
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Roles of conformational disorder and downhill folding in modulating protein–DNA recognition†
Xiakun
Chu
a and
Victor
Muñoz
 *abc
*abc
aIMDEA Nanosciences, Faraday 9, Campus de Cantoblanco, Madrid, 28049, Spain
bNational Biotechnology Center, Consejo Superior de Investigaciones Científicas, Darwin 3, Campus de Cantoblanco, Madrid, 28049, Spain
cDepartment of Bioengineering, School of Engineering, University of California, 95343 Merced, CA, USA. E-mail: vmunoz@cnb.csic.es
First published on 18th October 2017
Abstract
Transcription factors are thought to efficiently search for their target DNA site via a combination of conventional 3D diffusion and 1D diffusion along the DNA molecule mediated by non-specific electrostatic interactions. This process requires the DNA-binding protein to quickly exchange between a search competent and a target recognition mode, but little is known as to how these two binding modes are encoded in the conformational properties of the protein. Here, we investigate this issue on the engrailed homeodomain (EngHD), a DNA-binding domain that folds ultrafast and exhibits a complex conformational behavior consistent with the downhill folding scenario. We explore the interplay between folding and DNA recognition using a coarse-grained computational model that allows us to manipulate the folding properties of the protein and monitor its non-specific and specific binding to DNA. We find that conformational disorder increases the search efficiency of EngHD by promoting a fast gliding search mode in addition to sliding. When gliding, EngHD remains loosely bound to DNA moving linearly along its length. A partially disordered EngHD also binds more dynamically to the target site, reducing the half-life of the specific complex via a spring-loaded mechanism. These findings apply to all conditions leading to partial disorder. However, we also find that at physiologically relevant temperatures EngHD is well folded and can only obtain the conformational flexibility required to accelerate 1D diffusion when it folds/unfolds within the downhill scenario (crossing a marginal free energy barrier). In addition, the conformational flexibility of native downhill EngHD enables its fast reconfiguration to lock into the specific binding site upon arrival, thereby affording finer control of the on- and off-rates of the specific complex. Our results provide key mechanistic insights into how DNA-binding domains optimize specific DNA recognition through the control of their conformational dynamics and folding mechanism.
Introduction
Specific DNA recognition by regulatory proteins is fundamental to gene expression. These DNA-binding proteins must efficiently recognize their specific target sites among the millions of alternative non-specific sites present in genomic DNA. An intriguing implication is that the rate by which these proteins bind to their DNA target greatly exceeds the theoretical limit imposed by the occurrence of random collisions between the protein and the DNA specific site.1,2 To solve this paradox, a “facilitated diffusion” mechanism for DNA binding has been proposed.3,4 Such a mechanism involves standard three-dimensional (3D) diffusion combined with non-specific DNA binding5 followed by one-dimensional (1D) diffusion along the DNA molecule.4,6 The reduced dimensionality of facilitated diffusion is thought to greatly enhance the search and thus increase the rate. The phenomenon of 1D diffusion of DNA-binding proteins on DNA has been observed using single-molecule experiments,7–11 and analyzed by coarse-grained molecular simulations.12–18 The theoretical framework describing facilitated diffusion on protein–DNA interactions is also well established.6,19–21During 1D diffusion, the protein remains in contact with DNA by virtue of non-specific binding promoted by electrostatic interactions.22,23 This raises the second paradox of how to simultaneously maximize speed and stability.3,24–28 Non-specific binding should be processive to guarantee an efficient 1D search. However, the stronger the binding the slower the diffusion coefficient because the protein needs to break strong interactions to move forward.3,24–28 DNA-binding domains typically carry a net positive charge and thus bind to the polyanionic DNA molecule in a sequence independent manner.29
As a simple solution to this problem, the DNA-binding domain could just switch between two modes: a “search” mode in which the protein binds to any DNA and undergoes 1D-diffusion and a “recognition” mode in which the protein locks into the specific target site once encountered. This two-mode binding mechanism normally involves separate search and recognition protein domains, as it occurs for zinc finger based transcription factors.16,30–33 According to previous computational studies, the search domain facilitates 1D diffusion by smoothing the energy landscape of the DNA–protein interactions, but engaging the recognition mode involves crossing a kinetic barrier that necessarily lowers the rate of locking into the specific site thus increasing the chance to miss the target.34,35 Therefore, the optimization of DNA recognition requires that the conformational dynamics of the protein are coordinated with the specific binding event. It is, however, unclear how such a dual-mode binding mechanism can be implemented on many DNA-binding proteins that have just one structural domain rather than two. In that respect, it is interesting to note that DNA-binding domains exhibit partial structural disorder under native conditions36–40 and often fold with ultrafast kinetics that are characteristic of the downhill folding scenario.41–44
One interesting possibility is that the conformational pliability of downhill folding enables the implementation of search and recognition modes in a single domain together with nimble switching between them.45,46 In the downhill folding scenario, the free energy barrier to folding–unfolding is very small (<3kT),47 which results in ultrafast dynamics and minimally cooperative unfolding.42,44,48,49 It has been in fact proposed that downhill folders can operate as molecular rheostats, dynamically adjusting their characteristically broad conformational ensembles in response to cues.50 The molecular rheostat concept has been effectively exploited to develop ultra-high performance biosensors,51 but its potential role in controlling biological processes remains unknown.
To test this hypothesis, we focus on the binding to DNA of EngHD, a three-helix bundle DNA-binding domain from the Drosophila melanogaster transcription factor engrailed. The folding properties of EngHD have been thoroughly characterized in experiments and simulations.52–56 These studies highlight that EngHD folds/unfolds very rapidly, approaching the folding speed limit.42,44,57 EngHD readily changes its conformational properties in response to environmental changes or interactions,55,58,59 and is partially disordered at physiological temperatures.49 Quantitative analysis of thermodynamic and kinetic data, including differential scanning calorimetry, as well as long-timescale atomistic simulations, indicate that EngHD does indeed fold under the downhill scenario.49,60,61 The structural bases for the binding to DNA of homeodomains have also been thoroughly investigated using X-ray crystallography62 and NMR.63 In addition, NMR paramagnetic relaxation enhancement techniques have shown that homeodomains interact with DNA through the same binding interface whether they are bound specifically or non-specifically,64–66 which points to any differences between search and recognition modes being of dynamical rather than structural origin.
We investigate the coupling between folding and DNA binding using a coarse-grained structure-based model (CGSBM) that gives us the opportunity to manipulate the folding mechanism and stability of EngHD as well as its binding to DNA in the specific and non-specific modes. Our analysis reveals a concerted interplay between the conformational flexibility and folding mechanism of EngHD and the search and recognition binding modes. We find that intrinsic disorder and downhill folding increase the DNA searching capabilities of EngHD via kinetic and thermodynamic effects. These results strongly support the idea that the highly dynamic conformational ensembles of DNA-binding domains play a key role in the DNA recognition process. Moreover, our work provides a simple theoretical framework for the design and optimization of protein–DNA recognition.
Computational procedures and methods
To investigate the folding and DNA binding properties of EngHD, we use a structure-based model (SBM)67–70 or native-centric Go model in which the molecular complexity of both protein and DNA is coarse grained.Protein model
The EngHD model represents each amino acid with two beads (except for glycine): one representing the backbone and positioned at the Cα and another one representing the side-chain and positioned at its center of mass. The Hamiltonian for EngHD is expressed as:| VEngHDSBM = Vbond + VDihedral + VNative + VNon-native |
To fine tune the conformational disorder and stability of EngHD, we modified the strength of native contacts by changing the pre-factor εf of the LJ term in VNative. A small εf leads to large conformational disorder and low folding stability, and vice versa. We thus generated a series of EngHD models with different native stabilities by varying εf. These models cover the entire range from completely unfolded at all relevant temperatures (IDP-like chain) to the folding midpoint (i.e. equal populations of native and unfolded states) and to a stable folded state. The middle condition produced a folding free energy barrier of ∼1.3kT separating unfolded and native ensembles, which is fully consistent with the experimental estimates of the folding barrier of EngHD.49,61,74
To control the folding mechanism of EngHD (i.e. barrier height at the denaturation midpoint), we altered the energetic balance between local (close in sequence) and non-local (far away in sequence) interactions.75,76 On the mostly helical EngHD,77 this was simply achieved by changing the relative strength (R) of each individual contribution to VDihedral relative to the strength of each contribution to VNative, with the latter being the term that includes tertiary contacts. Changes in R from 0.1 to 3.0, that is, increasing εϕ (pre-factor for VDihedral) relative to εf (pre-factor for VNative), R = εϕ/εf, generate folding scenarios for EngHD ranging from apparent 2-state (i.e. barrier of ∼4.5kT at the denaturation midpoint) to global downhill (i.e. barrier of only ∼0.3kT). For clarity, we introduce the parameter downhillness that corresponds to R normalized according to the expression: downhillness = (R − 0.1)/(3 − 0.1). Thus, downhillness ranges from 0 (two-state) to 1 (one-state). Increasing R lowers the free energy barrier and also increases the degree of residual helical structure in the unfolded ensemble (Fig. S1, ESI†). The implication is that under downhill folding conditions, the α-helices are well formed in the unfolded ensemble resulting in a folding mechanism similar to the diffusion–collision model.78 Under two-state-like conditions, the folding of EngHD is close to the nucleation–condensation mechanism.79 As indicated above, the default parameter for the SBM results in a folding barrier of ∼1.3kT at the denaturation midpoint, low folding cooperativity and an unfolded state in which the helices are mostly formed.
DNA model
In our CGSBM, each nucleotide of the DNA molecule is represented by three beads. One bead represents the phosphate group (negatively charged), another bead the sugar and the third one the nitrogenous base. The double stranded DNA structure was kept rigid during the simulations.Simulations
The overall potential used for the simulations has the form:| V = VEngHDSBM + VEngHD–DNASBM + VEngHD–DNAEle |
Simulations were performed on a straight rigid 100 bp-long DNA molecule placed within a 20 × 20 × 40 nm3 simulation box aligned along the Z-axis. This DNA molecule includes one extended specific-binding site (10 bp long, same as in the crystal structure62) located in the center of the DNA molecule. EngHD is able to bind non-specifically to any potential binding site within the 100 bp DNA duplex through electrostatic interactions. The specific binding site includes additional stabilization energy from the contact interactions observed in the X-ray structure of the complex. Langevin dynamics simulations were performed using the GROMACS software with reduced units applied.80 We used a salt concentration of 0.01 M (low ionic strength) to maximize the probability of EngHD moving in the vicinity of DNA. For each independent set of parameters defining the EngHD folding scenario (varying εf or R), a set of 60 independent simulations of 1 × 105 reduced time units were performed to monitor the DNA binding properties. For the standard parameters (εf = 1.0 and R = 1), the folding temperature of EngHD is found at kT = 1.40. More details can be found in the ESI.†
Results and discussion
Our CGSBM includes a description of the conformational ensemble of the protein, non-specific protein binding to any segment of the dsDNA and specific binding to the target site located in the center of the DNA molecule. The folding behavior of EngHD and the specific binding to DNA are modeled by a standard SBM, which only takes into account interactions observed in the native crystal structures.62,77 Non-specific interactions with the DNA are modeled as pure electrostatics using a simple Debye–Hückel model. In a first step, we investigated the folding behavior of EngHD without DNA. The standard parameters for the SBM protein model rendered a marginal folding barrier of ∼1.3kT at the folding temperature (Fig. S2 and S3, ESI†). Therefore, according to these simulations EngHD folds in the downhill regime, consistent with the conclusions derived from the analysis of multiple experimental data.49,61,74 With these parameters, EngHD maintains a large degree of helical structure in the unfolded ensemble (Fig. S1, ESI†), again consistent with the expectation for a downhill folder.74 The helical content in the unfolded ensemble is almost as much as in the native state, indicating that the folding process can be roughly described as docking of the three pre-formed helices to form the bundle. This description is closely similar to the diffusion–collision mechanism.78 Overall, our results are consistent with previous experiments and also with atomistic simulations,54–56,81,82 supporting the significance of our CGSBM.67 To determine the coupling between the folding of EngHD and the binding to DNA, we performed molecular binding simulations at the folding temperature of EngHD starting from different non-associated states (see Computational procedures and methods and the ESI† for details).From these simulations, we could dissect the molecular details of the DNA recognition process and its coupling to the conformational dynamics of EngHD. We found that non-specific DNA binding takes place using a hybrid mechanism consisting of three- and one-dimensional (3D and 1D) diffusion modes. We could also observe the binding to the specific site, which can be divided into two steps. The first one involves the formation of the transition complex (TC), which occurs when EngHD reaches the specific binding site but has not formed the specific binding interactions yet. The second step (specific binding, or SB) involves EngHD locking into the specific binding site by forming all the interactions involved in the specific EngHD–DNA complex. We considered the protein performing pure 3D diffusion when it is >3 nm away from the DNA molecule to guarantee the absence of interactions between the two molecules. In contrast, we define 1D diffusion along the DNA (sliding) as a process by which the protein remains in constant contact with DNA, a definition that is similar to the facilitated diffusion mode used by other authors.4 We find that 1D diffusion significantly reduces the dimensionality of the search and thus accelerates the process, as expected.25 A close inspection of the motions undergone by the protein while performing 1D diffusion reveals two sub-categories of 1D diffusion. In the first one, the recognition α-helix of EngHD remains inserted into the major groove, resulting in a spiraling displacement along the DNA length (i.e. rotation around and translation along the Z-axis of DNA) (Fig. S4A and C, ESI†). We term this type of 1D diffusion a sliding search mode. In the second type of 1D diffusion, EngHD is not interacting tightly with the DNA and the displacement along the DNA long axis does not occur coupled to rotation (Fig. S4B and D, ESI†). During this type of motion, the protein remains more loosely associated with DNA but the displacement is still unidirectional along its length. Accordingly, we term this type of motion “gliding”. We also observed hopping, defined as events in which the protein becomes completely, but transiently, detached from DNA followed by rebinding to a nearby region in the DNA. Hopping events were observed rarely in our simulations, probably due to the low salt concentration used to increase non-specific binding. Likewise, we did not see many jumping events in which the protein dissociates from DNA, undergoes 3D diffusion and rebinds at a distant position in the DNA. We therefore combined hopping and jumping events together into the 3D diffusion mode. It is worth noting that our gliding mode is in some ways similar to the 2D hopping mode described in previous work by Levy and coworkers.12,14,15,18,39 However, in the limit of strong non-specific binding to DNA (low ionic strength) that we explore here, the protein moves along the DNA without detaching, and thus the term 1D gliding represents this search mode more accurately than the original definition of 2D hopping.4,6
From the folding free energy landscapes, we can see that EngHD has different conformational distributions for the various binding scenarios (Fig. 1C). When EngHD is sliding and/or gliding, and thus associated (even if somewhat loosely) with the DNA, its conformational distribution deviates from that of the free state. The gliding mode favors EngHD conformations that are either fully or partially unfolded, resulting in a net destabilization and also in the lowering of the folding free energy barrier. In contrast, the sliding mode favors the folded conformation, which implies that this mode requires EngHD to be fully folded. At the TC, which defines the transition from non-specific to specific binding, the EngHD conformational ensemble is similar to that of the protein performing sliding but with lower bias towards the native state, indicating that at the TC the protein is more weakly associated with DNA than when bound non-specifically to other DNA regions. Finally, during specific binding (SB), the folded state becomes highly stabilized by the strong specific interactions formed with the DNA target site and thus EngHD is locked into its native state. However, we should emphasize that these binding modes are highly dynamic and in constant exchange, as observed in individual trajectories (Fig. 1A and B). In other words, at the folding temperature (i.e. when is half unfolded), EngHD binds to DNA in a highly dynamical fashion in which binding modes and EngHD conformations are coupled and in constant exchange. Such dynamic folding–binding behavior may have interesting implications for the kinetic efficiency of protein–DNA recognition.37,83
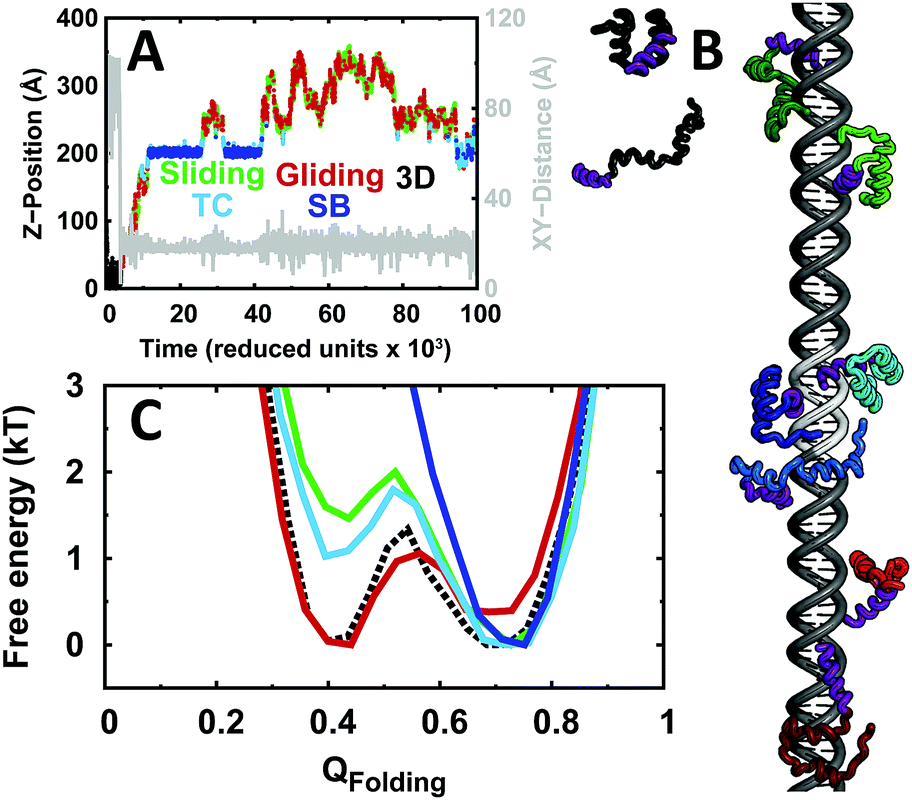 | ||
| Fig. 1 DNA recognition trajectory and folding free energy landscape of EngHD in the different binding stages. (A) Trajectory for EngHD–DNA recognition. XY-Distance is the distance of the EngHD centroid to the DNA main axis and Z-position its position along the long DNA axis. One specific binding site is placed at the center of the DNA molecule, corresponding to Z-positions between 185 Å and 215 Å. The search process is composed of segments in which the protein performs 3D diffusion, 1D sliding (bound non-specifically to the major groove of the DNA) and gliding (moving along the DNA axis while loosely associated with DNA) modes. The specific binding process is divided into two steps: the formation of the transition complex (TC) and locking into the specific binding site (SB). Trajectory segments corresponding to different binding modes are color-coded: 3D (black), gliding (red), sliding (green), TC (cyan) and SB (dark blue). (B) Snapshots of the EngHD interaction with DNA at different stages with EngHD color-coded as in A. Examples of folded (lighter) and unfolded (darker) conformations are provided. The DNA-interacting helix of EngHD is highlighted in purple and the specific binding site on DNA is shown in light gray. (C) Folding free energy landscape of EngHD along QFolding (i.e. fraction of folding native contacts) for the different binding modes. The data corresponding to 3D diffusion are shown with dashed lines since they were obtained from simulations of EngHD alone due to very low occurrence of 3D diffusion in the presence of DNA. | ||
To examine the interactions formed between EngHD and the DNA during non-specific binding, we calculated the minimum distance between each EngHD residue and the closest DNA atoms (Disti, where i is the index of the residue in EngHD). This analysis shows that during sliding and gliding, folded EngHD interacts with DNA via the canonical binding interface observed in the crystal structure (Fig. S5A, ESI†). This is consistent with previous theoretical investigations and experiments performed on other homeodomains.12,64–66 To evaluate the effects of non-specific binding on the EngHD conformational ensemble, we calculated Disti as a function of the folding order parameter QFolding for the sliding and gliding modes (Fig. 2A). In the gliding mode, unfolded EngHD manages to get its helix II, and especially residues R29, R30 and R31, significantly closer to DNA than folded EngHD (Fig. 2A and Fig. S5B, ESI†). This is so because in the EngHD native structure helix II is at the farthest end of the DNA specific binding interface, but once EngHD is unfolded this region can readily get into contact with DNA by making additional non-specific electrostatic interactions (Table S1, ESI†). These extra electrostatic interactions with DNA favor EngHD to be structurally disordered during gliding, thus biasing the folding free energy landscape slightly towards unfolded conformations (red in Fig. 1C). That is, the gliding mode favors the structural disorder of EngHD because unfolded conformations can make more non-specific interactions with DNA. In the sliding mode, Disti, electrostatic energy, and the number of protein–DNA salt bridges are independent of the EngHD conformation (Table S1, ESI†), indicating that there are not energetic biases for specific EngHD conformations in this binding mode. Therefore, the strong stabilization of the native state observed during sliding (green line in Fig. 1C) must come from entropic contributions. This entropic effect appears to arise from geometrical constraints since helix III of EngHD must remain inserted into the major groove of DNA during sliding, which impedes the unfolding of the protein without dissociation. The effect is in fact reminiscent of the stabilization of proteins in highly confined spaces.84 The structural preferences for the different binding modes are likely to have significant kinetic implications for DNA recognition. For instance, when the protein arrives at the specific site through gliding, it may be unfolded and thus it would need to refold at the TC before it is able to lock into the target (SB). For a sliding EngHD, the transition from TC to SB should not require conformational readjustments.
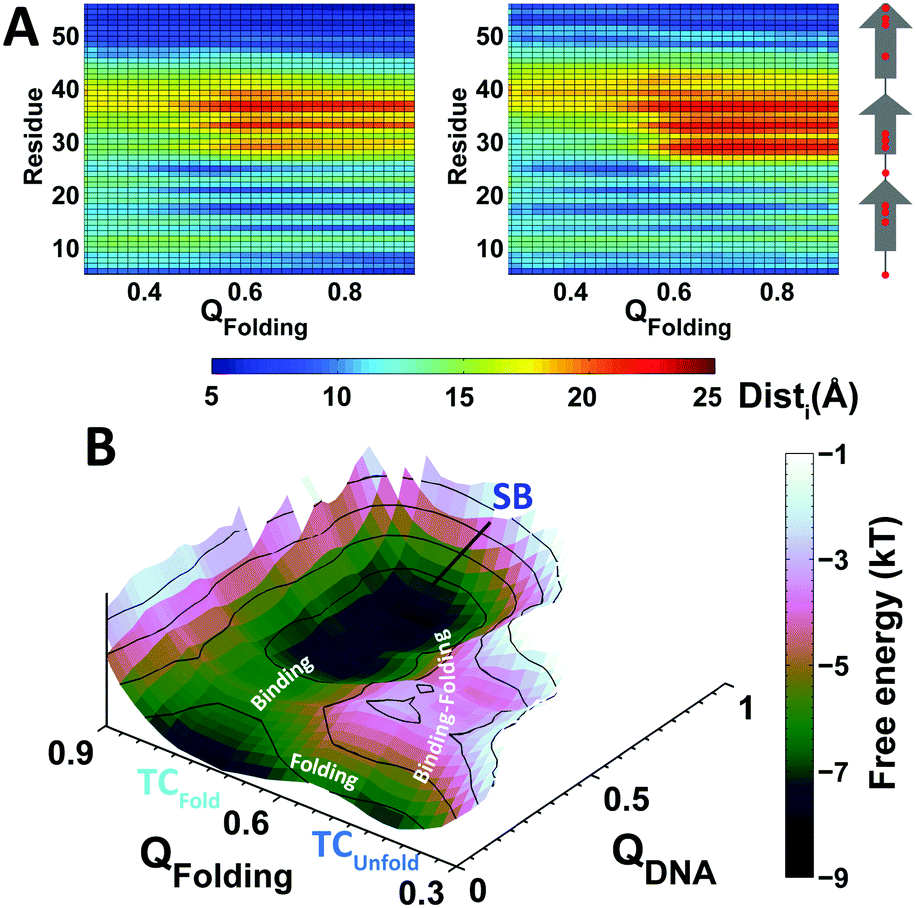 | ||
| Fig. 2 Structural analysis and free energy landscape of the interactions between DNA and EngHD as a function of QFolding. (A) Non-specific binding: mean minimum distance from each EngHD residue to the closest DNA atom during sliding (left) and gliding (right) as a function of QFolding. Red dots in the secondary structure assignment (rightmost) represent positively charged residues. (B) Specific binding: 2D free energy landscape of specific binding showing the TC and SB stages. QFolding and QDNA are the fraction of native contacts for EngHD folding and for specific DNA binding, respectively. | ||
To investigate the mechanistic implications that partial disorder on EngHD may have on DNA recognition, we extracted all of the transitions observed between the TC and SB from the trajectories and computed a free energy landscape for specific binding (Fig. 2B). The landscape highlights two possible pathways to go from TC to SB. The first pathway is a sequential process in which unfolded EngHD reaches the TC, folds up, and then locks into SB. This pathway corresponds to a conformational selection scenario in which the specific interactions select the folded structure from the broad conformational ensemble that EngHD populates while is at the TC.85,86 In the other pathway, EngHD folds and binds specifically in a concerted fashion resulting in an induced-fit binding scenario.87 In our simulations, the conformational selection pathway occurs with much higher probability than induced-fit. However, the coexistence of the two pathways is a manifestation of mechanistic complexity in line with what has been proposed for processes that involve binding coupled to the folding of a downhill folding protein.45,46,88
This is an interesting observation since EngHD is indeed a very fast folding protein classified as a downhill folder,60,61,74,89 and it is also conformationally flexible at its physiological temperature.49 To further investigate these possible effects, we performed binding simulations at varying degrees of unfolding, but without changing the folding scenario. This was achieved by simply tuning the strength of the native contacts of EngHD in our CGSBM (i.e. εf). εf controls the stability of the native state resulting in increasingly disordered conformational ensembles the smaller its value (Fig. S3, ESI†). However, tuning εf does not affect the magnitude of the free energy barrier at the folding temperature. We thus performed all DNA binding simulations at a common temperature (i.e. the folding temperature for εf = 1.0) to focus exclusively on the effects of structural disorder.
This analysis reveals that conformational disorder increases the probability of gliding at the expense of sliding (Fig. 3A). Under the strong non-specific binding conditions of our simulations (low ionic strength), 3D diffusion remains a minor component of the search motions regardless of the level of conformational disorder. Conformational disorder favors gliding because an unstructured EngHD exposes a larger effective electrostatic interaction surface. Moreover, the enhanced conformational dynamics inherent to a more disordered ensemble facilitates the transient binding–release events that also favor gliding over sliding motions. The effect of disorder on specific binding is the decrease of the relative population of SB and the increase of that of the TC (Fig. 3B). The latter reflects the extra penalty in binding free energy that must be paid to fold up the protein simultaneously with binding at small values of εf (Fig. 2C). The destabilization of SB vanishes as soon as εf is higher than 1 (even though EngHD may still be partly disordered). This is so because the slightly unfolded ensemble that EngHD populates under conditions of marginal stability and minimal folding barrier (i.e. 1.3kT) is able to bind specifically as much as the fully folded state (Fig. S3 and S6, ESI†).
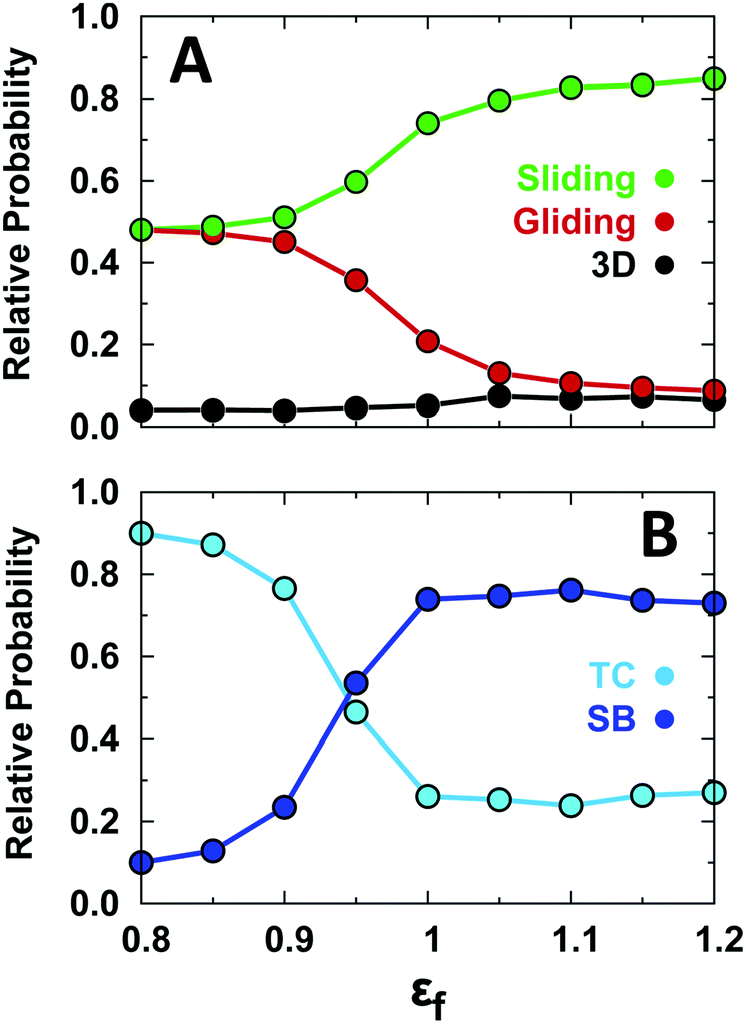 | ||
| Fig. 3 Relative probability of different binding modes as a function of the degree of conformational disorder. (A) Relative probabilities for the various non-specific binding modes: 3D diffusion, gliding, and sliding. (B) Relative probabilities for being at the TC or SB stage during specific binding. εf is the strength of the EngHD native contacts. | ||
We then evaluated the DNA search speed of EngHD by obtaining the 1D diffusion coefficient (D1) from the mean squared displacement (MSD) of the protein along the main DNA length (Z-axis).9,12,83,90,91 In our simulation, gliding is frequent but short-lived, and it quickly alternates with sliding and 3D diffusion modes. The short gliding half-life makes it impractical to calculate D1 for pure gliding with sufficient accuracy. Instead, we calculate a composite D1 that integrates sliding and gliding onto a global 1D diffusion mode. This integrated 1D mode becomes significantly faster (2.5 fold) as the degree of disorder on EngHD increases (Fig. 4A). The increase in diffusion coefficient mostly comes from gliding because at εf < 1 sliding becomes very short lived. That is, the more disordered the EngHD the faster it diffuses along DNA via gliding. The reason for this acceleration is that the gliding mode is still one-dimensional but the inherent flexibility of EngHD results in weaker binding to DNA and thus in faster motion.
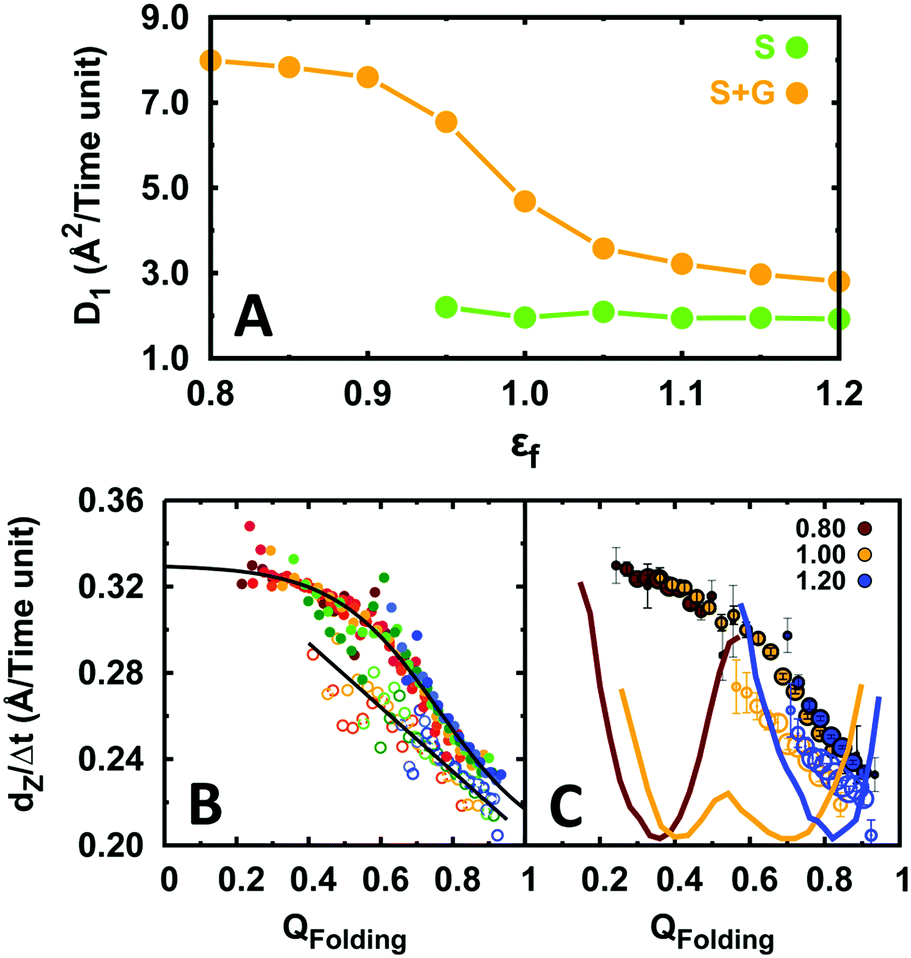 | ||
| Fig. 4 The effect of conformational disorder on the diffusion of EngHD along DNA. (A) 1D diffusion coefficient (in Angström2 per reduced time unit) as a function of conformational disorder. S corresponds to only sliding and S + G to integrated sliding and gliding. Sliding becomes very transient for εf < 0.95, impeding further determination of its diffusion coefficient. (B) Displacement along DNA for different conformational sub-ensembles of EngHD as a function of εf. The color code is from red to blue for εf increasing from 0.8 to 1.2. Examples of sliding are shown as empty circles and fitted to a straight line; examples of 1D integrated diffusion (gliding plus sliding) are shown as solid circles and fitted to a sigmoidal function. (C) Displacement along DNA for different conformational sub-ensembles of EngHD at three values of εf and with the size of the circles representing the population of the conformer. The corresponding folding free energy landscapes are also shown. | ||
Our results show that overall 1D diffusion speeds up as EngHD increases its structural disorder (i.e. always within the marginal folding barrier regime). To analyze the molecular basis of this observation, we introduce a quantity, which we term displacement (dZ) and that corresponds to the distance traveled by EngHD along the Z-axis between two consecutive frames separated by time interval Δt. This quantity is indicative of the 1D diffusive speed (dZ/Δt) and can be determined for individual conformations within the EngHD ensemble. Fig. 4B shows such data as a function of the folding order parameter (QFolding). These data show that the 1D diffusive speed does indeed increase as EngHD populates more open or unstructured conformations (lower QFolding). Interestingly, the speed up happens both for the integrated 1D mode and for pure sliding. Therefore, the presence of disorder on EngHD accelerates the 1D DNA search process. In Fig. 4C, we plot the same type of data but including the population of each of the conformational sub-ensembles. This graph highlights how the net acceleration of 1D diffusion is proportional to the population weighted degree of conformational disorder present in the EngHD ensemble. Sliding is faster for more disordered conformations but only occurs when EngHD is sufficiently folded (QFolding > 0.7), and thus decreasing εf has a marginal effect on the sliding speed. However, during gliding, EngHD can unfold completely without detaching from DNA, and thus at low εf, gliding is highly accelerated by disorder and eventually becomes the predominant 1D mode.
Another issue with functional significance is the kinetics of specific binding to the target site (SB). When the search occurs via 1D diffusion, the kinetic mechanism to form SB can be described by 4 basic rates (Fig. 5A): the rate of formation of the TC from adjacent non-specific binding sites (kS); the rate of locking into the SB (klock); the rate of escape from SB onto TC (k*); and the rate of escape from the TC to adjacent non-specific binding sites (kescape).
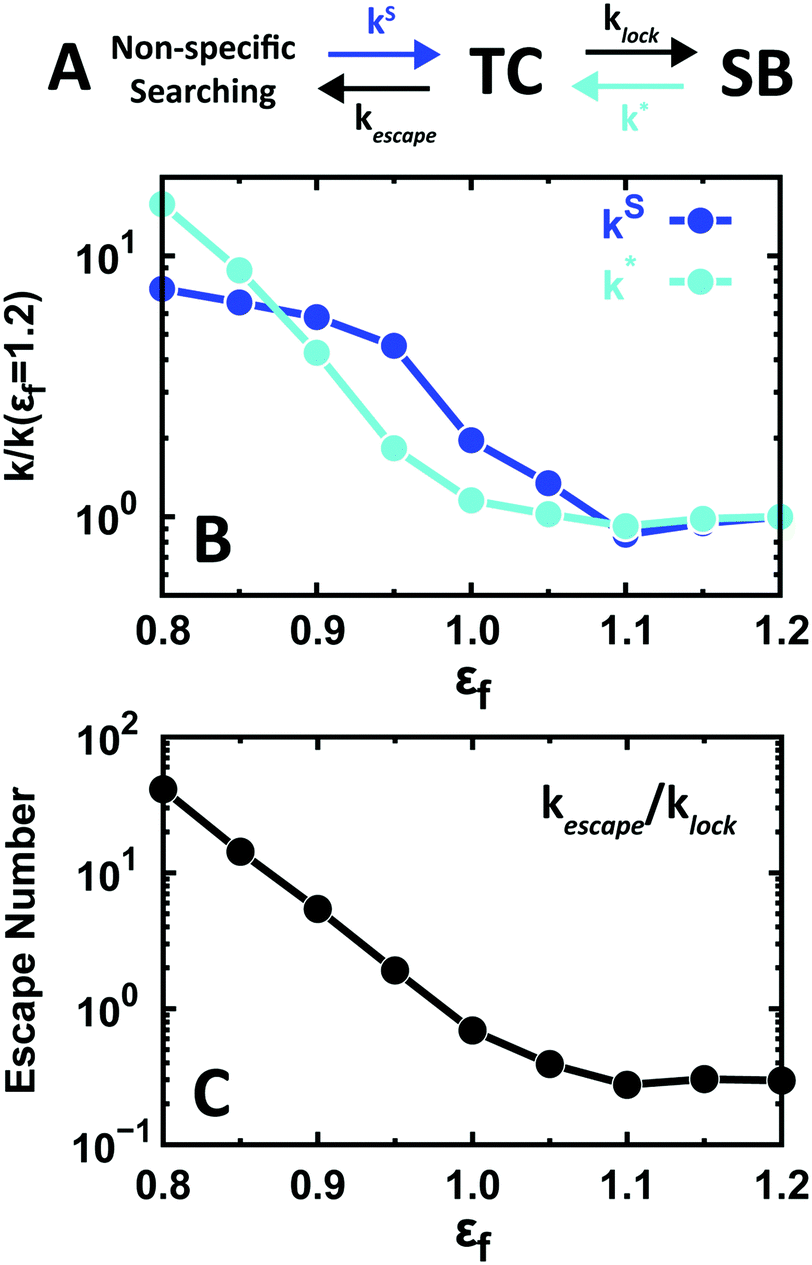 | ||
| Fig. 5 Microscopic kinetics of binding to the specific site as a function of conformational disorder. (A) Kinetic scheme of the different steps involved in specific binding. (B) Relative effects of conformational disorder on the rate of arrival at the TC (kS) and the off-rate from SB to TC (k*). (C) Escape number of EngHD (ratio between the specific binding rate (klock) and the rate of escape from the TC (kescape)). | ||
The effects of conformational disorder on these rates are significant. For instance, Fig. 5B highlights that conformational disorder increases the two rates that lead to the TC (by factors of 7 and 15), thus resulting in an increase of the TC population. Therefore, the presence of conformational disorder facilitates the arrival of the protein to the specific-binding basin of attraction (TC–SB), but it also decreases the dwell time on the specific binding site (1/k*). The latter corresponds to the time EngHD remains functionally active.
Another noteworthy effect that conformational disorder has on the overall kinetics is the increase of the propensity to be released from the specific binding basin of attraction (TC + SB). To quantify this effect, we use the ratio between kescape and klock, which we term the escape number. The escape number increases drastically (up to 150-fold) as a function of the population of unstructured conformations in the EngHD ensemble (i.e. εf < 1.1) (Fig. 5C). The increase in escape number is caused by the large cost of conformational entropy associated with specific binding when EngHD is partially unfolded and needs to fold up to lock into SB (Fig. 2B). This entropic penalty reduces the overall time EngHD spends within the TC–SB basin of attraction and thus decreases the specific binding affinity. At a glance, such an effect may seem to be functionally detrimental, but it has been previously pointed out that nimble control of gene expression requires that transcription factors bind to the target site very dynamically, and thus with fast on- and off-rates.92 From that viewpoint, our results indicate that the presence of conformational disorder may be functionally advantageous by a combination of: (1) implementation of a faster search by 1D diffusion and (2) facilitation of fast release from the specific binding site.
So far, we have investigated the effects of conformational disorder while EngHD was maintained in the downhill regime (folding barrier of 1.3kT at Tf). To investigate the effects of the folding scenario on DNA recognition, we changed the relative strength of local and non-local interactions (R is their ratio) in EngHD. A range of R between 0.1 and 3 varies the free energy barrier at the folding temperature of EngHD from ∼4.5kT to ∼0.3kT (Fig. S7, ESI,† Computational procedures and methods section), thus allowing us to explore the entire transition from nearly two-state to one-state downhill folding45,46,89 (or downhillness from 0 to 1).
We first performed binding simulations for the indicated range of EngHD folding scenarios, each one at its folding temperature (i.e. εf = 1). The analysis of these simulations showed relatively small changes in both 1D diffusion dynamics (sliding and gliding) and specific binding (Fig. S8, ESI†). Therefore, as a first approximation, DNA recognition is mostly insensitive to the folding mechanism of the DNA-binding domain once its thermodynamic bias results in partial disorder. It is apparent in Fig. 6A that the dynamics for sliding and for integrated 1D diffusion (sliding and gliding) are essentially unaffected by changes in the folding mechanism that maintain an intermediate degree of disorder on EngHD. The relative contributions of gliding and sliding to 1D diffusion change only very slightly. The same can be said for the kinetics of specific binding. The search speed (dZ/Δt) as a function of QFolding is similarly unaffected (Fig. 6B). However, the analysis of the motions for individual conformations of EngHD (Fig. 6C) reveals that the unresponsiveness of 1D diffusion to the folding mechanism comes from compensatory effects. Both gliding and sliding speeds increase as the protein becomes more unstructured. In the presence of a folding barrier, the conformational distribution is split into equally populated folded and unfolded ensembles, which experience slow and fast 1D diffusion, respectively (blue in Fig. 6C). On the other hand, a barrierless folding landscape results in 100% population of partially folded conformations, but these conformations also happen to diffuse at intermediate speeds (red in Fig. 6C). Therefore, the net balance remains essentially unaltered.
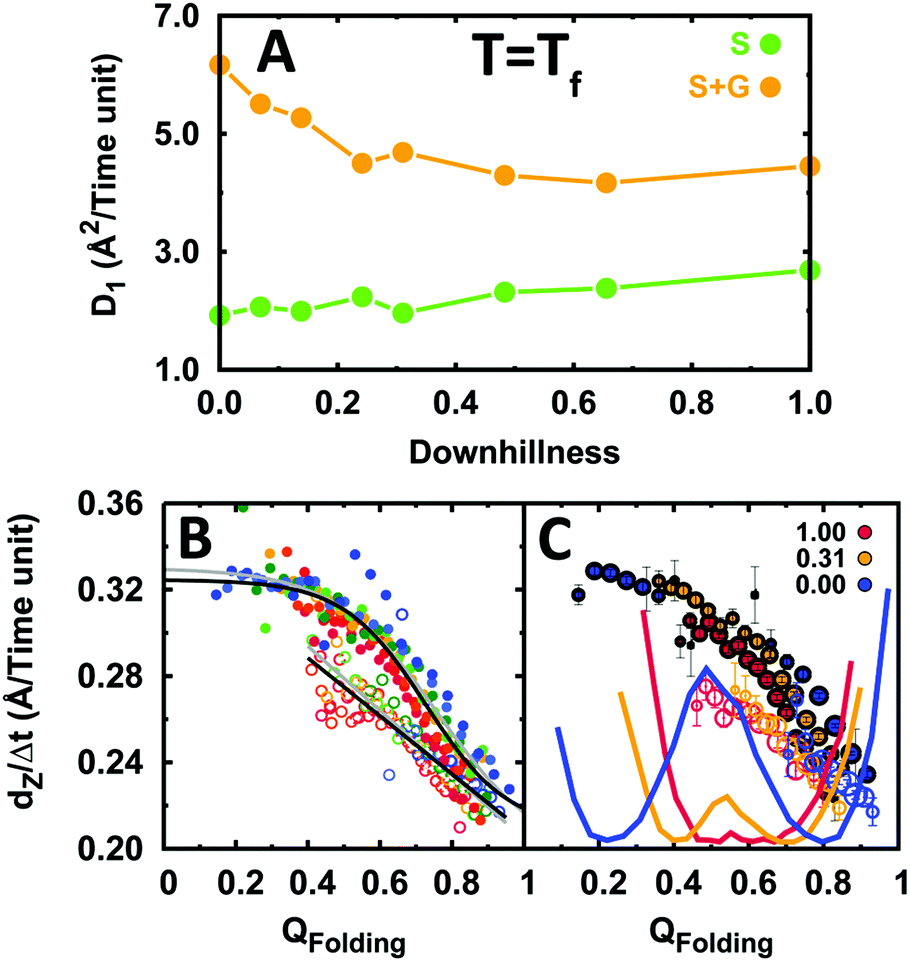 | ||
| Fig. 6 The effects of folding scenario on the diffusion of EngHD along DNA. (A) 1D diffusion coefficient as a function of folding downhillness for sliding and the integrated sliding and gliding modes. (B) Displacement along DNA for different conformational sub-ensembles of EngHD as a function of downhillness at the folding temperature. The color code is from red to blue for downhillness decreasing from 1 to 0. The black lines are fits to the data. Grey lines are fits to the data in Fig. 4 for comparison. (C) Displacement along DNA for different conformational sub-ensembles of EngHD at three downhillness levels with size of the circles representing the population of the conformer. The corresponding folding free energy landscapes are also shown. | ||
Our analysis indicates that the combination of significant structural disorder and a marginal folding barrier on the DNA binding domain produces a very dynamic DNA recognition process with nimble 1D diffusion towards the target site and fast release from it. From a general standpoint, these results shed light onto how the interplay between conformational disorder and folding mechanism of the DNA-binding domain optimizes the search for and release from the target DNA site. This conclusion has mechanistic implications for DNA recognition and gives practical clues for the design and optimization of DNA-binding proteins.
From a functional viewpoint, a more relevant question is whether EngHD exploits these features while operating in its biological environment. The analysis of differential scanning calorimetry data for EngHD has shown that this protein crosses a marginal folding barrier and has non-cooperative unfolding behavior, which is consistent with the results of our folding simulations using the standard SBM parameters.49,61,74 However, its physiological (i.e. room) temperature is lower than its experimentally determined folding temperature (∼325 K),54 which in principle suggests a limited amount of intrinsic disorder in its functional state. To explore conditions that may be more significant biologically, we performed binding simulations at a temperature below the folding temperature. We could do this simply by increasing the interaction strength in our CGSBM (εf > 1) (see Computational procedures and methods and the ESI†).
The results from these simulations are summarized in Fig. 7. In contrast to what occurs at the folding temperature, the data at room temperature show that the EngHD folding mechanism affects the efficiency of DNA recognition. In particular, we find that under these conditions the increase in downhillness speeds up both the sliding and gliding 1D diffusion modes (Fig. 7A), although the effect is relatively small (about a 25% increase). The analysis of individual conformations reveals that such acceleration arises from the fact that in the downhill scenario the protein experiences conformational fluctuations out of the native state even under native conditions (Fig. 7B and C). At room temperature, the two-state-like scenario (downhillness = 0) has a free energy landscape with a narrow native basin of attraction (Fig. 7C). As a consequence, the protein remains rigidly folded and 1D diffusion is relatively slow. Increasing downhillness progressively broadens the native basin of attraction resulting in a more flexible ensemble with conformational excursions out of the folded state that grow in probability and amplitude (Fig. 7C). Partially structured conformations are able to glide more efficiently (see above), and thus the overall 1D diffusion coefficient increases.
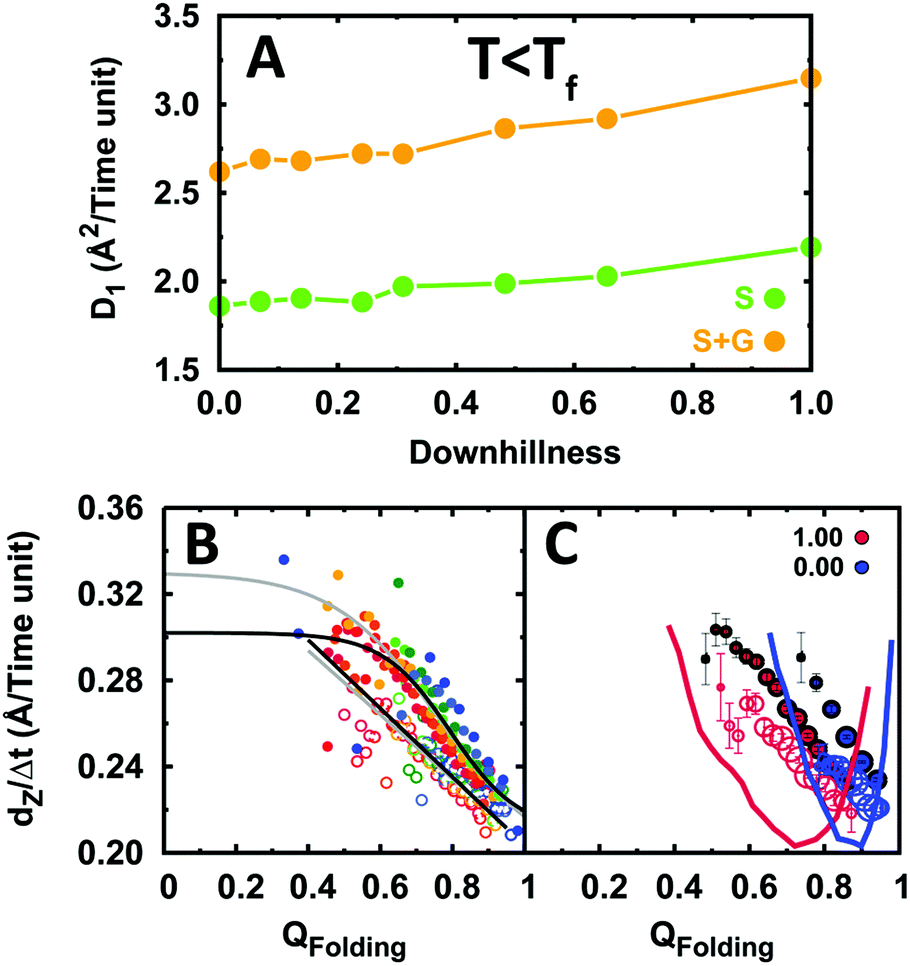 | ||
| Fig. 7 1D diffusion along DNA of EngHD under conditions of marginal native stability (T < Tf). (A) 1D diffusion coefficient as a function of folding downhillness at room temperature. (B) Displacement along DNA for different conformational sub-ensembles of EngHD as a function of downhillness at room temperature. The color code is from red to blue for downhillness decreasing from 1 to 0. The black lines are fits to the data. Grey lines are fits to the data in Fig. 4 for comparison. (C) Displacement along DNA for different conformational sub-ensembles of EngHD at the two extremes in downhillness with the size of circles representing the population of the conformer. The corresponding folding free energy landscapes are also shown. | ||
The effects of the EngHD folding mechanism on the kinetics of specific binding (kinetic scheme of Fig. 5A) are also minor in magnitude compared to the effects of structural disorder. However, it is interesting to note that the trends at the folding temperature and at room temperature are reversed (Fig. 8). At the folding temperature, the rate of formation of the TC from neighboring non-specific sites (kS) decreases with downhillness, whereas the rate of formation of the TC from SB (k*) increases. At room temperature, the more downhill the folding mechanism the more the kS increases and k* decreases (Fig. 8A). Therefore, at room temperature the one-state downhill scenario (downhillness = 1) results in stronger specific binding and longer residence times in SB, whereas the opposite is true at the folding temperature. Likewise, the one-state downhill scenario decreases the escape number at the folding temperature and increases it at room temperature (Fig. 8B).
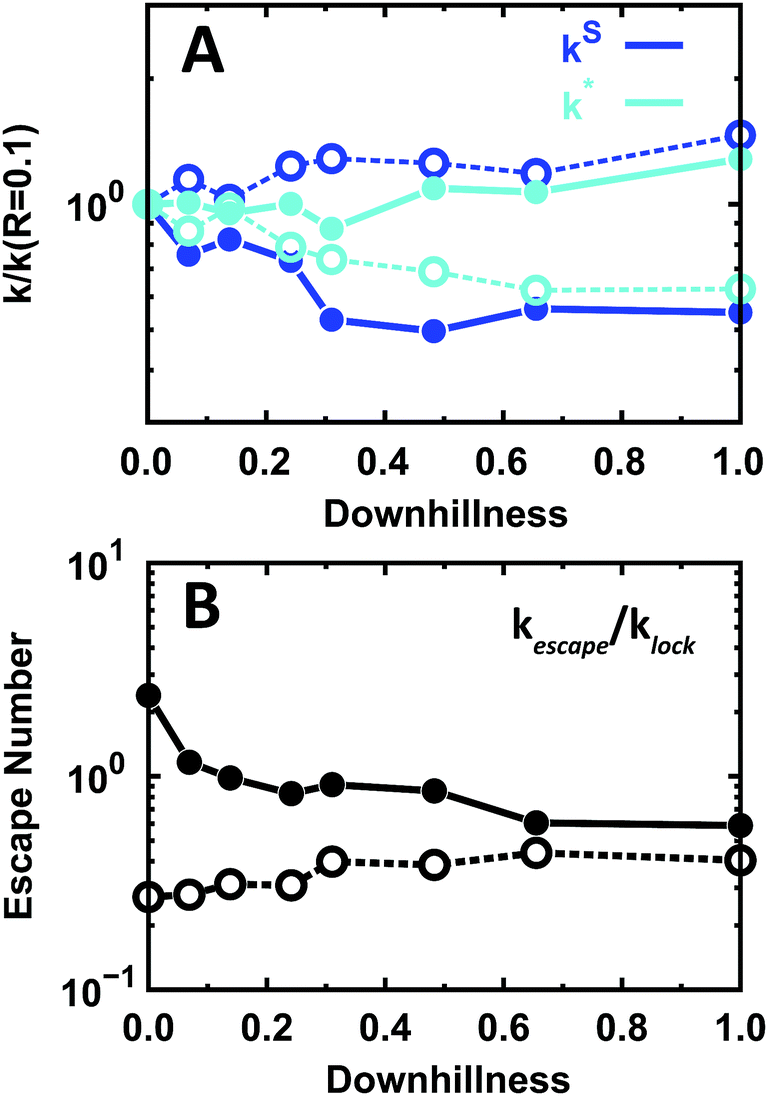 | ||
| Fig. 8 Microscopic kinetics of binding to the specific site for the one-state downhill scenario at folding and room temperature. The color code is the same as in Fig. 5. (A) Relative effects on the rate of arrival at the TC (kS) and the off-rate from SB to TC (k*). (B) Escape number of EngHD. Solid and open circles indicate folding and room temperature, respectively. | ||
How can these results be reconciled? The trend reversal at room temperature suggests a temperature dependent switch in the mechanism of specific binding coupled to folding. The simulations indicate that this mechanism does in fact involve dynamic selection between alternative pathways (Fig. 9). A fully folded EngHD exchanges between TC and SB exclusively via a conventional lock-and-key process (bottom pathway in Fig. 9). But EngHD can also be partially unfolded at the TC (see Fig. 2C), opening a second pathway to SB in which folding and binding occur concertedly via an induced-fit process (middle pathway in Fig. 9). Most of the flux in the induced-fit pathway is directed towards binding because the binding free energy is larger than the entropic penalty of fixing the chain. These two processes are dominant at low temperature at which EngHD populates a highly native-like ensemble. However, at higher temperature the EngHD ensemble is more disordered and thus excursions towards more extensively unfolded conformations become much more common. The largely unfolded conformations are marginally compatible with the TC, and the entropic penalty of fixing them into SB is then higher than the binding free energy. Under these conditions a new pathway emerges by which EngHD is highly restrained when at SB and thus acts as a loaded spring that eventually triggers its induced release (top pathway in Fig. 9). In contrast to the middle pathway, the flux of the top pathway is predominantly in the direction of release both from SB to TC and from TC to a free or non-specifically bound EngHD.
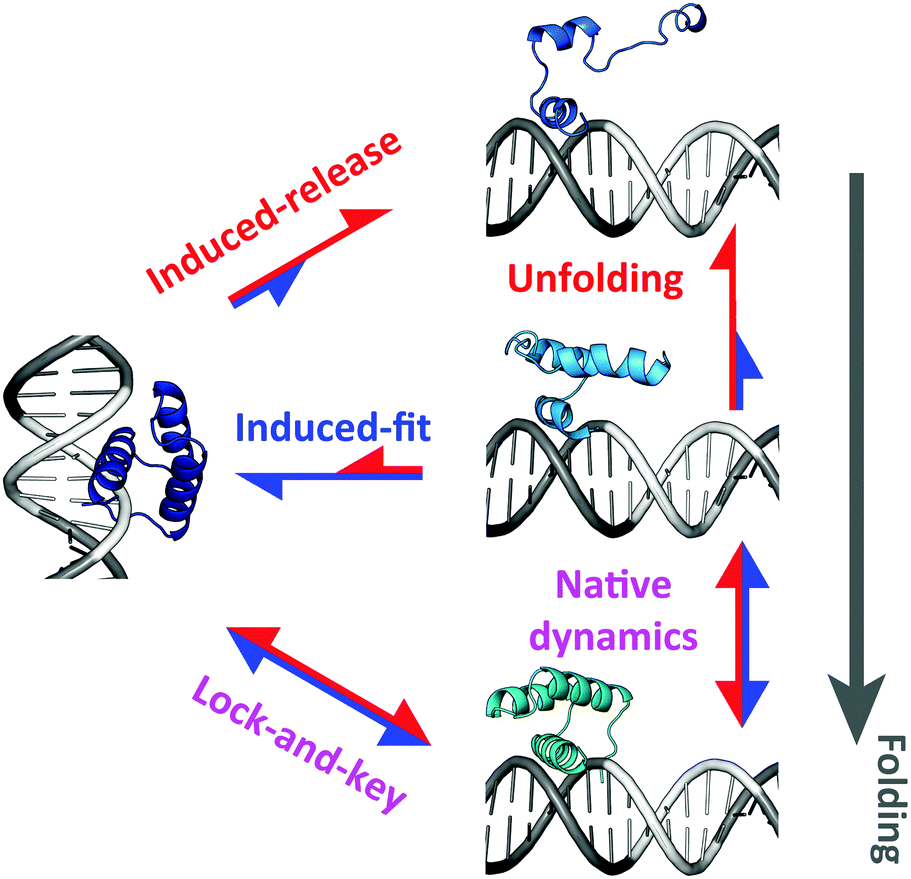 | ||
| Fig. 9 Scheme illustrating the different kinetic transitions occurring between TC and SB for a marginally stable DNA-binding domain. Red and blue arrows signify folding and room temperature. Single headed arrows indicate preferential flux and two headed arrows indicate bidirectional steps. The arrow length reflects the relative population of the pathway at each temperature: red for high temperature and blue for low temperature. | ||
The pathway selection mechanism nicely explains the switch in behavior at different temperatures that we observe when a marginally stable DNA-binding protein folds/unfolds within the downhill scenario. Moreover, it also explains why this phenomenon is not observed when the protein folds two-state (downhillness close to 0). The reason is that in the two-state folding regime the protein needs to cross a free energy barrier to exchange conformations. The barrier crossing event results in a separation of timescales that decouples folding from binding. Accordingly, in the two-state scenario the protein only uses the lock-and-key specific binding pathway (bottom in Fig. 9), regardless of whether it populates only native (low temperature) or both native and highly unfolded conformations (high temperature).
Some of the key aspects of this mechanism for specific binding to DNA use controlled conformational disorder to facilitate binding to, and release from, the specific DNA site. This effect is similar to the fly-casting mechanism proposed to accelerate biomolecular recognition.93,94 In this regard, it has been recently reported that the acceleration of conventional 3D-diffusion-mediated binding through fly-casting is strongly dependent on the interaction strength at the binding site (the quality of the “fly lure”).95 Likewise, we find that a downhill folding DNA-binding domain with marginal stability can either be induced-fitted onto the target site (e.g. low temperature or high fly lure) or induced-released off it (e.g. high temperature or low fly lure) by modulation of its conformational ensemble. Such modulation is mediated by temperature as we investigate here, or alternatively it could be mediated by binding to effectors, such as other components of the transcription complex.96,97 Therefore, the interplay between disorder, folding mechanism and binding free energy produces a sophisticated palette of control mechanisms. Such a control palette is likely to be instrumental for achieving highly dynamic on- and off-switching of gene expression required for a rapid response to cellular environments and stimuli.98,99
Conclusions
Interest in the role that conformational disorder plays in biomolecular function was sparked by the discovery of intrinsically disordered proteins,100–106 and has since then become a major focus of biophysical chemical research.107 Parallel efforts have shown that many single-domain proteins fold in a few microseconds42,44,108 and cross minimal or no barriers to folding, falling in the downhill folding scenario.49 Downhill folding is interesting because it results in gradual, non-cooperative unfolding50,109,110 that could have functional significance, for example by expanding functional diversity through binding to multiple targets,45,46 or via a molecular rheostat mechanism in which the conformational ensemble is subtly manipulated by an effector resulting in allosteric signals.50,51,110 Moreover, intrinsic disorder and downhill folding are closely related phenomena to the extent that partially structured IDPs have the conformational properties of the one-state downhill folding regime.41,45,46,111Here, we focused on the connections between intrinsic disorder and folding scenario in determining the mechanism by which DNA-binding domains efficiently find and bind to their target site. The connection between folding and DNA recognition is supported by the realization that DNA-binding domains exhibit conformational flexibility under native conditions.38,41 Our working hypothesis was that the specific properties of the one-state downhill folding scenario can enable fast conformational exchange between search competent and recognition competent (specific binding) DNA binding modes. Such fast exchange would thus solve the speed-stability paradox that emerges from the facilitated diffusion mechanism that has been proposed for efficient DNA recognition.
We thus investigated the interplay between folding mechanism, disorder and DNA binding of EngHD using simulations with a CGSBM. The standard parameterization of this model67,112 results in a folding mechanism for EngHD characterized by a marginal folding free energy barrier at the denaturation midpoint (i.e. 1.3kT) and a minimally cooperative unfolding process. These results are fully consistent with the folding properties of EngHD derived from experiments54,55,82 and their quantitative analysis,49,61,74 as well as from long-timescale MD simulations.60 To explore the potential effects of conformational disorder, we modified the interaction strength in the model, which allowed us to simulate conditions ranging from: (1) fully native, to (2) denaturation midpoint (QFolding ∼ 0.5), and to (3) unfolding-like. Finally, we also modified the folding mechanism of EngHD by tuning the relative balance between non-local and local interactions in stabilizing the native structure, a factor that is well known to be a major determinant of folding cooperativity.45,46,108,113–115 Practically, we achieved this modulation by changing the strength of the native contacts (non-local) and the dihedral term in the model rather than adding desolvation and/or many-body terms.116–121 This approach is simple and recapitulates existing experimental procedures to modify folding barriers through site-directed mutations.43,52,58,115,122–127
DNA recognition of EngHD is a complex process involving standard 3D diffusion, non-specific binding through electrostatic interactions, 1D diffusive search along the DNA length via various types of modes, and lock into the target site. Our simulations on a CGSBM reproduce all these processes thus permitting us to dissect how each of them is affected by the conformational properties of EngHD. We find that there is a strong coupling between the conformational status of the protein and the various modes by which it interacts with DNA. Such coupling is mediated by a combination of energetic and entropic factors that plays out in differential ways for the various binding modes.
The effect that intrinsic structural disorder has on the efficiency of DNA recognition of EngHD is very apparent. For instance, while binding to the specific site only occurs when EngHD is well folded, a partially unstructured EngHD is capable of binding non-specifically to DNA, and it does so forming additional electrostatic interactions with protein regions that are far from the DNA backbone in the canonical binding site. These delocalized long-range interactions facilitate a gliding mode in which the protein interacts loosely with the DNA resulting in fast 1D diffusion. The gliding mode, which is typical (although not exclusive) of unstructured conformations, is fast and results in linear displacements along the DNA length. In contrast, in the sliding mode the well-folded protein remains inserted into the DNA major groove performing a slower, spiral displacement around the DNA length. As a consequence, the presence of partial disorder on EngHD speeds up the 1D-diffusive search by facilitating gliding, which is nearly 3-times faster than sliding. Our results add to previous studies of conformational disorder that have reported acceleration of 3D diffusion via the “fly-casting” mechanism93,94,128 and enhancement of intersegment transfer between two different DNA fragments via a “monkey bar” mechanism.14,39,129 From all of these findings combined, we conclude that partially disordered conformations are key components of the “search competent” mode of DNA-binding domains.
The effects that conformational disorder has on specific binding are the opposite: structural disorder accelerates the rate of release from SB and greatly increases the escape from the specific basin of attraction. When EngHD populates unstructured conformations its residence time in the specific binding site is thus shortened. This effect is purely entropic, arising from the penalty that the protein pays to fold up while locking into the target site. A shorter SB residence time may be functionally advantageous to a certain extent because it can facilitate dynamical control of gene expression.92 However, binding to the specific site cannot be too weak, or dynamic, at the risk of becoming incompetent to trigger the assembly of the transcription complex and/or of making the protein miss its target site when searching by 1D diffusion. These results highlight the double-edged sword of protein conformational disorder in DNA recognition. The implication is that the functional response of the DNA-binding domain must thus involve a certain (non-zero) level of intrinsic disorder that optimizes these multivariate tradeoffs. The optimal level of structural disorder is presumably specific for each transcription factor and gene.
The other factor that we have investigated here is the folding scenario of the DNA-binding domain. Obviously, the folding properties of the DNA-binding domain can only have relevance in as much as the protein exhibits a certain degree of disorder (for a rigid native structure the folding mechanism has no functional relevance). Therefore, any potential role of the folding scenario must be by definition subtle. Our analysis indicates that in the presence of large degree of structural disorder (e.g. at the folding temperature) the folding mechanism has a negligible effect on DNA recognition. This is so because the thermodynamic conditions already guarantee a significant population of efficient gliders (i.e. partially to completely unfolded conformations) and favor quick release from the specific binding site by a spring-loaded mechanism.
However, the folding scenario becomes really important for DNA recognition under native conditions. This appreciation is functionally significant because the physiological temperature of Drosophila melanogaster is lower than the folding temperature of EngHD.54 For a barrier-crossing folding scenario, the native-like thermodynamic conditions that are biologically relevant imply the absence of structural disorder because partially folded conformations are inherently unstable (i.e. conform the barrier). Under native conditions, a two-state folder is locked into its specific recognition mode, not being able to search efficiently. On the other hand, the downhill scenario guarantees some degree of conformational disorder even under stabilizing native conditions (e.g. red profile in Fig. 7C). These partially folded conformations are able to glide efficiently (Fig. S9, ESI†), making the implementation of a 1D search mode under native conditions possible. Moreover, the absence of the folding barrier allows downhill folding domains to reconfigure with very fast (microsecond) dynamics. The implication is that a partially folded downhill domain can quickly reconfigure while it stays at the TC, and thus efficiently locks into SB through the induced-fit pathway of Fig. 9. The same native conditions guarantee a negligible population of unfolded conformations (QFolding < 0.5), thus effectively blocking the pathway for induced-release off SB.
Our theoretical predictions can be potentially assessed via targeted biophysical experiments. In that respect, it would be highly informative to determine the effect that certain degrees of intrinsic disorder in EngHD or different folding scenarios have on the apparent binding affinity to the specific binding site and to non-specific DNA sequences. Likewise, single-molecule experiments could be used to resolve the sliding and gliding 1D search modes of EngHD on long DNA molecules, thus to measure the relative effects on each different binding mode. Changing the level of intrinsic disorder on EngHD is relatively straightforward, as one could perform the experiments at different temperatures, or destabilizing EngHD via mutation. An attractive mutation in that regard is the single-point L16A mutation, which makes EngHD partially unfolded under physiological conditions.52 Optimized mutations that lead to thermostable versions of EngHD have also been achieved.130,131 Engineering the folding scenario of EngHD (and in particular its folding barrier) is also experimentally feasible by introducing mutations designed to enhance the helical propensity of the native helices,43,127,132,133 remove specific long-range interactions134 and/or modify electrostatic interactions.135
Summarizing, we can conclude that the fast-folding kinetics and downhill folding mechanism of EngHD enable this protein to swiftly interconvert between a (partially unfolded) search efficient mode and its well-folded target recognition mode even under physiological conditions in which the domain is native-like. These properties fulfill the requirements of the two-binding mode mechanism for efficient DNA recognition26,136 in which a conformationally dynamic EngHD performs fast 1D search via non-specific binding, while is able to quickly change conformation to lock into the specific binding site upon arrival.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Advanced Grant ERC-2012-ADG-323059 from the European Research Council to V. M. V. M. also acknowledges support from the Keck foundation and the CREST Center for Cellular and Biomolecular Machines (NSF-CREST-1547848).References
- A. D. Riggs, S. Bourgeois and M. Cohn, J. Mol. Biol., 1970, 53, 401–417 CrossRef CAS PubMed.
- O. G. Berg and P. H. von Hippel, Annu. Rev. Biophys. Biophys. Chem., 1985, 14, 131–158 CrossRef CAS PubMed.
- R. B. Winter, O. G. Berg and P. H. Von Hippel, Biochemistry, 1981, 20, 6961–6977 CrossRef CAS PubMed.
- P. H. von Hippel and O. G. Berg, J. Biol. Chem., 1989, 264, 675–678 CAS.
- H. C. Berg, Random walks in biology, Princeton University Press, 1993 Search PubMed.
- S. E. Halford and J. F. Marko, Nucleic Acids Res., 2004, 32, 3040–3052 CrossRef CAS PubMed.
- N. Shimamoto, J. Biol. Chem., 1999, 274, 15293–15296 CrossRef CAS PubMed.
- P. C. Blainey, A. M. van Oijen, A. Banerjee, G. L. Verdine and X. S. Xie, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5752–5757 CrossRef CAS PubMed.
- Y. M. Wang, R. H. Austin and E. C. Cox, Phys. Rev. Lett., 2006, 97, 048302 CrossRef CAS PubMed.
- J. Elf, G.-W. Li and X. S. Xie, Science, 2007, 316, 1191–1194 CrossRef CAS PubMed.
- P. Hammar, P. Leroy, A. Mahmutovic, E. G. Marklund, O. G. Berg and J. Elf, Science, 2012, 336, 1595–1598 CrossRef CAS PubMed.
- O. Givaty and Y. Levy, J. Mol. Biol., 2009, 385, 1087–1097 CrossRef CAS PubMed.
- A. Marcovitz and Y. Levy, Biophys. J., 2009, 96, 4212–4220 CrossRef CAS PubMed.
- D. Vuzman, M. Polonsky and Y. Levy, Biophys. J., 2010, 99, 1202–1211 CrossRef CAS PubMed.
- D. Vuzman and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21004–21009 CrossRef CAS PubMed.
- T. Terakawa, H. Kenzaki and S. Takada, J. Am. Chem. Soc., 2012, 134, 14555–14562 CrossRef CAS PubMed.
- T. Ando and J. Skolnick, PLoS Comput. Biol., 2014, 10, e1003990 Search PubMed.
- A. Bhattacherjee, D. Krepel and Y. Levy, Wiley Comput. Mol. Sci., 2016, 6, 515–531 CrossRef CAS.
- O. G. Berg, R. B. Winter and P. H. Von Hippel, Biochemistry, 1981, 20, 6929–6948 CrossRef CAS PubMed.
- K. V. Klenin, H. Merlitz, J. Langowski and C.-X. Wu, Phys. Rev. Lett., 2006, 96, 018104 CrossRef PubMed.
- A. B. Kolomeisky, Phys. Chem. Chem. Phys., 2011, 13, 2088–2095 RSC.
- V. K. Misra, J. L. Hecht, A.-S. Yang and B. Honig, Biophys. J., 1998, 75, 2262–2273 CrossRef CAS PubMed.
- M. T. Record, J.-H. Ha and M. A. Fisher, Methods Enzymol., 1991, 208, 291–343 CAS.
- M. Slutsky and L. A. Mirny, Biophys. J., 2004, 87, 4021–4035 CrossRef CAS PubMed.
- S. E. Halford, Biochem. Soc. Trans., 2009, 37, 343–348 CrossRef CAS PubMed.
- L. Mirny, M. Slutsky, Z. Wunderlich, A. Tafvizi, J. Leith and A. Kosmrlj, J. Phys. A: Math. Theor., 2009, 42, 434013 CrossRef.
- O. Bénichou, Y. Kafri, M. Sheinman and R. Voituriez, Phys. Rev. Lett., 2009, 103, 138102 CrossRef PubMed.
- A. Veksler and A. B. Kolomeisky, J. Phys. Chem. B, 2013, 117, 12695–12701 CrossRef CAS PubMed.
- U. Gerland, J. D. Moroz and T. Hwa, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12015–12020 CrossRef CAS PubMed.
- A. Tafvizi, F. Huang, A. R. Fersht, L. A. Mirny and A. M. van Oijen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 563–568 CrossRef CAS PubMed.
- L. Zandarashvili, A. Esadze, D. Vuzman, C. A. Kemme, Y. Levy and J. Iwahara, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5142–E5149 CrossRef CAS PubMed.
- L. Zandarashvili, D. Vuzman, A. Esadze, Y. Takayama, D. Sahu, Y. Levy and J. Iwahara, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1724–E1732 CrossRef CAS PubMed.
- J. Iwahara and Y. Levy, Transcription, 2013, 4, 58–61 CrossRef PubMed.
- A. Marcovitz and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 17957–17962 CrossRef CAS PubMed.
- A. Marcovitz and Y. Levy, J. Phys. Chem. B, 2013, 117, 13005–13014 CrossRef CAS PubMed.
- J. Liu, N. B. Perumal, C. J. Oldfield, E. W. Su, V. N. Uversky and A. K. Dunker, Biochemistry, 2006, 45, 6873–6888 CrossRef CAS PubMed.
- M. Fuxreiter, I. Simon and S. Bondos, Trends Biochem. Sci., 2011, 36, 415–423 CrossRef CAS PubMed.
- P. L. Privalov and A. I. Dragan, Biophys. Chem., 2007, 126, 16–24 CrossRef CAS PubMed.
- D. Vuzman, A. Azia and Y. Levy, J. Mol. Biol., 2010, 396, 674–684 CrossRef CAS PubMed.
- H. J. Dyson, Mol. BioSyst., 2012, 8, 97–104 RSC.
- A. N. Naganathan, R. Perez-Jimenez, V. Muñoz and J. M. Sanchez-Ruiz, Phys. Chem. Chem. Phys., 2011, 13, 17064–17076 RSC.
- J. Kubelka, J. Hofrichter and W. A. Eaton, Curr. Opin. Struct. Biol., 2004, 14, 76–88 CrossRef CAS PubMed.
- W. Y. Yang and M. Gruebele, Biophys. J., 2004, 87, 596–608 CrossRef CAS PubMed.
- H. Gelman and M. Gruebele, Q. Rev. Biophys., 2014, 47, 95–142 CrossRef CAS PubMed.
- V. Muñoz, L. A. Campos and M. Sadqi, Curr. Opin. Struct. Biol., 2016, 36, 58–66 CrossRef PubMed.
- V. Muñoz and M. Cerminara, Biochem. J., 2016, 473, 2545–2559 CrossRef PubMed.
- J. D. Bryngelson, J. N. Onuchic, N. D. Socci and P. G. Wolynes, Proteins: Struct., Funct., Bioinf., 1995, 21, 167–195 CrossRef CAS PubMed.
- F. Liu and M. Gruebele, Chem. Phys. Lett., 2008, 461, 1–8 CrossRef CAS.
- A. N. Naganathan, U. Doshi and V. Muñoz, J. Am. Chem. Soc., 2007, 129, 5673–5682 CrossRef CAS PubMed.
- M. M. Garcia-Mira, M. Sadqi, N. Fischer, J. M. Sanchez-Ruiz and V. Munoz, Science, 2002, 298, 2191–2195 CrossRef CAS PubMed.
- M. Cerminara, T. M. Desai, M. Sadqi and V. Muñoz, J. Am. Chem. Soc., 2012, 134, 8010–8013 CrossRef CAS PubMed.
- T. L. Religa, J. S. Markson, U. Mayor, S. M. V. Freund and A. R. Fersht, Nature, 2005, 437, 1053–1056 CrossRef CAS PubMed.
- S. Gianni, N. R. Guydosh, F. Khan, T. D. Caldas, U. Mayor, G. W. N. White, M. L. DeMarco, V. Daggett and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13286–13291 CrossRef CAS PubMed.
- U. Mayor, C. M. Johnson, V. Daggett and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13518–13522 CrossRef CAS PubMed.
- U. Mayor, N. R. Guydosh, C. M. Johnson, J. G. Grossmann, S. Sato, G. S. Jas, S. M. V. Freund, D. O. V. Alonso, V. Daggett and A. R. Fersht, Nature, 2003, 421, 863–867 CrossRef CAS PubMed.
- A. R. Fersht and V. Daggett, Cell, 2002, 108, 573–582 CrossRef CAS PubMed.
- W. A. Eaton, V. Muñoz, S. J. Hagen, G. S. Jas, L. J. Lapidus, E. R. Henry and J. Hofrichter, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 327–359 CrossRef CAS PubMed.
- U. Mayor, J. G. Grossmann, N. W. Foster, S. M. V. Freund and A. R. Fersht, J. Mol. Biol., 2003, 333, 977–991 CrossRef CAS PubMed.
- T. L. Religa, C. M. Johnson, D. M. Vu, S. H. Brewer, R. B. Dyer and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9272–9277 CrossRef CAS PubMed.
- K. Lindorff-Larsen, S. Piana, R. O. Dror and D. E. Shaw, Science, 2011, 334, 517–520 CrossRef CAS PubMed.
- A. N. Naganathan, J. M. Sanchez-Ruiz and V. Munoz, J. Am. Chem. Soc., 2005, 127, 17970–17971 CrossRef CAS PubMed.
- E. Fraenkel, M. A. Rould, K. A. Chambers and C. O. Pabo, J. Mol. Biol., 1998, 284, 351–361 CrossRef CAS PubMed.
- T. Sprules, N. Green, M. Featherstone and K. Gehring, Biochemistry, 2000, 39, 9943–9950 CrossRef CAS PubMed.
- J. Iwahara and G. M. Clore, Nature, 2006, 440, 1227–1230 CrossRef CAS PubMed.
- J. Iwahara and G. M. Clore, J. Am. Chem. Soc., 2006, 128, 404–405 CrossRef CAS PubMed.
- J. Iwahara, M. Zweckstetter and G. M. Clore, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15062–15067 CrossRef CAS PubMed.
- C. Clementi, H. Nymeyer and J. N. Onuchic, J. Mol. Biol., 2000, 298, 937–953 CrossRef CAS PubMed.
- J. K. Noel, P. C. Whitford, K. Y. Sanbonmatsu and J. N. Onuchic, Nucleic Acids Res., 2010, 38, W657–W661 CrossRef CAS PubMed.
- Y. Levy, S. S. Cho, J. N. Onuchic and P. G. Wolynes, J. Mol. Biol., 2005, 346, 1121–1145 CrossRef CAS PubMed.
- Y. Levy, P. G. Wolynes and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 511–516 CrossRef CAS PubMed.
- M. S. Cheung, J. M. Finke, B. Callahan and J. N. Onuchic, J. Phys. Chem. B, 2003, 107, 11193–11200 CrossRef CAS.
- A. Azia and Y. Levy, J. Mol. Biol., 2009, 393, 527–542 CrossRef CAS PubMed.
- X. Chu, Y. Wang, L. Gan, Y. Bai, W. Han, E. Wang and J. Wang, PLoS Comput. Biol., 2012, 8, e1002608 CAS.
- A. N. Naganathan, U. Doshi, A. Fung, M. Sadqi and V. Muñoz, Biochemistry, 2006, 45, 8466–8475 CrossRef CAS PubMed.
- M. Knott, H. Kaya and H. S. Chan, Polymer, 2004, 45, 623–632 CrossRef CAS.
- G. Zuo, J. Wang and W. Wang, Proteins: Struct., Funct., Bioinf., 2006, 63, 165–173 CrossRef CAS PubMed.
- N. D. Clarke, C. R. Kissinger, J. Desjarlais, G. L. Gilliland and C. O. Pabo, Protein Sci., 1994, 3, 1779–1787 CrossRef CAS PubMed.
- M. Karplus and D. L. Weaver, Protein Sci., 1994, 3, 650–668 CrossRef CAS PubMed.
- A. R. Fersht, Curr. Opin. Struct. Biol., 1997, 7, 3–9 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- I. A. Hubner, E. J. Deeds and E. I. Shakhnovich, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17747–17752 CrossRef CAS PubMed.
- M. E. McCully, D. A. C. Beck and V. Daggett, Biochemistry, 2008, 47, 7079–7089 CrossRef CAS PubMed.
- X. Chu, F. Liu, B. A. Maxwell, Y. Wang, Z. Suo, H. Wang, W. Han and J. Wang, PLoS Comput. Biol., 2014, 10, e1003804 Search PubMed.
- H.-X. Zhou and K. A. Dill, Biochemistry, 2001, 40, 11289–11293 CrossRef CAS PubMed.
- H. R. Bosshard, Physiology, 2001, 16, 171–173 CAS.
- C.-J. Tsai, B. Ma, Y. Y. Sham, S. Kumar and R. Nussinov, Proteins: Struct., Funct., Bioinf., 2001, 44, 418–427 CrossRef CAS PubMed.
- D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 98–104 CrossRef CAS.
- A. N. Naganathan and M. Orozco, J. Am. Chem. Soc., 2011, 133, 12154–12161 CrossRef CAS PubMed.
- D. De Sancho and V. Muñoz, Phys. Chem. Chem. Phys., 2011, 13, 17030–17043 RSC.
- A. Granéli, C. C. Yeykal, R. B. Robertson and E. C. Greene, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1221–1226 CrossRef PubMed.
- H. Qian, M. P. Sheetz and E. L. Elson, Biophys. J., 1991, 60, 910 CrossRef CAS PubMed.
- G. L. Hager, J. G. McNally and T. Misteli, Mol. Cell, 2009, 35, 741–753 CrossRef CAS PubMed.
- B. A. Shoemaker, J. J. Portman and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS PubMed.
- E. Trizac, Y. Levy and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2746–2750 CrossRef CAS PubMed.
- K. Umezawa, J. Ohnuki, J. Higo and M. Takano, Proteins: Struct., Funct., Bioinf., 2016, 84, 1124–1133 CrossRef CAS PubMed.
- M.-Y. Tsai, B. Zhang, W. Zheng and P. G. Wolynes, J. Am. Chem. Soc., 2016, 138, 13497–13500 CrossRef CAS PubMed.
- J. S. Graham, R. C. Johnson and J. F. Marko, Nucleic Acids Res., 2011, 39, 2249–2259 CrossRef CAS PubMed.
- N. Hao and E. K. O’shea, Nat. Struct. Mol. Biol., 2012, 19, 31–39 CAS.
- D. E. Levy and J. E. Darnell, Nat. Rev. Mol. Cell Biol., 2002, 3, 651–662 CrossRef CAS PubMed.
- A. K. Dunker, J. D. Lawson, C. J. Brown, R. M. Williams, P. Romero, J. S. Oh, C. J. Oldfield, A. M. Campen, C. M. Ratliff, K. W. Hipps, J. Ausio, M. S. Nissen, R. Reeves, C. Kang, C. R. Kissinger, R. W. Bailey, M. D. Griswold, W. Chiu, E. C. Garner and Z. Obradovic, J. Mol. Graphics Modell., 2001, 19, 26–59 CrossRef CAS PubMed.
- H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS PubMed.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS PubMed.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS PubMed.
- J. Habchi, P. Tompa, S. Longhi and V. N. Uversky, Chem. Rev., 2014, 114, 6561–6588 CrossRef CAS PubMed.
- D. Eliezer, Curr. Opin. Struct. Biol., 2009, 19, 23–30 CrossRef CAS PubMed.
- A. K. Dunker, I. Silman, V. N. Uversky and J. L. Sussman, Curr. Opin. Struct. Biol., 2008, 18, 756–764 CrossRef CAS PubMed.
- S. DeForte and V. N. Uversky, RSC Adv., 2016, 6, 11513–11521 RSC.
- V. Muñoz, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 395–412 CrossRef PubMed.
- V. Muñoz, Int. J. Quantum Chem., 2002, 90, 1522–1528 CrossRef.
- M. Sadqi, D. Fushman and V. Muñoz, Nature, 2006, 442, 317–321 CrossRef CAS PubMed.
- Y. Wang, X. Chu, S. Longhi, P. Roche, W. Han, E. Wang and J. Wang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3743–E3752 CrossRef CAS PubMed.
- P. C. Whitford, J. K. Noel, S. Gosavi, A. Schug, K. Y. Sanbonmatsu and J. N. Onuchic, Proteins: Struct., Funct., Bioinf., 2009, 75, 430–441 CrossRef CAS PubMed.
- V. I. Abkevich, A. M. Gutin and E. I. Shakhnovich, J. Mol. Biol., 1995, 252, 460–471 CrossRef CAS PubMed.
- E. Shakhnovich, Chem. Rev., 2006, 106, 1559–1588 CrossRef CAS PubMed.
- V. Muñoz and L. Serrano, Folding Des., 1996, 1, R71–R77 CrossRef.
- A. Badasyan, Z. Liu and H. S. Chan, J. Mol. Biol., 2008, 384, 512–530 CrossRef CAS PubMed.
- H. S. Chan, Z. Zhang, S. Wallin and Z. Liu, Annu. Rev. Phys. Chem., 2011, 62, 301 CrossRef CAS PubMed.
- M. S. Cheung, A. E. García and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 685–690 CrossRef CAS PubMed.
- S. S. Cho, P. Weinkam and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 118–123 CrossRef CAS PubMed.
- M. R. Ejtehadi, S. P. Avall and S. S. Plotkin, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15088–15093 CrossRef CAS PubMed.
- Z. Liu and H. S. Chan, Phys. Biol., 2005, 2, S75 CrossRef CAS PubMed.
- N. Ferguson, P. J. Schartau, T. D. Sharpe, S. Sato and A. R. Fersht, J. Mol. Biol., 2004, 344, 295–301 CrossRef CAS PubMed.
- N. Ferguson, T. D. Sharpe, P. J. Schartau, S. Sato, M. D. Allen, C. M. Johnson, T. J. Rutherford and A. R. Fersht, J. Mol. Biol., 2005, 353, 427–446 CrossRef CAS PubMed.
- A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1525–1529 CrossRef CAS.
- F. Liu and M. Gruebele, J. Mol. Biol., 2007, 370, 574–584 CrossRef CAS PubMed.
- V. Muñoz, M. Sadqi, A. N. Naganathan and D. de Sancho, HFSP J., 2008, 2, 342–353 CrossRef PubMed.
- W. Y. Yang and M. Gruebele, Nature, 2003, 423, 193–197 CrossRef CAS PubMed.
- Y. Levy, J. N. Onuchic and P. G. Wolynes, J. Am. Chem. Soc., 2007, 129, 738–739 CrossRef CAS PubMed.
- D. Vuzman and Y. Levy, Isr. J. Chem., 2014, 54, 1374–1381 CrossRef CAS.
- M. E. McCully, D. A. Beck and V. Daggett, Protein Eng., Des. Sel., 2012, 26, 35–45 CrossRef PubMed.
- P. S. Shah, G. K. Hom, S. A. Ross, J. K. Lassila, K. A. Crowhurst and S. L. Mayo, J. Mol. Biol., 2007, 372, 1–6 CrossRef CAS PubMed.
- M. B. Prigozhin, Y. Liu, A. J. Wirth, S. Kapoor, R. Winter, K. Schulten and M. Gruebele, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8087–8092 CrossRef CAS PubMed.
- E. Larios, J. W. Pitera, W. C. Swope and M. Gruebele, Chem. Phys., 2006, 323, 45–53 CrossRef CAS.
- F. Liu, D. Du, A. A. Fuller, J. E. Davoren, P. Wipf, J. W. Kelly and M. Gruebele, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2369–2374 CrossRef CAS PubMed.
- Ø. Halskau, R. Perez-Jimenez, B. Ibarra-Molero, J. Underhaug, V. Muñoz, A. Martinez and J. M. Sanchez-Ruiz, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8625–8630 CrossRef PubMed.
- H.-X. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8651–8656 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Models and methods, and tables and additional figures. See DOI: 10.1039/c7cp04380e |
| This journal is © the Owner Societies 2017 |