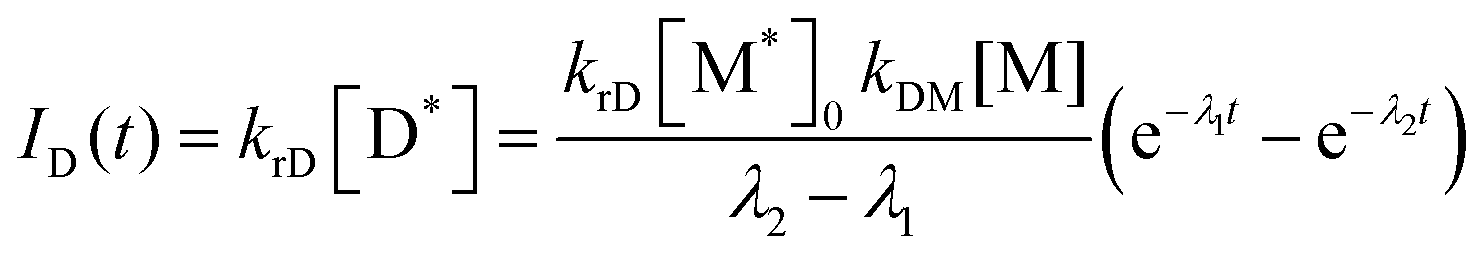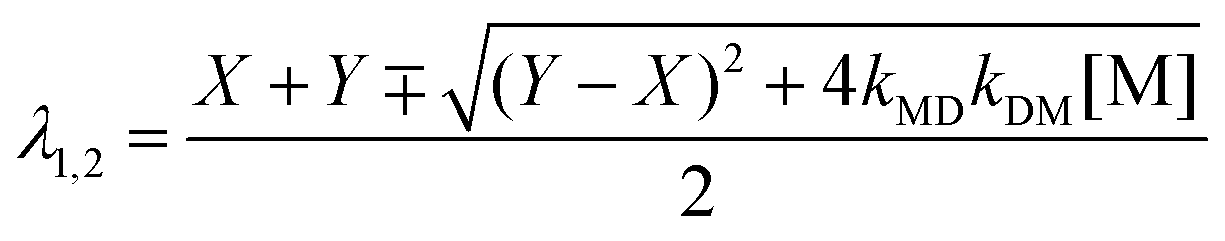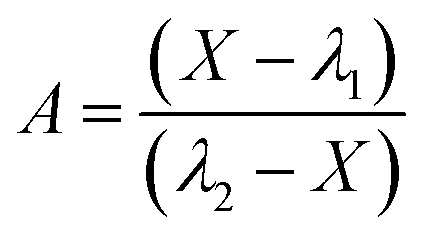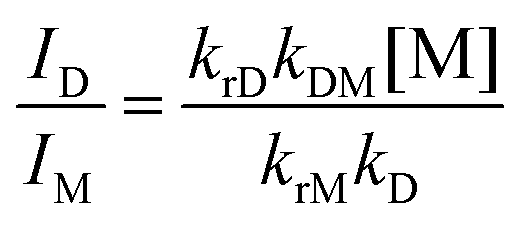Photokinetic study on remarkable excimer phosphorescence from heteroleptic cyclometalated platinum(II) complexes bearing a benzoylated 2-phenylpyridinate ligand†
Received
11th October 2017
, Accepted 24th November 2017
First published on 24th November 2017
Abstract
Novel heteroleptic cyclometalated platinum(II) complexes consisting of 5′-benzoylated 2-phenylpyridinate (ppy) cyclometalated and acetylacetonate ancillary ligands were synthesized, and their photoluminescence (PL) properties were investigated. The 5′-benzoylated complex without any other substituents exhibited phosphorescence-based monomer emission at 479 nm in dichloromethane (10 μM, rt) with a PL quantum yield of 0.28. On the other hand, in poly(methyl methacrylate) (PMMA) film, remarkable excimer emission additionally emerged at ca. 600 nm with a relatively high PL quantum yield of 0.47 as the doping level increased to 0.20 mmmol g−1, which was comparably intense in comparison with the monomer emission. In the case of the complexes with unsubstituted, 4′-benzoylated, and 5′-fluorinated ppy cyclometalated ligands, excimer emission was modestly generated at the same doping level, and thus the introduction of a benzoyl group to the 5′-position is effective to obtain remarkable excimer emission. The combination of benzoyl and fluoro groups was more effective at inducing excimer emission, and the intensity of excimer emission of the 2-(5-benzoyl-4,6-difluorophenyl)pyridinate-based complex was 3.5 times larger than that of monomer emission at a doping level of 0.20 mmmol g−1 in PMMA. From the analysis of PL lifetimes at varying concentrations, photokinetic profiles were fully analyzed according to the model system for the irreversible excimer formation, and the excimer formation rate constant of the 5′-benzoylated complex was determined in dichloromethane as 2.2 × 109 M−1 s−1, which is 4.4 times larger than that of the unsubstituted complex. We also fabricated an organic light-emitting diode using the 2-(5-benzoyl-4,6-difluorophenyl)pyridinate-based complex as a single emitter. The device exhibited pseudo-white EL with the Commission internationale de l’éclairage chromaticity coordinates of (0.42, 0.42).
Introduction
Organic light-emitting diodes (OLEDs) have been attracting much attention due to their applicability to flat-panel displays, illumination devices, electronic papers, and so on.1–4 In OLEDs, the excitons of an emitting material generated by charge recombination are divided into singlet and triplet states at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 according to the spin statistics theorem. Thus, when a fluorescent emitter is used, the device can achieve an internal quantum efficiency (ηint) of at most 25%. On the other hand, phosphorescent emitters can achieve ηint as high as 100% in theory, taking account of the intersystem crossing (ISC) from the singlet excited state to the triplet state.1–7 Hence, organometallic compounds including platinum(II)2,7–26 and iridium(III)27–33 have been enthusiastically developed as phosphorescent emitters because the strong spin–orbit coupling due to the heavy metal center facilitates the ISC to provide efficient phosphorescence under ambient conditions. Actually, high current efficiencies of more than 50 cd A−1 have been achieved by employing phosphorescent platinum(II) complexes.34,35
:3 according to the spin statistics theorem. Thus, when a fluorescent emitter is used, the device can achieve an internal quantum efficiency (ηint) of at most 25%. On the other hand, phosphorescent emitters can achieve ηint as high as 100% in theory, taking account of the intersystem crossing (ISC) from the singlet excited state to the triplet state.1–7 Hence, organometallic compounds including platinum(II)2,7–26 and iridium(III)27–33 have been enthusiastically developed as phosphorescent emitters because the strong spin–orbit coupling due to the heavy metal center facilitates the ISC to provide efficient phosphorescence under ambient conditions. Actually, high current efficiencies of more than 50 cd A−1 have been achieved by employing phosphorescent platinum(II) complexes.34,35
Heteroleptic cyclometalated platinum(II) complexes, consisting of 2-phenylpyridinate cyclometalated (C^N) and β-diketonate ancillary (O^O) ligands, adopt four-coordinated square-planar structures that enhance intermolecular interactions to yield excimer phosphorescence emerging in longer wavelength regions along with the monomer one.2,8,15,17,22,24,36 In the case of the blue phosphorescent complex such as (dfppy)Pt(acac) (dfppy, 2-(4,6-difluorophenyl)pyridinate-N,C2′; acac, acetylacetonate-O,O), an optimized balance of monomer and excimer emissions yielded a broadened electroluminescence (EL) spectrum covering the whole visible region, and it allows us to fabricate a white OLED using this complex as a single emitting dopant.8 This type of white OLED is important for potential application in the low-cost and facile fabrication of room lighting and backlights of displays because precise tuning of the balance of primary color emitting materials is avoided and a smaller amount of emitter is consumed.2 From this viewpoint, a variety of cyclometalated platinum(II) complexes showing excimer emission have so far been developed.2,8,11,14,16–18,24,26,37,38 Recently, we also reported a heteroleptic cyclometalated platinum(II) complex consisting of dibenzo[b,d]furan-2-ylpyridinate-N,C2′ and 1,3-bis(3,4-dibutoxyphenyl)propane-1,3-dionate as cyclometalated and ancillary ligands, respectively, which showed remarkable excimer emission in polymer matrices due to its expanded π-planes of the ligands enhancing the intermolecular interaction.17 However, there are few reports of the structure–function relationships and photokinetic features of excimer emission of cyclometalated platinum(II) complexes.14,17,24 Upon development of excimer—emissive complexes, it has been extensively discussed how intensely the excimer emission emerges relative to that of the monomer.11,14,17,18,24 To discuss how the excimer emission is generated, the photokinetic analysis on the basis of the structural factors is of particular importance. In this regard, Shinozaki and coworkers estimated the excimer and excited trimer formation rate constants of tridentate cyclometalated platinum(II) complexes by analysing their photoluminescence (PL) decays.39 However, the excimer formation kinetics of (C^N)Pt(O^O)-type complexes has not been sufficiently discussed, although the kinetic aspects of the excimer emission of this type of organoplatinum(II) complexes should provide valuable insights to manage their electroluminescence behaviour in OLED applications.
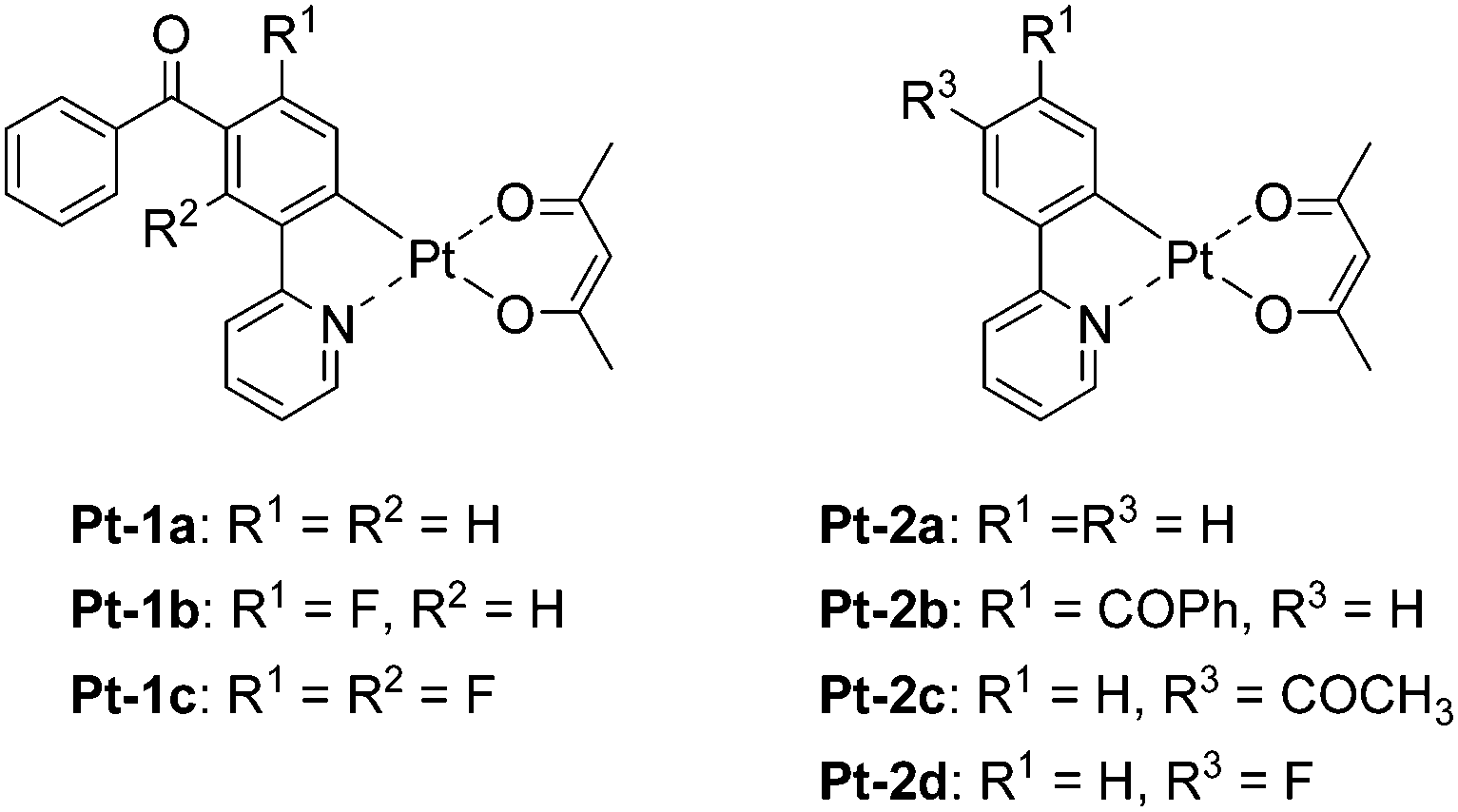
To obtain white luminescence through the optimized balance of the monomer and excimer emissions from a (C^N)Pt(O^O)-type complex, ligand design to yield monomer emission in the blue region is essential.14 We recently reported bis- and tris-cyclometalated iridium(III) complexes bearing benzoylated dfppy ligands.33 The introduction of a benzoyl group to the 5′-position of each dfppy ligand is effective to obtain a more blue-shifted PL wavelength (λPL) than those of the corresponding dfppy-based complexes. In particular, tris[2-(5-benzoyl-4,6-difluorophenyl)pyridinate-N,C2′]iridium(III) exhibited blue PL at 463 nm with an excellent PL quantum yield (ΦPL) of 0.90 in deaerated dichloromethane (10 μM) at rt. Taking into consideration that the impact of the cyclometalated ligand on λPL is comparable between organoiridium(III) and organoplatinum(II) complexes, the benzoylated dfppy ligand should be a good candidate to obtain (C^N)Pt(O^O)-type complexes emitting in the blue region. Here, we report novel cyclometalated platinum(II) complexes Pt-1a–c (Fig. 1) bearing the 5′-benzoylated 2-phenylpyridinate (ppy) ligands with or without fluorine substituent(s), and their excimer phosphorescence behaviours are investigated in detail, especially focusing on the impacts of the introduced substituents on the radiative excimer formation. We investigate the photokinetics of these complexes to clarify how the excimer formation is facilitated. In addition, the fabrication of a white OLED employing Pt-1c as a single emitting dopant is demonstrated.
 |
| | Fig. 1 Structures of platinum(II) complex Pt-1 bearing a benzoyl group on the 5′-position of its 2-phenylpyridinate ligand and its reference complex Pt-2. | |
Experimental
General
The starting materials, HC^N-1a–c,33HC^N-2b31 and HC^N-2c,40 and the reference platinum(II) complexes (dfppy)Pt(acac)9 and Pt-2a9 were prepared according to the literature. The other reagents were used as obtained from Wako Pure Chemicals, Tokyo Chemical Industry, Sigma-Aldrich, or Kanto Chemicals. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained on a JEOL ECS-400 or a JEOL ECX-400 spectrometer using TMS (0.00 ppm) as an internal standard. 19F NMR spectra were obtained on a JEOL ECX-400 (376 MHz) or a Varian-500 (470 MHz) spectrometer, using CFCl3 (0.00 ppm) as an external standard. Laser desorption/ionization time-of-flight (LDI-TOF) mass spectra were recorded on a Shimadzu–Kratos AXIMA-CFR PLUS TOF mass spectrometer. Elemental analyses were carried out on a J-Science MICRO CORDER JM10 analyzer.
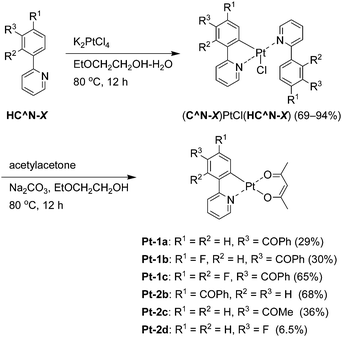
General procedure for the synthesis of the precursor (C^N-X)PtCl(HC^N-X)
These compounds were prepared according to the conventional procedure.41,42 A mixture of the 2-phenylpyridine derivative HC^N-X (X = 1a–c, 2b, or 2c, 1.50 mmol) and potassium tetrachloridoplatinate (0.754 mmol) in a mixture of 2-ethoxyethanol and water (3:1, v/v, 33 mL) was stirred at 80 °C for 12 hours. After cooling, the reaction mixture was concentrated on a rotary evaporator, and then the residue was dissolved in dichloromethane (50 mL) and washed with 10% NaClaq (50 mL × 2). The organic solution was dried over anhydrous sodium sulfate and it was concentrated on a rotary evaporator. Hexane was added to the concentrated solution to afford a yellow solid of (C^N-X)PtCl(HC^N-X) (X = 1a–c, 2b, and 2c; 69, 84, 86, 85, and 94%, respectively). These materials were used in the next reaction without further purification.
General procedure for the synthesis of the platinum(II) complex Pt-X.
A mixture of (C^N-X)PtCl(HC^N-X) (X = 1a–c, 2b, 2c, or 2d, 1.72 mmol), acetylacetone (0.873 g, 8.72 mmol), and sodium carbonate (1.80 g, 17.0 mmol) in 2-ethoxyethanol (34 mL) was stirred at 80 °C for 12 hours. After cooling, the reaction mixture was concentrated on a rotary evaporator. Then the residue was dissolved in dichloromethane (100 mL) and washed with 10% NaClaq (100 mL × 2). The organic solution was dried over anhydrous sodium sulfate and evaporated to dryness. The residue was purified by silica gel column chromatography using chloroform as eluent. Further purification by recrystallization from ethyl acetate or acetonitrile gave a yellow solid of Pt-X.
Pt-1a (yield: 29%). 1H NMR (400 MHz, CD2Cl2) δ 2.00 (s, 3H), 2.02 (s, 3H), 5.52 (s, 1H), 7.20 (ddd, J = 7.3, 5.9 and 1.4 Hz, 1H), 7.46–7.51 (m, 2H), 7.54 (dd, J = 8.2 and 1.8 Hz, 1H), 7.58 (tt, J = 7.3 and 1.4 Hz, 1H), 7.67–7.74 (m, 2H), 7.75–7.79 (m, 2H), 7.86 (ddd, J = 7.7, 7.3 and 1.8 Hz, 1H), 7.94 (d, J = 1.8 Hz, 1H), 9.02 (ddd, J = 5.9, 0.9 and 0.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 27.19, 28.31, 102.73, 119.00, 122.00, 124.47, 128.27, 129.94, 130.18, 131.60, 131.96, 133.08, 138.57, 138.63, 145.30, 147.50, 147.72, 167.31, 184.56, 186.22, 196.82. LDI-TOF MS: m/z [M]+ calcd for C23H19NO3Pt: 552; found: 552. Anal. calcd for C23H19NO3Pt: C, 50.00; H, 3.47; N, 2.54. Found: C, 49.92; H, 3.68; N, 2.52.
Pt-1b (yield: 30%). 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 3H), 2.04 (s, 3H), 5.52 (s, 1H), 7.18 (ddd, J = 7.3, 6.0 and 1.4 Hz, 1H), 7.36 (d, J = 11.0 Hz, 1H), 7.43–7.49 (m, 2H), 7.58 (tt, J = 7.3 and 1.4 Hz, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 6.9 Hz, 1H), 7.82–7.87 (m, 3H), 9.01 (dd, J = 6.0 and 0.9 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ −105.37 (1F). LDI-TOF MS: m/z [M]+ calcd for C23H18FNO3Pt: 570; found: 570. Anal. calcd for C23H18FNO3Pt: C, 48.42; H, 3.18; N, 2.46. Found: C, 48.49; H, 3.22; N, 2.55.
Pt-1c (yield: 65%). 1H NMR (400 MHz, CD2Cl2) δ 2.01 (s, 3H), 2.03 (s, 3H), 5.53 (s, 1H), 7.18–7.24 (m, 2H), 7.45–7.50 (m, 2H), 7.61 (tt, J = 7.3 and 1.4 Hz, 1H), 7.84–7.90 (m, 3H), 7.93 (d, J = 8.2 Hz, 1H), 9.04 (ddd, J = 5.9, 0.9 and 0.9 Hz, 1H). 19F NMR (376 MHz, CDCl3) δ −114.84 (1F), −107.49 (1F). LDI-TOF MS: m/z [M]+ calcd for C23H17F2NO3Pt: 588; found: 588. Anal. calcd for C23H17F2NO3Pt: C, 46.94; H, 2.91; N, 2.38. Found: C, 47.15; H, 3.15; N, 2.36.
Pt-2b (yield: 68%). 1H NMR (400 MHz, CDCl3) δ 1.89 (s, 3H), 2.01 (s, 3H), 5.46 (s, 1H), 7.20 (ddd, J = 7.3, 6.0 and 1.4 Hz, 1H), 7.44–7.49 (m, 2H), 7.51–7.59 (m, 3H), 7.70 (d, J = 8.2 Hz, 1H), 7.84–7.90 (m, 3H), 8.00 (d, J = 1.4 Hz, 1H), 9.05 (dd, J = 6.0 and 0.9 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 27.05, 28.30, 102.62, 119.35, 122.36, 122.57, 125.42, 128.11, 130.28, 132.18, 132.21, 137.42, 138.10, 138.46, 138.78, 147.76, 148.57, 167.19, 184.47, 186.00, 197.42. LDI-TOF MS: m/z [M + H]+ calcd for C23H20NO3Pt: 553; found: 553. Anal. calcd for C23H19NO3Pt: C, 50.00; H, 3.47; N, 2.54. Found: C, 49.97; H, 3.48; N, 2.38.
Pt-2c (yield: 36%). 1H NMR (400 MHz, CDCl3) δ 2.03 (s, 3H), 2.04 (s, 3H), 2.59 (s, 3H), 5.50 (s, 1H), 7.19 (ddd, J = 7.3, 6.0 and 1.4 Hz, 1H), 7.72–7.79 (m, 3H), 7.86 (ddd, J = 7.3, 7.3 and 1.4 Hz, 1H), 8.07 (s, 1H), 9.03 (d, J = 6.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 26.56, 27.18, 28.27, 102.69, 118.94, 121.98, 122.31, 129.50, 130.56, 133.16, 138.59, 145.32, 147.45, 148.25, 167.30, 184.53, 186.19, 198.09. LDI-TOF MS: m/z [M]+ calcd for C18H17NO3Pt: 490; found: 490. Anal. calcd for C18H17NO3Pt: C, 44.08; H, 3.49; N, 2.86. Found: C, 44.01; H, 3.72; N, 2.87.
Pt-2d (yield: 6.5%). 1H NMR (400 MHz, CDCl3) δ 1.991 (s, 3H), 1.987 (s, 3H), 5.46 (s, 1H), 6.98 (ddd, J = 9.7, 8.2 and 2.7 Hz, 1H), 7.11–7.17 (m, 2H), 7.50–7.57 (m, 2H), 7.81 (ddd, J = 7.5, 7.5 and 1.4 Hz, 1H), 9.00 (ddd, J = 6.3, 6.3 and 0.9 Hz, 1H). 19F NMR (470 MHz, CDCl3) δ −121.55 (1F). LDI-TOF MS: m/z [M]+ calcd for C16H14FNO2Pt: 466; found: 466. Anal. calcd for C16H14FNO2Pt: C, 41.21; H, 3.03; N, 3.00. Found: C, 40.83; H, 2.91; N, 2.94.
X-ray crystallography
The single crystals of Pt-1a, Pt-1b, and Pt-1c suitable for X-ray crystallography were grown by the slow diffusion of their dichloromethane solutions to hexane. Diffraction data were collected on a Rigaku AFC-7 Mercury CCD diffractometer, using graphite monochromated Mo-Kα radiation (λ = 0.71075 Å). The cell parameters were collected to maximum 2θ values of 61.2–61.5° at temperatures of 25 ± 1 °C for Pt-1a and 20 ± 1 °C for Pt-1b and Pt-1c. The structures were solved by direct methods using the SIR9243 program and expanded using Fourier techniques with the DIRDIF9944 program. All calculations were performed using the Crystal Structure 3.845 software packages. The crystal data and refinement details of the crystal structure determination are given in Table S1 (ESI†).
UV-vis and PL spectroscopic measurements
UV-vis absorption and PL spectra were recorded on a Shimadzu UV-3600 and a Horiba Jobin Yvon Fluorolog-3 spectrophotometer, respectively. ΦPLs were measured on a Hamamatsu Photonics C9920-12 absolute PL quantum yield measurement system. PL lifetimes (τPLs) were obtained on a Horiba Jobin Yvon FluoroCube spectroanalyzer using a 390 nm nanosecond-order LED light source. The sample solutions for PL measurements were bubbled with nitrogen gas (flow rate; 50 mL min−1) to remove oxygen. Poly(methyl methacrylate) (PMMA) films (thickness; ca. 60 nm) were fabricated on quartz substrates using a spin-coating method at a rate of 1500 rpm (2 s) followed by 3000 rpm (60 s) using toluene solutions of PMMA (14 mg ml−1) and platinum(II) complexes. The obtained films were annealed for 1 h at 120 °C. Their spectra were measured under a nitrogen atmosphere.
Fabrication and characterization of OLEDs
A pre-coated indium tin oxide (ITO; thickness, 150 nm; sheet resistance, ca. 10 Ω sq−1) substrate, 1,3-bis(carbazol-9-yl)benzene (mCP), lithium fluoride, and aluminium were purchased from Kintec Company, Tokyo Chemical Industry, Sigma-Aldrich, and Kurt J. Lesker Company, respectively. 2,3,5,6-Tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) and tris(8-hydroxyquinolinato)aluminium (Alq3) were purchased from e-Ray Optoelectronics Technology. N,N′-Di(1-naphthyl)-N,N′-diphenyl-(1,10-biphenyl)-4,4′-diamine (NPB), 4,4′-cyclohexylidenebis[N,N-bis(4-methylphenyl)benzenamine] (TAPC), and 1,3,5-tris(1-phenyl-1H-benzimidazol-2-yl)benzene (TPBi) were purchased from Luminescence Technology.
Patterned ITO substrates were cleaned with acetone to remove any residual photoresists and brushed with a liquid detergent. After soaking in hot water, they were consecutively sonicated in 2-propanol and de-ionized water. Then the dried substrates were treated by UV-Ozone. The cleaned substrates were loaded in a deposition chamber. Successive layers of F4TCNQ, NPB, TAPC, mCP:Pt-1c, TPBi, Alq3, lithium fluoride and aluminium were sequentially deposited using a thermal evaporator under a reduced pressure of 10−8 Torr. The fabricated devices were encapsulated using UV-curable epoxy resin in a nitrogen environment. The OLED performance was studied at room temperature, using a Keithley 2400 source-meter and a spectrascan PR655 spectroradiometer.
Results and discussion
Synthesis and molecular structures of Pt-1a–c
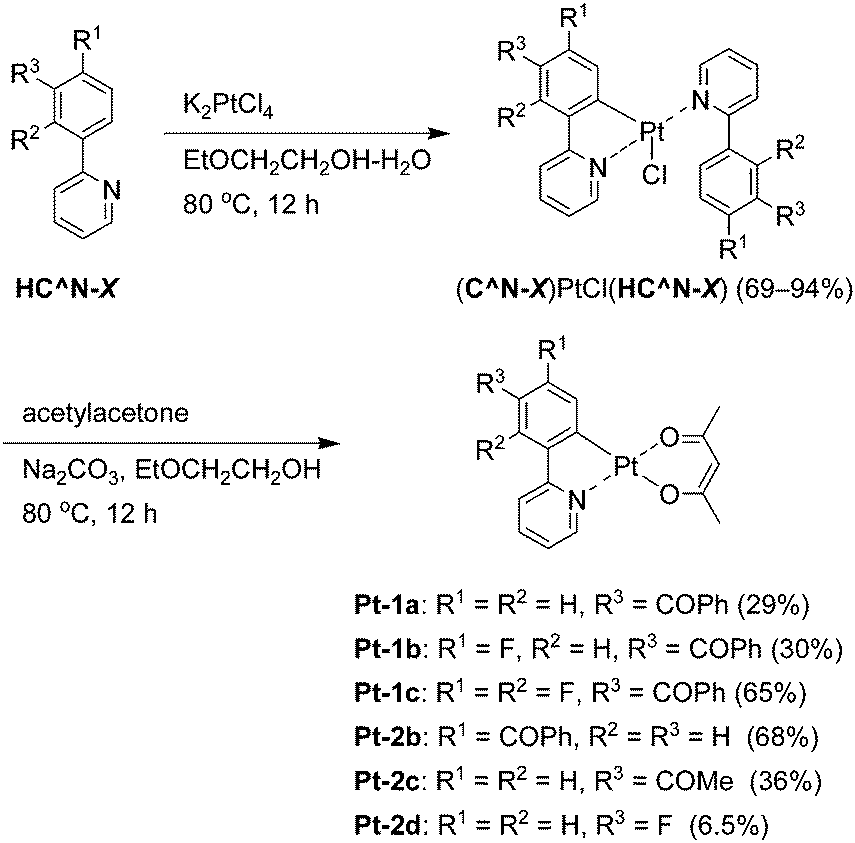
The novel benzoylated platinum(II) complexes Pt-1a–c were synthesized according to Scheme 1. The 5′-benzoylated 2-phenylpyridine derivatives HC^N-1a–c were reacted with potassium tetrachloridoplatinate to afford the corresponding mononuclear complexes (C^N-1)PtCl(HC^N-1) in 69–94% yields, which were subjected to the ligand exchange reaction to yield Pt-1a–c in 29, 30 and 65% yields, respectively. We found that recrystallization from hot ethyl acetate solution afforded highly pure Pt-1a–c. The low synthetic yields of Pt-1a and Pt-1b were due to their relatively high solubility in ethyl acetate at rt. On the other hand, Pt-1c was obtained in a higher yield because of its lower solubility in ethyl acetate at ambient temperature. Reference complexes Pt-2b–d were also synthesized in the same procedure.
 |
| | Scheme 1 Synthesis of Pt-1a–c and Pt-2b–2d. | |
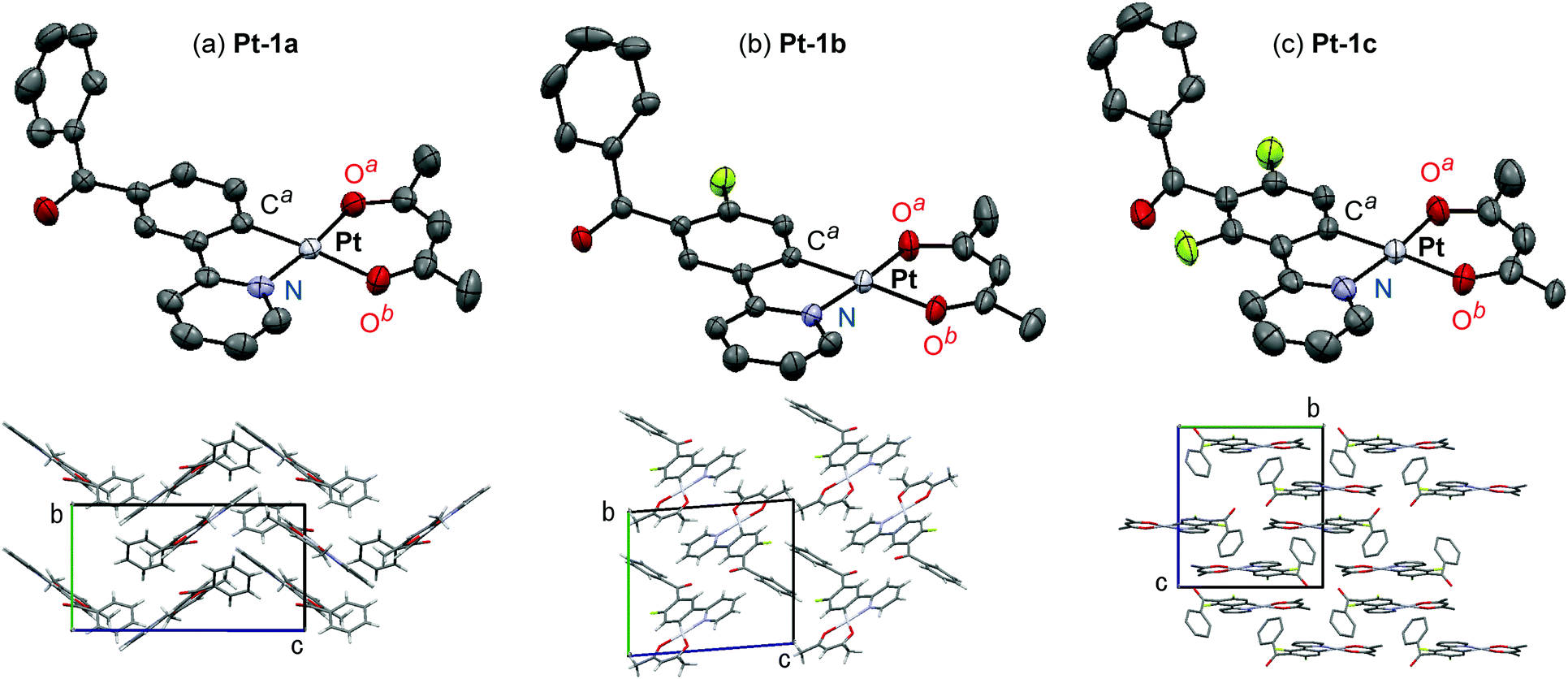
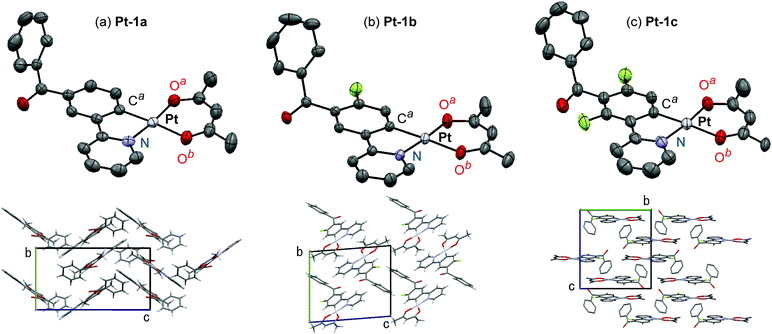
Molecular structures of Pt-1a–c were characterized by X-ray crystallography using their single crystals. The ORTEP drawings of Pt-1a–c are shown in Fig. 2, and the crystal data are summarized in Table S1 (ESI†). As seen in typical heteroleptic cyclometalated platinum(II) complexes so far reported, Pt-1a–c adopt a four-coordinated square–planar coordination geometry.9,10,17,23,46–49 The mean plane deviations of a mean plane consisting of the (ppy)Pt(acac) skeleton except the benzoyl group and the hydrogen atoms were 0.044, 0.015, and 0.021 Å for Pt-1a, Pt-1b, and Pt-1c, respectively, suggesting their quite high planarities. In Pt-1a, the bond lengths of Pt–Ca, Pt–N, Pt–Oa, and Pt–Ob were 1.965, 1.988, 1.997, and 2.09 Å, respectively, and the angles of N–Pt–Ca, Ca–Pt–Oa, Oa–Pt–Ob, and Ob–Pt–N were 81.7°, 92.8°, 92.3°, and 93.3°, respectively. The bond lengths and angles of Pt-1a are similar to those of Pt-1b, Pt-1c, and other reported platinum(II) complexes.9,10,47,49 The benzoyl groups of Pt-1a–c were distorted with respect to the mean plane of the ppy skeleton: the angles between the ppy mean plane and a mean plane of the phenyl group in the benzoyl group were 64.9°, 61.3° and 69.1° for Pt-1a–c, respectively. The π-stacking distances between neighbor molecules were 3.39, 3.46 and 3.41 Å in the crystals of Pt-1a–c, respectively, indicating their strong π–π interactions essential to excimer formation.36
 |
| | Fig. 2 ORTEP drawings and crystal packings of (a) Pt-1a, (b) Pt-1b, and (c) Pt-1c. The hydrogen atoms are omitted for clarity, and the thermal ellipsoids are presented at the 50% probability level. | |
Photoluminescence properties
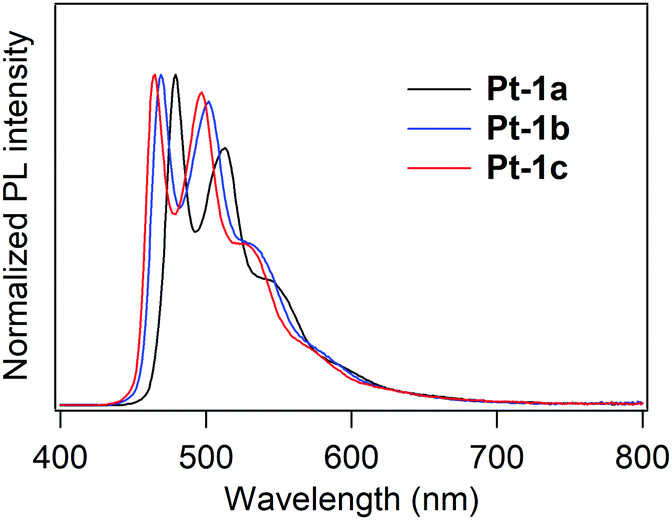
PL spectra of Pt-1a–c in deaerated dichloromethane (10 μM) at rt are shown in Fig. 3, and the spectral and photophysical data are summarized in Table 1. All the complexes showed PL lifetimes in a micro-second or a sub-micro-second order ranging from 0.295 to 1.37 μs, indicating that the observed emission is phosphorescence. Pt-1a exhibits bluish green PL with a spectrum that has two peaks at 479 and 513 nm and two shoulders at ca. 550 and ca. 600 nm. This type of spectral shape is characteristic of the monomer emission of (C^N)Pt(O^O)-type complexes.9,13,15,17,24 The lowest energy emission peak was blue-shifted by 5 nm in comparison with the unsubstituted complex Pt-2a (λPL; 484 nm in dichloromethane,50Fig. 1). Thus, the introduction of the benzoyl group is actually effective in inducing a blue shift of PL. The fluorinated complexes Pt-1b and Pt-1c exhibited greenish blue PL with the lowest energy emission peaks of 469 and 465 nm, respectively, further blue-shifted in comparison with Pt-1a. Thompson and co-workers reported that the λPL of (dfppy)Pt(acac) was 466 nm in 2-methyltetrahydrofuran (ΦPL; 0.02),9 and it also shows the same λPL in dichloromethane.50 Thus, the combination of benzoyl and fluorine substituents is slightly effective in inducing a blue shift of λPL. The ΦPL decreased according to the increase in the number of fluorine substituents: the ΦPLs of Pt-1a–c were 0.28, 0.13 and 0.063, respectively. One might see that this is due to facilitation of the non-radiative relaxation process in accordance with the increase in the lowest triplet energy, as previously reported for (C^N)Pt(O^O)-type complexes.9 The radiative rate constant (kr) and non-radiative rate constant (knr) of Pt-1a–c were calculated from their ΦPLs and τPLs according to the following eqn (1) and (2):
 |
| | Fig. 3 PL spectra of Pt-1a–c in deaerated dichloromethane (10 μM, rt). | |
Table 1 PL properties of Pt-1a–c in deaerated dichloromethane (10 μM, rt), where the excited wavelength was 390 nm
| Compound |
λ
PL (nm) |
Φ
PL
|
τ
PL
(μs) |
k
r (μs−1) |
k
nr (μs−1) |
|
Detected at the shortest λPL. The value of χ2 was 1.0–1.1.
|
|
Pt-1a
|
479, 513 |
0.28 |
1.37 |
0.20 |
0.53 |
|
Pt-1b
|
469, 502 |
0.13 |
1.03 |
0.13 |
0.84 |
|
Pt-1c
|
465, 497 |
0.063 |
0.295 |
0.21 |
3.18 |
Almost similar krs (0.13–0.21 μs−1) are obtained for Pt-1a–c, whereas knr increases with the increase in the number of introduced fluoro groups (0.53, 0.84, and 3.18 μs−1, respectively). This result clearly shows that the increase in the lowest triplet energy gives rise to facilitation of the non-radiative decay process.
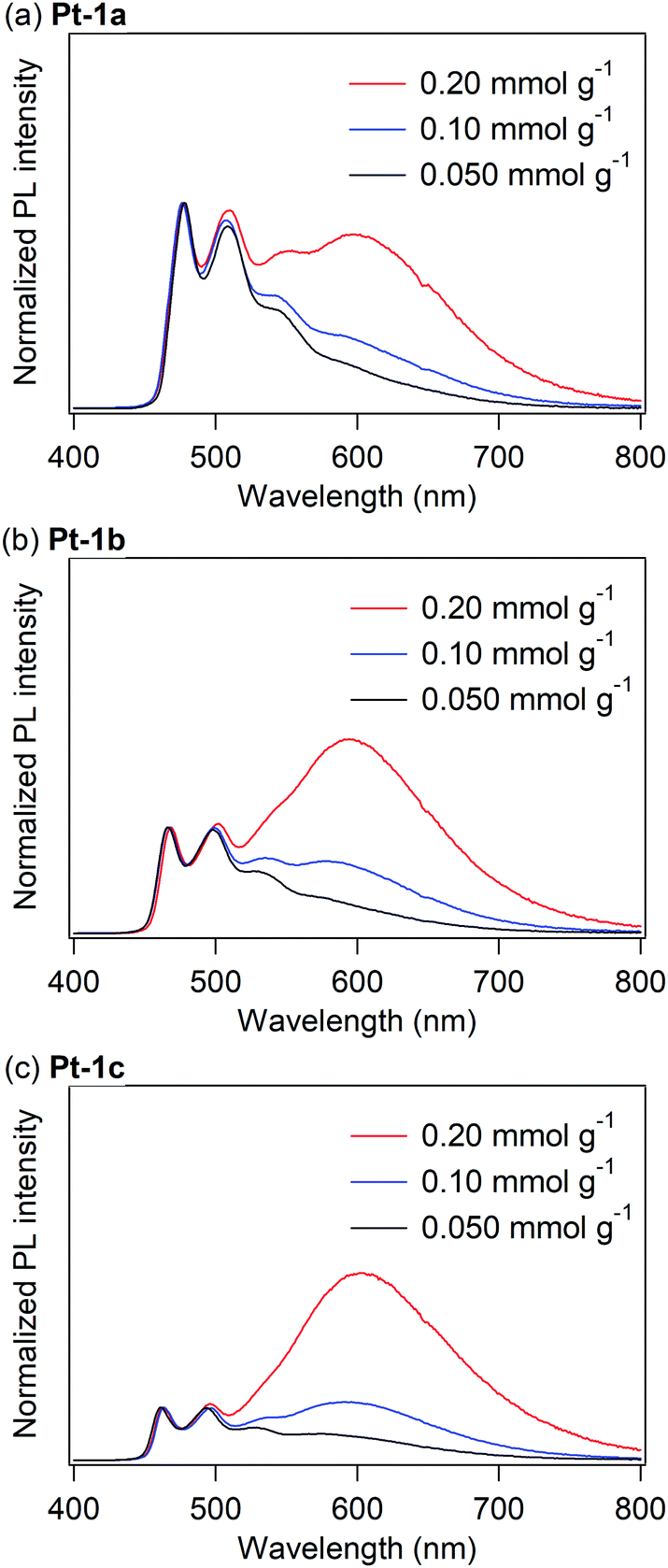
Fig. 4 shows the PL spectra of PMMA films doped with Pt-1a–c, where the doping levels were varied as 0.050, 0.10, and 0.20 mmol g−1, approximately corresponding to 3, 5, and 10 wt%, respectively. The PL spectral and photophysical data of the doped PMMA films are summarized in Table 2. The PMMA film doped with 0.050 mmol g−1 of Pt-1a showed an almost identical PL spectrum to that in dichloromethane. As the doping level of Pt-1a increased, a new emission band appeared at ca. 600 nm with increasing intensities (Fig. 4a), assignable to excimer emission because the spectral shape of the UV-vis absorption spectrum of Pt-1a in the PMMA film is identical regardless of the doping level (Fig. 5 and Fig. S1, ESI†).17,51 In addition, the PMMA film at the doping level of 0.20 mmol g−1 showed the excitation spectrum detected at the excimer emission band to be identical to the spectra detected at the monomer ones (Fig. S2, ESI†). If the new emission band originates from an excited aggregate, a new absorption band such as a metal–metal-to-ligand charge transfer transition band should appear at the longer wavelength region.52 On the other hand, emissive excimer molecules do not show any absorption bands in their steady-state absorption spectra because they form the dimeric structure only in the excited state.51 In addition, the significantly broad spectral shape of the new emission band is characteristic of the excimer: in general, the excimer emission shows a broad structureless band generated by a radiative relaxation process of the excimer to a dissociated ground state.53 Therefore, the new emission band of Pt-1a is not aggregate-based emission but excimer emission. For Pt-1b and Pt-1c, remarkable excimer emission was also observed at ca. 600 nm as the doping level increased. Obviously, the excimer emission was enhanced in comparison with Pt-1a with the increase in the number of introduced fluoro groups. The ΦPL of Pt-1a in the PMMA film at the doping level of 0.050 mmol g−1 was 0.62, which was much higher than that in dichloromethane. It has been reported that the triplet metal-centered (3MC) states of (C^N)Pt(O^O)-type complexes adopt highly distorted structures,54,55 and thus non-radiative relaxation is facilitated through the 3MC state. On the other hand, the non-radiative decay through structural relaxation should be suppressed in rigid media. This should be the reason why Pt-1a is highly emissive in the PMMA film. This is also the case with Pt-1b and Pt-1c, with ΦPLs of 0.45 and 0.51 at a doping level of 0.050 mmol g−1, respectively. In this regard, the ΦPL is not drastically changed in the range of the doping level of 0.050–0.20 mmol g−1 for each of Pt-1a–c, with values of 0.40–0.62, 0.45–0.47, and 0.51–0.62 for Pt-1a–c, respectively. That is, the present complexes are still emissive even when remarkable excimer formation occurs, although excimer emission is often associated with concentration quenching.17,24,51,56 In particular, Pt-1c shows a relatively high ΦPL of 0.53 at a doping level of 0.20 mmol g−1, where the relative intensity of the excimer emission is more than three times as high as that of the monomer emission.
 |
| | Fig. 4 PL spectra of PMMA films doped with (a) Pt-1a, (b) Pt-1b and (c) Pt-1c at rt under a nitrogen atmosphere. | |
Table 2 PL properties of Pt-1 in PMMA film under a nitrogen atmosphere, where the excited wavelength was 390 nm
| Compound |
Doping level (mmol g−1) |
λ
PL (nm) |
Φ
PL
|
|
Pt-1a
|
0.050 |
478, 508 |
0.62 |
| 0.10 |
476, 507, 583 |
0.40 |
| 0.20 |
477, 510, 576 |
0.47 |
|
Pt-1b
|
0.050 |
466, 498 |
0.45 |
| 0.10 |
467, 498, 534, 577 |
0.47 |
| 0.20 |
468, 501, 597 |
0.46 |
|
Pt-1c
|
0.050 |
461, 493, 527, 572 |
0.51 |
| 0.10 |
464, 497, 539, 591 |
0.62 |
| 0.20 |
463, 496, 603 |
0.53 |
 |
| | Fig. 5 UV-vis absorption spectra of Pt-1a in PMMA at varying doping levels. | |
In order to investigate the substituent effect of the benzoyl group on the excimer emission, the PL spectra of Pt-1a were compared with those of the reference complexes Pt-2a–d: Pt-2a has no substituent on the cyclometalated ligand, and Pt-2b, Pt-2c, and Pt-2d have a 4′-benzoyl, a 5′-acetyl, and a 5′-fluoro group, respectively. The PL spectra of Pt-2a–d in dichloromethane (10 μM) and in PMMA film (0.20 mmol g−1) are shown in Fig. 6. For Pt-2a and Pt-2b, almost the same PL spectra were obtained in both dichloromethane and PMMA (Fig. 6a and b). This clearly shows that the remarkable excimer emission of Pt-1a is brought about by the substituent effect of the 5′-benzoyl group, and the substituted position is also important. In the case of Pt-2c, remarkable excimer emission was obtained in the PMMA film, the spectrum of which was very similar to that of Pt-1a although the excimer-to-monomer emission ratio was somewhat smaller than that of Pt-1a (Fig. 6c). This indicates that the remarkable excimer emission of Pt-1a is not based on π-extension by introduction of the benzoyl group, but predominantly on the electronic effect of the carbonyl moiety. In fact, the phenyl group of the benzoyl group is distorted with respect to the ppy mean plane as discussed in the X-ray crystallography section (Fig. 1), and thus the benzoylated cyclometalated ligand does not facilitate the intramolecular stacking interaction. Although Pt-2d showed a new emission band at ca. 600 nm, its intensity was quite modest in comparison with the monomer emission. Therefore, not all electron-withdrawing groups at the 5′-position facilitate remarkable excimer emission. Further investigation was carried out for the effect of combination of the benzoyl and fluoro groups, and the PL spectra of poly(9-vinylcarbazole) films doped with Pt-1c and (dfppy)Pt(acac) were measured. In this case, as (dfppy)Pt(acac) is not compatible with PMMA, poly(9-vinylcarbazole) was used as a matrix polymer. The excimer-to-monomer emission ratio of Pt-1c is much higher than that of (dfppy)Pt(acac) (Fig. S1 in the ESI†). Thus the introduction of the benzoyl group to the fluorinated ppy ligand is effective to obtain considerably enhanced excimer emission.
 |
| | Fig. 6 PL spectra of (a) Pt-2a, (b) Pt-2b, (c) Pt-2c and (d) Pt-2d in dichloromethane (10 μM) and PMMA films (0.20 mmol g−1) under deaerated conditions. | |
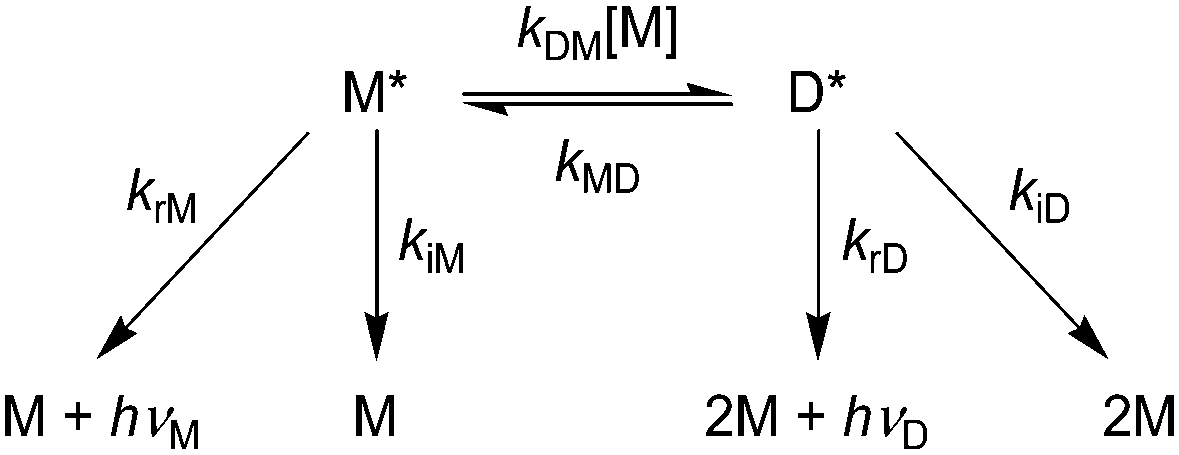
Photokinetic studies
To investigate how the benzoylated cyclometalated ligand facilitates the excimer emission, we carried out the kinetic analysis of the PL decay process. Birks showed a general kinetic scheme for excimer formation and decay as shown in Scheme 2, where M, M*, and D* are a ground-state monomer, an excited monomer, and an excimer, respectively.57krM and krD are the radiative rate constants for M* and D*, respectively, and kiM and kiD are the non-radiative rate constants for M* and D*, respectively. kDM is the second-order rate constant for excimer formation, and kMD is the first-order rate constant for the regeneration of M* from D*. Eqn (3) and (4) represent the PL intensities of monomer emission (IM(t)) and excimer emission (ID(t)) at the time t in accordance with this kinetic model:| |  | (3) |
| |  | (4) |
where kM, kD, λ1, λ2, and A are defined by eqn (5)–(8) (the minus (plus) sign corresponds to λ1 (λ2) in eqn (7));| | 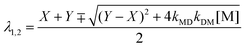 | (7) |
| |  | (8) |
withandUnder the initial conditions of [M*] = [M*]0 and [D*] = 0 at t = 0, suppose that the number of M* generated by photoexcitation is much smaller than that of M and the concentration change of M is ignorable ([M]; constant). In general, the PL decay profiles of both monomer and excimer emissions should be doubly exponential as represented by eqn (3) and (4) when the excimer formation reaction is reversible (kMD > 0).58 However, when the excimer formation process is irreversible (kMD = 0), eqn (3) and (4) are represented by eqn (11) and (12), respectively:| | | IM(t) = krM[M*]0e−(kM + kDM[M]t) | (11) |
| |  | (12) |
 |
| | Scheme 2 The general mechanism of excimer formation and monomer (hνM) and excimer emissions (hνD). | |
Therefore, the PL decay profiles of monomer and excimer emissions should be singly and doubly exponential, respectively, in the case of the irreversible excimer formation.
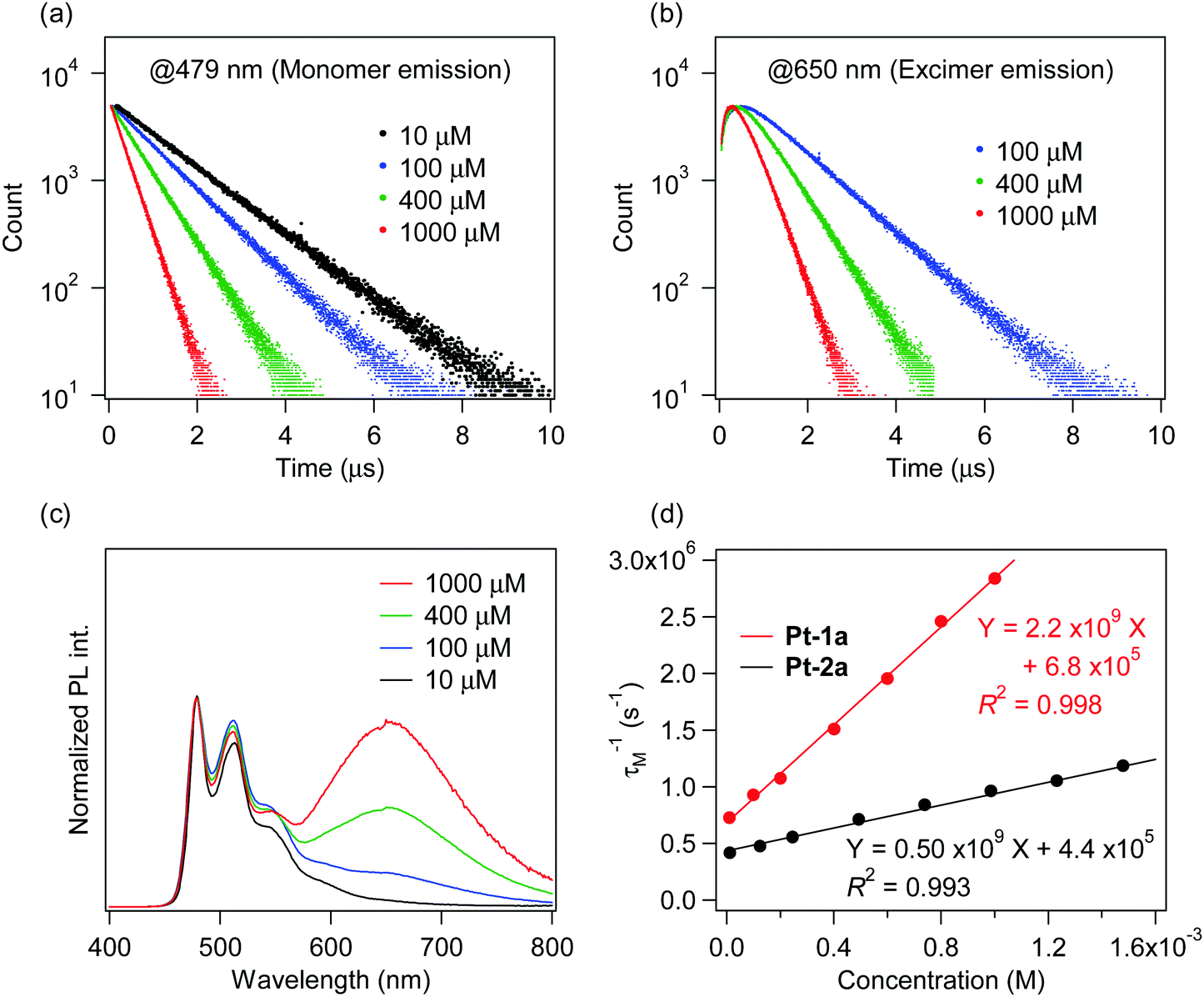
The investigation of the kinetics of the excimer emission is more favorable in solution than in PMMA film because the diffusion rate of molecules in PMMA is low59 and thus competes with the excimer formation rate. So the PL decay profiles of Pt-1a were obtained in dichloromethane at varying concentrations from 10 μM to 1.0 mM (Fig. 7). The excimer emission was observed at ca. 650 nm in dichloromethane when the concentration was more than 100 μM. In Fig. 7a and 7b, the PL decay profiles of monomer emission (479 nm) and excimer emission (650 nm) are shown, respectively. The decays of both monomer and excimer emissions were accelerated as the concentration increased. The decay profiles of monomer emission were singly exponential at any investigated concentration. On the other hand, those of excimer emission were doubly exponential, showing rise- and decay-type curves. These results correspond to the decay profiles represented by eqn (11) and (12). The decay profiles of monomer emission were well fitted at any concentration to the singly exponential decay (i.e., eqn (13)) with χ2 values less than 1.1, where the fitting parameters aM, τM and bM are the coefficient (>0), the lifetime, and the background intensity, respectively, for monomer emission:
| |  | (13) |
 |
| | Fig. 7 PL decay profiles detected at (a) 479 nm (monomer emission) and (b) 650 nm (excimer emission) for Pt-1a, (c) PL spectra of Pt-1a, and (d) plots and approximate lines of τM−1 against concentration for Pt-1a and Pt-2a in dichloromethane at rt. All samples were excited at 390 nm. | |
Next, the decay profiles of excimer emission were also well fitted to doubly exponential eqn (14) with χ2 values less than 1.2, where the fitting parameters aD, τD1, τD2, and bD are the coefficient (>0), the lifetime of the positive component, the lifetime of the negative component, and the background intensity, respectively, for excimer emission:
| |  | (14) |
The obtained τM, τD1, and τD2 are summarized in Table 3. Fig. 7d shows the plots of τM−1 against the concentration of the prepared solution, i.e. [M]. τM−1 had a good linear relationship to [M] with an R2 value of 0.998. This indicates that the decay profiles of monomer emission of Pt-1a obey eqn (11) at any investigated concentration, and thus τM−1 is represented using eqn (15) according to eqn (11) and (13):
Table 3 Fitted values of τM, τD1 and τD2 of Pt-1a
| [M] (mM) |
τ
M (μs) |
τ
D1 (μs) |
τ
D2 (μs) |
|
Not determined due to the weak excimer emission.
|
| 0.010 |
1.4 |
—a |
—a |
| 0.10 |
1.1 |
—a |
—a |
| 0.20 |
0.93 |
—a |
—a |
| 0.40 |
0.66 |
—a |
—a |
| 0.60 |
0.51 |
0.53 |
0.22 |
| 0.80 |
0.41 |
0.42 |
0.20 |
| 1.0 |
0.35 |
0.38 |
0.20 |
Therefore, the intercept and slope of the approximate line correspond to the kM and the kDM, respectively, which were provided as 6.8 × 105 s−1 and 2.2 × 109 M−1 s−1, respectively. The τD1s were almost identical to the τMs at the same concentrations as shown in Table 3. The τD2s were ca. 0.2 μs and independent of the concentration. These results show that the decay profiles of excimer emission of Pt-1a obey eqn (12) and the kD (= τD2−1) was 4.8 × 106 s−1.60 Therefore, the experimental results are consistent with eqn (11) and (12), indicating that the present excimer emission behaviour includes the irreversible or pseudo-irreversible excimer formation reaction. On the other hand, Shinozaki and co-workers reported that tridentate cyclometalated platinum(II) complexes showed the reversible process of excimer and excited trimer formation, although the values of kDMs (2.8–4.5 × 109 M−1 s−1) were similar to that of Pt-1a.39 Next, radiative and non-radiative decay rate constants of M* and D* are investigated. It was supposed that the krM is the same as kr obtained at a concentration of 10 μM; 2.0 × 105 s−1 (Table 1). Then the kiM was determined as 4.8 × 105 s−1 according to eqn (5). The ratio of the excimer-to-monomer emission intensity (ID/IM) is represented using eqn (16).57
| | 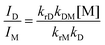 | (16) |
I
D/IM was 1.4 at a concentration of 0.60 mM, which was determined from the relative areas obtained by the deconvolution of the PL spectrum. krD was estimated at 1.0 × 106 s−1 from these values, and kiD was estimated at 3.9 × 106 s−1 according to eqn (6). From the above, the rate constants for Pt-1a were fully characterized and they are summarized in Table 4.
Table 4 The rate constants for Pt-1a and Pt-2a
|
|
Pt-1a
|
Pt-2a
|
|
k
r at 10 μM.
Calculated using the PL spectrum at 0.60 mM.
|
|
k
DM (109 M−1 s−1) |
2.2 |
0.50 |
|
k
M (105 s−1) |
6.8 |
4.4 |
|
k
rM (105 s−1)a |
2.0 |
2.1 |
|
k
iM (105 s−1) |
4.8 |
2.9 |
|
k
D (106 s−1) |
4.9 |
6.4 |
|
k
rD (106 s−1)b |
1.0 |
5.2 |
|
k
iD (106 s−1)b |
3.9 |
1.2 |
The unsubstituted complex Pt-2a also showed similar behaviour to Pt-1a, which also exhibited excimer emission at around 650 nm. The plots of τM−1 against concentration for Pt-2a are also shown in Fig. 7d, and the obtained rate constants for Pt-2a are also summarized in Table 4. The kDM of Pt-2a was 0.50 × 109 M−1 s−1, a quarter of that of Pt-1a. This result indicates that introduction of the benzoyl group facilitates the excimer formation reaction.
Electroluminescence properties
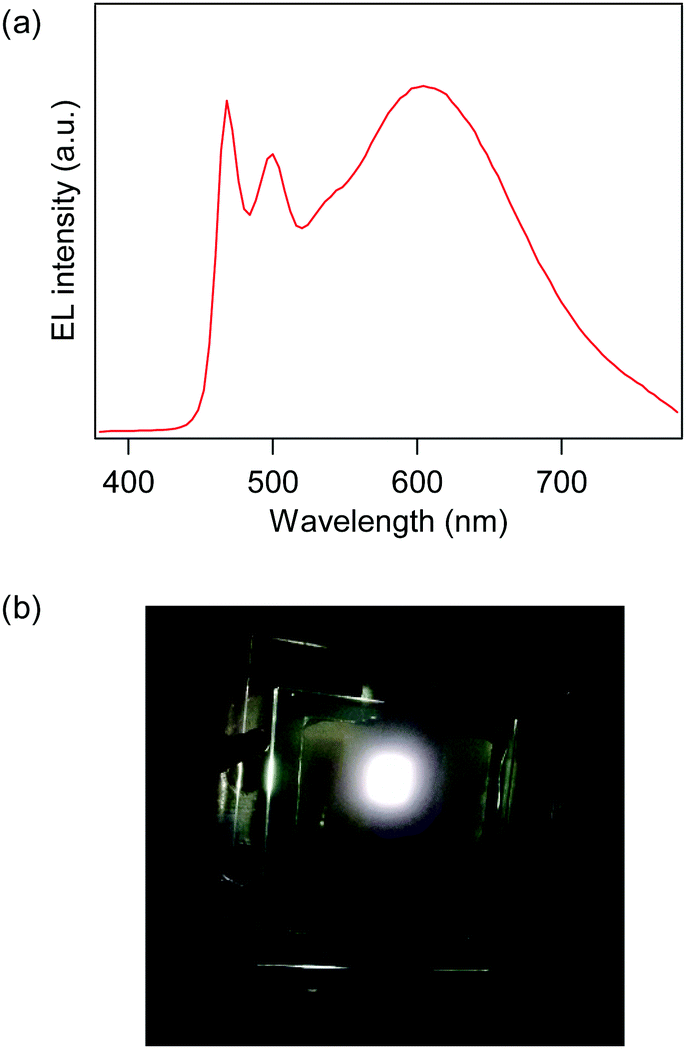
From the PL properties investigated above, Pt-1c is the best white emitter among Pt-1a–c because it exhibits the most blue-shifted monomer emission and remarkable excimer emission suitable to obtain emission covering the whole visible region. Therefore, we fabricated an OLED employing Pt-1c, where the device structure is as follows: ITO (150 nm, anode)/F4TCNQ (3 nm)/NPB (50 nm)/TAPC (15 nm)/mCP:Pt-1c (30 nm)/TPBi (15 nm)/Alq3 (20 nm)/LiF (1.5 nm)/Al (100 nm, cathode), where the doping level of Pt-1c was 15 wt%. The electroluminescence (EL) spectrum at the maximum luminance (Lmax) and the photograph of the emitting device are shown in Fig. 8. The device exhibited pseudo-white EL with the Commission internationale de l’éclairage (CIE) chromaticity coordinates of (0.42, 0.42) and a correlated color temperature of 3430 K at the Lmax, and the EL spectrum covered the visible region from 460 to over 700 nm due to the combination of monomer and excimer emissions, as shown in Fig. 8a. The EL was obtained at more than 4.0 V, and the maximum values of luminance, current efficiency and power efficiency were 11500 cd m−2 (at 8.8 V), 16.0 cd A−1 (at 7.8 V) and 6.68 lm W−1 (at 7.4 V). We also fabricated some OLEDs bearing an emitting layer doped with 2–10% of Pt-1c.61 The EL colour changed from sky-blue to pseudo-white as the doping level increased just like the PL of the doped PMMA films (Fig. S3 in the ESI†).
 |
| | Fig. 8 (a) The EL spectrum at the Lmax and (b) the photograph of the device employing Pt-1c. | |
Conclusions
In conclusion, we found that (ppy)Pt(acac)-type complexes, Pt-1a–c, bearing a benzoyl group at the 5′-position of the ppy ligand exhibit remarkable excimer emission at ca. 600 nm in PMMA film along with monomer emission at 461–478 nm. The X-ray structural analysis revealed the distortion of the benzoyl group from the ppy skeleton, and that the enhancement of excimer emission is not due to π-extension of the cyclometalated ligand by introduction of the benzoyl group, but likely due to an electronic effect caused by the carbonyl moiety of the benzoyl group. The kinetic analysis of PL decays of Pt-1a revealed that excimer formation from the excited and ground-state monomers was an irreversible or a pseudo-irreversible process. From the comparison with the analysis for the reference unsubstituted complex, the excimer formation is obviously facilitated, indicating that the introduced benzoyl group plays a crucial role in the formation of the radiative excimer. We also found that the introduction of additional fluoro group(s) together with the benzoyl group gives rise to enhancement of excimer emission. In particular, the 2-(5-benzoyl-4,6-difluorophenyl)pyridinate-based complex exhibited considerably enhanced excimer emission at ca. 600 nm along with greenish blue monomer emission when doped into PMMA film at the doping level of 0.20 mmol g−1. Using this excimer–emissive complex as a single emitting dopant, a pseudo-white OLED with CIE chromaticity coordinates of (0.42, 0.42) was successfully fabricated, showing the maximum luminance of 11500 cd m−2 and the maximum current efficiency of 16.0 cd A−1. Although the electronic effect of the benzoyl group on excimer emission enhancement is still not clarified at this point, we believe that it is important to elucidate such a substituent effect towards the molecular design to control the excimer behaviour of phosphorescent organometallic complexes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by the JSPS Grant-in-Aid for Scientific Research (B) (JSPS KAKENHI Grant Number JP16H04194). AS acknowledges the UGC, India. The authors are grateful to Tomoki Watanabe for helpful discussions.
Notes and references
- X. Yang, G. Zhou and W.-Y. Wong, Chem. Soc. Rev., 2015, 44, 8484 RSC.
- T. Fleetham and J. Li, J. Photonics Energy, 2014, 4, 040991 CrossRef.
- G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467 RSC.
- M. Aparna, K. Pankaj, M. N. Kamalasanan and C. Subhas, Semicond. Sci. Technol., 2006, 21, R35 CrossRef.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS.
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048 CrossRef CAS.
- S. Huo, J. Carroll and D. A. K. Vezzu, Asian J. Org. Chem., 2015, 4, 1210 CrossRef CAS.
- V. Adamovich, J. Brooks, A. Tamayo, A. M. Alexander, P. I. Djurovich, B. W. D'Andrade, C. Adachi, S. R. Forrest and M. E. Thompson, New J. Chem., 2002, 26, 1171 RSC.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS PubMed.
- W.-Y. Wong, Z. He, S.-K. So, K.-L. Tong and Z. Lin, Organometallics, 2005, 24, 4079 CrossRef CAS.
- Y. Wang, X. Deng, Y. Liu, M. Ni, M. Liu, H. Tan, X. Li, W. Zhu and Y. Cao, Tetrahedron, 2011, 67, 2118 CrossRef CAS.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed.
- T. Sato, H. Awano, O. Haba, H. Katagiri, Y.-J. Pu, T. Takahashi and K. Yonetake, Dalton Trans., 2012, 41, 8379 RSC.
- S. C. F. Kui, P. K. Chow, G. S. M. Tong, S.-L. Lai, G. Cheng, C.-C. Kwok, K.-H. Low, M. Y. Ko and C.-M. Che, Chem. – Eur. J., 2013, 19, 69 CrossRef CAS PubMed.
- D. Kourkoulos, C. Karakus, D. Hertel, R. Alle, S. Schmeding, J. Hummel, N. Risch, E. Holder and K. Meerholz, Dalton Trans., 2013, 42, 13612 RSC.
- J. Luo, Y. Liu, Q. Chen, D. Shi, Y. Huang, J. Yu, Y. Wang, Z. Zhang, G. Lei and W. Zhu, Dalton Trans., 2013, 42, 1231 RSC.
- T. Shigehiro, S. Yagi, T. Maeda, H. Nakazumi, H. Fujiwara and Y. Sakurai, J. Phys. Chem. C, 2013, 117, 532 CAS.
- T. Fleetham, L. Huang and J. Li, Adv. Funct. Mater., 2014, 24, 6066 CrossRef CAS.
- G. Li, T. Fleetham and J. Li, Adv. Mater., 2014, 26, 2931 CrossRef CAS PubMed.
- H. Li, J. Li, J. Ding, W. Yuan, Z. Zhang, L. Zou, X. Wang, H. Zhan, Z. Xie, Y. Cheng and L. Wang, Inorg. Chem., 2014, 53, 810 CrossRef CAS PubMed.
- A. Poloek, C.-W. Lin, C.-T. Chen and C.-T. Chen, J. Mater. Chem. C, 2014, 2, 10343 RSC.
- P. Pinter, H. Mangold, I. Stengel, I. Münster and T. Strassner, Organometallics, 2016, 35, 673 CrossRef CAS.
- M. Z. Shafikov, D. N. Kozhevnikov, M. Bodensteiner, F. Brandl and R. Czerwieniec, Inorg. Chem., 2016, 55, 7457 CrossRef CAS PubMed.
- T. Shigehiro, Q. Chen, S. Yagi, T. Maeda, H. Nakazumi and Y. Sakurai, Dyes Pigm., 2016, 124, 165 CrossRef CAS.
- F. K.-W. Kong, M.-C. Tang, Y.-C. Wong, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 6351 CrossRef CAS PubMed.
- G.-Z. Lu, Y.-M. Jing, H.-B. Han, Y.-L. Fang and Y.-X. Zheng, Organometallics, 2017, 36, 448 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS PubMed.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed.
- C.-H. Yang, S.-W. Li, Y. Chi, Y.-M. Cheng, Y.-S. Yeh, P.-T. Chou, G.-H. Lee, C.-H. Wang and C.-F. Shu, Inorg. Chem., 2005, 44, 7770 CrossRef CAS PubMed.
- S. Ikawa, S. Yagi, T. Maeda, H. Nakazumi, H. Fujiwara and Y. Sakurai, Dyes Pigm., 2012, 95, 695 CrossRef CAS.
- K. H. Lee, J. S. Hwang, D. H. Chae, S. J. Lee, Y. K. Kim and S. S. Yoon, Mol. Cryst. Liq. Cryst., 2012, 563, 185 CrossRef CAS.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- N. Okamura, T. Nakamura, S. Yagi, T. Maeda, H. Nakazumi, H. Fujiwara and S. Koseki, RSC Adv., 2016, 6, 51435 RSC.
- Q. Wang, I. W. H. Oswald, X. Yang, G. Zhou, H. Jia, Q. Qiao, Y. Chen, J. Hoshikawa-Halbert and B. E. Gnade, Adv. Mater., 2014, 26, 8107 CrossRef CAS PubMed.
- K.-H. Kim, J.-L. Liao, S. W. Lee, B. Sim, C.-K. Moon, G.-H. Lee, H. J. Kim, Y. Chi and J.-J. Kim, Adv. Mater., 2016, 28, 2526 CrossRef CAS PubMed.
- D. Kim and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 11371 CrossRef CAS PubMed.
- L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. G. Williams, Chem. Commun., 2012, 48, 5817 RSC.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401 CrossRef CAS.
- T. Kayano, S. Takayasu, K. Sato and K. Shinozaki, Chem. – Eur. J., 2014, 20, 16583 CrossRef CAS PubMed.
- A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS PubMed.
- J.-Y. Cho, K. Y. Suponitsky, J. Li, T. V. Timofeeva, S. Barlow and S. R. Marder, J. Organomet. Chem., 2005, 690, 4090 CrossRef CAS.
- H. Tsujimoto, S. Yagi, Y. Honda, H. Terao, T. Maeda, H. Nakazumi and Y. Sakurai, J. Lumin., 2010, 130, 217 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
-
P. T. Beurskens, G. Admiraal, G. Beuskens, W. P. Bosman, R. d. Gelder, R. Israel and J. M. M. Smits, The DIRDIF-99 program system; technical report of crystallography laboratory, University of Nijimegen, Nijimegen, The Netherlands, 1999 Search PubMed.
-
Crystal structure 3.8, Rigaku and Rigaku Americas, Woodlands, TX, USA, 2000.
- M. Ebina, A. Kobayashi, T. Ogawa, M. Yoshida and M. Kato, Inorg. Chem., 2015, 54, 8878 CrossRef CAS PubMed.
- Y. Xing, C. Liu, X. Song and J. Li, J. Mater. Chem. C, 2015, 3, 2166 RSC.
- C.-H. Chen, F.-I. Wu, Y.-Y. Tsai and C.-H. Cheng, Adv. Funct. Mater., 2011, 21, 3150 CrossRef CAS.
- C. Li, S. Wang, Y. Huang, B. Zheng, Z. Tian, Y. Wen and F. Li, Dalton Trans., 2013, 42, 4059 RSC.
- Measured in deaerated dichloromethane at rt.
- T. Förster, Angew. Chem., Int. Ed., 1969, 8, 333 CrossRef.
- P. Brulatti, V. Fattori, S. Muzzioli, S. Stagni, P. P. Mazzeo, D. Braga, L. Maini, S. Milita and M. Cocchi, J. Mater. Chem. C, 2013, 1, 1823 RSC.
- B. Stevens and M. I. Ban, Trans. Faraday Soc., 1964, 60, 1515 RSC.
- D. Escudero and W. Thiel, Inorg. Chem., 2014, 53, 11015 CrossRef CAS PubMed.
- Y. Xu, Y. Luo, M. Li, R. He and W. Shen, J. Phys. Chem. A, 2016, 120, 6813 CrossRef CAS PubMed.
- E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501 RSC.
- J. B. Birks, D. J. Dyson and I. H. Munro, Proc. R. Soc. London, Ser. A, 1963, 275, 575 CrossRef CAS.
- B. D’Andrade and S. R. Forrest, Chem. Phys., 2003, 286, 321 CrossRef.
- J. Zhang and C. H. Wang, J. Phys. Chem., 1986, 90, 2296 CrossRef CAS.
- According to eqn (12), kM + kDM[M] should be smaller than kD when τD1−1 and τD2−1 correspond to kM + kDM[M] and kD, respectively. We verified the large/small relationship between kM + kDM[M] and kD. When [M] is lower than 1.0 mM, the value of kM + kDM[M] is actually smaller than 2.9 × 106 s−1, which is smaller than the kD (4.9 × 106 s−1).
- Upon varying the doping level
of Pt-1c, the device structure was somewhat modified as follows; ITO (150 nm, anode)/m-MTDATA (60 nm)/F4TCNQ (3 nm)/NPB (50 nm)/TAPC (15 nm)/mCP:Pt-1c (30 nm)/TPBi (15 nm)/Alq3 (20 nm)/LiF (1.5 nm)/Al (100 nm), where the m-MTDATA is 4,4′,4′′-tris[phenyl(m-tolyl)amino]triphenylamine.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of novel compounds, normalized UV-vis and excitation spectra of Pt-1a, PL spectra of Pt-1c and (dfppy)Pt(acac) in the PVCz film, current density–voltage–luminance (J–V–L) curves and efficiency characteristics of OLEDs, CIE chromaticity coordinates of EL from OLEDs doped with various concentrations of Pt-1c, and the crystal data of Pt-1a–c. CCDC 1578228–1578230. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp06944h |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Takeshi
Maeda
a,
Takeshi
Maeda