
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceValence orbitals and local bond dynamics around N atoms of histidine under X-ray irradiation†
Sebastian
Eckert
 *ab,
Johannes
Niskanen
b,
Raphael M.
Jay
a,
Piter S.
Miedema
b,
Mattis
Fondell
b,
Brian
Kennedy
b,
Wilson
Quevedo
b,
Marcella
Iannuzzi
c and
Alexander
Föhlisch
ab
*ab,
Johannes
Niskanen
b,
Raphael M.
Jay
a,
Piter S.
Miedema
b,
Mattis
Fondell
b,
Brian
Kennedy
b,
Wilson
Quevedo
b,
Marcella
Iannuzzi
c and
Alexander
Föhlisch
ab
aInstitut für Physik und Astronomie, Universität Potsdam, Karl-Liebknecht-Str. 24/25, 14476 Potsdam, Germany. E-mail: sebeckert@uni-potsdam.de
bInstitute for Methods and Instrumentation for Synchrotron Radiation Research, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Albert-Einstein-Str. 15, 12489 Berlin, Germany
cPhysical-Chemistry Institute, University of Zürich, Winterthurerstr. 190, 8057 Zürich, Switzerland
First published on 28th November 2017
Abstract
The valence orbitals of aqueous histidine under basic, neutral and acidic conditions and their X-ray induced transformations have been monitored through N 1s resonant inelastic X-ray scattering. Using density functional ab initio molecular dynamics simulations in the core-hole state within the Z + 1 approximation, core-excitation-induced molecular transformations are quantified. Spectroscopic evidence for a highly directional X-ray-induced local N–H dissociation within the scattering duration is presented for acidic histidine. Our report demonstrates a protonation-state and chemical-environment dependent propensity for a molecular dissociation, which is induced by the absorption of high energy photons. This case study indicates that structural deformations in biomolecules under exposure to ionizing radiation, yielding possible alteration or loss of function, is highly dependent on the physiological state of the molecule upon irradiation.
1 Introduction
With the advent of long lasting space missions and the planned inhabitation of the planet mars1 with its drastically different atmospheric composition, the impact of ionizing cosmic radiation on living organisms on the macroscopic and on biomolecules on the microscopic scale has become a research topic of public interest. A controlled exposure to ionizing radiation and detection of its impact on molecular systems on their intrinsic ultrafast timescale in laboratory scale experiments is difficult to achieve. As a case study, we monitor the reaction of the amino acid histidine to N 1s core excitations using resonant inelastic X-ray scattering (RIXS). Histidine is a building block of proteins with a rich structure-function relation2–5 and contains three N sites which exhibit a pH-dependent protonation6–10 altering the activity of the molecule in biological processes. Especially the protonation of the imidazole ring of histidine allows for participation of histidine in acid–base catalytic processes, like the catalytic triad,11 and enables histidine to form active side chains in carbonic anhydrase enzymes.12Molecular local valence electronic structure is accessed in X-ray spectroscopy through transitions between strongly localized core levels and valence orbitals often spanning large parts of the investigated molecular system. This approach provides element specific access to selected sites in the system with orbital as well as chemical state selectivity.13 RIXS combines the chemical state selectivity through characteristic shifts of X-ray absorption resonances with access to the local occupied valence electronic structure through the detection of emission lines.14 The transition intensity and energy of these lines is strongly dependent on the overlap between the probed valence orbitals and the core hole. The finite lifetime of the core excited state in the RIXS mechanism introduces an intrinsic temporal component to the detection of the occupied valence states.15,16 Although the lifetimes of core-excited states of light elements are usually below 10 fs, the excitation of electrons from a localized core orbital and the resulting reduced screening of the nucleus can induce drastic deformations of molecular geometries on this ultrashort timescale.17–23 Thus, RIXS intrinsically provides the unique opportunity to monitor the ultrafast initial molecular deformations induced by the absorption ionizing radiation in biologically relevant systems containing multiple atomic sites of the same element, before the dynamics occur along decay channel dependent paths.
We establish N 1s X-ray spectroscopic signatures of local protonation at the imidazole ring of histidine and link them directly to altered local occupied and unoccupied electronic structure of the molecule. In addition, N 1s RIXS transitions are used to access to the chemical-state-dependent propagation of the molecule on the core excited state potential. We identify site-specific character of molecular orbitals of the imidazole ring of histidine and their X-ray induced transformation in basic and neutral environments. These spectroscopic fingerprints are used to unravel a highly directional dissociation of the system under acidic conditions which we find to be independent of the excited N site.
2 Experimental and computational details
For every shift of the beamtime at the synchrotron Bessy II (Helmholtz-Zentrum Berlin) the 80 mM histidine solution was prepared by dissolving L-histidine, 98+% ordered from Alfa Aesar in deionized water. The solution was degassed and sprayed into the high vacuum chamber of the liquid flexRIXS experiment24 within a liquid jet. The jet had a diameter of 20 micrometer and was run with a JASCO High-Performance Liquid Chromatography pump at a flow rate of 0.5 ml per minute. The pH-value of the solution was adjusted using hydrochloric acid and potassium hydroxide. The jet was excited with horizontally polarized X-ray photons at the N 1s absorption resonance with a bandwidth of 250 meV using the beam line U49-2_PGM-1.25 K-edge absorption lines of N2 gas were used for photon energy calibration. We detected the incident photon energy dependent resonant inelastic X-ray emission spectra of the sample in a 90° scattering geometry. The soft X-ray emission spectrometer Scienta XES 35026 was used in slitless operation to acquire the emission spectra. The N 2p emission intensity in the acquired data was integrated as a measure of the NEXAFS signal. The RIXS data was recorded with 100 meV steps in incident energy.The molecular structures for the simulations of histidine in the different protonation states and surrounding water molecules were optimized with the ORCA quantum chemistry software27 using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional28 and TZVP basis sets.29 The spectrum calculations were performed in the framework presented in ref. 30, now for optimized structures. The PBE28 exchange–correlation functional was used together with the aug-cc-pV5Z basis set31 for the excited site and TZV2P-MOLOPT basis sets32 with Goedecker–Teter–Hutter pseudopotentials33 for other atoms. Structural relaxation in the core-excited state was approximated by propagating the nuclei within the ionized-molecule Z + 1 approximation starting from the optimized structure in rest. In this calculation and in the calculation for ground state charges, pseudopotentials were used for all atoms. The simulated dynamics thus account for the reduced screening of the core at an excited site, neglecting core excited state specific bond dissociation mechanisms. The elastic line results in from summation over all excited states, using the dipole matrix element for absorption (the same for emission) in the optimized structure only, due to complications rising from changing ordering of the orbitals during structural changes. All calculations except geometry optimization were carried out by using the CP2K software.34 The energy axis of the simulated spectra was adapted for good overlap between the experimentally detected and the simulated π* absorption resonances. A gaussian broadening of 0.25 eV on the incident energy and 1 eV on the emission energy axis was applied to the simulated spectra.
3 Results and discussion
Fig. 1 presents the pH-dependent N 1s partial fluorescence yield (PFY) Near Edge X-ray Absorption Fine Structure (NEXAFS) signal of histidine (aq) in comparison to simulations. Solvent water molecules were added to the N sites that are capable of forming hydrogen bonds with the solvent. The simulated structures correspond to the pH dependent structural changes reported by Li and Hong,8 but especially for neutral conditions a histidine tautomer with opposite protonation of the furthest N (tele-N, Nτ) and the closest N (pros-N, Nπ) in the imidazole ring can be expected to coexist in a tautomeric equilibrium in the solution. The results of simulations for the tautomers are summarized in the ESI† and are in agreement with the conclusions drawn throughout the article.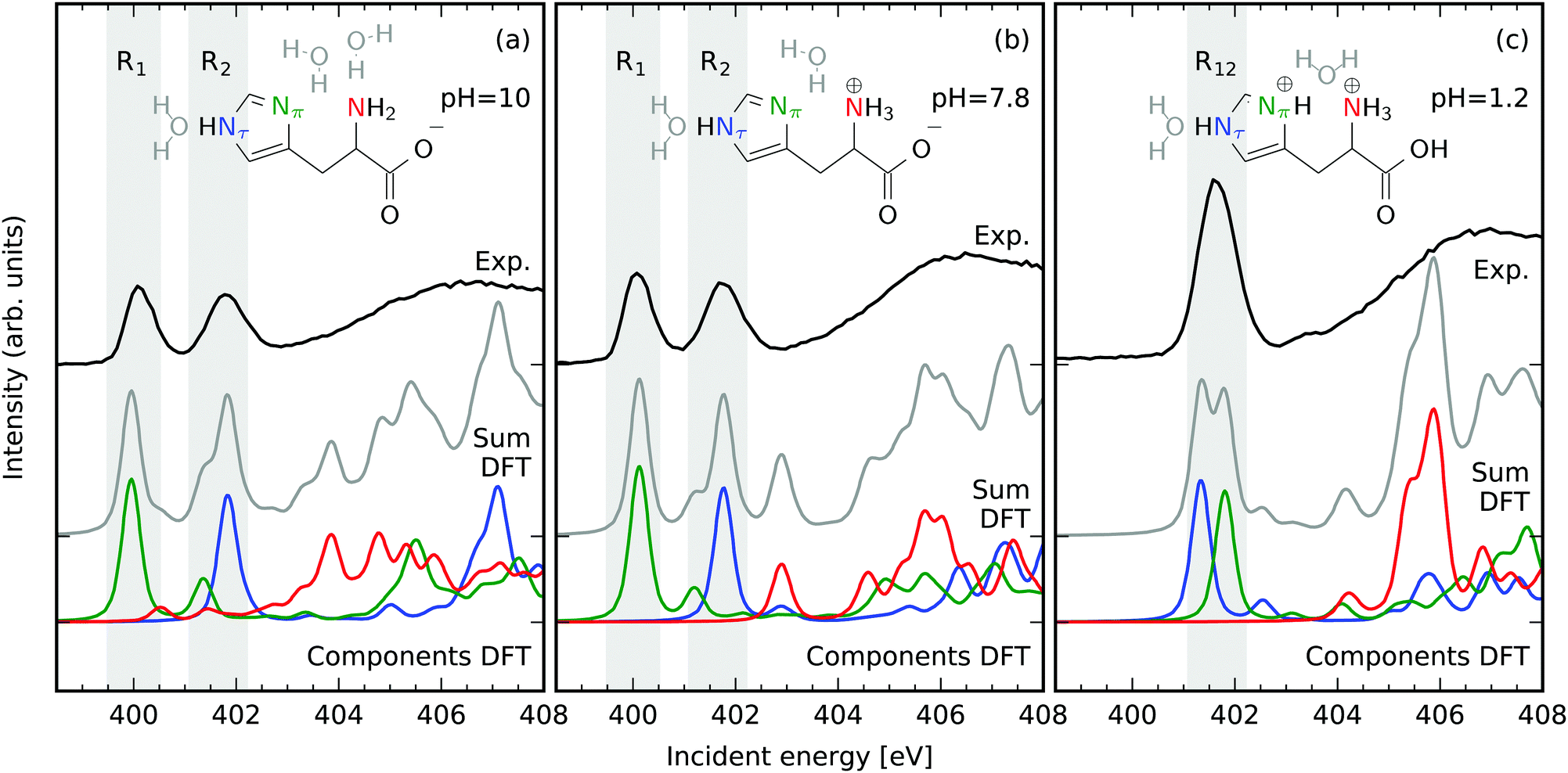 | ||
| Fig. 1 Different N protonation reflected in N 1s NEXAFS of histidine (aq, 80 mM) in dependence on the pH value in comparison to simulations. Different protonation of the N atoms in the imidazole-ring in basic pH = 10 (a) and neutral pH = 7.8 (b) conditions is reflected in two well separated X-ray absorption resonances. The resonances of the amine group are shifted towards higher energies. Protonation of both N atoms in the imidazole ring in acidic conditions pH = 1.2 (c) yields a single resonance. Relevant regions for the latter extraction of resonant emission spectra are marked with grey background and labeled R1, R2 for basic and neutral and R12 for acidic conditions. | ||
The NEXAFS spectra of both histidine in basic Fig. 1a and neutral Fig. 1b conditions exhibit two distinct resonances in the range of photon energies below the onset of the continuum absorption at approximately 403 eV. These have also been reported to be present for histidine powder samples.35 Comparing to the simulated spectra each of these resonances can be attributed to N 1s absorption of dominantly one N atom in the imidazole ring of histidine. The resonance at 400 eV is dominated by N 1s X-ray absorption at the deprotonated Nπ, whereas the resonance at 401.5 eV can be associated with absorption at the protonated Nτ. This finding agrees well with the N 1s NEXAFS signature of chemical (de-)protonation detected for conjugated solvated systems.36–38 In an acidic environment the resonance at 400 eV vanishes in exchange for an intensity increase of the second resonance at 401.5 eV resembling the protonation of both the Nπ and the Nτ atoms. It is important to note that the resonances of the amine group (red in Fig. 1) are located energetically above the ionization threshold of the 1s levels of the N atoms in the imidazole ring. Hence, spectral signatures of the amine N atom overlap with the non resonant X-ray emission from the imidazole N-sites. Even though attempts have been made to separate spectral contributions from the different groups,39 we rely on the well separated bound Nπ and the Nτ resonances and do not discuss the amine N atom throughout this work. The large chemical shift induced by a N deprotonation within the imidazole ring is present even though hydrogen bonding to neighboring water molecules is taken into account in the PFY NEXAFS simulations. Consequently, the energy of the N 1s π* absorption line is a clear indicator for the chemical state of the imidazole nitrogen sites. As these resonances can be associated with specific N atoms in the imidazole ring, their selective excitation provides access to the local occupied valence electronic structure at these sites in the subsequent decay.
3.1 Neutral and basic solution
The X-ray emission spectra of the Nπ and Nτ in basic and neutral environments, displayed in Fig. 2, were generated by integrating the detected intensity in the intervals of incident energies R1 and R2 marked in Fig. 1a and b. The resonant X-ray emission spectra in the regions were simulated for different times after the X-ray excitation taking X-ray induced dynamics within the Z + 1 equivalent core approximation into account. Within this approximation the core excited N* atom is regarded to be equivalent to an ionized oxygen atom (ignoring the excited electron). All nuclei in the system are propagated on the core excited potential energy surface using ab initio molecular dynamics as described in the experimental and computational details section. The best agreement between the simulated spectra and the experimental data is found after the longest simulated propagation of the system in the potential of the core excited state for 10 fs. This corresponds to twice the lifetime of the N 1s core hole. An underestimation of the speed of the nuclei can be ascribed to the semiclassical treatment of the motion from a frozen molecular configuration. Hence, the slower simulated dynamics originate from the disregard of the finite temperature and the quantum-mechanical nature of the system. Additionally, only a single molecular conformation is simulated whereas an ensemble of different configurations in the fluctuating H-bonding solvent network are probed in the experiment. Having these uncertainties in mind, we interpret the shifts and intensity changes of emission lines and their the excitation-induced dynamics with characteristic molecular orbitals. These simplifications are justified by the qualitative agreement between experimentally detected spectral signatures and simulated spectra for long lifetimes of the core excited state.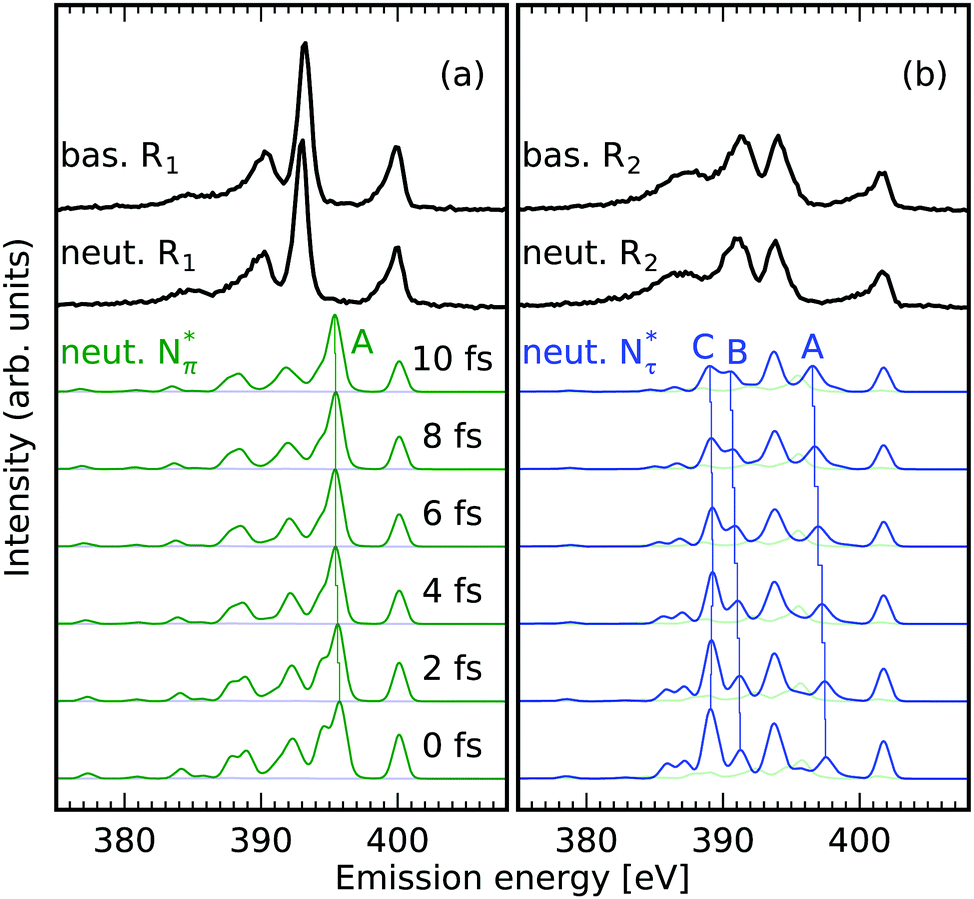 | ||
| Fig. 2 N 1s core-excited-state dynamics in the imidazole-ring of histidine in basic and neutral environments; the dependence on the position of the core hole. Experimental (black) and theoretical (Nπ green and Nτ blue) RIXS spectra integrated in the incident energy intervals R1 (a) and R2 (b) marked in Fig. 1a and b of the NEXAFS. Capital letters mark explicitly discussed transitions. (a) Only minor impact of core excited state dynamics on RIXS spectra of the deprotonated Nπ atom is detected in the region of incident energies 399.5 eV to 400.5 eV R1. (b) Elongations of the chemical bonds of the protonated Nτ atom upon core excitation induces shifts and intensity variations of emission lines probed in the region of incident energies 401.1 eV to 402.2 eV R2. | ||
The shape of the emission spectra in the two regions R1 and R2 does not exhibit a strong dependence on the pH value between the basic and neutral solution, because only the amine group is affected in the pH change (see molecular structures in Fig. 1a and b). We study the orbitals for conditions upon core excitation at the Nπ and the Nτ atom at 0 fs and after 10 fs of core-excited-state propagation, to associate spectral features with the local valence electronic structure changes induced by the core excitation. Orbital ordering does not remain the same during the dynamics, and we adapt the convention of subindexing the orbital number with the corresponding time after excitation: 0 fs or 10 fs. We will focus on molecular orbitals for the highlighted transitions in the simulated spectra in Fig. 2, for the neutral pH only. The orbitals are numbered according to the associated transition energy to the core hole, which can be altered through the dynamics in the core excited state. We hence characterise orbitals according to their spectral contribution which can result in a mismatch of numbering in the compared orbitals.
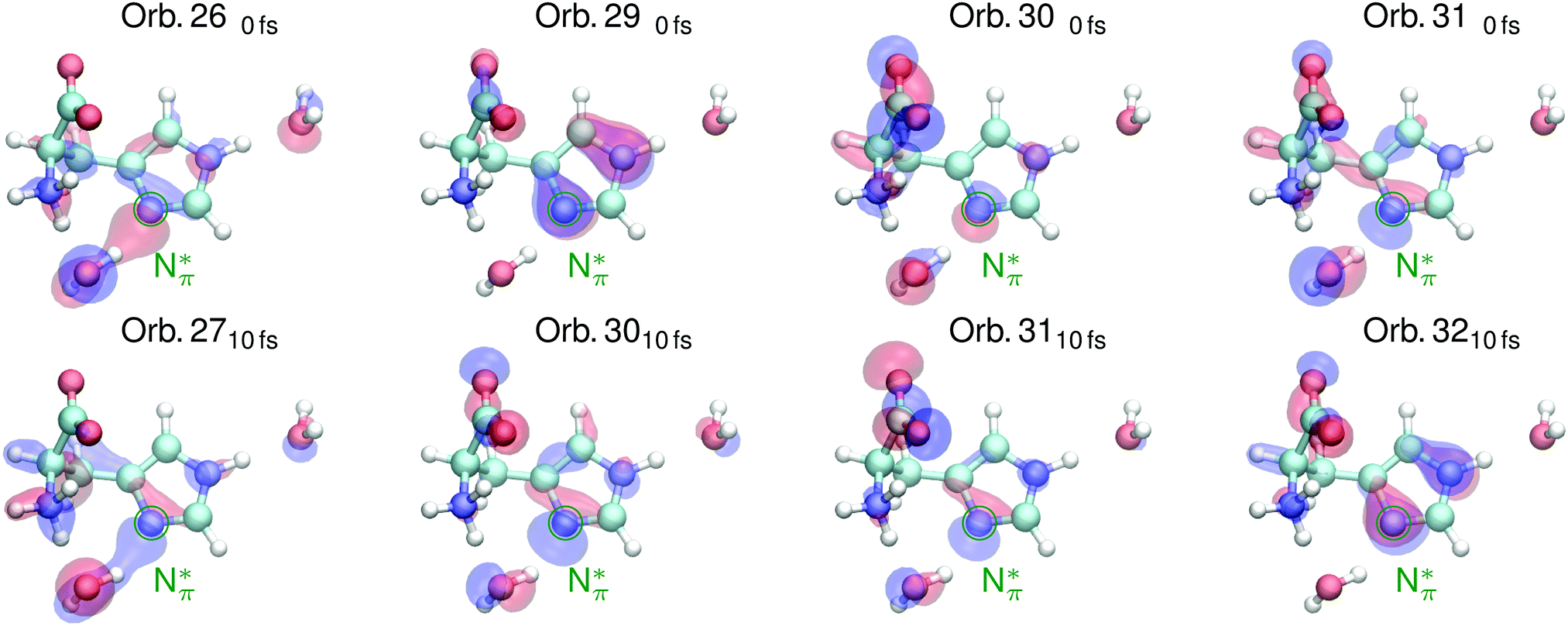 | ||
| Fig. 3 Valence orbitals attributed to transitions in simulated RIXS spectra of the deprotonated Nπ atom in the imidazole ring of histidine in Fig. 2 under neutral conditions upon core excitation (top) and after 10 fs of core excited state dynamics (bottom). | ||
 | ||
| Fig. 4 Valence orbitals attributed to transitions in simulated RIXS spectra of the protonated Nτ atom in the imidazole ring of histidine in Fig. 2 under neutral conditions upon core excitation (top) and after 10 fs of core excited state dynamics (bottom). | ||
The highlighted emission line B in Fig. 2b, lower in emission energy, reduces in emission energy within the simulated time frame of core excited state propagation. The corresponding orbital 140fs which has N–H σ character as well as π character with respect to the N–C bond transforms into a dominantly N–H σ orbital which increases the local bonding character of the orbital, lowering the emission energy.
The marked emission line C for Nτ in Fig. 2b, lowest in energy, reduces drastically in intensity due to the core excited state dynamics. The associated orbital 110fs in Fig. 4 for the neutral conditions represents an N–H σ bonding orbital with dominantly N 2p character, which is locally similar to the in-plane lone pair orbital of the deprotonated Nπ. After 10 fs dynamics, this feature is dominated by emission from orbital 1210fs which does not have the same lone pair character and thus results in lower emission intensity.
3.2 Acidic solution
The acidic histidine exhibits drastically different dynamics for Nπ and Nτ hole states, with stronger alteration of bond lengths, compared to basic or neutral environments. As the NEXAFS data and simulations in Fig. 1c show, both protonated Nπ and Nτ exhibit their π* absorption resonance in the incident energy region R12 ranging from 401.1 eV to 402.2 eV and a single N site can not be selected by the choice of the excitation energy. The emission spectra integrated over this π* region are shown in Fig. 5a. Due to overlap of the corresponding emission spectra, the experimental spectrum does not exhibit as well-separated emission features as for basic and neutral conditions. Instead, a broad inelastic emission band with a shoulder towards high emission energies at 394 eV is present. This loss in substructure severely complicates direct assignment with the site-specific occupied orbitals of resonant X-ray emission spectrum. | ||
| Fig. 5 Loss of access to occupied local valence electronic structure due to site dependent core excited state dynamics. (a) Smearing of emission lines upon excitation on the common resonance of the two protonated N sites in imidazole ring of histidine in the region of incident energies 401.1 eV to 402.2 eV R12 marked in Fig. 1c. The spectrum of the Nτ atom develops a intense emission line (blue) whereas the Nπ spectrum undergoes only slight changes (green). (b and c) Orbitals associated with the marked transitions in (a) for core holes at the Nπ (b) and Nτ (c) sites. The core excited state lone pair orbital formation is independent of the excited site but reflected differently in the spectra. | ||
The Nπ atom (deprotonated in basic and neutral environments) generates an emission spectrum with scattering-duration-dependent changes similar to the ones detected for the protonated Nτ under neutral conditions (green in Fig. 5a). Spectral features for the site undergo only slight emission energy and intensity alterations. Upon the core excitation the emission spectra of the two sites contain features which are very similar in both emission energy and intensity. Within the simulated timeframe of core excited state dynamics, the emission spectrum for the Nτ atom (blue in Fig. 5a) develops an intense emission line. The spectrum simulated after 10 fs fully resembles the spectra of the deprotonated N sites under neutral conditions in Fig. 2a.
Blum et al. found that aqueous glycine in the protonated NH3+ form undergoes rapid dynamics before X-ray emission after core-level ionization, whereas in the deprotonated NH2 such behaviour is not observed.22 As we detect similar trends in the conjugated imidazole ring of histidine, we studied the correlation between the degree of core excited state dynamics and the local charge at the N sites by calculating the charges from Mulliken population analysis in the ground state (see Table 1). For all simulated structures, the degree of core excited state dynamics can be correlated with the local charge of the investigated N atom. Core excited state dynamics has the most severe impact on the emission spectra of the protonated imidazole N atom, which have a higher local charge than the deprotonated imidazole N-atoms, in the neutral and basic species (see ESI† for simulated dynamics of the additional basic and neutral tautomers). A higher local charge in the ground state yields a reduced possibility to screen the N 1s core hole upon X-ray excitation, which induces a stronger repulsion of the nuclei in the core excited state.
| N atom | Nτ | Nπ |
|---|---|---|
| Molecular species | Mulliken charge | |
| Basic Nτ-prot./Nπ-deprot. | −0.032267 | −0.337711 |
| Basic Nπ-prot./Nτ-deprot. | −0.293876 | −0.029615 |
| Neutral Nτ-prot./Nπ-deprot. | −0.000632 | −0.299764 |
| Neutral Nπ-prot./Nτ-deprot. | −0.032901 | −0.267361 |
| Acidic | 0.044248 | 0.010467 |
The detected correlation between increased local charge and the degree of core excited state dynamics extracted from the Mulliken charge analysis also holds for the acidic species. Both, the Nπ and the Nτ atom exhibit a positive charge and the more positive Nτ atom exhibits the most drastic core excited state dynamics, analogous to the other pH values. The site-independent X-ray induced N–H dissociation of the Nτ atom under acidic conditions can be attributed to an overall bond destabilization through charge increase on the imidazole ring, which is also reflected in the positive Mulliken charges on both the Nτ and the Nπ atom. Screening of any of the N 1s holes at the imidazole N sites causes a destabilization of the weakest bond in the system.
4 Summary and conclusions
In this study the local valence electronic structure at the N atoms of aqueous histidine and its X-ray-induced transformation accessible through RIXS have been presented in dependence on the inspected chemical state of the system. The occupied valence states of the Nπ and Nτ atoms in the imidazole ring of histidine could be probed selectively by excitation at their well separated bound absorption resonances in basic and neutral solutions. Under these conditions the occupied local valence electronic structure at these N atoms was also accessible through distinct emission channels. These emission channels were only mildly affected by core excited state dynamics, yielding the possibility to assign characteristic molecular orbitals to the detected transitions and correlate them to N protonation states. In this way the detected electronic transitions highest in emission energy could be attributed to lone pair orbitals in the plane defined by the imidazole ring and to N–C bonding π orbitals for the deprotonated Nπ atoms. In contrast, orbitals with local non-bonding out-of-plane N 2p character contribute for the protonated Nτ atoms. Under acidic conditions, an excitation-site-independent X-ray induced Nτ–H dissociation makes the local occupied valence electronic structure inaccessible through N 1s emission spectra. Here, the dissociation-induced orbital transformations have a different impact on the spectra dependent on the excited N site, which consequently smears out the intensity over a wide range of emission energies at the common resonance of the two N atoms. The degree of core-excited-state dynamics was found to correlate with the local charge on the investigated N atoms through Mulliken charge analysis on the simulated molecular structures. Our findings of stronger core-excited-state dynamics through local charge increase and thus reduced possibility of N 1s core hole screening are in agreement with the data by Blum et al.22 who detect the largest dynamic effects upon core-level excitation in aqueous glycine at N sites with the most positively charged surroundings.Conclusively, our results demonstrate that the inherent temporal component of the RIXS process can be utilized to locally probe bond deformations induced by ionizing radiation by signatures in resonant N 1s resonant X-ray emission spectra. The identification of the highly directional, excitation site independent dissociation of the Nτ–H bond in histidine exclusively under the acidic conditions demonstrates that the resistance of biomolecular samples against ionizing radiation can depend largely on their physiological state and the local chemical environment.
Conflicts of interest
The authors have no conflict of interest to declare.Acknowledgements
We thank HZB for the allocation of synchrotron radiation beamtime. We thank Christian Weniger for technical support during the measurements. Data acquisition software support by Martin Beye is thankfully acknowledged. S. E., R. M. J., and A. F. acknowledge funding from the ERC-ADG-2014 – Advanced Investigator Grant No. 669531 EDAX under the Horizon 2020 EU Framework Programme for Research and Innovation.References
- E. Musk, New Space, 2017, 5, 46–61 CrossRef.
- A. L. Hansen and L. E. Kay, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E1705–E1712 CrossRef CAS PubMed.
- J. Puttick, E. N. Baker and L. T. Delbaere, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 100–105 CrossRef CAS PubMed.
- L. Hedstrom, Chem. Rev., 2002, 102, 4501–4523 CrossRef CAS PubMed.
- J. Pelton, D. Torchia, N. Meadow and S. Roseman, Protein Sci., 2008, 2, 543–558 CrossRef PubMed.
- C. N. Pace, G. R. Grimsley and J. M. Scholtz, J. Biol. Chem., 2009, 284, 13285–13289 CrossRef CAS PubMed.
- G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2009, 18, 247–251 CAS.
- S. Li and M. Hong, J. Am. Chem. Soc., 2011, 133, 1534–1544 CrossRef CAS PubMed.
- S. P. Edgcomb and K. P. Murphy, Proteins: Struct., Funct., Genet., 2002, 49, 1–6 CrossRef CAS PubMed.
- N. Shimba, Z. Serber, R. Ledwidge, S. M. Miller, C. S. Craik and V. Dötsch, Biochemistry, 2003, 42, 9227–9234 CrossRef CAS PubMed.
- G. Dodson, Trends Biochem. Sci., 1998, 23, 347–352 CrossRef CAS PubMed.
- D. N. Silverman and S. Lindskog, Acc. Chem. Res., 1988, 21, 30–36 CrossRef CAS.
- J. Stöhr, NEXAFS Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992, vol. 25 Search PubMed.
- L. J. P. Ament, M. van Veenendaal, T. P. Devereaux, J. P. Hill and J. van den Brink, Rev. Mod. Phys., 2011, 83, 705–767 CrossRef CAS.
- F. Gelmukhanov and H. Ågren, Phys. Rev. A: At., Mol., Opt. Phys., 1996, 54, 379–393 CrossRef CAS.
- J.-E. Rubensson, J. Electron Spectrosc. Relat. Phenom., 2000, 110–111, 135–151 CrossRef CAS.
- L. Weinhardt, M. Blum, O. Fuchs, A. Benkert, F. Meyer, M. Bär, J. Denlinger, W. Yang, F. Reinert and C. Heske, J. Electron Spectrosc. Relat. Phenom., 2013, 188, 111–120 CrossRef CAS.
- S. Schreck, A. Pietzsch, K. Kunnus, B. Kennedy, W. Quevedo, P. S. Miedema, P. Wernet and A. Föhlisch, Struct. Dyn., 2014, 1, 054901 CrossRef PubMed.
- M. Odelius, J. Phys. Chem. A, 2009, 113, 8176–8181 CrossRef CAS PubMed.
- L. Weinhardt, E. Ertan, M. Iannuzzi, M. Weigand, O. Fuchs, M. Bär, M. Blum, J. D. Denlinger, W. Yang, E. Umbach, M. Odelius and C. Heske, Phys. Chem. Chem. Phys., 2015, 17, 27145–27153 RSC.
- S. Schreck, A. Pietzsch, B. Kennedy, C. Såthe, P. S. Miedema, S. Techert, V. N. Strocov, T. Schmitt, F. Hennies, J.-E. Rubensson and A. Föhlisch, Sci. Rep., 2016, 7, 20054 CrossRef PubMed.
- M. Blum, M. Odelius, L. Weinhardt, S. Pookpanratana, M. Bär, Y. Zhang, O. Fuchs, W. Yang, E. Umbach and C. Heske, J. Phys. Chem. B, 2012, 116, 13757–13764 CrossRef CAS PubMed.
- R. C. Couto, V. V. Cruz, E. Ertan, S. Eckert, M. Fondell, M. Dantz, B. Kennedy, T. Schmitt, A. Pietzsch, F. F. Guimarães, H. Ågren, F. Gel'mukhanov, M. Odelius, V. Kimberg and A. Föhlisch, Nat. Commun., 2017, 8, 14165 CrossRef CAS PubMed.
- K. Kunnus, I. Rajkovic, S. Schreck, W. Quevedo, S. Eckert, M. Beye, E. Suljoti, C. Weniger, C. Kalus, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, K. J. Gaffney, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, S. Techert, P. Wernet and A. Föhlisch, Rev. Sci. Instrum., 2012, 83, 123109 CrossRef PubMed.
- T. Kachel, J. Large-Scale Res. Facil., 2016, 2, A72 CrossRef.
- J. Nordgren, G. Bray, S. Cramm, R. Nyholm, J.-E. Rubensson and N. Wassdahl, Rev. Sci. Instrum., 1989, 60, 1690 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- J. Niskanen, K. Kooser, J. Koskelo, T. Käämbre, K. Kunnus, A. Pietzsch, W. Quevedo, M. Hakala, A. Föhlisch, S. Huotari and E. Kukk, Phys. Chem. Chem. Phys., 2016, 18, 26026–26032 RSC.
- J. T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- J. V. de Vondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 1703–1710 CrossRef CAS.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. V. de Vondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- Y. Zubavichus, A. Shaporenko, M. Grunze and M. Zharnikov, J. Phys. Chem. A, 2005, 109, 6998–7000 CrossRef CAS PubMed.
- M. J. Thomason, C. R. Seabourne, B. M. Sattelle, G. A. Hembury, J. S. Stevens, A. J. Scott, E. F. Aziz and S. L. M. Schroeder, Faraday Discuss., 2015, 179, 269–289 RSC.
- S. Eckert, J. Norell, P. S. Miedema, M. Beye, M. Fondell, W. Quevedo, B. Kennedy, M. Hantschmann, A. Pietzsch, B. E. Van Kuiken, M. Ross, M. P. Minitti, S. P. Moeller, W. F. Schlotter, M. Khalil, M. Odelius and A. Föhlisch, Angew. Chem., Int. Ed., 2017, 56, 6088–6092 CrossRef CAS PubMed.
- S. Eckert, P. Miedema, W. Quevedo, B. O'Cinneide, M. Fondell, M. Beye, A. Pietzsch, M. Ross, M. Khalil and A. Föhlisch, Chem. Phys. Lett., 2016, 647, 103–106 CrossRef CAS.
- F. Meyer, M. Blum, A. Benkert, D. Hauschild, Y. L. Jeyachandran, R. G. Wilks, W. Yang, M. Bär, C. Heske, F. Reinert, M. Zharnikov and L. Weinhardt, J. Phys. Chem. B, 2017, 121, 6549–6556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp05713j |
| This journal is © the Owner Societies 2017 |