
A ring polymer molecular dynamics study of the OH + H2(D2) reaction
 *a
and
*a
and
Abstract
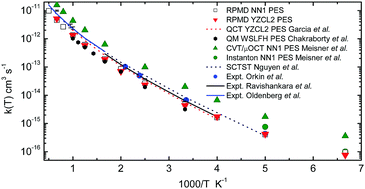
In this work we have performed a ring polymer molecular dynamics (RPMD) study of the OH + H2 and OH + D2 reactions at temperatures ranging from 150 K to 2000 K using two available ab initio potential energy surfaces (PESs) that have been termed as the YZCL2 and NN1 PES, respectively. The YZCL2 PES was developed by Yang et al. [J. Chem. Phys., 2001, 115(1), 174] which is based on points fitted by a modified Shephard interpolation method and calculated with unrestricted coupled-cluster theory with all single and double excitations and a perturbative account of the triple excitations (UCCSD(T)) method with an aug-cc-pVQZ basis. The NN1 PES was constructed by Chen et al. [J. Chem. Phys., 2013, 138(15), 154301] using a neural networks method to fit ab initio energies calculated at the UCCSD(T)-F12a/AVTZ level of theory. We show that both techniques provide reliable PESs. The RPMD thermal rate coefficients and the kinetic isotope effects (KIEs) calculated using these two PESs are in very good agreement with each other as well as with previous experimental values available to date. Besides, we have shown that these two procedures for fitting PESs can yield even more similar RPMD rate coefficients when the same level of ab initio theory is employed, at least for the present OH + H2 reaction. Comparison of the previous theoretical calculations on the NN1 PES, namely, instanton theory and canonical variational theory with microcanonical optimized multidimensional tunneling, shows that the present RPMD results are more consistent and accurate. Future experimental measurements of the KIEs and accurate quantum mechanical calculations on these PESs are highly desirable, especially at low temperatures.
 Please wait while we load your content...
Please wait while we load your content...

