients without a laser pulse). In all compounds, the time evolution of the polarization is very similar to that observed in CW transient EPR (Fig. 2A) and consists of three components: (1) a fast initial emission, (2) signal inversion emission-to-absorption and (3) a decay. According to the analysis of ref. 21, these changes of radical polarization in the DS0 state are due to the so-called reversed-quartet mechanism RQM, a process by which reverse intersystem crossing between QT1 and DT1 is fully competitive with the decay of these states to the ground state DS0 and to spin lattice relaxation back to a Boltzmann distribution (see also Section Kinetics and radical polarization).
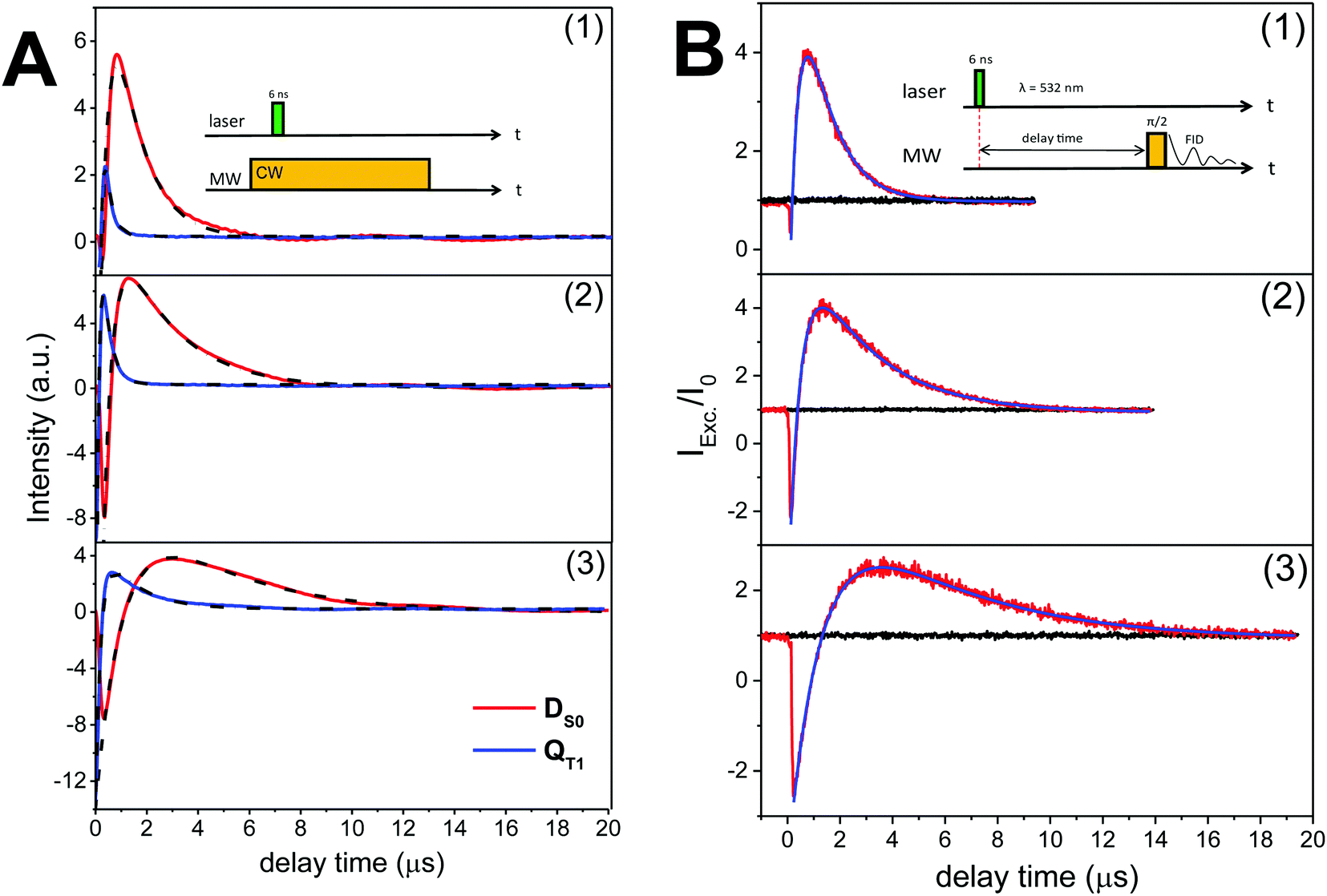 | ||
| Fig. 2 (A) Time evolutions of the CW transient X-band (9.8 GHz) EPR signals of FN (1), FN-1a (2) and FN-2a (3) after laser pulse at 532 nm. Red and blue traces are the DS0 state detected at the low-field hyperfine line and QT1 detected at the central hyperfine line, respectively. Black traces are the bi-exponential fits; the rates are shown in Fig. 3. The field-dependent spectra are shown in Fig. S2 (ESI†). (B) Normalized FID intensities (Iexc/I0) of 0.8 mM (1) FN, (2) FN-1a and (3) FN-2a in toluene (20 μl) as a function of the delay time after a laser pulse of 4 mJ energy. Red and black traces are FID intensities with or without laser pulse, respectively. Blue traces are fits with two exponential decays. Time constants are summarized in Fig. 3. Inset: Pulse sequences. νMW = 9.8 GHz; shot rep. time: 20 ms; FID signals were collected at the low-field hyperfine nitroxide transition of the DS0 state (see Fig. 1) after a dead time of 200 ns from the MW pulse. | ||
The data in Fig. 2B show that at the time point of the maximal, absorptive FID signal the nitroxide FID reaches an intensity (Imax) of about 4-fold (FN and FN-1a) to 3-fold (FN-2a) the Boltzmann equilibrium value (I0). The detected signal is proportional to the sample magnetization M = c·P, where c is the spin concentration and P is the polarization defined here as magnetization per unit volume and spin. P is in turn proportional to 〈SZ〉 and thus to the difference in population of the detected transition.28 If all molecules were photo-excited (excitation efficiency α = 1), the maximal transient polarization Pmax from Fig. 2B would be 4·P0 (for FN and FN-1a) and 3·P0 for FN-2a, where P0 is the Boltzmann equilibrium value. However, the excitation efficiency is usually much less than 1 and the detected signal results to:
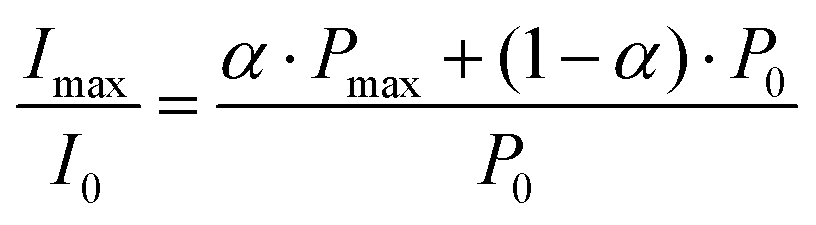 | (2) |
Estimate of FN polarization using 3C60
We know from previous studies that the transient 3C60 signal in solution is proportional to its thermal equilibrium polarization (Fig. S4, ESI†).12,29 Thus α(3C60) can be determined by comparing the FID intensity of the 3C60 triplet state signal in solution with a reference signal, for instance here 4-oxo-TEMPO-d16-15N.3C60 in solution consists of a single narrow EPR line with an electron spin–spin relaxation time (T2 ≈ 450 ns)29 that permits FID detection by pulse EPR at room temperature. The 3C60 signal after a laser pulse was detected by integrating the FID over a short time window (<10 ns) right after the spectrometer detection dead time (tdead), Fig. S4 (ESI†). The delay time between the laser pulse and the microwave detection pulse was incremented in 20 ns steps. According to the experiments and analysis in ref. 12 and 30, 3C60 shows no detectable magnetization immediately after light excitation. Because of the fast rotation in solution, both triplet state EPR transitions collapse within the same resonance line and the observed build-up signal is due to spin–lattice relaxation (T1), with the maximum signal corresponding to the triplet Boltzmann magnetization. Therefore, the intensity of this maximal signal, if calibrated against the Boltzmann signal of reference samples with known concentration, can be used to estimate the number of photo-excited 3C60 molecules.
Under the same experimental conditions, the Boltzmann FID signal of a 15N-labeled nitroxide radical (4-oxo-TEMPO-d16-15N) and the Boltzmann FID signal of 3C60 are proportional to the spin concentration c and their respective Boltzmann polarisation P0, i.e. IFID ∝ c·P0, with the same proportionality constant. For 3C60 the concentration c of the molecules in their triplet state depends on α, i.e. c(3C60) = α·c(C60). The FID intensities of 3C60 and 15N nitroxide in Boltzmann equilibrium can be compared as
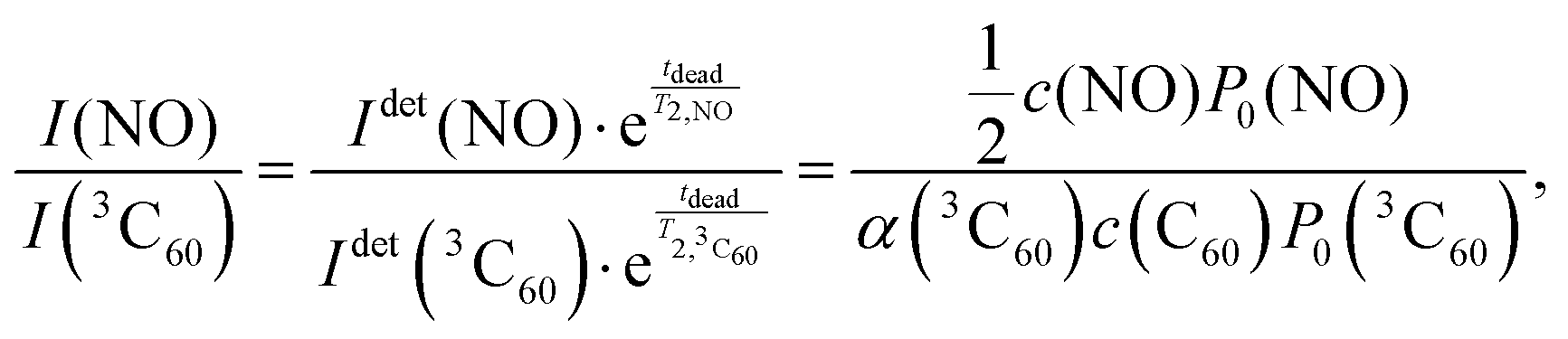 | (3) |
Here Idet(NO) and Idet(3C60) are the detected transient FID signals after a π/2 mw pulse and the detection dead time. T2,NO and 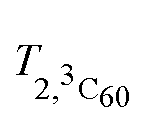 are the spin–spin relaxation times of 15N nitroxide and 3C60, respectively. The factor 1/2 takes into account that only one of the two 15N nitroxide hyperfine lines is detected. P0 is defined here as the equilibrium magnetization per unit volume and spin:12
are the spin–spin relaxation times of 15N nitroxide and 3C60, respectively. The factor 1/2 takes into account that only one of the two 15N nitroxide hyperfine lines is detected. P0 is defined here as the equilibrium magnetization per unit volume and spin:12
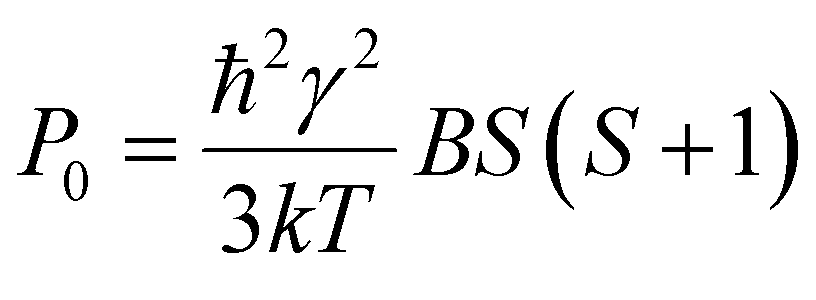 | (4) |
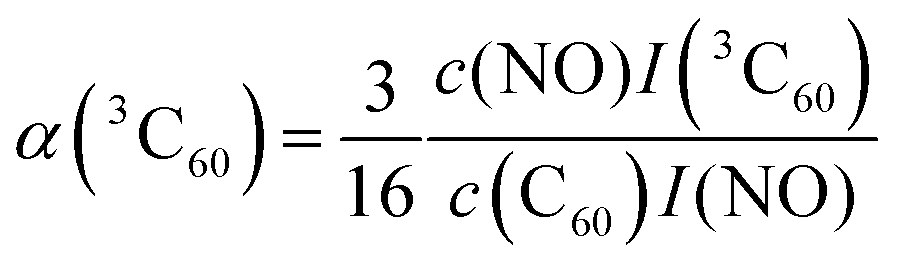 | (5) |
For this purpose, a calibration curve of the 15N nitroxide FID signal (recorded at the low-field EPR line) was measured, and α(3C60) was extracted from eqn (5) for different concentrations of C60, as shown in Tables S3 and S4 (ESI†). We obtain α(3C60) on the order of 0.05–0.1, depending on the considered sample concentration. For comparison, the calculated value while taking into account our laser parameters (laser pulse length and energy, beam spot area, absorption cross section and quantum yield of 3C60) amounts to ≈0.15 (see SI3, ESI†). Obviously, the latter value does not include losses through the optical path.
Finally, the excitation efficiency of FN can be estimated by a comparison to α(3C60) under the same experimental conditions (according to eqn (1) of SI3, ESI†):
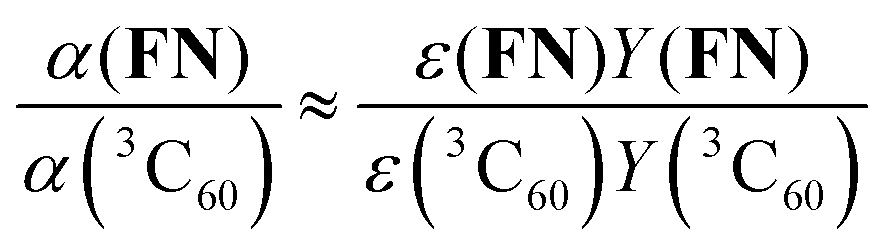 | (6) |
Kinetics of radical polarization
From the transient signals in Fig. 2 we note that the molecular size has an effect on kinetics. The build-up of the QT1 and DS0 EPR signals and their decay become systematically slower from FN to FN-2a. We expect that the lifetime of radical polarization has an impact on steady-state polarization required for DNP; therefore we examined the polarization decays in more detail. The kinetics for the formation and decay of the spin-polarized states QT1 and DS0 is a complex mix of population changes between the spin-sublevels due to exchange coupling of the DT1 and QT1 states and due to spin lattice relaxation. Corvaja et al. treated this problem using a rate equation approach including kinetic terms connecting the spin sublevels of DT1, DQ1 and DS0. This model leads to a complex series of equations with rate constant that are not readily accessible. A later work by Wasielewski et al.32 corroborated a semi-quantitative description of the RQM kinetics. Within this model the initial fast emissive polarization of QT1 arises from a fast spin selective decay of DT1 |(−1/2)〉 to the QT1 |(3/2)〉 spin state, as these two levels have the smallest energy gap (Scheme 1). Optical data indicated that the decay of QT1 matches the emissive built up of DS0.32 A subsequent RQM mechanism leads to a slower polarization inversion (emission-to-absorption) and slower decays to Boltzmann equilibrium.As the initial signal emission is fast as compared to the time scale of our experiments, we considered only the kinetics of polarization inversion and decay. For comparison, we fitted the kinetics of the pulsed and the CW transient EPR signals. We note that the CW transient EPR spectra (Fig. 2A), which were measured at low MW power (here ω1/2π ≈ 0.2 MHz) and in on-resonance EPR fields, are proportional to the difference in the populations of the detected transitions, as illustrated by Corvaja et al.17
The curves in Fig. 2 were fitted using a simple bi-exponential function, which reproduces the effective kinetics of the population difference in the DS0 S = 1/2 spin state (we recall that the radical concentration is constant). The fits are displayed in Fig. 2 superimposed to the data. The rates obtained from both FID and CW transient EPR were quite similar, and are plotted in Fig. 3. As a major result we found that the time constants for the radical polarization decay τDS0,decay are consistent – within the error – with the radical T1e measured by saturation recovery ELDOR.18 Deviations might be due to small differences in temperature or concentrations due to different experimental conditions. The observation of a T1e-limited polarization lifetime of DS0 is consistent with the fact that the polarization of the excited QT1 state decays faster and rather matches the DS0 build up constant (Fig. 2A), supporting the RQM mechanism proposed by Corvaja et al.22 We note that the present data on FN-1a and FN-2a illustrate this trend even more clearly than those on FN because of the slower kinetics. Overall, the decay time constant τDS0,decay of radical polarization increases with the size of the molecule, according to an increasing rotational correlation time τc and T1e.18
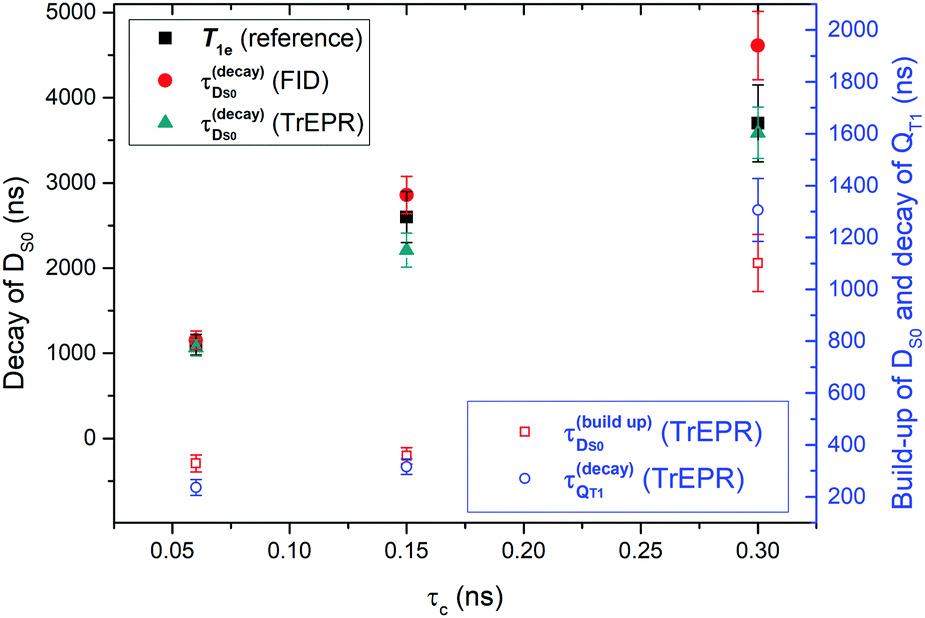 | ||
| Fig. 3 Summary of fitted time constants for polarization build-up and decay of DS0 and the QT1 states as a function of the rotational correlation times (τc)18 for the three different FNs. Plotted are decay rates of DS0 as compared to the spin lattice relaxations from ref. 18 and build-up rates of DS0 as compared to the decay rate of QT1. | ||
Optically-driven DNP
Finally, we examined whether the photo-induced polarization of the nitroxide radical can be transferred to the protons of the toluene solvent using steady-state illumination with a 3 W CW laser at 445 nm. For this purpose, the 1H-NMR signals of toluene at different concentrations of FN derivatives with/without illumination were measured. Fig. 4 collects the 1H-NMR signal enhancements of toluene at 0.34 Tesla after a laser illumination of 3–5 s, depending on the sample size and concentration. Representative time-domain NMR data for FN-1a are shown in Fig. S5 (ESI†). Although the enhancements are small (ε = I/I0 ≤ 1.5), several observations indicate that the DNP effect follows the expected behaviour from the Overhauser mechanism eqn (1) and are correlated with the polarization profiles shown in Fig. 2. The observed DNP enhancements of toluene are positive, in contrast to the common expectation from dipolar relaxation with a positive coupling factor. Indeed, according to Fig. 2, the long-lived positive radical polarization Pexc > P0 gives rise to a negative DNP saturation factor seff = (S0 − 〈SZ〉)/S0. Thus, the resulting sign of the enhancement in eqn (1) must be positive. We also observe a pronounced concentration dependence, with an optimal concentration around 1.5 mM, likely due to deactivation of the excited states at concentrations ≥1 mM.29 We note that we do not observe a DNP enhancement with FN. The build-up curve of the DNP enhancements is shown in Fig. 5. Importantly, it follows expected kinetics according to T1n that is on the time scale of seconds.33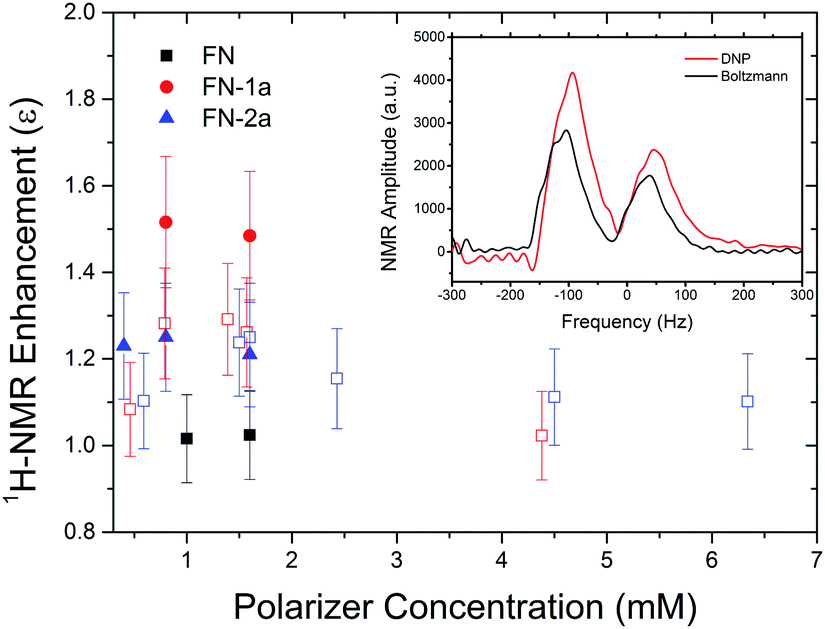 | ||
| Fig. 4 1H-NMR signal enhancements of toluene doped with FN (black), FN-1a (red) or FN-2a (blue) in a 4 mm O.D. quartz tube (volume 20 μl) at different concentrations, illuminated to a 3 W 445 nm laser for 5 s. The unfilled red and blue squares are samples in a reduced volume of 10 μl (1.1 mm O.D. tubes), exposed for 3 s. The inset shows the 1H-NMR spectrum of toluene mixed with 0.8 mM FN-1a with (red) and without laser (black), respectively. The two peaks are contributed by methyl and ring protons, respectively. | ||
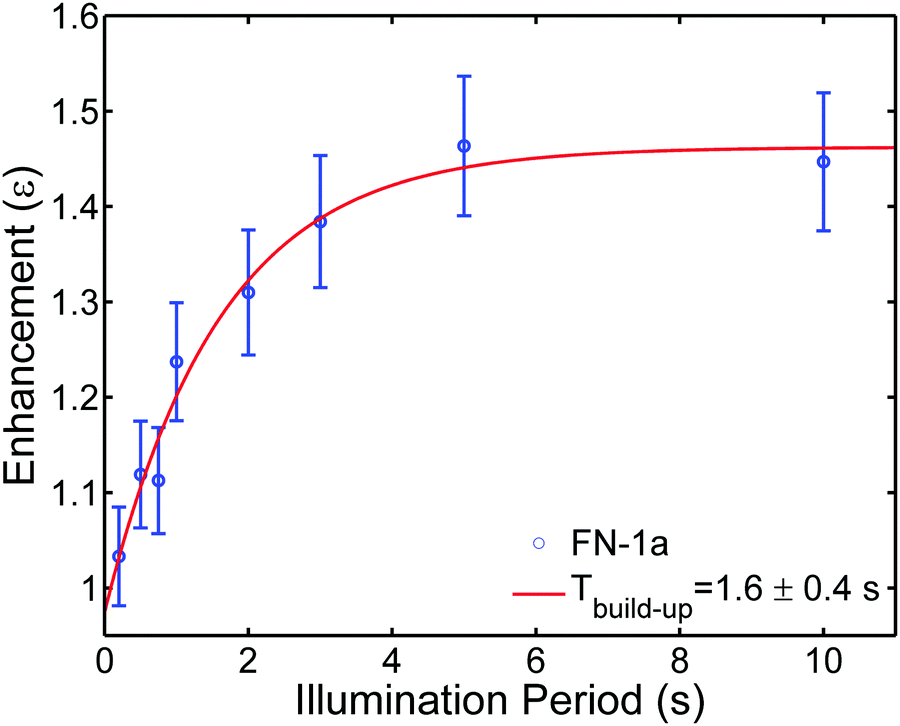 | ||
| Fig. 5 Build-up of 1H-NMR signal enhancements of toluene doped with 0.8 mM FN-1a at different laser (2.8 W) illumination periods. The red trace is the fitted curve with a build-up time Tbuild-up = 1.6 ± 0.4 s. The T1n for FN-1a is 3.2 ± 0.3 s, the difference between T1n and Tbuild-up is likely due to the heating effects. | ||
Our DNP results appear to be consistent with the previous report of a photo-induced 1H-NMR DNP signal up to −4 of water in a mixture of Rose-Bengal and TEMPO.16 As compared to our enhancements, the reported larger negative enhancement is consistent with the completely emissive (negative) transient polarization of TEMPO mixed with Rose-Bengal.13 The occurrence of emission and absorption in the traces of FN-1a and FN-2a (Fig. 2) might attenuate the steady-state total polarization and seems to be a disadvantage. Moreover, the photo-excited polarization of Rose-Bengal was estimated to be Pmax/P0 ≈ −150,13 which is one order of magnitude larger than our estimate for the FN derivatives. On the other hand, Rose-Bengal is unstable under light excitation and the experiments in ref. 16 were performed under flow-conditions,16 whereas we observed no degradation of the polarizers over several cycles. Another interesting observation is the absence of photo-induced DNP with FN (Fig. 4), which – at this stage – can be only rationalized by a shorter lifetime of photo-induced radical polarization (Fig. 3).
Conclusions
All these considerations and the comparison with the Rose-Bengal/nitroxide system shed light on some key parameters to optimize optically-driven liquid DNP. These are the excitation efficiency of the dye, the photo-excited polarization of the radical, its time evolution and lifetime. The stability of the dye is also a critical aspect. It might be possible to improve the performance of the polarizers through rational molecular design; however, the complexity of the whole mechanism still demands an experimental approach based on systematic dye-polarizer screening. The successful optically-driven DNP experiments suggest that this method could potentially become an attractive way to enhance NMR in a liquid state.Conflicts of interest
The authors declare that there are no conflicts of interest.Acknowledgements
We thank Dr. Vladimir Belov, Jürgen Bienert, Jens Schimpfhauser and Jan Seikowski from MPIBPC Chemical Synthesis facility for synthesizing fullerene nitroxide and its derivative. This work was funded by the Max Planck Society and the COST Action TD1103 (European Network on Hyperpolarization). G. L. thanks the Alexander von Humboldt Foundation for a fellowship. N. E. acknowledges the Graduate School for Neurosciences, Biophysics and Molecular Biosciences in Göttingen (GGNB) for financial support. Open Access funding provided by the Max Planck Society.References
- J. H. Ardenkjaer-Larsen, G. S. Boebinger, A. Comment, S. Duckett, A. S. Edison, F. Engelke, C. Griesinger, R. G. Griffin, C. Hilty, H. Maeda, G. Parigi, T. Prisner, E. Ravera, J. van Bentum, S. Vega, A. Webb, C. Luchinat, H. Schwalbe and L. Frydman, Angew. Chem., Int. Ed., 2015, 54, 9162–9185 CrossRef CAS PubMed.
- G. Liu, M. Levien, N. Karschin, G. Parigi, C. Luchinat and M. Bennati, Nat. Chem., 2017, 9, 676–680 CrossRef CAS PubMed.
- D. Hausser and D. Stehlik, Adv. Magn. Reson., 1968, 3, 79–139 Search PubMed.
- W. Müller-Warmuth and K. Meise-Gresch, in Adv. Magn. Opt. Reson., ed J. S. Waugh, Academic Press, 1983, vol. 11, pp. 1–45 Search PubMed.
- M. Bennati, I. Tkach and M. T. Türke, Electron Paramagn. Reson., 2011, 22, 155–182 CAS.
- M.-T. Turke and M. Bennati, Phys. Chem. Chem. Phys., 2011, 13, 3630–3633 RSC.
- C. Griesinger, M. Bennati, H. M. Vieth, C. Luchinat, G. Parigi, P. Hofer, F. Engelke, S. J. Glaser, V. Denysenkov and T. F. Prisner, Prog. Nucl. Magn. Reson. Spectrosc., 2012, 64, 4–28 CrossRef CAS PubMed.
- A. Henstra, T. S. Lin, J. Schmidt and W. T. Wenckebach, Chem. Phys. Lett., 1990, 165, 6–10 CrossRef CAS.
- K. Tateishi, M. Negoro, A. Kagawa and M. Kitagawa, Angew. Chem., Int. Ed., 2013, 52, 13307–13310 CrossRef CAS PubMed.
- K. Tateishi, M. Negoro, S. Nishida, A. Kagawa, Y. Morita and M. Kitagawa, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7527–7530 CrossRef CAS PubMed.
- P. W. Atkins and G. T. Evans, Mol. Phys., 1974, 27, 1633–1644 CrossRef CAS.
- G.-H. Goudsmit, H. Paul and A. I. Shushin, J. Phys. Chem., 1993, 97, 13243–13249 CrossRef CAS.
- H. Takahashi, M. Iwama, N. Akai, K. Shibuya and A. Kawai, Mol. Phys., 2013, 112, 1012–1020 CrossRef.
- F. Arena, F. Bullo, F. Conti, C. Corvaja, M. Maggini, M. Prato and G. Scorrano, J. Am. Chem. Soc., 1997, 119, 789–795 CrossRef CAS.
- F. Conti, C. Corvaja, C. Gattazzo, A. Toffoletti, P. Bergo, M. Maggini, G. Scorrano and M. Prato, Phys. Chem. Chem. Phys., 2001, 3, 3526–3531 RSC.
- M. W. Dale and C. J. Wedge, Chem. Commun., 2016, 52, 13221–13224 RSC.
- C. Corvaja, M. Maggini, M. Prato, G. Scorrano and M. Venzin, J. Am. Chem. Soc., 1995, 117, 8857–8858 CrossRef CAS.
- N. Enkin, G. Liu, C. Gimenez-Lopez Mdel, K. Porfyrakis, I. Tkach and M. Bennati, Phys. Chem. Chem. Phys., 2015, 17, 11144–11149 RSC.
- C. F. Richardson, D. I. Schuster and S. R. Wilson, Org. Lett., 2000, 2, 1011–1014 CrossRef CAS PubMed.
- N. Enkin, G. Liu, I. Tkach and M. Bennati, Phys. Chem. Chem. Phys., 2014, 16, 8795–8800 RSC.
- F. Conti, C. Corvaja, M. Maggini, F. Piu, G. Scorrano and A. Toffoletti, Appl. Magn. Reson., 1997, 13, 337–346 CrossRef CAS.
- V. Rozenshtein, A. Berg, E. Stavitski, H. Levanon, L. Franco and C. Corvaja, J. Phys. Chem. A, 2005, 109, 11144–11154 CrossRef CAS PubMed.
- H. Moons, E. Goovaerts, V. P. Gubskaya, I. A. Nuretdinov, C. Corvaja and L. Franco, Phys. Chem. Chem. Phys., 2011, 13, 3942–3951 RSC.
- C. Corvaja, M. Maggini, M. Ruzzi, G. Scorrano and A. Toffoletti, Appl. Magn. Reson., 1997, 12, 477–493 CrossRef.
- J. Brickmann and G. Kothe, J. Chem. Phys., 1973, 59, 2807–2814 CrossRef CAS.
- A. Bencini and D. Gatteschi, Electron Paramagnetic Resonance of Exchange Coupled systems, Springer, Berlin, 1990 Search PubMed.
- N. J. Turro, I. V. Koptyug, H. Vanwilligen and A. K. A. Mclauchlan, J. Magn. Reson., Ser. A, 1994, 109, 121–123 CrossRef CAS.
- M. Mehring and V. Weberruss, Object-Oriented Magnetic Resonance: Classes and Objects, Calculations and Computations, Academic Press, 1st edn, 2001 Search PubMed.
- M. Bennati, A. Grupp, P. Bäuerle and M. Mehring, Chem. Phys., 1994, 185, 221–227 CrossRef CAS.
- M. Bennati, A. Grupp, M. Mehring, K. P. Dinse and J. Fink, Chem. Phys. Lett., 1992, 200, 440–444 CrossRef CAS.
- K. Kordatos, T. Da Ros, M. Prato, S. Leach, E. J. Land and R. V. Bensasson, Chem. Phys. Lett., 2001, 334, 221–228 CrossRef CAS.
- E. M. Giacobbe, Q. Mi, M. T. Colvin, B. Cohen, C. Ramanan, A. M. Scott, S. Yeganeh, T. J. M. M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 3700–3712 CrossRef CAS PubMed.
- M. T. Turke and M. Bennati, Appl. Magn. Reson., 2012, 43, 129–138 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06073d |
| ‡ Current address: State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing 100191, China. |
| This journal is © the Owner Societies 2017 |