Structural stability and magnetic-exchange coupling in Mn-doped monolayer/bilayer MoS2†
Received
2nd September 2017
, Accepted 30th November 2017
First published on 8th December 2017
Abstract
Ferromagnetic (FM) two-dimensional (2D) transition metal dichalcogenides (TMDs) have potential applications in modern electronics and spintronics and doping of TMDs with transition metals can enhance the magnetic characteristics. In this work, the structural stability, electronic states, and magnetic properties of Mn-doped monolayer/bilayer MoS2 are studied systematically by first-principles calculations. Substitutional Mn dopants at the Mo sites are energetically favorable in both monolayer and bilayer MoS2 under the S-rich condition which is common in the synthesis of MoS2 nanosheets. Two Mn dopants participate in the FM interaction in monolayer MoS2 and magnetic coupling of two Mn dopants via the double-exchange mechanism can be mediated by the nearest neighboring S. Magnetic coupling can be ascribed to the competition between the double-exchange, direct-exchange, and super-exchange interactions, which take place between two Mn dopants in bilayer MoS2 with the MniMnMo, MniMnS and MnMo–MnMo configurations. Our results reveal the geometrical dependence of magnetic-exchange coupling suggesting that Mn-doped monolayer/bilayer MoS2 has large potential in spintronic devices.
1. Introduction
The discovery of graphene and its unique properties have stimulated extensive research in two-dimensional (2D) materials1–4 and efforts have been made to fabricate high-quality 2D sheets to study the novel physical properties and design new devices.5–9 However, the gapless feature of pristine graphene limits its integration into microelectronic and nanoelectronic devices.1 On the other hand, many transition metal dichalcogenides (TMDs) possessing interesting semiconducting, metallic, superconducting, and magnetic properties have potential applications in photovoltaics,10,11 optical detection,12–14 photocatalysis,15,16 field-effect transistors,17,18 as well as lithium-ion batteries.19–22 These compounds are composed of MX2 (M = Mo, W, Nb, Re, Ta; X = S, Se, Te) layers with covalent bonding within the layers and weak van der Waals (vdW) bonding between layers enabling easy exfoliation. Semiconducting TMDs such as MoS2 are potential candidates in next-generation electronic and optoelectronic devices.23,24 Recently, a spin lifetime of 1 ns or longer exceeding that of graphene has been observed by polarization-resolved photoluminescence measurements.25 The spin–orbit coupling and absence of inversion symmetry induce spin splitting at the valence band maximum thus suppressing spin relaxation and enhancing the spin lifetime.26–28
In spintronics, attention has been paid to strategies that induce and manipulate the magnetic properties in semiconductors and stable magnetism in semiconducting 2D materials. Doping is a simple technique to produce stable magnetic properties in 2D materials.29,30 Theoretical and experimental studies have been conducted to explore magnetism in MoS2 nanosheets doped with transition metals.31–34 For example, substitutional replacement of Mo with TM atoms (V, Mn, Fe, Co, Cu, Ni, Zn, Cd and Hg) can induce significant magnetism in monolayer MoS235–38 if the dopants are separated from each other at a certain distance.39 Substitutional replacement of Mo with Mn can even produce FM properties via the double-exchange mechanism.37,38 Since this interaction is short-ranged, percolation is a key factor in controlling the long-range magnetic order.37 Shu et al.40 have observed FM coupling in Fe-doped monolayer MoS2 through magnetic double-exchange coupling but antiferromagnetic (AFM) coupling in bilayer and multilayer samples by magnetic super-exchange coupling. The electronic states and magnetic properties can be manipulated by the ‘pizza’ and ‘sandwich’ configurations of Mn clusters in MoS2 nanosheets41 thereby suggesting that the magnetism as well as magnetic-exchange coupling among dopants can be manipulated by altering the doping configuration and distribution, which are sensitive to the growth conditions and parameters such as the S/Mo ratio and temperature. Hence, the magnetic coupling behavior and effects of different doping configurations are interesting from the perspective of fundamental understanding and spintronic applications. In this work, based on first-principles calculations, the structural stability and magnetic exchange interactions in Mn-doped MoS2 nanosheets are systematically studied and applications to spintronic devices are discussed thermodynamically and kinetically.
2. Computational method and model
The calculation is performed using the Vienna ab initio simulation package based on density functional theory.42,43 The electron–ion core interaction is described by the projector augmented wave potentials44 which are more accurate than the ultra-soft pseudo-potentials. The Perdew–Burke–Ernzerhof45 formulation of the generalized gradient approximation is chosen to describe the exchange–correlation interaction. The DFT-D2 approach within the Grimme scheme is adopted to correct the contribution of the vdW interactions between different layers46 and the cutoff energy for the plane waves is 400 eV. To avoid the interactions between dopants in neighboring unit cells, a 4 × 4 × 1 supercell of the MoS2 primitive cell in which the separation between dopants is larger than 12 Å is adopted. A 15 Å vacuum layer is added in the out-of-plane direction to inhibit the interactions of neighboring layers. The Brillouin–zone integration is performed by using an 11 × 11 × 1 k-mesh according to the Monkhorst–Pack scheme and a Gaussian smearing broadening of 0.2 eV is introduced. A conjugate-gradient algorithm is implemented to relax the ions to the ground states with an energy convergence of 1.0 × 10−5 eV and force convergence of 0.02 eV Å−1 on each ion, respectively. The supercells and magnetic moment distribution are visualized by the VESTA software.47
In addition, the GGA functional fails to describe partially occupied 3d orbitals of transition metal elements. Therefore using a model of GGA with the correction of a Hubbard U term (GGA+U) to describe the 3d orbitals of impurity transition metal elements seems to be necessary.36,40 However, several on site Hubbard U parameters have been tested for Mo (4d) orbitals, which yield almost the same results as the GGA calculations.31,48 Similarly, Cheng et al. studied the magnetism of monolayer MoS2 doped with Mn by using GGA+U, which agrees with that of the theoretical results within GGA. In order to obtain a reasonable U parameter, the role of U parameter in the range of 3–5 eV (U = 3, 3.5, 4, 4.5, and 5 eV) is investigated. Taking two Mn dopants substitutionally inserted into monolayer MoS2 as a test example, we find that adjusting the U value does not change the magnetic moment (total and local magnetic moments) and FM characteristic upon comparing the result calculated with the GGA method (Fig. S1 in the ESI†). Thus, the GGA+U method is not considered to describe the Mn dopant in MoS2 nanosheets in this work.
The thermodynamic stability of the Mn dopant in MoS2 nanosheets is evaluated by calculating the formation energy (Ef) as shown in the following equation:
| | | Ef = ET − EP − ΔnMoμMo − ΔnSμS − ΔnMnμMn, | (1) |
where
ET and
EP are the energies of the Mn-doped and perfect MoS
2 sheets, respectively, and
μi and Δ
ni are the chemical potential and difference of atomic species
i (
i = Mo, S, and Mn) in the Mn-doped and perfect MoS
2 sheets.
μMnμMn and
μMo are the respective chemical potentials of a single Mn and Mo atom. The energy per atom of α-Mn is taken as
μMn.
38μMo is defined within a range of values corresponding to Mo-rich or S-rich growth conditions. For the Mo-rich condition,
μMo is taken as the energy of a Mo atom within its stable fcc lattice.
μMo is determined from the difference in the energy between a diatomic S
2 molecule and one formula unit of MoS
2, corresponding to the S-rich condition. In order to maintain thermodynamic equilibrium of the crystalline phase (
μMo + 2
μS =
μMoS2), the value of
μS should satisfy the requirement of
μS(bulk) − Δ
Hf <
μS <
μS(bulk), in which the upper (lower) limit corresponds to the S-rich (Mo-rich) condition and Δ
Hf is the formation heat of MoS
2. The computed formation heat is 1.44 eV which is in good agreement with reported results.
40,49
3. Results and discussion
3.1 Mn dopant in monolayer MoS2
The structural stability and magnetic properties of the isolated Mn dopant in monolayer MoS2 are first investigated. Five doping configurations are considered, that is, Mn adsorbed on top of the Mo, S or hollow sites (AMo, AS, and AH) and Mn replacing Mo or S (MnMo and MnS) as schematically illustrated in Fig. 1a. Metal-doped MoS2 has been synthesized50,51 and Au on top of the S, Mo, and hollow sites of MoS2 has been observed.52 Furthermore, Cr/V/Nb dopants directly substitute Mo atoms,53,54 and Pt atoms are localized at S vacancies exhibiting dynamic hopping to the nearby surface layers.55 Up to now, the position of Mn dopants in MoS2 has been ambiguous. The formation energies of the five possible doping configurations as a function of the chemical potential difference for different S concentrations (ΔμS = μS − μbulk) are calculated and shown in Fig. 1b. The formation energy of the MnMo doping configuration is the lowest one and negative in most cases. In particular, under the S-rich growth condition, the formation energy of MnMo doping in monolayer MoS2 is −1.702 eV which is in good agreement with the reported value of −1.8 eV.42 Therefore, Mn dopants tend to replace Mo atoms in monolayer MoS2 and it has been observed experimentally.54,56
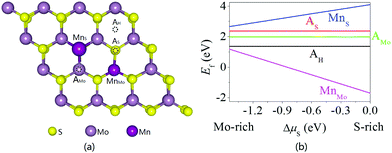 |
| | Fig. 1 (a) Atomic model and (b) formation energy of a single Mn dopant in monolayer MoS2. | |
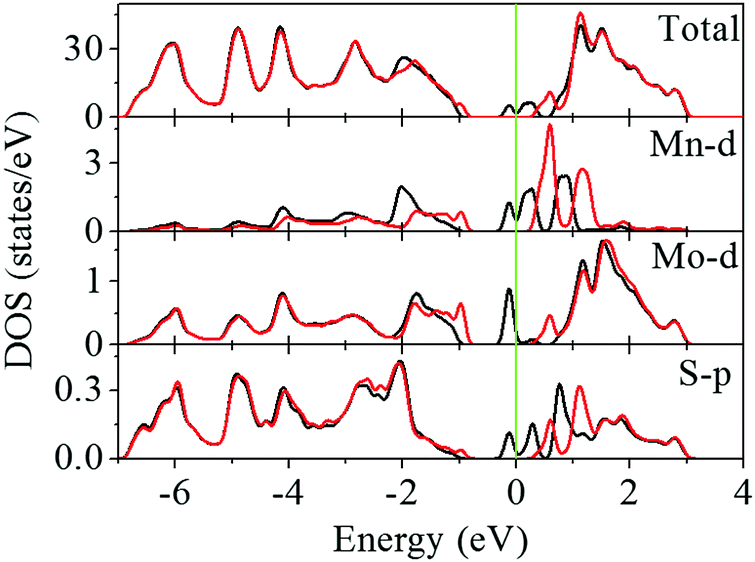
To understand the spin splitting, the electronic states of monolayer MoS2 with the MnMo doping configurations are calculated. The trigonal prismatic coordination of Mo atoms leads to degeneracy of the Mo 4d levels. The lowest energy band of the Mo dz2 orbital is fully occupied, the degenerate energy bands of the Mo dxy and Mo dx2−y2 orbitals are unoccupied, and those of Mo d and Mo dyz bands are the highest ones. Hybridization among the Mo dz2, Mo dxy, Mo dx2−y2, and S 3p orbitals dominates the conduction and valence band edges of MoS2.23–27 As shown in Fig. 2, spin polarization occurs in monolayer MoS2 with the MnMo doping configuration leading to highly localized states in the band gap. The partial density of states (PDOS) suggests that the mid-gap states mainly arise from the d states of the Mn dopant and its neighboring spin-polarized 3d states of Mo atoms and 3p states of S atoms. Strong hybridization between the 3d states of Mn and 3p states of adjacent S atoms produces mid-gap states in the spin-up channel (Fig. 3b and c). Moreover, no band gap is observed from the spin-up channel, although a considerable band gap is kept in the spin-down channel, indicating the half-metallic feature of the Mn-doped monolayer MoS2. Thus, it is expected to provide 100% spin polarization current and favorable spintronic applications. Some valence electrons (3d64s1) of the Mn dopant transfer to the nearest-neighboring S atoms and the residual valence electrons produce spin polarization and a magnetic moment of 1.035 μB (Table S1, ESI†). This is in good agreement with the previously reported value of ∼1 μB.32 The Mn 3d orbitals undergo crystal-field splitting (Δcf) and interatomic Hund's exchange splitting (Δex). The five Mn 3d states are split into three groups, anti-bonding states, e1 (dxy and dx2−y2), e2 (dxz and dyz), and a1 (dz2) due to the trigonal prismatic coordination of S atoms. As schematically shown in Fig. 3d, the energies of the spin-down states a1 and e1 are lower than those of the spin-up states e2, suggesting Δcf > Δex.
 |
| | Fig. 2 Total and partial density of states of Mn substituting Mo (MnMo) in monolayer MoS2. The black and red lines represent the spin-up and spin-down channels, respectively, and the Fermi level which is set at zero is indicated by the green line. | |
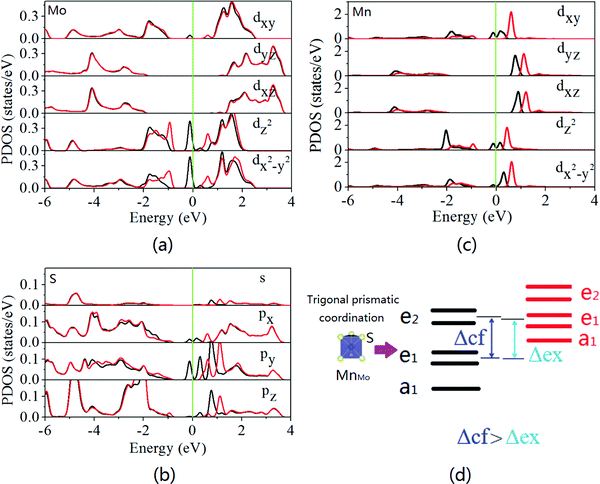 |
| | Fig. 3 Orbital decomposed density of states of Mn substituting Mo (MnMo) in monolayer MoS2: (a) Mo 4d states, (b) S 3s and 3p states, and (c) Mn 3d states. The Mo and S atoms are the nearest to the Mn dopant. The black and red lines represent the spin-up and spin-down channels, respectively. The Fermi level is set at zero and indicated by the green line. (d) Schematic plot of the splitting of Mn d orbitals due to crystal-field splitting (Δcf) and interatomic Hund's exchange (Δex). | |
The long-range ferromagnetism is mediated by the AFM coupling between the localized Mn 3d states and delocalized S 3p states in monolayer MoS2.37,38 It has been predicted that the Curie temperature of Mn-doped monolayer MoS2 can reach room temperature if the Mn dopant concentration is 10–15%.38 It has been demonstrated by experiments that an inert substrate (i.e., graphene) allows only 2 at% doping of Mn in MoS2, otherwise it is difficult to retain the 2D morphology.57,58 To further study the effects of the dopant concentration on magnetic-exchange coupling, two Mn dopants are substitutionally inserted into monolayer MoS2 and the magnetic-exchange coupling is evaluated by comparing the energies of the FM and AFM states:
where
EFM and
EAFM are the energies of the Mn-doped MoS
2 in the FM and AFM states, respectively. The energy of the FM state is less than that of the AFM state when two Mn dopants are close to each other. That is, the FM state is preferred. The magnetic coupling interactions in Mn-doped monolayer MoS
2 are further examined by the spin density distribution (
Fig. 4). When two Mn
Mo dopants are separated by a distance less than 16.53 Å (Fig. S2, ESI
†), the Mn dopants are highly spin-polarized with the same spin-polarization direction and the nearest neighboring S atoms between two Mn dopants are AFM coupled with the S atoms (
Fig. 4a–c). But it is different if the separation between two Mn dopants is increased to 22.05 Å and the energy of the FM state is equal to that of the AFM state (
Fig. 4d). The results indicate long-range FM coupling of two Mn dopants in monolayer MoS
2.
38 The results are different from those of Co-doped monolayer MoS
2 that is nonmagnetic when the dopants are close to each other.
59 However, the system becomes magnetic with a magnetic moment of 6.00
μB if the two dopants are separated by 5.50 Å. Based on the mean-field approximation (MFA), the Curie temperature (
TC) is estimated from the energy difference between the FM and AFM states with the following equation:
| |  | (3) |
where
kB is the Boltzmann constant and
n is the number of dopants in a supercell. The estimated
TC is 224 K and lower than room temperature and it is in good agreement with the reported value of ∼200 K obtained by Monte Carlo simulation.
37
 |
| | Fig. 4 Spin-density isosurface distributions for FM and AFM configurations of Mn-doped monolayer MoS2 with the 2MnMo configuration in (a) 4 × 4, (b) 5 × 5, (c) 6 × 6, and (d) 7 × 7 supercells. The red and green isosurfaces represent the positive and negative spin densities, respectively. The isosurface value is 0.003 e Å−3. | |
3.2 Single Mn dopant in bilayer MoS2
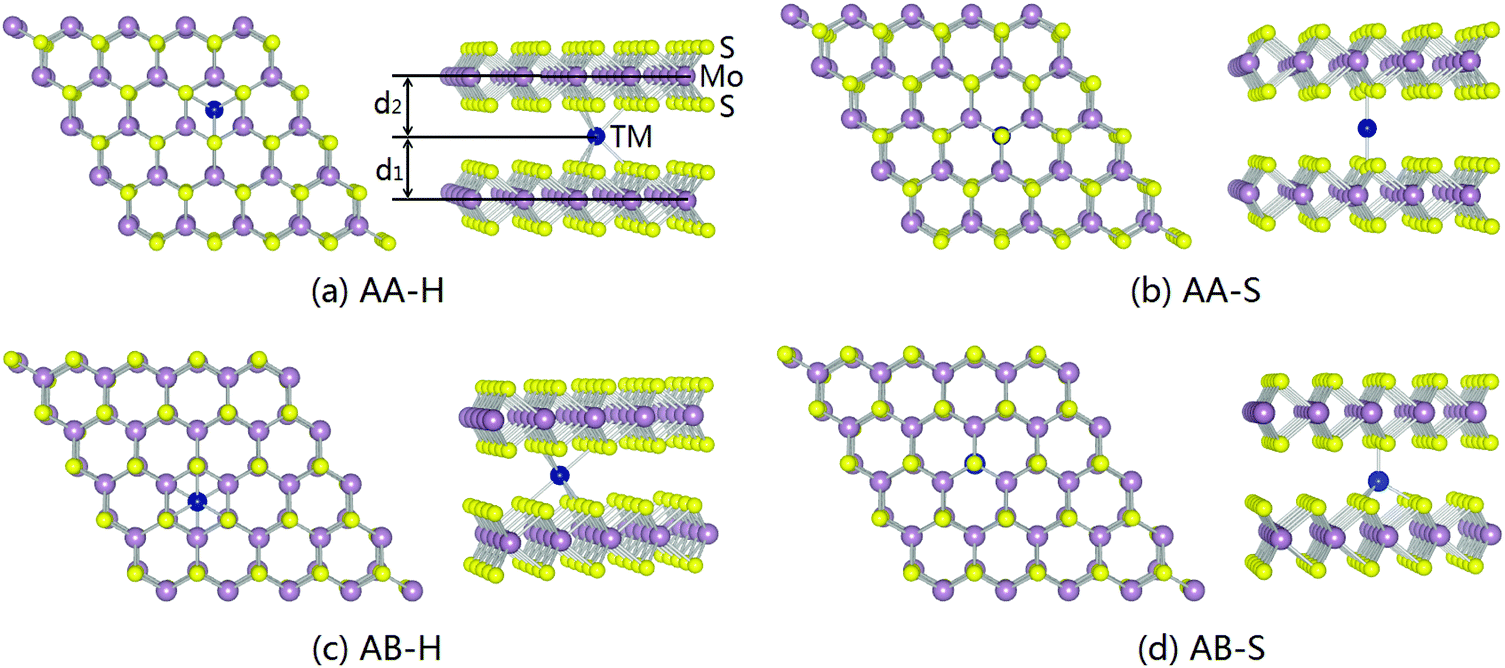
In 2D materials, surface doping is commonly implemented to manipulate the physical properties, but oxidization and migration may take place on these exposed atoms producing structural and functional instability. It may be overcome if the dopants can be fixed in the interlayer of bilayer MoS2. The two stacking sequences, AA and AB, exist in bilayer MoS2. In AA (AB) stacking, the Mo atoms in one layer are on top of the Mo (S) atoms in the other layer (Fig. S3, ESI†). The AB stacking configuration is more stable than the AA one, but both AA and AB stacking configurations are obtained by CVD.60 Here, both are considered and two typical interlayer doping sites are considered: (1) dopant just above the hollow site of the hexagonal rings in bilayer MoS2 (H) and (2) dopant directly under the S atom in the upper layer (S). As shown in Fig. 5a and c, for the Mn dopant at the H site in bilayer MoS2 with the AA and AB stacking configurations (AA-H and AB-H), the Mn dopant is bonded to three S atoms in each sublayer. For the Mn dopant directly under the S atom in the upper layer of the AA stacking configuration (AA-S), the Mn dopant is bonded to one S atom in the upper sublayer and one S atom in the lower sublayer (Fig. 5b). For the Mn dopant directly under the S atom in the upper layer in the AB configuration (AB-S), the Mn dopant is bonded to one S atom in the upper sublayer and three S atoms in the lower sublayer (Fig. 5d). As shown in Table S2 (ESI†), for the Mn dopant in the AA-H (AA-S) stacking configuration, the interlayer distance decreases (increases) to 3.665 Å (3.872 Å) from the original value of 3.720 Å, whereas the interlayer distance in the AB stacking configuration is between 3.112 Å and 3.121 Å, which is close to the ideal interlayer distance of bilayer MoS2 (3.110 Å). In the AA-S and AB-S configurations, the bond lengths between the Mn dopant and S atoms are between 2.113 Å and 2.220 Å, which is shorter than that of the covalent Mn–S bond (∼2.37 Å). However, in the AA-H and AB-H configurations, the bond lengths between the Mn dopant and S atoms increase to the range of 2.421–2.563 Å. The interlayer Mn dopant and S atoms of bilayer MoS2 in all the configurations are covalent bonded. Fig. 6 shows the charge density difference of the Mn-doped bilayer MoS2 with the AA and AB configurations. The electron density accumulates between the Mn dopants and adjacent S atoms characteristic of covalent bonding.
 |
| | Fig. 5 Atomic models of a single Mn dopant in bilayer MoS2 with the (a and b) AA and (c and d) AB stacking configurations. | |
 |
| | Fig. 6 Charge density difference of a single Mn dopant in bilayer MoS2 with the (a and b) AA and (c and d) AB stacking configurations. The red and turquoise isosurfaces represent the gain and depletion of charge density, respectively. The isosurface value is 0.005 e Å−3. | |
Owing to the different doping environments, the AA-H, AA-S, AB-H, and AB-S doping configurations exhibit completely distinct magnetic moments of 5.07 μB, 3.03 μB, 3.87 μB, and 3.00 μB, respectively. The overall magnetic moments can be ascribed to spin polarization of excess d electrons on the Mn dopant. Each Mn atom possesses 5 electrons in the 3d orbital. When Mn is doped at the H site in bilayer MoS2 with both the AA and AB stacking configurations, the interaction between the Mn dopant and S is weak because of the larger bond length. Strong interactions between Mn and S are induced, accompanied by substantial electron transfer from the 4s to 3d states of the Mn dopant if Mn is doped at the S site in bilayer MoS2 with the AA and AB stacking configurations. This reduces the unpaired electrons and magnetic moment.40,61
To again investigate magnetic coupling of a single Mn dopant in bilayer MoS2, the orbital decomposed density of states and splitting of Mn 3d orbitals are presented in Fig. 7. The magnetic moment is sensitive to the doping environment. In the AA-H doping configuration, the trigonal prismatic coordination of S atoms around the Mn dopant is similar to that of the MnMo dopant in monolayer MoS2, but the bond length between Mn and S atoms is larger and as a result, crystal-field splitting is reduced and Δcf < Δex (Fig. 7b). The magnetism is mainly due to spin polarization around the Mn atom (Fig. 7a and Table S2, ESI†). In spite of some differences in the electronic states, similar results are observed from the AA-S doping configuration (Fig. 7c and d). The octahedral and tetrahedral coordination of S atoms around the Mn dopant exist in the AB-H and AB-S doping configurations. The bond length between Mn and S atoms is equal and the antibonding states e1 and e2 have the same energy, so that crystal-field splitting disappears (Fig. 7e–h). Similar to the Mn doped bilayer MoS2, the doping environment dependent electronic states of Cr, Fe, and Co in bilayer MoS2 are also observed (Fig. S4–S6, ESI†).61
 |
| | Fig. 7 Orbital decomposed density of states and splitting of 3d orbitals in a single Mn dopant in bilayer MoS2 with the (a, b and c, d) AA and (e, f and g, h) AB stacking configurations. | |
3.3 Double Mn dopants in bilayer MoS2
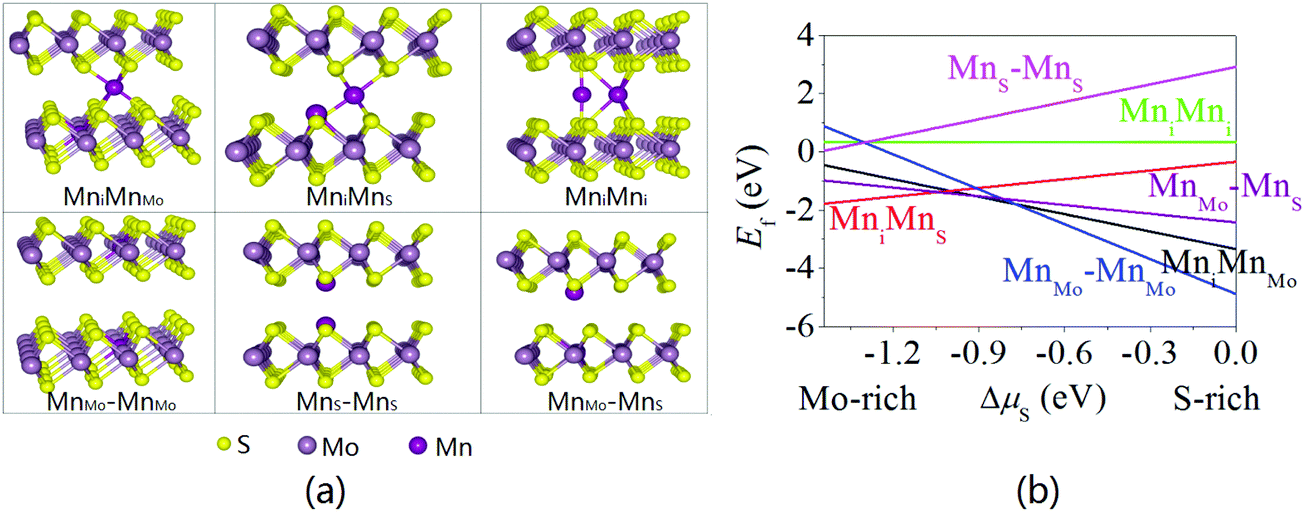
The structural and magnetic properties of the 4 × 4 supercell in bilayer MoS2 are investigated by considering six doping configurations (Fig. 8a): (i) an intercalated Mn atom (Mni) and MnMo dopant (MniMnMo), (ii) an intercalated Mn atom (Mni) and MnS dopant (MniMnS), (iii) two intercalated Mn atoms (MniMni), (iv) two MnMo dopants in different sublayers (MnMo–MnMo), (v) two MnS dopants in different sublayers (MnS–MnS), and (vi) MnMo and MnS dopants in different sublayers (MnMo–MnS). Fig. 8b shows the formation energies of the six doping configurations as a function of chemical potentials. It can be seen that MniMnS is the most stable doping configuration under the Mo-rich condition (ΔμS < −1.024 eV), but MnMo–MnMo becomes the most stable one in the S-rich regime (ΔμS > −0.75 eV) and MniMnMo and MnMo–MnS are stable if ΔμS is in the range between −1.024 and −0.812 eV. The doping configurations of MniMni, and MnS–MnS are energetically unfavorable. Although experimental results demonstrate that S vacancies are energetically favorable in most cases,49,62,63 Mn dopants prefer to substitutionally replace Mo under the S-rich condition and the MnMo–MnMo doping configurations can be obtained more easily. The results are different from the recently reported results in which the FeMoFei doping configuration is observed to be stable under the S-rich condition.40
 |
| | Fig. 8 (a) Atomic models and (b) formation energy of two Mn dopants in bilayer MoS2. | |
The magnetic ground states of the stable doping configurations are evaluated according to eqn (2). The calculated ΔE values are −823, 42, −241 and 5 meV for the MniMnMo, MniMnS, MnMo–MnMo, and MnMo–MnS doping configurations, respectively. Consequently, the MniMnMo and MnMo–MnMo doping configurations should exhibit the FM state, whereas the MniMnS and MnMo–MnS doping configurations should have the AFM state. However, it is hard to obtain the MniMnMo and MnMoMnS doping configurations in bilayer MoS2 due to the small stable region of the chemical potential. Fig. 9 shows the spin density (ρ↑–ρ↓) distribution in the bilayer MoS2 with the MniMnMo, MniMnS, MnMo–MnMo, and MnMo–MnS doping configurations. In bilayer MoS2 with the MniMnMo doping configuration, the two Mn dopants exhibit double-exchange coupling and the same spin polarization direction. Moreover, the Mn dopants are AFM coupled with the nearest neighboring S atoms (Fig. 9a), which is similar to the case of monolayer MoS2 with FM coupling. In bilayer MoS2 with the MniMnS doping configuration, direct chemical bonding and exchange coupling take place between the two Mn dopants with opposite spin polarization resulting in the AFM state. Although two Mn dopants in bilayer MoS2 with the MnMo–MnS doping configuration also have the opposite polarization direction, the magnetic exchange coupling is different from that in bilayer MoS2 with the MniMnS doping configuration, and super-exchange coupling takes place between the two Mn atoms with opposite spin polarization (Fig. 9d). Owing to AFM coupling between the Mn dopants, bilayer MoS2 with the MniMnS and MnS–MnS doping configurations possesses negligible magnetism. Meanwhile, the opposite polarization of the Mn dopant and nearest neighboring Mo atoms or S atoms leads to AFM coupling. In bilayer MoS2 with the MnMo–MnMo doping configuration, the distance between two Mn dopants is 6.788 Å which is so large that the interaction between them is weak and they are independent. Hence, the result is the same as that of the Mn-doped monolayer MoS2. The estimated TC of bilayer MoS2 with the MnMo–MnMo doping configuration is 931 K and well above room temperature. Therefore, room-temperature ferromagnetism is expected from bilayer MoS2 with different Mn doping configurations spurring potential application in spintronic devices.
 |
| | Fig. 9 Spin-density isosurface distributions of two Mn dopants in bilayer MoS2 with different doping configurations: (a) MniMnMo, (b) MniMnS, (c) MnMo–MnMo, and (d) MnMo–MnS. The red and green isosurfaces represent the positive and negative spin densities, respectively, and the isosurface value is 0.02 e Å−3. | |
The results indicate that the MnMo doping configuration is energetically stable in the S-rich regime of the MoS2 nanosheet, while the MniMnS doping configuration is more favorable in the S-deficient environment. The S-rich environment is generally adopted to synthesize MoS2 nanosheets. Thus, the MnMo doping configuration is energetically favorable in Mn-doped MoS2 and similar behaviors occur in Fe-, Cr-, V-, and Nb-doped MoS2 nanosheets.52,53 For instance, in the Fe-doped MoS2 layered crystals prepared by the chemical vapor transport method, the replacement of Mo by Fe takes place in the S-rich regime, while the replacement of S by Fe takes place under the Mo-rich condition.40 However, Pt atoms are preferentially localized at S vacancies and usually hop to the nearby surface layers,54 and Au atoms are preferentially on the top of S, Mo, and hollow sites of MoS2.51 Additionally, Co adsorption on the Mo site was evidenced from the high angle annular dark field image, which is confirmed as the energetically favourable geometry by density functional theory calculations.
So far, a large number of theoretical studies have been done to explore the stable magnetism in TM-doped MoS2 nanosheets. However, they mainly focused on studying the influences of a single dopant on the magnetic properties of MoS2 nanosheets and the magnetic-exchange coupling among dopants is commonly ignored. In fact, the magnetic properties are sensitive to magnetic-exchange coupling among dopants in the host materials. Shu et al.40 showed that the layer number is crucial in controlling the magnetic properties of Fe-doped MoS2, that is, FM coupling occurs in Fe-doped monolayer MoS2, but AFM coupling in Fe-doped bi- and trilayer MoS2. The direct and mediated bonding interactions between the intercalated and substitutional Fe dopants follow the superexchange mechanism. As for Mn-doped MoS2 nanosheets in this work, it is found that the doping sites are crucial in controlling the magnetic properties. Particularly, AFM coupling takes place in monolayer MoS2 with MnMo–MnS or MniMnS doping configurations, and otherwise, FM coupling occurs in the MnMo–MnMo or MniMnMo doping configurations. Since the doping sites depend on the synthesis conditions, the precise control of doping sites is required to generate ferromagnetism.
4. Conclusion
The structural stability, electronic states, and magnetic properties of Mn-doped monolayer/bilayer MoS2 are studied by first-principles calculations. The structural stability of Mn-doped MoS2 depends on the chemical potential and layer number. In the S-rich environment, Mn replacement of Mo in monolayer MoS2 (MnMo) is stable but in the S-deficient environment, the MniMnS doping configuration is more favorable in bilayer MoS2. Mn dopants lead to FM coupling in monolayer MoS2 and magnetic coupling of Mn dopants via the double-exchange mechanism can be mediated by the nearest neighboring S. In bilayer MoS2, the magnetic-exchange coupling is related to the competition between the double-exchange, direct-exchange, and super-exchange interactions. Specifically, in the MniMnMo doping configuration, double-exchange coupling takes place between the two Mn dopants and it is the same as that of the Mn-doped monolayer MoS2 with FM coupling. Although the two Mn dopants have opposite spin polarization, the magnetic-exchange mechanisms of the MniMnS and MnMo–MnS doping configurations are different. The findings provide insights into the development of intrinsic diluted magnetic semiconductors based on 2D MoS2 nanosheets.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was jointly supported by the National Natural Science Foundation of China (Grant No. 51471130, 51771144, 51501012), the Natural Science Foundation of Shaanxi Province (No. 2017JZ015), Fund of the State Key Laboratory of Solidification Processing in NWPU (SKLSP201708), Fundamental Research Funds for the Central Universities, City University of Hong Kong Applied Research Grant (ARG) No. 9667122, as well as City University of Hong Kong Strategic Research Grant (SRG) No. 7004644. This work was carried out at the HPCC Platform in Xian Jiaotong University.
References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, S. C. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef CAS PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stormer and P. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS PubMed.
- C. Lee, Q. Li, W. Kalb, X.-Z. Liu, H. Berger, R. W. Carpick and J. Hone, Science, 2010, 328, 76 CrossRef CAS PubMed.
- A. H. C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217 CrossRef CAS PubMed.
- B. W. H. Baugher, H. O. H. Churchill, Y. Yang and P. J. Herrero, Nat. Nanotechnol., 2014, 9, 262 CrossRef CAS PubMed.
- A. Pospischil, M. M. Furchi and T. Mueller, Nat. Nanotechnol., 2014, 9, 257 CrossRef CAS PubMed.
- O. Lopez-Sanchez, D. Lembke, M. Kayci, A. Radenovic and A. Kis, Nat. Nanotechnol., 2013, 8, 497 CrossRef CAS PubMed.
- J. Xia, X. Huang, L. Z. Liu, M. Wang, L. Wang, B. Huang, D. D. Zhu, J. J. Li, C. Z. Gu and X. M. Meng, Nanoscale, 2014, 6, 8949 RSC.
- W. Zhang, C. P. Chu, J. K. Huang, C. H. Chen, M. L. Tsai, Y. H. Chang, C. T. Liang, Y. Z. Chen, Y. L. Chueh and J. H. He, Sci. Rep., 2014, 4, 3826 CrossRef PubMed.
- H. L. Zhuang and R. G. Hennig, Chem. Mater., 2013, 25, 3232 CrossRef CAS.
- A. B. Laursen, S. Kegnæs, S. Dahl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 5577 CAS.
- B. Radisavljevic, A. Radenovic, J. Brivio, V. Giacometti and A. Kis, Nat. Nanotechnol., 2011, 6, 147 CrossRef CAS PubMed.
- J. S. Ross, P. Klement, A. M. Jones, N. J. Ghimire, J. Yan, D. G. Mandrus, T. Taniguchi, K. Watanabe, K. Kitamura, W. Yao, H. C. David and X. D. Xu, Nat. Nanotechnol., 2014, 9, 268 CrossRef CAS PubMed.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826 CrossRef CAS PubMed.
- X. Zhao, C. M. Hayner, M. C. Kung and H. H. Kung, ACS Nano, 2011, 5, 8739 CrossRef CAS PubMed.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170 CrossRef CAS PubMed.
- M. Chhowalla, H. S. Shin, G. Eda, L. J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- H. S. Lee, S.-W. Min and S. Im, Nano Lett., 2012, 12, 3695 CrossRef CAS PubMed.
- R. S. Sundaram, M. Engel, A. Lombardo, R. Krupke, A. C. Ferrari, P. Avouris and M. Steiner, Nano Lett., 2013, 13, 1416 CrossRef CAS PubMed.
- K. F. Mak, K. L. He, J. Shan and T. F. Heinz, Nat. Nanotechnol., 2012, 7, 494 CrossRef CAS PubMed.
- D. Xiao, G. B. Liu, W. X. Feng, X. D. Xu and W. Yao, Phys. Rev. Lett., 2012, 108, 196802 CrossRef PubMed.
- Z. Y. Zhu, Y. C. Cheng and U. Schwingenschlögl, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 153402 CrossRef.
- Y. Ohno, R. Terauchi, T. Adachi, F. Matsukura and H. Ohno, Phys. Rev. Lett., 1999, 83, 4196 CrossRef CAS.
- Q. Chen, X. Ouyang, S. Yuan, R. Li and J. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 16835 CAS.
- S. Y. Wang, T. S. Ko, C. C. Huang, D. Y. Lin and Y. S. Huang, Jpn. J. Appl. Phys., 2014, 53, 04EH07 CrossRef.
- A. N. Andriotis and M. Menon, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 125304 CrossRef.
- W. S. Yun and J. D. Lee, Phys. Chem. Chem. Phys., 2014, 16, 8990 RSC.
- H.-P. Komsa, N. Berseneva, A. V. Krasheninnikov and R. M. Nieminen, Phys. Rev. X, 2014, 4, 031044 Search PubMed.
- M. Kan, S. Adhikari and Q. Sun, Phys. Chem. Chem. Phys., 2014, 16, 4990 RSC.
- Q. Yue, S. Chang, S. Qin and J. Lin, Phys. Lett. A, 2013, 377, 1362 CrossRef CAS.
- Y. C. Chen, Z. Y. Zhu, W. B. Mi, Z. B. Guo and U. Schwingenschlögl, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 100401(R) CrossRef.
- A. Ramasubramaniam and D. Naveh, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195201 CrossRef.
- R. Mishra, W. Zhou, S. J. Pennycook, S. T. Pantelides and J.-C. Idrobo, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 144409 CrossRef.
- Y. R. Wang, S. Li and J. B. Yi, Sci. Rep., 2016, 6, 24153 CrossRef CAS PubMed.
- H. B. Shu, P. F. Luo, P. Liang, D. Cao and X. S. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 7534 CAS.
- M. Zhang, Z. J. Huang, X. Wang, H. Y. Zhang, T. H. Li, Z. L. Wu, Y. H. Luo and W. Cao, Sci. Rep., 2016, 6, 19504 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653 CrossRef CAS.
- Y. Wang, S. S. Wang, Y. H. Lu, J. Z. Jiang and S. Y. A. Yang, Nano Lett., 2016, 16, 4576 CrossRef CAS PubMed.
- W. Zhou, X. L. Zou, S. Najmaei, Z. Liu, Y. M. Shi, J. Kong, J. Luo, P. M. Ajayan, B. I. Yakobson and J.-C. Idrobo, Nano Lett., 2013, 13, 2615 CrossRef CAS PubMed.
- B. Li, L. Huang, M. Z. Zhong, N. J. Huo, Y. T. Li, S. X. Yang, C. Fan, J. H. Yang, W. P. Hu, Z. M. Wei and J. B. Li, ACS Nano, 2015, 9, 1257 CrossRef CAS PubMed.
- G. L. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y. C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810 CrossRef CAS PubMed.
- Y. C. Lin, D. O. Dumcenco, H. P. Komsa, Y. Niimi, A. V. Krasheninnikov, Y. S. Huang and K. Suenaga, Adv. Mater., 2014, 26, 2857 CrossRef CAS PubMed.
- A. W. Robertson, Y. C. Lin, S. S. Wang, H. Sawada, C. S. Allen, Q. Chen, S. Lee, G. D. Lee, J. Lee, S. Han, E. Yoon, A. I. Kirkland, H. Kim, K. Suenaga and J. H. Warner, ACS Nano, 2016, 10, 10227 CrossRef CAS PubMed.
- J. Suh, T.-E. Park, D. Y. Lin, D. Fu, J. Park, H. J. Jung, Y. B. Chen, C. Ko, C. Jang, Y. H. Sun, R. Sinclair, J. Chang, S. Tongay and J. Q. Wu, Nano Lett., 2014, 14, 6976 CrossRef CAS PubMed.
- H. S. Li, S. S. Wang, H. Sawada, G. G. D. Han, T. Samuels, C. S. Allen, A. I. Kirkland, J. C. Grossman and J. H. Waener, ACS Nano, 2017, 11, 3392 CrossRef CAS PubMed.
- J. Gao, Y. D. Kim, L. B. Liang, J. C. Idrobo, P. Chow, J. W. Tan, B. C. Li, L. Li, B. G. Sumpter, T.-M. Lu, V. Meunier, J. Hone and N. Koratkar, Adv. Mater., 2016, 28, 9735 CrossRef CAS PubMed.
- K. H. Zhang, S. Feng, J. J. Wang, A. Azcatl, N. Lu, R. Addou, N. Wang, C. J. Zhou, J. Lerach, V. Bojan, M. J. Kim, L. Q. Chen, R. M. Wallace, M. Terrones, J. Zhu and J. A. Robinson, Nano Lett., 2015, 15, 6586 CrossRef CAS PubMed.
- H. Tan, W. Hu, C. Wang, C. Ma, H. L. Duan, W. S. Yan, L. Cai, P. Guo, Z. H. Sun, Q. H. Liu, L. Cai, P. Guo, Z. H. Sun, Q. H. Liu, X. S. Zheng, F. C. Hu and S. Q. Wei, Small, 2017, 340, 1701389 CrossRef PubMed.
- Y. R. Wang, S. A. Li and J. B. Yi, Sci. Rep., 2016, 6, 24153 CrossRef CAS PubMed.
- J. X. Yan, J. Xia, X. L. Wang, L. Liu, J. L. Kuo, B. K. Tay, S. S. Chen, W. Zhou, Z. Liu and Z. X. Shen, Nano Lett., 2015, 15, 8155 CrossRef CAS PubMed.
- Z. Y. Huang, X. Y. Peng, H. Yang, C. Y. He, L. Xue, G. L. Hao, C. X. Zhang, W. L. Liu, X. Qi and J. X. Zhong, RSC Adv., 2013, 3, 12939 RSC.
- A. M. Van der Zande, P. Y. Huang, D. A. Chenet, T. C. Berkelbach, Y. M. You, G.-H. Lee, T. F. Heinz, D. R. Reichman, D. A. Muller and J. C. Hone, Nat. Mater., 2013, 12, 554 CrossRef CAS PubMed.
- S. Najmaei, Z. Liu, W. Zhou, X. Zou, G. Shi, S. Lei, B. I. Yakobson, J.-C. Idrobo, P. M. Ajayan and J. Lou, Nat. Mater., 2013, 12, 754 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp05988d |
|
| This journal is © the Owner Societies 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Xumei
Zhao
b,
Yuhong
Huang
c,
Kewei
Xu
ad,
Tai
Min
a,
Xumei
Zhao
b,
Yuhong
Huang
c,
Kewei
Xu
ad,
Tai
Min