
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAxial–equatorial isomerism and semiexperimental equilibrium structures of fluorocyclohexane†
Marcos
Juanes
 a,
Natalja
Vogt
*bc,
Jean
Demaison
b,
Iker
León
a,
Alberto
Lesarri
*a and
Heinz Dieter
Rudolph
b
a,
Natalja
Vogt
*bc,
Jean
Demaison
b,
Iker
León
a,
Alberto
Lesarri
*a and
Heinz Dieter
Rudolph
b
aDepartamento de Química Física y Química Inorgánica, Facultad de Ciencias, Universidad de Valladolid, 47011 Valladolid, Spain. E-mail: lesarri@qf.uva.es; Web: http://www.uva.es/lesarri
bSection of Chemical Information Systems, University of Ulm, Albert-Einstein-Allee 47, 89081 Ulm, Germany. E-mail: natalja.vogt@uni-ulm.de
cDepartment of Chemistry, Lomonosov Moscow State University, 119991 Moscow, Russia
First published on 31st October 2017
Abstract
An experimental-computational methodology combining rotational data, high-level ab initio calculations and predicate least-squares fitting is applied to the axial–equatorial isomerism and semiexperimental equilibrium structure determination of fluorocyclohexane. New supersonic-jet microwave measurements of the rotational spectra of the two molecular conformations, together with all 13C isotopologues of both isomeric forms are reported. Equilibrium rotational constants are obtained from the ground-state rotational constants corrected for vibration–rotation interactions and electronic contributions. Equilibrium structures were determined by the mixed estimation (ME) method. Different computational methods were tested for the evaluation of predicate values of the structural parameters, and a computationally effective procedure for estimating reliable dihedral angles is proposed. Structural parameters were fitted concurrently to predicate parameters and moments of inertia, affected with appropriate uncertainties. The new structures of the title compound are regarded as accurate to 0.001 Å and 0.2°, illustrating the advantages of this methodology. Structural comparisons are offered with the cyclohexane prototype, revealing subtle substituent effects. For comparison purposes the equilibrium structures for the two fluorocyclohexane isomers and cyclohexanone are computed from high-level ab initio theory with inclusion of adjustments for basis set dependence and correlation of the core electrons.
Introduction
The semiexperimental (SE) method is an experimental-computational procedure where equilibrium rotational constants are determined from the experimental ground-state rotational constants and theoretical rovibrational corrections (based principally on an ab initio cubic force field). Since the pioneering work of Pulay et al.1 the SE method is reputed to deliver very accurate estimates to equilibrium structures2 of (mostly) semirigid molecules of small and medium molecular sizes,3–6 reaching bond length accuracy to about 0.001 Å, and similar four-digit accurate values for bond angles. However, difficulties for the application of the SE method increase rapidly with the molecular size. From the experimental point of view, determination of rotational parameters for full sets of isotopologues is hampered by spectral congestion and weak intensities due to larger rotational partition functions, distributing the rotational energy among a larger number of states. Moreover, it was observed that, in some cases, the rotational constants of deuterated species do not improve the fits.7 Furthermore, the system of normal equations of the least-squares method may easily become ill-conditioned (i.e. sensitive to small errors of the input data), in particular for atoms close to the center of mass (in other words, their isotopic substitution does not bring new information).These difficulties may be circumvented by the use of the predicate observations or mixed estimation (ME) method,8,9 where auxiliary information called predicates is added with appropriate weights to the data matrix during the least-squares fit.10 Estimates of the predicates (bond lengths and bond angles) are generally (but not necessarily) obtained from quantum chemical (QC) calculations at an accessible level of theory, eventually improved by corrections based on comparisons between QC predictions and known equilibrium structures for various types of chemical bonds. An important advantage of the mixed estimation method is that no specific constraints are introduced, which avoids biased results. The determination of the predicates for different organic molecules has already been discussed.11 It is easy to obtain reliable predicates for the CH bond lengths, the single CC bond lengths and related bond angles. However, as observed in fructose,12 deoxyribose,12 proline,13 succinic anhydrid,14 pseudopelletierine15 and several other molecules the prediction of accurate dihedral angles is a problem, the error being sometimes as large as several degrees. Indeed, a provisional criticism of the ME method is the difficulty to obtain reliable estimates for a full set of internal coordinates and to simultaneously estimate their accuracy. In this paper, we plan to study two aspects of this problem: (1) how to obtain predicates of the CF bond lengths and of dihedral angles and (2) how to achieve computational accuracy without having recourse to expensive methods such as the coupled–cluster methods. Then, these results will be applied to the determination of the semiexperimental structure of the axial and equatorial conformers of fluorocyclohexane. Fluorocyclohexane is an appropriate candidate to compare the small structural and energetic effects caused by axial/equatorial substituents in the canonical cyclohexane chair. Moreover, the small conformational energies between the axial and equatorial forms anticipate the possibility of detecting all 13C isotopologues in natural abundance for both species, improving the empirical data for the structural analysis. The structure of cyclohexanone was additionally optimized ab initio to compare with fluorocyclohexane and in order to have a larger variety of torsional angles.
The first study of the structure of fluorocyclohexane by Andersen in 1962 used gas-phase electron diffraction.16 He determined the ratio between the axial and equatorial conformations and found that the conformation with fluorine in equatorial position is more stable by about 170 cal mol−1. Slightly later, the microwave spectra of both forms were analyzed by Pierce et al.17,18 In 1971 Scharpen measured the relative intensities of rotational transitions of both conformers and determined an energy difference of 259(28) cal mol−1.19 Empirical structures for both conformers were independently determined more recently by Bialkowska-Jaworska et al.20 and Durig et al.21 The latter authors also determined an accurate value of the enthalpy difference, ΔH = 137(14) cal mol−1 from the analysis of the infrared spectra in gas and xenon solution. The intensities of several well-isolated and well-shaped conformational bands were measured as a function of temperature (at 5.0 °C intervals between −60 and −100 °C) and ΔH was determined by application of the van’t Hoff equation assuming that the conformational enthalpy differences are not a function of temperature in the range studied. The conformational equilibria were also studied theoretically by Storz.22
Experimental and computational methods
The jet-cooled rotational spectrum of fluorocyclohexane was measured in the frequency range 6–20 GHz with a Fourier transform microwave (FT-MW) spectrometer based on the Balle-Flygare23 design. The sample was vaporized on an external liquid reservoir inserted in the carrier gas line (ice temperature). A stream of argon (backing pressures ca. 1 bar) was flowed over the sample and expanded through a solenoid valve (Parker, nozzle diameter 1.0 mm), creating a pulsed supersonic jet. The jet was probed within a Fabry–Perot microwave resonator, formed by two spherical mirrors in near confocal position (diameter 33 cm). The injection valve was located near the center of one of the mirrors, resulting in a collinear arrangement of the jet and resonator axis.24 The gas pulses (∼500 μs) expanding the vaporized sample were followed by short microwave impulses (∼1 μs, <100 mW), polarizing the polar molecules. Up to 4 microwave pulses were used per gas pulse. The emitted free-induction decay was recorded in the time-domain (410 μs), amplified and recorded with a heterodyne receiver centered at 30 MHz. The digital signal was processed with the FTMW++ program developed at the Leibniz Universität Hannover.24 Typically, hundreds/thousands of experiments were coadded in the time domain for signal averaging. All frequency oscillators in the system were locked to a rubidium standard, providing frequency accuracies of the rotational transitions below 5 kHz. Line transitions appear as Doppler doublets, so the rest frequencies were taken from the averaged frequencies.Different ab initio calculations were required for this work. The geometry optimizations were performed at the level of coupled-cluster method25 with a perturbative treatment of connected triples26 (CCSD(T)) using the cc-pwCVTZ basis set,27 all electrons being correlated (AE). The second-order Møller–Plesset perturbation theory (MP2)28 was also used at the frozen-core (FC) and all-electron (AE) levels using different basis sets: cc-pVTZ,29 6-311+G(3df,2pd),30 cc-pwCVTZ and cc-pwCVQZ.27 The density functional theory (B3LYP)31–33 was also employed with the 6-311+G(3df,2pd) basis set. In order to determine the rovibrational contributions to the rotational constants, the anharmonic force field up to semidiagonal quartic terms was calculated at the MP2_FC/cc-pVTZ level of theory.34 This calculation was repeated for each isotopologue, as different isotopes require distinct vibrational corrections. To avoid the nonzero force field dilemma,35 all force fields were evaluated at the corresponding optimized geometries. The MP2 and B3LYP calculations were performed with the Gaussian 09 package (Rev. A.02 or C.01),36 whereas the MolPro program37 was used for the CCSD(T) calculations. The charges on the atoms were calculated using the Atoms in Molecules (AIM) theory38 with its implementation in Gaussian03 by Cioslowski et al.39 The calculations were performed at the B3LYP/6-311+G(3df,2pd) level of theory at the equilibrium structure.
Results
1. Rotational spectra
We first measured and analyzed the rotational spectra for the parent isotopologue (12C) of both axial and equatorial conformers, extending previous experiments. Later on, we could assign all monosubstituted 13C isotopologues in natural abundance (ca. 1%) for the two conformers, as illustrated in Fig. 1. A centrifugal-distorted rotational Hamiltonian complete up to quartic terms in the asymmetric-top reduction and the Ir representation was used to fit the spectra.40 The quality of the structural fit is very sensitive to the true accuracy of the ground-state rotational constants.41,42 As the small number of transitions and their low angular momentum quantum numbers (J = 1–6) did not permit to obtain a full set of meaningful centrifugal distortion constants, the mixed regression method was used.9 The experimental frequencies were fitted together with appropriately weighted ab initio (predicate) values for the centrifugal distortion constants. The predicate constants were obtained from a harmonic force field calculated at the MP2_FC/cc-pVTZ level, the uncertainty used to determine their weight being 10% of their value. The experimental rotational frequencies are given in Tables S1–S10 (ESI†) and the derived rotational constants are collected in Tables 1 and 2. It is worth noting that the centrifugal distortion effects are larger in the axial form.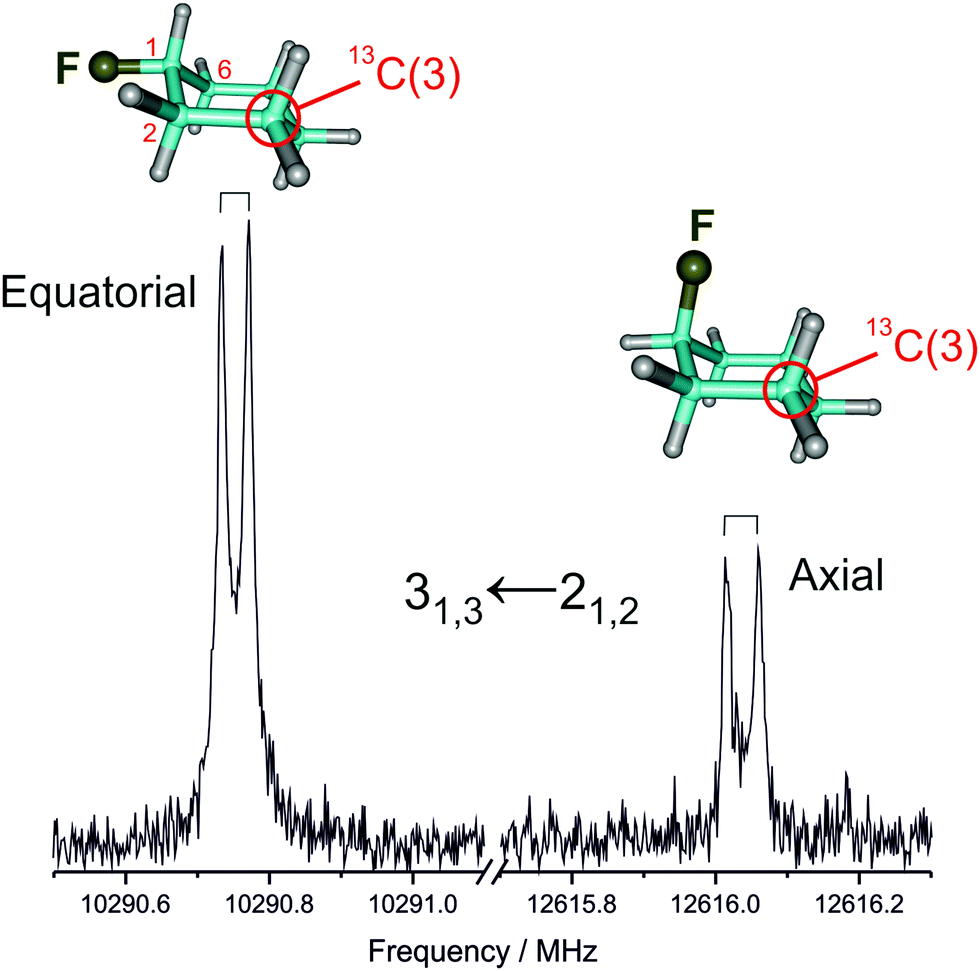 | ||
| Fig. 1 A typical rotational transition corresponding to one of the 13C monosubstituted isotopologues of equatorial and axial fluorocyclohexane, measured in natural abundance. | ||
| Parent | 13C(1) | 13C(2) | 13C(3) | 13C(4) | |
|---|---|---|---|---|---|
| a Rotational constants (A, B, C), centrifugal distortion constants (ΔJ, ΔJK, ΔK, δJ, δK), number of fitted transitions (N) and rms deviation of the fit (σ). b Standard errors in parentheses in units of the last digit. | |||||
| A /MHz | 4313.36670(41)b | 4309.5918(14) | 4255.1484(33) | 4255.0763(12) | 4310.5763(18) |
| B/MHz | 2188.78913(17) | 2179.702324(51) | 2187.94976(13) | 2174.164386(46) | 2153.373068(66) |
| C/MHz | 1591.61099(11) | 1587.368270(50) | 1583.48471(10) | 1576.365987(36) | 1573.224557(53) |
| Δ J/kHz | 0.13231(42) | 0.131836(85) | 0.13161(23) | 0.129916(77) | 0.12958(11) |
| Δ JK/kHz | 0.12551(40) | 0.120431(77) | 0.12367(22) | 0.128700(76) | 0.113250(97) |
| Δ K/kHz | 0.6283(20) | 0.63016(40) | 0.6128(11) | 0.60836(36) | 0.64220(55) |
| δ J/kHz | 0.03511(11) | 0.034951(22) | 0.035284(63) | 0.034637(21) | 0.034137(29) |
| δ K/kHz | 0.25114(81) | 0.24999(16) | 0.24702(44) | 0.24760(15) | 0.24672(21) |
| N | 30 | 8 | 12 | 10 | 9 |
| σ/kHz | 54.9 | 1.7 | 1.7 | 1.7 | 1.8 |
| Parent | 13C(1) | 13C(2) | 13C(3) | 13C(4) | |
|---|---|---|---|---|---|
| a Rotational constants (A, B, C), centrifugal distortion constants (ΔJ, ΔJK, ΔK, δJ, δK), number of fitted transitions (N) and rms deviation of the fit (σ). b Standard errors in parentheses in units of the last digit. | |||||
| A /MHz | 3562.96908(16)b | 3557.1443(26) | 3513.75817(37) | 3522.20266(13) | 3562.82765(62) |
| B/MHz | 2628.624997(95) | 2609.74750(24) | 2622.57510(28) | 2614.58544(10) | 2584.80434(12) |
| C/MHz | 1980.88163(11) | 1972.02062(24) | 1968.11042(22) | 1961.731079(86) | 1956.016082(83) |
| Δ J/kHz | 0.5543(35) | 0.5353(60) | 0.5515(66) | 0.5499(26) | 0.5398(18) |
| Δ JK/kHz | −1.043(12) | −1.019(16) | −1.019(19) | −1.0185(65) | −1.0307(36) |
| Δ K/kHz | 1.285(15) | 1.263(20) | 1.253(23) | 1.2453(80) | 1.2873(45) |
| δ J/kHz | 0.0877(10) | 0.0849(13) | 0.0861(15) | 0.08896(57) | 0.0947(20) |
| δ K/kHz | 0.0854(10) | 0.0792(12) | 0.0831(15) | 0.08676(56) | 0.08158(28) |
| N | 32 | 6 | 7 | 7 | 6 |
| σ/kHz | 26.1 | 0.93 | 0.89 | 1.3 | 1.09 |
2. Equilibrium structures
The Born–Oppenheimer (BO) equilibrium structures were optimized at the CCSD(T)_AE/cc-pwCVTZ level of theory for the equatorial and axial conformers of fluorocyclohexane. The small effect of further basis set enlargement (cc-pwCVTZ → cc-pwCVQZ) was then estimated at the MP2 level. The resulting estimate was:| rBOe = re[CCSD(T)_AE/cc-pwCVTZ] + re[MP2_AE/cc-pwCVQZ] + re[MP2_AE/cc-pwCVTZ] | (1) |
The accuracy of this equation, which is based on the additivity of small corrections, was confirmed many times, see for instance ref. 42–45. The BO structure of equatorial cyclohexanone was computed similarly (see Table S11, ESI†) with details of the calculations given in Table S12 (ESI†). The BO structures of equatorial and axial conformers of fluorocyclohexane are presented in Tables 3 and 4, respectively, and the details of the calculations are given in Tables S13 and S14 (ESI†).
| Method Basis set | MP2_FC/cc-pVTZ | B3LYP/6-311+a | r SEe |
r
BO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e e
|
|---|---|---|---|---|
| a 6-311+G(3df,2pd). b Estimated according to eqn (1) (see text). | ||||
| C1C2 | 1.5125 | 1.5189 | 1.51218(43) | 1.5131 |
| C2C3 | 1.5276 | 1.5347 | 1.53121(71) | 1.5282 |
| C3C4 | 1.5260 | 1.5324 | 1.52554(25) | 1.5262 |
| C1Fq | 1.3954 | 1.4063 | 1.39260(62) | 1.3945 |
| C1Ha | 1.0940 | 1.0945 | 1.0942(15) | 1.0939 |
| C2Hq | 1.0899 | 1.0910 | 1.0893(24) | 1.0900 |
| C2Ha | 1.0925 | 1.0935 | 1.0934(15) | 1.0930 |
| C3Hq | 1.0900 | 1.0912 | 1.0892(22) | 1.0901 |
| C3Ha | 1.0937 | 1.0947 | 1.0946(13) | 1.0941 |
| C4Hq | 1.0906 | 1.0918 | 1.0905(17) | 1.0907 |
| C4Ha | 1.0934 | 1.0945 | 1.0941(15) | 1.0939 |
| C1C2C3 | 110.19 | 110.66 | 110.168(32) | 110.24 |
| C2C3C4 | 110.90 | 111.50 | 111.007(21) | 111.02 |
| C3C4C5 | 110.78 | 111.43 | 110.982(22) | 110.97 |
| C2C1C6 | 111.73 | 112.36 | 112.007(36) | 111.96 |
| FqC1C2 | 109.28 | 109.17 | 109.084(30) | 109.17 |
| FqC1Ha | 106.69 | 105.77 | 106.51(15) | 106.51 |
| C1C2Hq | 109.60 | 109.63 | 109.50(15) | 109.59 |
| C1C2Ha | 108.16 | 108.29 | 108.01(20) | 108.16 |
| C2C3Hq | 109.94 | 109.70 | 109.84(15) | 109.87 |
| C2C3Ha | 109.38 | 109.39 | 109.03(25) | 109.34 |
| C3C4Hq | 110.32 | 110.15 | 110.246(74) | 110.26 |
| C3C4Ha | 109.17 | 109.21 | 109.121(75) | 109.14 |
| HqC2Ha | 107.60 | 107.21 | 107.53(28) | 107.61 |
| HqC3Ha | 106.73 | 106.34 | 107.07(23) | 106.73 |
| HqC4Ha | 107.00 | 106.57 | 107.03(21) | 106.97 |
| C1C2C3C4 | 56.36 | 54.76 | 55.990(51) | 55.97 |
| FqC1C2C3 | −178.55 | −177.38 | −178.154(37) | −178.18 |
| FqC1C2Hq | 58.71 | 59.88 | 59.05(19) | 59.11 |
| FqC1C2Ha | −58.33 | −56.78 | −57.77(30) | −57.93 |
| C1C2C3Hq | 179.07 | 177.48 | 178.57(16) | 178.64 |
| C1C2C3Ha | −64.05 | −66.24 | −64.40(14) | −64.53 |
| C2C3C4Hq | −178.51 | −176.90 | −178.20(14) | −178.20 |
| C2C3C4Ha | 64.18 | 66.39 | 64.53(13) | 64.58 |
| Method Basis set | MP2_FC/cc-pVTZ | B3LYP/6-311+a | r SEe |
r
BOe
|
|---|---|---|---|---|
| a 6-311+G(3df,2pd). b Estimated according to eqn (1) (see text). | ||||
| C1C2 | 1.5159 | 1.5218 | 1.51423(94) | 1.5162 |
| C2C3 | 1.5259 | 1.5326 | 1.5257(13) | 1.5265 |
| C3C4 | 1.5260 | 1.5323 | 1.52690(71) | 1.5261 |
| C1Fa | 1.4015 | 1.4134 | 1.4036(13) | 1.4013 |
| C1Hq | 1.0909 | 1.0921 | 1.0908(25) | 1.0908 |
| C2Hq | 1.0901 | 1.0912 | 1.0901(27) | 1.0902 |
| C2Ha | 1.0932 | 1.0945 | 1.0938(32) | 1.0937 |
| C3Hq | 1.0903 | 1.0916 | 1.0902(24) | 1.0905 |
| C3Ha | 1.0915 | 1.0924 | 1.0921(38) | 1.0919 |
| C4Hq | 1.0906 | 1.0918 | 1.0905(46) | 1.0907 |
| C4Ha | 1.0944 | 1.0955 | 1.0949(39) | 1.0949 |
| C1C2C3 | 111.40 | 112.21 | 111.605(57) | 111.62 |
| C2C3C4 | 110.84 | 111.47 | 110.942(77) | 110.96 |
| C3C4C5 | 110.62 | 111.27 | 110.618(53) | 110.79 |
| C2C1C6 | 112.37 | 112.98 | 112.729(76) | 112.61 |
| C2C1Hq | 110.71 | 110.67 | 110.667(80) | 110.71 |
| FaC1Hq | 106.31 | 105.52 | 106.14(14) | 106.15 |
| C1C2Hq | 109.03 | 108.99 | 109.04(17) | 109.02 |
| C1C2Ha | 107.90 | 107.65 | 107.56(22) | 107.74 |
| C2C3Hq | 109.86 | 109.69 | 109.72(20) | 109.78 |
| C2C3Ha | 109.01 | 109.05 | 108.77(34) | 109.05 |
| C3C4Hq | 110.32 | 110.12 | 110.27(14) | 110.26 |
| C3C4Ha | 109.30 | 109.36 | 109.30(13) | 109.28 |
| HqC2Ha | 107.38 | 106.86 | 107.21(32) | 107.36 |
| HqC3Ha | 107.10 | 106.68 | 107.27(34) | 107.09 |
| HqC4Ha | 106.91 | 106.49 | 107.00(33) | 106.89 |
| C1C2C3C4 | 55.20 | 53.25 | 54.72(10) | 54.66 |
| C3C2C1Hq | −178.14 | −176.30 | −177.53(13) | −177.57 |
| HqC1C2Hq | 58.88 | 60.31 | 59.28(28) | 59.31 |
| HqC1C2Ha | −57.44 | −55.25 | −56.66(34) | −56.89 |
| C1C2C3Hq | 177.79 | 175.92 | 177.15(20) | 177.20 |
| C1C2C3Ha | −65.13 | −67.59 | −65.79(18) | −65.76 |
| C2C3C4Hq | −179.36 | −177.64 | −179.12(24) | −179.07 |
| C2C3C4Ha | 63.36 | 65.68 | 63.52(21) | 63.73 |
3. Determination of structural predicates
For the determination of the predicates, the MP2_FC/cc-pVTZ level of theory generally gives satisfactory results for the CH bond lengths, the single CC bond lengths and the bond angles.11 The accuracy is about 0.002 Å for the CH bonds and slightly better than 0.003 Å for the CC bonds. For the bond angles, the MP2_FC/6-311+G(3df,2pd) method gives slightly better results than MP2_FC/cc-pVTZ, the accuracy of the former being better than 0.3° in most cases instead of 0.4° for the latter.12 It is worth noting that, for large molecules, it may be advantageous to use DFT methods such as B3LYP or B2PLYP.46,47 This choice is at first sight somewhat arbitrary because there are many functionals and many basis sets available. However, the B3LYP method is broadly used and is known to give accurate results with the split-valence 6-311+G(3df,2pd) basis set.48As observed in fructose, deoxyribose,12 and pseudopelletierine,15 the MP2 method fails to deliver accurate dihedral angles, the error being sometimes as large as several degrees. This error may be easily explained by the fact it requires much less energy to modify a dihedral angle than a bond angle (it requires about 4.2 kJ mol−1 to distort a ∠(CCC) bond angle by 10° and only 0.8 kJ mol−1 to distort a τ(CCCC) dihedral angle by 10°).49 For the molecules investigated up to now, the accuracy was found to be much less sensitive to the basis set than to the method, and the CCSD/cc-pVTZ level of theory was found to be a significant improvement over the MP2 method.12,15 However, although the CCSD method can be easily used in the case of the cyclohexane derivatives investigated in this work, it is significantly more expensive than the MP2 method. For this reason, it would be useful to find a cheaper approximation.
We have examined the residuals of 103 dihedral angles τ of organic molecules analyzed so far and we observe a nice correlation between τe − τ[MP2_FC/cc-pVTZ] and τ[MP2_FC/cc-pVTZ] − τ[B3LYP/6-311+G(3df,2pd)], as seen in Fig. 2. This fact allows us to predict the torsional angles with a standard deviation of 0.37°, which is acceptable for predicates. The resulting equation (complete list of angles in Table S15, ESI†) is given by
| τe − τ[MP2_FC/cc-pVTZ] = −0.2916(15) × τ[MP2_FC/cc-pVTZ] − τ[B3LYP/6-311+G(3df,2pd)] | (2) |
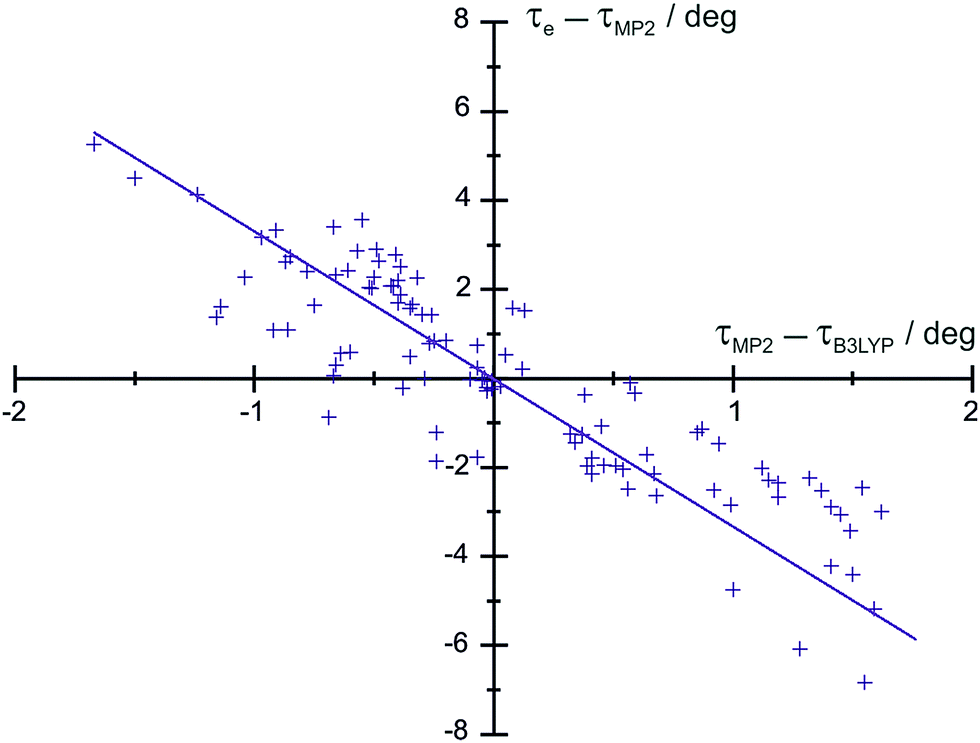 | ||
| Fig. 2 Plot of τe–τ[MP2_FC/cc-pVTZ] as a function of τ[MP2_FC/cc-pVTZ]–τ[B3LYP/6-311+G(3df,2pd)] for a selection of molecules in Table S15 (ESI†). | ||
As a further check the predicates for both conformers of fluorocyclohexane and cyclohexanone are compared in Table S16 (ESI†). Obviously, the angles involving the electronegative oxygen atom are less well reproduced but this might perhaps be improved by using the 6-311+G(3df,2pd) basis set instead of cc-pVTZ in the MP2 calculations.
For 15 molecules with the C(sp3)F single bond, we tried to estimate the CF bond lengths using the MP2 and B3LYP methods. The B3LYP/6-311+G(3df,2pd) level of theory gives rather unsatisfactory results, as seen in Table 5. The MP2_FC/cc-pVTZ level of theory gives a re − rcalc offset of −0.0022(21) Å. However, the problem is that this offset is not constant, as shown in Table 5. The main reason is that fluorine is extremely electronegative and, in such a case, diffuse functions are required. However, replacing the cc-pVTZ basis set by the much larger aug-cc-pVTZ does not significantly improve the situation, in particular the offset and its standard deviation are larger (−0.0054(28) Å). Replacing the basis set by Pople's 6-311+G(3df,2pd) gives much better results with an offset of −0.0042(10) Å, but again this offset is not constant. On the other hand, a linear fit of re as a function of r[MP2_FC/6-311+G(3df,2pd)] is very satisfactory (correlation coefficient of 0.999999, F-test as large as 24246876), the standard deviation of the fit being 0.0011 Å. In conclusion, to predict the C(sp3)F bond length, the following empirical equation was used
| re[CF] = 0.99827(20) × r[MP2_FC/6-311+G(3df,2pd)] | (3) |
| r e | MP2_FC/cc-pVTZ | MP2_FC/6a | B3LYP/6a | q(F) | q(C) | Δq | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a 6-311+G(3df,2pd) basis set. b N. Vogt, J. Demaison and H. D. Rudolph, Mol. Phys., 2014, 112, 2873–2883. c J. Breidung, J. Cosléou, J. Demaison, K. Sarka and W. Thiel, Mol. Phys., 2004, 102, 1827–1841. d N. C. Craig, D. Feller, P. Groner, H. Y. Hsin, D. C. McKean and D. J. Nemchick, J. Phys. Chem. A, 2007, 111, 2498–2506. e J. Demaison, J. Breidung, W. Thiel and D. Papoušek, Struct. Chem., 1999, 10, 129–133. | ||||||||
| CF4 | 1.3152 | 1.3194 | 1.3174 | 1.3240 | −0.641 | 2.555 | 3.196 | |
| CClF3 | 1.3208 | 1.3227 | 1.3232 | 1.3277 | −0.638 | 2.031 | 2.669 | |
| CF3CN | 1.3240 | 1.3288 | 1.3271 | 1.3334 | −0.631 | 1.991 | 2.622 | |
| CCl2F2 | 1.3274 | 1.3278 | 1.3303 | 1.3321 | −0.634 | 1.509 | 2.143 | |
| CHF3 | 1.3312 | 1.3338 | 1.3320 | 1.3384 | −0.651 | 1.870 | 2.522 | |
| CF3CCH | 1.3324 | 1.3369 | 1.3356 | 1.3428 | −0.639 | 1.917 | 2.556 | |
| CHClF2 | 1.3352 | 1.3384 | 1.3375 | 1.3417 | −0.647 | 1.387 | 2.034 | |
| CCl3F | 1.3361 | 1.3342 | 1.3406 | 1.3379 | −0.630 | 0.992 | 1.622 | |
| c-C3H4F2 | 1.3428 | 1.3453 | 1.3448 | 1.3528 | ||||
| CH2F2 | 1.3523 | 1.3544 | 1.3543 | 1.3604 | −0.664 | 1.158 | 1.822 | |
| CH2ClF | 1.3576 | 1.3598 | 1.3610 | 1.3631 | −0.651 | 0.772 | 1.423 | |
| CF3Li | 1.3803 | 1.3842 | 1.3827 | 1.3887 | −0.683 | 1.123 | 1.805 | |
| CH3F | 1.3827 | 1.3809 | 1.3830 | 1.3883 | −0.660 | 0.640 | 1.300 | |
| C6H11F eq. | 1.3945 | 1.3954 | 1.3971 | 1.4063 | −0.670 | 0.569 | 1.239 | This work |
| C6H11F ax. | 1.4013 | 1.4015 | 1.4025 | 1.4134 | −0.672 | 0.583 | 1.255 | This work |
Once this result was established, we checked whether it would be possible to improve the Schomaker–Stevenson equation replacing the difference of electronegativities by the difference of charges Δq calculated with the AIM theory.38 There is indeed a correlation between r(CF) and Δq with a Spearman rank correlation coefficient of −0.95. However, this correlation is not quantitative, indicating that other factors apart from the difference of charge are not negligible, as explained in ref. 50.
4. Semiexperimental structure
The semiexperimental equilibrium rotational constants for each direction of the principal axis system, Be, were calculated from the experimental ground state rotational constants, B0, using the following equation:| Be = B0 + ΔBvib | (4) |
It is possible to estimate the accuracy of the semiexperimental equilibrium rotational constants using the planar moment of inertia Pb = (Ia + Ic – Ib)/2, which should be constant for the parent species and the isotopologues 13C1 and 13C4. For the equatorial form, its range is 0.00075 u Å2 whereas for the axial form it is 0.00242 u Å2. In other words, the rotational constants of the axial form are about three times less accurate than those of the equatorial form, probably because non-rigidity effects are larger for this isomer.
For the equatorial form, the following uncertainties were used for the weighting of the rotational constants (in kHz): 10 for A, and 5 for B and C. For the axial form, the uncertainties are: 100 for A and 50 for B and C. For the predicates, the used uncertainties were 0.002 Å for the CH bond lengths, 0.003 Å for the other lengths, 0.2° for the bond angles and 0.5° for the dihedral angles. The compatibility of these weights was checked using statistical diagnostics as explained in ref. 9.
The rovibrational corrections, the semiexperimental equilibrium rotational constants, residuals of the fit and leverage values hii are given in Tables S17 and S18 (ESI†) for the equatorial and axial conformers, respectively.
For the equatorial conformer, the determined parameters in Table 3 are precise, and an examination of the residuals of the fit in Table S17 (ESI†), confirms that the rotational constants and the predicates are compatible. More importantly, inspection of the leverage values, h, shows that: (1) the rotational constants contribute significantly to the determination of the parameters, their median value being 0.71 and being larger than 0.88 for most A rotational constants (a leverage point, defined as hii = ∂ŷi/∂yi, where ŷi and yi are the fitted and measured observations, respectively, is high when a small change in the input value causes a large change in the solution, with 0 ≤ hii ≤ 1).
For the axial conformer in Table 4 and Table S18 (ESI†), the situation is less favorable, the determined parameters are slightly less precise and the residuals of the fit are one order of magnitude larger. The ground state rotational constants of the 13C isotopologues have been determined from very few lines. Thus, it is not possible to exclude that some rotational constants are less accurate. It is also possible that the calculated rovibrational corrections are less accurate.
The semiexperimental equilibrium Cartesian coordinates are given for both conformers in Tables S19 and S20 (ESI†).
Discussion
Accurate equilibrium structures have been determined for the chair forms of equatorial and axial cyclohexane, providing reference data for the molecule and revealing the subtle structural changes between both isomers and the consequences of the fluorine substitution in the cyclohexane ring. The bond parameters, regarded as accurate to 0.001 Å and 0.2°, are given in Tables 3 and 4. The structural determination relies in a mixed estimation fit to predicate (ab initio) bond parameters and equilibrium moments of inertia, each data set accompanied by appropriate uncertainties. The mixed estimation method thus provides a route to equilibrium structure determination at a reduced computational cost.The most noticeable result is the large values of the CF bond lengths for both conformers in Table 5, the axial value being still longer by almost 0.007 Å. The changes in the heavy atom structural parameters of cyclohexane with the substitution of the fluorine are significant for the CαCβ distances compared to their distance of 1.5258(6) Å in cyclohexane,52 where there is a clear reduction of 0.014 Å for the equatorial form and 0.010 Å for the axial form. The other CC bond lengths and the CH bond lengths are only slightly affected. The dihedral angle τ(CCCC) does not vary much (55.73° in cyclohexane vs. 55.97° and 54.66° for the equatorial and axial forms). Likewise, the bond angle ∠(C1C2C3) is nearly constant (111.11° in cyclohexane and 110.17° and 111.61° for the equatorial and axial forms, respectively). The most affected bond angle should be ∠(C6C1C2), but its value (112.0° in the equatorial form and 112.6° in the axial form) remains close to the cyclohexane value. Thus, the heavy atom ring parameters of cyclohexane appear to be only slightly affected by monosubstitution of the single fluorine atom. In cyclohexanone, the CαCβ bond length is shortened by 0.014 Å as in equatorial fluorocyclohexane and the CβCγ is lengthened by 0.007 Å and, contrary to fluorocyclohexanes, the angle ∠(C6C1C2) is significantly increased, up to 115°.
It is informative to compare the semiexperimental equilibrium structure, rSEe, with the empirical substitution structure, rs. The substitution method53 is widely used to determine molecular structures because it is believed to provide near-equilibrium values. A further advantage is that it is quite simple, the atomic coordinates being obtained from isotopic differences of moments of inertia by using equations formulated by Kraitchman.54 However, the key model assumption, i.e., constant rovibrational contributions upon isotopic substitution, is only approximate. Costain has proposed an empirical formula to estimate the coordinates uncertainties,55
| δz = 0.0015/|z| | (5) |
We first used Kraitchman's equations with the semiexperimental equilibrium rotational constants of the equatorial conformer (Table S17, ESI†), and found results in nice agreement with the rSEe structure, indicating that the rotational constants are accurate (or, more likely, that they have identical systematic errors). Then, we used the ground state rotational constants in Table 1 to determine the rs structure of the equatorial conformer. The results in Table S21 (ESI†) made evident that the rs coordinates are at least one order of magnitude less precise than the rSEe ones. Furthermore, the small rs coordinates are not as reliable as anticipated. Still, the most worrying aspect is that the rs method is not able to correctly predict the small changes in the bond lengths, the rs values being (in Å): C1C2 = 1.522(4); C2C3 = 1.514(7); C3C4 = 1.532(3) whereas the corresponding rSEe values are 1.5122(4); 1.5312(7) and 1.5255(3). In conclusion, for a molecule as fluorocyclohexane, the rs method does not provide a reliable structure, as already pointed out by Bialkowska-Jaworska et al.20 Unfortunately, this behavior seems to be rather general,11,12,57 evidencing that the accuracy of the substitution method is limited.
Conclusion
In conclusion, we have demonstrated that the mixed estimation method is operationally superior in terms of accuracy and computational cost to pure ab initio calculations or conventional semiexperimental methods for the determination of accurate equilibrium structures, exploiting the synergy between spectroscopic data and quantum chemical predictions. The application of the mixed estimation method to new chemical systems will expand our description of the factors controlling the molecular structure.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from MINECO-FEDER (CTQ2015-68148-C2-2-P) is gratefully acknowledged. N. V., J. D. and H. D. R. thank the Dr Barbara Mez-Starck Foundation (Germany) for support.References
- P. Pulay, W. Meyer and J. E. Boggs, J. Chem. Phys., 1978, 68, 5077–5085 CrossRef CAS.
- Equilibrium Molecular Structures: From Spectroscopy to Quantum Chemistry, ed. J. Demaison, J. E. Boggs and A. G. Császár, CRC Press, Boca Raton, 2011 Search PubMed.
- K. L. Bak, J. Gauss, P. Jørgensen, J. Olsen, T. Helgaker and J. F. Stanton, J. Chem. Phys., 2001, 114, 6548–6556 CrossRef CAS.
- F. Pawłowski, P. Jørgensen, J. Olsen, F. Hegelund, T. Helgaker, J. Gauss, K. L. Bak and J. F. Stanton, J. Chem. Phys., 2002, 116, 6482–6496 CrossRef.
- M. Mendolicchio, E. Penocchio, D. Licari, N. Tasinato and V. Barone, J. Chem. Theory Comput., 2017, 13, 3060–3075 CrossRef CAS PubMed.
- C. Puzzarini, J. F. Stanton and J. Gauss, Int. Rev. Phys. Chem., 2010, 29, 273–367 CrossRef CAS.
- N. Vogt, J. Demaison, J. Vogt and H. D. Rudolph, J. Comput. Chem., 2014, 35, 2333–2342 CrossRef CAS PubMed.
- L. S. Bartell, D. J. Romenesko and T. C. Wong, in Chemical Society Specialist Periodical Report No. 20, ed. G. A. Sim and L. E. Sutton, The Chemical Society, London, 1975, vol. 3, pp. 72–79 Search PubMed.
- J. Demaison, in Equilibrium Molecular Structures: From Spectroscopy to Quantum Chemistry, ed. J. Demaison, J. E. Boggs and A. G. Császár, CRC Press, Boca Raton, 2011, pp. 29–52 Search PubMed.
- D. A. Belsley, Conditioning Diagnostics, Wiley, New York, 1991, pp. 298–299 Search PubMed.
- J. Demaison, N. C. Craig, E. J. Cocinero, J.-U. Grabow, A. Lesarri and H. D. Rudolph, J. Phys. Chem. A, 2012, 116, 8684–8692 CrossRef CAS PubMed.
- N. Vogt, J. Demaison, E. J. Cocinero, P. Écija, A. Lesarri, H. D. Rudolph and J. Vogt, Phys. Chem. Chem. Phys., 2016, 18, 15555–15563 RSC.
- N. Vogt, J. Demaison, S. V. Krasnoshchekov, N. F. Stepanov and H. D. Rudolph, Mol. Phys., 2017, 115, 942–951 CrossRef CAS.
- N. Vogt, E. P. Altova, D. N. Ksenafontov and A. N. Rykov, Struct. Chem., 2015, 26, 1481–1488 CrossRef CAS.
- M. Vallejo-López, P. Écija, N. Vogt, J. Demaison, A. Lesarri, F. J. Basterretxea and E. J. Cocinero, Chem. – Eur. J., 2017 DOI:10.1002/chem.201702232 , in press.
- P. Andersen, Acta Chem. Scand., 1962, 16, 2337–2340 CrossRef CAS.
- L. Pierce and R. Nelson, J. Am. Chem. Soc., 1966, 88, 216–219 CrossRef CAS.
- L. Pierce and J. F. Beecher, J. Am. Chem. Soc., 1966, 88, 5406–5410 CrossRef CAS.
- L. H. Scharpen, J. Am. Chem. Soc., 1972, 94, 3737–3739 CrossRef CAS.
- E. Białkowska-Jaworska, M. Jaworski and Z. Kisiel, J. Mol. Struct., 1995, 350, 247–254 CrossRef.
- J. R. Durig, R. M. Ward, K. G. Nelson and T. K. Gounev, J. Mol. Struct., 2010, 976, 150–160 CrossRef CAS.
- C. A. Stortz, J. Phys. Org. Chem., 2010, 23, 1173–1186 CrossRef CAS.
- T. J. Balle and W. H. Flygare, Rev. Sci. Instrum., 1981, 52, 33–45 CrossRef CAS.
- (a) J.-U. Grabow and W. Stahl, Z. Naturforsch., A: Phys. Sci., 1990, 45, 1043–1044 CrossRef; (b) J.-U. Grabow, W. Stahl and H. Dreizler, Rev. Sci. Instrum., 1996, 67, 4072–4084 CrossRef CAS.
- G. D. Purvis, III and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- K. A. Peterson and T. H. Dunning, Jr., J. Chem. Phys., 2002, 117, 10548–10560 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- W. Kohn and L. J. Sham, Phys. Rev. A, 1965, 140, 1133–1138 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. G. Császár, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 273–289 CrossRef.
- W. D. Allen and A. G. Császár, J. Chem. Phys., 1993, 98, 2983–3015 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS; (b) H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, A. Mitrushenkov, G. Rauhut, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, G. Hetzer, T. Hrenar, G. Knizia, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, P. Palmieri, K. Pflüger, R. Pitzer, M. Reiher, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and A. Wolf, MOLPRO, 2009.
- R. F. W. Bader, Atoms In Molecules, Oxford University Press, New York, 1990 Search PubMed.
- (a) J. Cioslowski, A. Nanayakkara and M. Challacombe, Chem. Phys. Lett., 1993, 203, 137–142 CrossRef CAS; (b) J. Cioslowski and P. R. Surján, THEOCHEM, 1992, 255, 9–33 CrossRef; (c) J. Cioslowski and B. B. Stefanov, Mol. Phys., 1995, 84, 707–716 CrossRef CAS; (d) B. B. Stefanov and J. Cioslowski, J. Comput. Chem., 1995, 16, 1394–1404 CrossRef CAS; (e) J. Cioslowski and S. T. Mixon, J. Am. Chem. Soc., 1991, 113, 4142–4145 CrossRef CAS; (f) J. Cioslowski, Chem. Phys. Lett., 1992, 194, 73–78 CrossRef CAS; (g) J. Cioslowski and A. Nanayakkara, Chem. Phys. Lett., 1994, 219, 151–154 CrossRef CAS.
- J. K. G. Watson, in Vibrational Spectra and Structure, ed. J. R. Durig, Elsevier, Amsterdam, 1977, 6, pp. 1–89 Search PubMed.
- H. M. Jaeger, H. F. Schaefer, III, J. Demaison, A. G. Császár and W. D. Allen, J. Chem. Theory Comput., 2010, 6, 3066–3078 CrossRef CAS PubMed.
- H. D. Rudolph, J. Demaison and A. G. Császár, J. Phys. Chem. A, 2013, 117, 12969–12982 CrossRef CAS PubMed.
- J. Demaison, H. D. Rudolph and A. G. Császár, Mol. Phys., 2013, 111, 1539–1562 CrossRef CAS.
- C. Puzzarini and V. Barone, Phys. Chem. Chem. Phys., 2011, 13, 7189–7197 RSC.
- N. Vogt, L. S. Khaikin, O. E. Grikina and A. N. Rykov, J. Mol. Struct., 2013, 1050, 114–121 CrossRef CAS.
- E. Penocchio, M. Piccardo and V. Barone, J. Chem. Theory Comput., 2015, 11, 4689–4707 CrossRef CAS PubMed.
- M. Piccardo, E. Penocchio, C. Puzzarini, M. Biczysko and V. Barone, J. Phys. Chem. A, 2015, 119, 2058–2082 CrossRef CAS PubMed.
-
(a) J. M. L. Martin, J. El-Yazal and J.-P. Franc
![[o with combining cedilla]](https://www.rsc.org/images/entities/char_006f_0327.gif) is, Mol. Phys., 1995, 86, 1437–1450 CrossRef CAS;
(b) A. D. Boese, W. Klopper and J. M. L. Martin, Int. J. Quantum Chem., 2005, 104, 830–845 CrossRef CAS.
is, Mol. Phys., 1995, 86, 1437–1450 CrossRef CAS;
(b) A. D. Boese, W. Klopper and J. M. L. Martin, Int. J. Quantum Chem., 2005, 104, 830–845 CrossRef CAS. - I. Hargittai and J. B. Levy, Struct. Chem., 1999, 10, 387–389 CrossRef CAS.
- E. A. Robinson, S. A. Johnson, T.-H. Tang and R. J. Gillespie, Inorg. Chem., 1997, 36, 3022–3030 CrossRef CAS PubMed.
- V. Barone, J. Chem. Phys., 2005, 122, 014108 CrossRef PubMed.
- J. Demaison, N. C. Craig, P. Groner, P. Écija, E. J. Cocinero, A. Lesarri and H. D. Rudolph, J. Phys. Chem. A, 2015, 119, 1486–1493 CrossRef CAS PubMed.
- (a) C. C. Costain, J. Chem. Phys., 1958, 29, 864–874 CrossRef CAS; (b) B. P. Van Eijck, in Accurate Molecular Structures, ed. A. Domenicano and I. Hargittai, Oxford University Press, Oxford, 1992, pp. 47–64 Search PubMed.
- J. Kraitchman, Am. J. Phys., 1953, 21, 17–24 CrossRef CAS.
- C. C. Costain, Trans. Am. Crystallogr. Assoc., 1966, 2, 157–164 CAS.
- J. Demaison and H. D. Rudolph, J. Mol. Spectrosc., 2002, 215, 78–84 CrossRef CAS.
- N. C. Craig, J. Demaison, P. Groner, H. D. Rudolph and N. Vogt, J. Phys. Chem. A, 2015, 119, 195–204 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S21. See DOI: 10.1039/c7cp06135h |
| This journal is © the Owner Societies 2017 |