
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew insights into the solubility of graphene oxide in water and alcohols†
Vadim V.
Neklyudov
a,
Nail R.
Khafizov
a,
Igor A.
Sedov
b and
Ayrat M.
Dimiev
 *a
*a
aLaboratory for Advanced Carbon Nanomaterials, Kazan Federal University, 18 Kremlevkaya str., 420008 Kazan, Russian Federation. E-mail: AMDimiev@kpfu.ru
bDepartment of Physical Chemistry, Kazan Federal University, 18 Kremlevkaya str., Kazan 420008, Russian Federation
First published on 6th June 2017
Abstract
One of the main advantages of graphene oxide (GO) over its non-oxidized counterpart is its ability to form stable solutions in water and some organic solvents. At the same time, the nature of GO solutions is not completely understood; the existing data are scarce and controversial. Here, we demonstrate that the solubility of GO, and the stability of the as-formed solutions depend not just on the solute and solvent cohesion parameters, as commonly believed, but mostly on the chemical interactions at the GO/solvent interface. By the DFT and QTAIM calculations, we demonstrate that the solubility of GO is afforded by strong hydrogen bonding established between GO functional groups and solvent molecules. The main functional groups taking part in hydrogen bonding are tertiary alcohols; epoxides play only a minor role. The magnitude of the bond energy values is significantly higher than that for typical hydrogen bonding. The hydrogen bond energy between GO functional groups and solvent molecules decreases in the sequence: water > methanol > ethanol. We support our theoretical results by several experimental observations including solution calorimetry. The enthalpy of GO dissolution in water, methanol and ethanol is −0.1815 ± 0.0010, −0.1550 ± 0.0012 and −0.1040 ± 0.0010 kJ g−1, respectively, in full accordance with the calculated trend. Our findings provide an explanation for the well-known, but poorly understood solvent exchange phenomenon.
Introduction
The last decade witnessed unprecedented research on graphene and graphene oxide (GO). In particular, GO was successfully tested for numerous potential applications.1 One of the main advantages of GO over its non-oxidized counterpart is its ability to form stable solutions in several solvents, by exfoliating to single-atomic-layer sheets.2 The solution phase provides reactants easy and unimpeded access of to the GO surface, opening unlimited avenues for liquid phase processing. Such applications of GO as polymer composites, selective membranes, hydrogels, electrode materials, etc. are based on the liquid phase processing of GO. Thus, understanding the parameters controlling the solubility of GO and the properties of the as-prepared solutions is extremely important for successful implementation of GO. Against the expectations, the chemistry of GO solutions is significantly less studied, compared to that of the solid GO material.It is well-known that GO is very well soluble in water. The solubility of GO in water is limited only by the formation of a nematic phase and by the associated viscosity of solutions at high GO concentrations.3 It was also reported that GO is soluble in organic solvents such as DMF, N-methyl-2-pyrrolidone, and ethylene glycol.4,5 At the same time, it was found to be insoluble in methanol (MeOH) and ethanol (EtOH).4 Such selectivity is difficult to explain from perspectives of the old rule of thumb “like dissolves like”: GO contains numerous tertiary alcohol groups on the basal planes, and thus should be soluble in alcohols. Interestingly, despite the fact that GO does not directly disperse in alcohols, it can be transferred into the alcoholic phase from aqueous solutions by gradual replacement of water by alcohols.6,7 The amount of water remaining, as well as the nature of the GO/liquid interface in the resulting GO/alcohol solutions is basically unknown. In general, the experimental data on GO solubility in different solvents are not systematic and controversial.
Several attempts have been made recently to explain GO solubility and solution stability based on different solubility parameters. The authors of ref. 4 discuss these phenomena with respect to solvent parameters such as dipole moment, surface tension, Hansen and Hildebrand solubility parameters. They conclude that the most important factor in choosing appropriate solvents is the closeness of the Hildebrand parameters for GO and a solvent. The Hildebrand parameters for GO and MeOH and EtOH are close, however, solubility in these solvents, as noted above, is low. For water, this parameter is very different from that for GO, however, GO is well soluble in water. There is an apparent contradiction between this approach and experimental data.
Recently, Gudarzi discussed the stability of GO solutions with respect to the DLVO theory,8 and found a good correlation between experimental data and theory. However, in our opinion, the application of this theory to truly two-dimensional GO is not straightforward due to the well-known non-additivity principle for nanomaterials.9
In general, at present, there is little understanding of the real factors influencing the solubility of GO in different solvents.
Interestingly, more or less stable dispersions of GO in low-molecular weight alcohols can be prepared by sonication.4 This yields low-stability non-transparent dispersions suggesting the non-exfoliated state of GO in alcohols. However, mechanical agitation of GO with a magnetic stirrer does not result in any visible dissolution of GO in alcohols. Thus, we must differentiate between the dispersibility and solubility of GO. From the perspectives discussed above, we consider GO insoluble in alcohols. In this report, we do not study dispersions, but only solutions of GO, where GO is in the fully exfoliated state.
While GO flakes have lateral dimensions from hundreds of nanometers through tens of microns, their thickness is only one atom. From this point, GO solutions can be considered as pseudo-true solutions. The only difference is that GO flakes organize the solution into the liquid-crystalline phase, which is not the case for real true solutions. However, the solubility of GO must be guarded by the same factors as the solubility of substances yielding true solutions. This factor is the interaction between solute species and solvent molecules. We suggest that the solubility of GO and the stability of the as-formed solutions are a function of the chemical structure of the GO/solvent interface rather than a function of the formal macroscopic solvent parameters. Thus, one needs to investigate interactions between single GO functional groups and solvent molecules. This task is very difficult to accomplish experimentally. This is the case where the computational approach might be helpful to solve the problem. In GO field, theoretical studies have already demonstrated to be beneficial for understanding the fine chemical structure of GO10–14 and its decomposition upon annealing.15 In addition, the study of hydrogen bonding is of independent interest in theoretical chemistry for two-dimensional materials such as GO.
In this report, we investigate the interface between GO and three different solvents: water, methanol and ethanol. We assess the strength of hydrogen bonding of the three solvent molecules with GO functional groups. We also provide some experimental data supporting our theoretical findings.
Calculation details
The optimization of the initial GO structure (free of solvents) was performed as follows. First, the initial structure decorated by functional groups was preliminarily optimized, as described below, by the quantum mechanic (QM) method implemented in the PRIRODA package.16,17 Then, in vacuo DFT calculations were carried out in the same program by the sequential optimization of the obtained structure using the B3LYP18 hybrid functional, and the 6-31G(d,p) and 6-311++G(d,p) basis sets. Final optimization was performed at the B3LYP/6-311++G(d,p) level of theory in the GAUSSIAN 0919 program package.To obtain the structure of GO with bound solvent molecules (here and further called GO solvates), several initial GO structures were generated as described above, and randomly aligned solvent molecules were evenly allocated on both sides of the GO structural fragment. After that, the structures with the lowest energies were chosen for further calculations based on the results obtained by the QM optimization method. The initial structures, whose optimization leads to the most energetically favorable solvates, and the optimized structures used for further calculations are shown in Fig. S1 of the ESI.† Then, geometrical optimization of the GO solvates was performed as described for the free GO structure. The solvent effects were taken into account using the IEF-PCM solvent reaction field model20 implemented in the GAUSSIAN 09 software. Topological electronic density analysis was performed using the AIMAll software package21 (version 10.11.24).
Results and discussion
In the first step, it is very important to choose a realistic GO model that does not contradict the known experimental data on the topic. This is why in our report we paid special attention to this matter.GO planes consist of two different areas, often called domains: the areas on intact graphene and the oxidized areas. These domains have lateral dimensions from 2 to 8 nm, and irregularly distributed on the GO plane. This can be clearly seen from the HRTEM images of GO.22 According to the common view on the GO structure, the oxidized domains contain hydroxo (tertiary alcohols) and epoxy (1,2-ethers) groups.2,23,24 The flake edges might contain additional functional groups, but their contribution to the solubility is negligible due to their relatively small number.
Today, when the general structure of GO is more or less clear, the remaining challenge is to reveal the fine chemical structure of the oxidized domains, i.e. the arrangement of epoxy and alcohol groups relative to each other. Here, again, the theoretical methods of study had already demonstrated their effectiveness. The optimal arrangement of the functional groups with respect to each other was modeled by different methods.11–14 First, the arrangement of the functional groups in a small unit was modelled. Then, the arrangement of the elementary units was optimized. Thus, the most probable structures were determined. In general, the results obtained by the different groups, are in accordance with each other.
The model structure of GO, used in this work is shown in Scheme 1A. The cell of 4 × 4 benzene ring units contains 48 carbon atoms. The central part of the cell was functionalized in accordance with the optimal elementary units11 shown in Scheme 1B and C. The epoxide/hydroxyl ratio was chosen as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Some additional information about GO model structure is presented in Fig. S2 of the ESI.† In our model, the oxidized domain is fully situated inside the frame of non-oxidized carbon atoms; thus, the functional groups of the oxidized domain almost do not experience the influence of the edge atoms connected with hydrogens. By definition, any model structure is terminated by hydrogen atoms. Subsequently, if the functional group of interest is located on the edge carbon atom (when the model structure is too small), or in close proximity to it, this carbon atom in its essence is a part of a polycyclic aromatic molecule, but not GO. Due to the influence of the H-terminated edge atoms, the chemistry of the polycyclic aromatic molecules is very different from that of GO. In our structure (Scheme 1), the oxidized domain is the neighboring non-oxidized domain, not the edge atom. Thus, the chosen structure most effectively reflects the structure of real GO, for the given number of C atoms.
:2. Some additional information about GO model structure is presented in Fig. S2 of the ESI.† In our model, the oxidized domain is fully situated inside the frame of non-oxidized carbon atoms; thus, the functional groups of the oxidized domain almost do not experience the influence of the edge atoms connected with hydrogens. By definition, any model structure is terminated by hydrogen atoms. Subsequently, if the functional group of interest is located on the edge carbon atom (when the model structure is too small), or in close proximity to it, this carbon atom in its essence is a part of a polycyclic aromatic molecule, but not GO. Due to the influence of the H-terminated edge atoms, the chemistry of the polycyclic aromatic molecules is very different from that of GO. In our structure (Scheme 1), the oxidized domain is the neighboring non-oxidized domain, not the edge atom. Thus, the chosen structure most effectively reflects the structure of real GO, for the given number of C atoms.
 | ||
| Scheme 1 (A) The model structure of GO used in this work, (B) the elementary unit containing two hydroxyl and two epoxy groups, and (C) the elementary unit containing only two hydroxyl groups. | ||
As the next step, our model was optimized. To confirm the accuracy of the chosen calculation method, the obtained geometrical parameters of GO functional groups have been reproduced with sufficient accuracy (Table 1). The oxidized fullerenes have been chosen for comparison purposes, as those closest to the structure to GO, and for which the actual structure had been determined by X-ray crystallography diffraction. The optimized structure was then used to model its interaction with solvent molecules.
| Structure | Bond/angle | Value |
|---|---|---|
| epEpoxy group. hHydroxyl group. | ||
| GO model (this work) | C–Oep | 1.46 Å |
| C–Cep | 1.48 Å | |
| C–O–Cep | 61.1° | |
| C–Oh | 1.44 Å | |
| O–Hh | 0.99 Å | |
| C–O–Hh | 104.3° | |
| Oxidized fullerene (experimental data)25 | C–Oep | 1.43 Å |
| C–Cep | 1.48 Å | |
| C–O–Cep | 62.0° | |
| C–Oh | 1.44 Å | |
| O–Hh | 1.00 Å | |
| C–O–Hh | 93.2° | |
| Oxidized fullerene (calculated by DFT LDA (VASP))14 | C–Oep | 1.44 Å |
| C–Cep | 1.51 Å | |
| C–Oh | 1.47 Å | |
| O–Hh | 0.98 Å | |
| C–O–Hh | 107.9° | |
The structure of GO solvates
Scheme 2 represents the three basic types of hydrogen bonding between GO functionalities and solvent molecules. In addition, different combinations of these three types can be realized, where one solvent molecule bonds simultaneously with more than one functional group. In the first step, we attempted to optimize the orientation of a single solvent molecule in proximity to a chosen functional group. Using this approach, we could not obtain a structure, where a solvent molecule would bond with GO by a single hydrogen bond: the solvent molecule tends to establish multiple bonds simultaneously with several neighboring GO functionalities.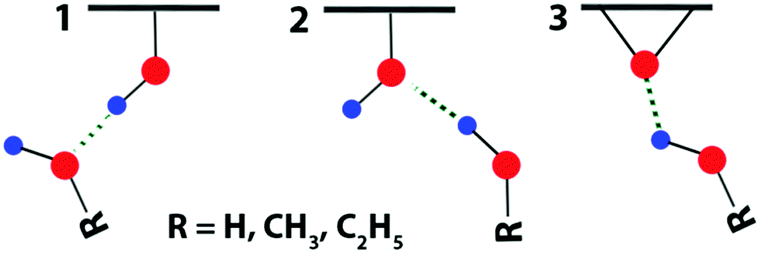 | ||
| Scheme 2 Schematic representation of the three different types of hydrogen bonding in the solvates of GO. Red balls denote the O atoms and blue balls the H atoms. (1) and (2) are the bonding to the tertiary alcohol. (3) is the bonding to the epoxide group. | ||
This might not be the case in the real system, where several solvent molecules compete for the binding sites at the GO interface. This is why in the next step, the model structure was surrounded by 20 molecules, 10 from each side of the flake, to attain maximal solvation. The chosen number of solvent molecules is based on the geometrical considerations. The diameter of a water molecule is ∼3 Å; the surface area of the oxidized domain is ∼30 Å. The molecules of the alcohols are larger; this affects their number.
Topological analysis of the optimized structures
To analyze the type of bonding between the solvent molecules and GO, realized in the solvated structures, and to calculate the energy of the as-formed bonds we used the topological theory of R. Bader, often referred to as QTAIM.26 This theory is based on the analysis of the electron density distribution in the system. The areas of space, where the gradient of electron density turns to zero, are called critical points (CPs) that in this work were determined by analyzing the Hessian eigenvalues. The CPs that correspond to the saddle points between two atoms are called the bond critical points (BCPs). The existence of BCPs is the necessary and sufficient condition for the existence of a chemical bond, including the hydrogen bond. Only this type of CP will be discussed further. Note, the distribution of CPs for a given conformation of the molecule is unequivocal.By the common approach, the strength of a bond can be estimated as the difference between the full energy of the structure and the energies of its constituent parts. For our system, using this approach is very difficult due to the large number of participating molecules, and respectively, the large number of possible conformations. Back in 1998, based on Bader's quantum theory, E. Espinosa27,28 proposed an alternative approach to calculate hydrogen bond energies (1):
| E(kcal mol−1) = 313.754·v(r) (a.u.), | (1) |
 | ||
| Fig. 1 Optimized structures of the solvates of graphene oxide with water (A), methanol (B) and ethanol (C). Hydrogen atoms are marked in blue, oxygen in red, carbon in black; BCPs are marked in green. | ||
We assume that these solvate structures are established mostly via hydrogen bonding. However, this statement requires more detailed analysis. The actual nature of hydrogen bonding is still under question. In our study, we use some of the most accepted approaches based on the QTAIM, and related quantitative parameters. Thus, Koch and Popelier29 developed eight criteria for a chemical bond to be classified as the hydrogen bond. Nakanishi et al.30 discussed three major criteria: existence of a critical point, the values of electron density, and the Laplacian of electron density at the critical point. According to Koch and Popelier,29 for hydrogen bonding, the value of the electron density at the critical point must be from 0.002 to 0.035 a.u., and the value of Laplacian must be from 0.024 to 0.139 a.u.
To analyze the bonds, established between solvent molecules and GO functionalities, we consider four main topological parameters: (1) distribution of critical points, (2) electron density at the critical point, (3) electron density Laplacian values, and (4) potential energy density at the critical point (represented as the bond energy calculated according to eqn (1)). All the four parameters were calculated in the AIMAll program package, based on the wave functions for the optimized solvated structures.
In Fig. 1, we show some examples of selected topological parameters for the discussed bonds. We also show the electron density distribution diagrams for some solvent molecules situated in the centre of the GO structure. The selected molecules represent the different types of bonding. Fig. 2 represents the values of electron densities for all the bonds realized in the optimized solvate structures of GO with water, methanol and ethanol. The topological parameters of all the formed bonds are provided in Tables S1–S3 of the ESI.†
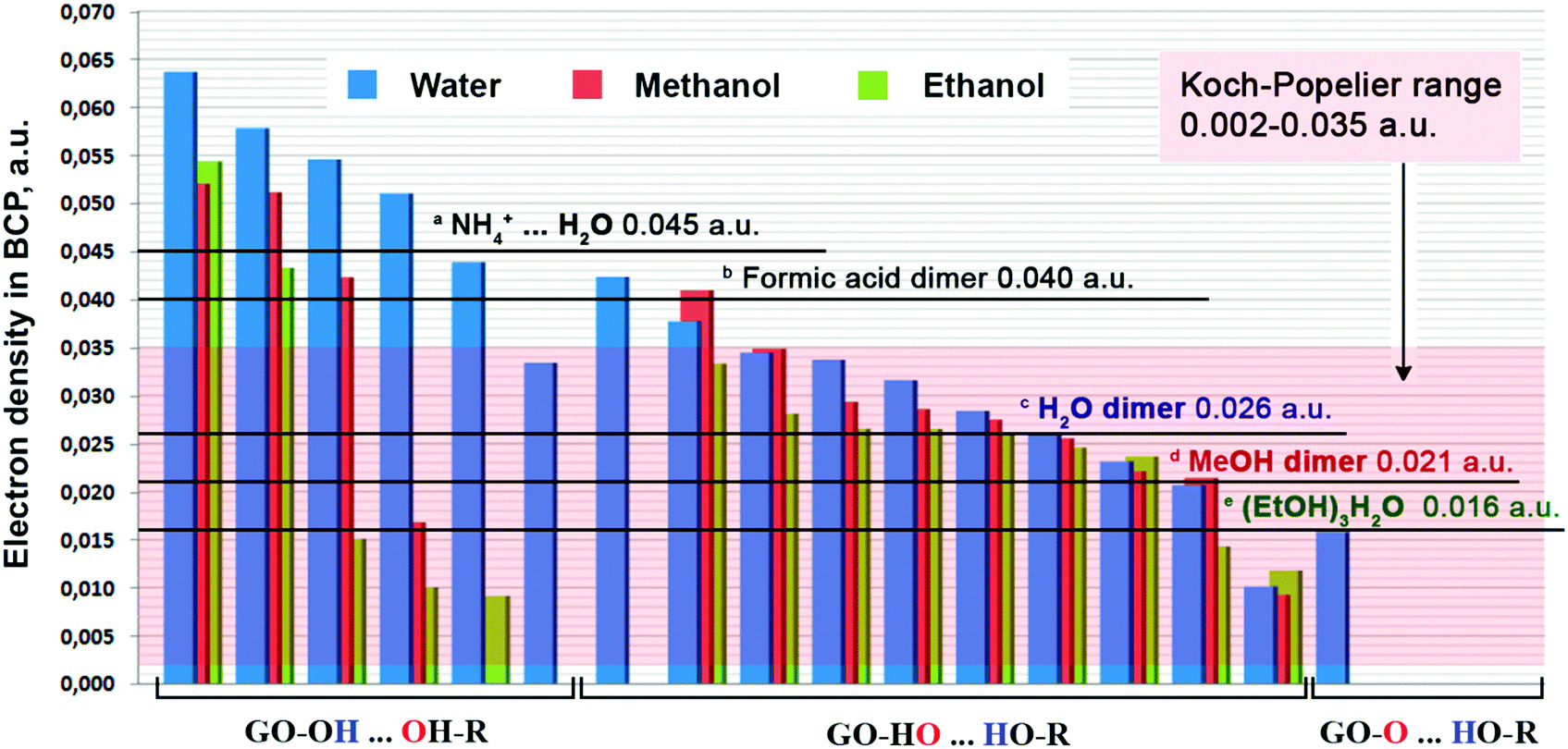 | ||
| Fig. 2 Electronic density ρ(r) in BCPs for the bonds established in water, methanol and ethanol solvates, classified by the bonding types. Some literature data are given for comparison purposes: (a) – ref. 31, (b) – ref. 32, (c) – ref. 33, (d) – ref. 34, and (e) – ref. 35 (EtOH⋯HOEt bond in (EtOH)3H2O). | ||
Weak interactions with energies less than 5 kJ mol−1 have been excluded from considerations.
As evident from Fig. 2, for most of the forming bonds, the electron density values lie in the Koch–Popelier range.29 For the bonds formed between the hydrogen atom of GO and the oxygen atom of the solvent molecule, the electron density values exceed the upper limits of the Koch–Popelier range.
This is especially true for water molecules as the solvent. For comparison purposes, in Fig. 2 we included the calculated values of electron densities ρ(r) for intermolecular interactions in the dimers of water, methanol, ethanol, and methanoic acid, and in the associates of ammonium cations with water molecules. The calculated electron density values for the last two are higher than the corresponding upper limits of the Koch–Popelier range; still the authors of ref. 31 and 32 qualify these interactions as hydrogen bonding.
For all the interactions in the optimized GO solvates, the Laplacian values at critical points lie in the 0.019–0.153 a.u. region (see Tables S1–S3 of the ESI†). The same is true for the distribution of the Laplacian values and for the electron density values in general: for the majority of the established intercations, they fit into the Koch–Popelier range. Thus, we conclude that the solvent–GO interaction can be classified as hydrogen bonding. Interestingly, the electron density values for almost all of the type 1 interactions, are higher than those for the methanoic acid dimers, and even for the charged NH4+–H2O pairs.
The hydrogen bonding in the formic acid dimer is relatively strong due to the high positive charge on the H atom in carboxyl groups. The same is true for the charged ammonium cation. Our model structure contains neither carboxyl groups nor charged functional groups. Still, the electron density between the interacting atoms is higher than that for the two discussed pairs. This fact points at the very strong character of the hydrogen bonding between GO and the solvent molecules, especially for water. Fig. 3 represents the distribution of hydrogen bond energies for all the bonds established between GO and the solvent molecules.
 | ||
| Fig. 3 H-bond energy for water, methanol and ethanol solvates classified by the bonding types. (a) – Ref. 36 and (b) – ref. 34. | ||
Similar to the electron densities, the most frequent type of bonding is type 2 (Scheme 2); however, the highest energy densities are registered for type 1 bonding.
Apparently, in solutions, GO flakes form independent species, i.e. the solvates of non-stoichiometric contents. As evident from Fig. 3, water molecules establish stronger bonds than molecules of methanol and ethanol. Thus, the average bond energies for type 1 bonding are 15.4, 12.0, and 7.3 kcal mol−1 for water, methanol and ethanol, respectively. For type 2 bonding, these values are 7.4, 6.7, and 5.7 kcal mol−1, respectively. Interestingly, the role of epoxide groups in interaction with solvent molecules (type 3 H-bonding) is quite insignificant. It is well-known that epoxy groups hardly form hydrogen bonds, and this is the reason for the poor solubility of ethers in water. The ability of the O atom to share electrons in an R–O–R group is considerably weaker than in an R–O–H group, which can additionally take part in hydrogen bonding through its own H atom. Indeed, the bonding with epoxy groups (type 3) in the studied GO solvates, is accomplished only with one water molecule, and characterized by an interaction energy of less than 4 kcal mol−1. It should be noted that the absolute values of the concerned H-bonding energies may contain errors of the calculation method. Owing to this, their direct comparison with the values obtained by other methods should be done with caution. However, this is not the case when the above-mentioned values are compared to each other, since they are obtained at the same level of theory and using the same method of calculation.
As the reference, in Fig. 3, we provide the ranges for weak, moderate and strong hydrogen bonds, according to the classification made by Parthasarathi et al.31 The majority of the bond energy values fall in the moderate bond region. At the same time, several type 1 bonds fall in the strong bond region. Note, most of the established bonds are stronger than the hydrogen bonds in the methanol dimers, or even in the water dimers. Type 1 bonds are extremely strong. The magnitude of these energy values (Fig. 3) suggests that the water molecules constituting the first contact layer with GO, should be considered as an integral part of GO not only in solution, but also for solid GO obtained from water solution. It was shown earlier by the TGA MS analysis,37 that solid GO loses water in the two main temperature regions: 70–100 °C and 140–160 °C. The lower temperature interval is associated with water weakly physisorbed to GO. The origin of the second temperature interval was not very clear: theoretically, there should not be any more water, if it is already evaporated at ∼100 °C. Now, we can speculate that this is the water, constituting the first contact layer with GO. The loss of this water occurs simultaneously with the decomposition of the oxygen functional groups. Here, we can further speculate that water molecules, constituting the first contact layer, serve as protecting groups, protecting solid GO from decomposition. This might be the case for other solvents capable of forming strong H-bonds with functional groups of GO.
Next, we analyze the dependence of the bond energy on the bond length (Fig. 4). It is well established that these parameters are in the opposite proportion; the dependence might be described by the second-order polynomial.38 The regression equations and curves, presented by solid lines, are shown in Fig. 4. Note, the bond length between GO functional groups and water molecules is shorter than those for methanol and ethanol molecules.
 | ||
| Fig. 4 The H-bond energy versus H⋯O distance curve. The solid lines are the regression curves. | ||
In addition to the topological analysis discussed above, we assessed the strength of H-bonds by geometrical parameters. The structural parameters of the previously discussed H-bonds are given in Table 2 (complete data are given in the ESI† in Tables S4–S6 and in Fig. S3–S5). The analysis of their geometrical characteristics allows one to classify these H-bonds as of a medium strength, in accordance with the Jeffrey classification.39 The average values of the H-bond geometrical parameters in the solvates of GO with water, methanol, and ethanol are shown in Fig. 5. It is evident that the distances and angles of the type 2 H-bond do not change significantly upon changing the solvent. In the case of the type 1 H-bond, the angles and distances decrease and increase, respectively, from water to ethanol. This indicates a decrease in the strength of the H-bond in the water–methanol–ethanol sequence, which is in full agreement with the analysis of the H-bond energy characteristics discussed above.
| Bonding type | H⋯O distance, Å | O⋯HO angle, ° | O⋯O distance, Å |
|---|---|---|---|
| GO–H2O solvate | |||
| 1 | 1.59–1.83 | 152.40–167.95 | 2.59–2.77 |
| 2 | 1.73–2.37 | 145.51–170.77 | 2.67–3.23 |
| GO–MeOH solvate | |||
| 1 | 1.64–2.16 | 130.91–170.78 | 2.62–2.91 |
| 2 | 1.72–2.43 | 142.13–175.64 | 2.71–3.26 |
| GO–EtOH solvate | |||
| 1 | 1.62–2.42 | 117.20–160.45 | 2.58–3.21 |
| 2 | 1.81–2.28 | 147.13–177.86 | 2.80–3.15 |
 | ||
| Fig. 5 Average values of the (A) H⋯O and (B) O⋯O distances, and (C) the OH⋯O angle in the solvates of GO with water, methanol, and ethanol. The inset demonstrates the types of H-bonds. The lines are drawn as a guide to the eye. | ||
In aqueous solutions, the water molecules from the first contact layer can establish additional bonds with “free” water molecules from the bulk solvent (see Fig. 1). For the solvates with methanol and ethanol, the available outer surface of the solvate is formed by hydrophobic alkyl groups. This renders the entire solvated structure hydrophobic, and prevents further interactions with the alcohol molecules from the bulk of the solvent. This must be the main reason inhibiting the solubility of GO in the chosen alcohols. The commonly used Hildebrand and Hansen parameters do not take into account the actual structure of the GO/solvent interface, which is critical for understanding the stability of GO solutions.
In addition, calculations with Grimme's dispersion correction were performed in the GAUSSIAN 09 program. The results of these calculations are represented in Fig. S6 and in Table S7 of the ESI.† The analysis of these data allows us to conclude that the dispersion correction for the present structures does not affect the H-bond energy characteristics.
Preparation of GO solvates
The same order of the bonding energy values for water and alcohols suggests that, in principle, water molecules at the GO/water interface can be replaced by the alcohol molecules if the latter are taken in excess. One only needs to break the strong hydrogen bonds between GO and water molecules, or to overcome some activation barrier. This, if true, must result in the coagulation and precipitation of the alcohol-solvated GO, due to the hydrophobicity of the as-formed solvate surface, and a lowering of the affinity of the solvate surface to the solvent.To confirm this hypothesis, in the next experiment, 29.2 mg paste of GO, containing 33 wt% pure GO, was dissolved in 1.5 mL of DI water (Ω 18 MOm m−1), resulting in an absolutely homogeneous and stable solution with 6 mg mL−1 GO content (see Fig. 6). Next, 10 mL of methanol was added dropwise under constant stirring to obtain a fully homogeneous solution. This solution was transferred to the round-bottom flask, and another 38.5 mL of methanol was added with stirring. This resulted in a GO–water–methanol solution with 3 vol% water and 97 vol% methanol; the GO concentration was 0.1 mg mL−1. A small part of this solution, kept as the control, stayed on the shelf for several weeks without any signs of precipitation (Fig. 6a). The rest of the mixture was refluxed at 67 °C under constant stirring for 5 h. The first signs of coagulation were visible in 1 h. Fig. 6b shows a sample of the coagulated solution. The coagulation of the heated solution and the stability of the non-heated solution confirm our hypothesis. For more evidence, we further separated the methylated GO from the solvent by centrifugation. The introduction of the still-wet GO precipitate into benzene resulted in the dissolution of MeOH–GO in benzene (Fig. 6c). The solution was stable for about 1 min, after which it precipitated. For comparison, GO from the control sample (transferred into methanol, but not heated), does not dissolve in benzene at all (Fig. 6d) (also see the movie in the ESI†). This observation additionally confirms the hydrophobic surface of the methanol-solvated GO.
 | ||
| Fig. 6 Photographs of GO dispersions in methanol–water solution before (a) and after (b) heating. Methylated GO disperses in benzene (c) and the control GO sample does not disperse in benzene (d). Photos (c) and (d) are taken 30 s after agitation. | ||
Interestingly, a similar experiment with ethanol did not result in the coagulation of the solution even after 7 h refluxing. This might be due to the notably lower energy values of hydrogen bonding with ethanol than that with methanol. Also, the kinetic control might play a role: the bulkier and more hydrophobic ethanol molecules are less efficient in replacing water molecules.
The enthalpy of solution of GO in water, methanol and ethanol was measured at 298 K using a semi-adiabatic TAM III solution calorimeter by breaking a sealed ampoule containing a solid GO sample into 100 ml of solvent inside a calorimetric cell. Methanol and ethanol were 99.9% and 99.8% pure, respectively. GO was dried and brought to a constant weight in the presence of P2O5 under reduced pressure (10 mm Hg) for 3 days. The dried GO was transferred to ampoules in a glove box. The measured enthalpy of dissolution of GO in water, methanol and ethanol was found to be −0.1815 ± 0.0010, −0.1550 ± 0.0012 and −0.1040 ± 0.0010 kJ g−1, respectively. Apparently, the observed exothermic effect is associated with the formation of hydrogen bonds between solvent molecules and GO functional groups. These values of the enthalpy of dissolution are in good agreement with our calculation data. The enthalpy values are in the same sequence as the hydrogen bond energies: water > methanol > ethanol.
Based on our findings, now we can explain the solubility of GO in alcohols and some other hydrophilic organic solvents. First of all, alcohols cannot dissolve solid GO. This might happen because the energy gain in the case of alcohols is less than that for water, and to a lesser extent compensates the energy expenditure for breaking the bond between the layers. Secondly, if the direct hydrogen bonding between GO and an alcohol molecule is still established, the latter is oriented by its alkyl part to the exterior, rendering the surface of the as-formed solvate hydrophobic, and preventing the formation of the hydrogen bonding between the GO solvate and the solvent molecules from the bulk. This all renders solid GO insoluble in the alcohols. However, if GO is already in the dissolved state in an aqueous solution, then alcohols interact with water–GO solvates and GO remains in the dissolved state. This explains the solvent exchange phenomena described in ref. 7 and 8. Note that this solvent exchange must occur only in the bulk of solution, but not at the GO/water interface, which most likely remains unchanged. Without heating, the alcohol molecules establish hydrogen bonding with water molecules of the first contact layer, but not directly with GO functionalities. These bonds are relatively weak, and the solvent molecules bonded to the water molecules of the first contact layer are in constant dynamic equilibrium with the molecules in the bulk solvent. This helps to keep the GO–H2O solvate suspended, or dissolved, in the alcohol solvents.
Conclusions
(1) In the systems GO–water, GO–methanol and GO–ethanol, hydrogen bonding is established between GO oxygen functional groups and solvent molecules. The strongest bonding is formed in the GO solvates with water; the bonding with methanol is weaker, and with ethanol is the weakest.(2) The epoxide groups of GO play a very minor role in hydrogen bonding with solvent molecules. All the interaction of GO with solvent occurs mostly via tertiary alcohol groups. The most frequently established type of bonding is the ROH⋯OH–GO type. The less frequent is the RHO⋯HO–GO type; however, this type of bonding is significantly stronger.
(3) The energy of hydrogen bonding between GO and solvent molecules is higher than that of a typical hydrogen bonding. This is especially true for the interactions between GO and water, confirming the extremely highly hydroscopic nature of GO. The magnitudes of bond energies let us consider the water molecules of the first contact layer as an integral part of GO not only in solution, but also for solid GO obtained from water solution.
(4) As for the GO solvates with methanol and ethanol, their outer surface is formed by hydrophobic alkyl groups. This prevents the formation of hydrogen bonding between the hydrophobic solvate surface and solvent molecules in bulk solution, rendering GO insoluble in alcohols. In alcoholic GO solutions prepared by gradual solvent exchange, the solvent molecules do not interact directly with GO functionalities, but with the water molecules constituting the first coordination layer. These bonds are weak, and the dynamic equilibrium between the bonded solvent molecules and those in bulk render GO soluble in alcohols.
Acknowledgements
The work was performed with the support of the Russian Government Program for Competitive Growth of Kazan Federal University.Notes and references
- A. M. Dimiev and S. Eigler, Graphene Oxide: Fundamentals and Applications, John Wiley & Sons, Ltd., United Kingdom, 2016 Search PubMed.
- A. M. Dimiev, Mechanism of formation and chemical structure of graphene oxide, in Graphene Oxide: Fundamentals and Applications, ed. A. M. Dimiev and S. Eigler, John Wiley & Sons, Ltd., United Kingdom, 2016, pp. 36–84 Search PubMed.
- C. Valles, Rhelogy of graphene oxide dispersions, in Graphene Oxide: Fundamentals and Applications, ed. A. M. Dimiev and S. Eigler, John Wiley & Sons, Ltd., United Kingdom, 2016, pp. 121–146 Search PubMed.
- D. Konios, M. M. Stylianakis, E. Stratakis and E. Kymakis, J. Colloid Interface Sci., 2014, 430, 108–112 CrossRef CAS PubMed.
- J. I. Paredes, S. Villar-Rodil, A. Martinez-Alonso and J. M. D. Tascon, Langmuir, 2008, 24, 10560–10564 CrossRef CAS PubMed.
- M. M. Gudarzi, M. H. M. Moghadam and F. Sharif, Carbon, 2013, 63, 403–415 CrossRef.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, K. Konstantinov, E. Moulton, J. M. Razal and G. G. Wallace, ACS Nano, 2013, 7, 3981–3990 CrossRef CAS PubMed.
- M. M. Gudarzi, Langmuir, 2016, 32, 5058–5068 CrossRef CAS PubMed.
- C. A. Batista, R. G. Larson and N. A. Kotov, Science, 2015, 350, 12424771 Search PubMed.
- D. W. Boukhvalov and M. I. Katnelson, J. Am. Chem. Soc., 2008, 130, 10697–10701 CrossRef CAS PubMed.
- L. Wang, Y. Y. Sun, K. Lee, D. West, Z. F. Chen, J. J. Zhao and S. B. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1614061 Search PubMed.
- L. Wang, K. Lee, Y.-Y. Sun, M. Lucking, Z. Chen, J. J. Zhao and S. B. Zhang, ACS Nano, 2009, 3, 2995–3000 CrossRef CAS PubMed.
- J.-A. Yan, L. Xian and M. Y. Chou, Phys. Rev. Lett., 2009, 103, 0868021 Search PubMed.
- J.-A. Yan and M. Y. Chou, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 1254031 Search PubMed.
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed.
- D. N. Laikov, Chem. Phys. Lett., 1997, 281, 151–156 CrossRef CAS.
- D. N. Laikov and Yu. A. Ustynyuk, Russ. Chem. Bull., 2005, 54, 820–826 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- J. Tomasi, B. Mennucci and E. Cances, J. Mol. Struct., 1999, 464, 211–226 CrossRef CAS.
- T. A. Keith, AIMAll program Version 10.11.24, 2010, http://aim. tkgristmill.com.
- C. Erickson, R. Erni, Z. Lee, N. Alem, W. Gannett and A. Zettl, Adv. Mater., 2010, 22, 4467–4472 CrossRef PubMed.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53–56 CrossRef CAS.
- W. Cai, R. D. Piner, F. J. Stadermann, S. Park, M. A. Shaibat, Y. Ishii, D. Yang, A. Velemakanni, S. J. An, M. Stoller, J. An, D. Chen and R. S. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS PubMed.
- S. Huang, Z. Xiao, F. Wang, L. Gan, X. Zhang, X. Hu, S. Zhang, M. Lu, Q. Pan and L. Xu, J. Org. Chem., 2004, 69, 2442–2453 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules: A quantum theory, Oxford University Press, Oxford, 1990 Search PubMed.
- E. Espinosa, I. Alkorta, I. Rozas, J. Elguero and E. Molins, Chem. Phys. Lett., 2001, 336, 457–461 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, J. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747–9754 CrossRef CAS.
- W. Nakanishi, S. Hayashi and K. Narahara, J. Phys. Chem. A, 2008, 112, 13593–13599 CrossRef CAS PubMed.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2006, 110, 3349–3351 CrossRef CAS PubMed.
- O. Galvez, P. C. Gomez and L. F. Pacios, J. Chem. Phys., 2003, 118, 4878–4895 CrossRef CAS.
- J. M. Guevara-Vela, R. Chavez-Calvillo, M. Garcia-Revilla, J. H. Trujillo, O. Christiansen, E. Francisco, K. N. Pendas and T. Rocha-Rinza, Chem. – Eur. J., 2013, 19, 14304–14315 CrossRef CAS PubMed.
- C. D. Keefe and Z. Istvankova, Can. J. Chem., 2011, 89, 34–46 CrossRef CAS.
- S. M. Mejia, M. J. L. Mills, M. S. Shaik, F. Mondragon and P. L. A. Popelier, Phys. Chem. Chem. Phys., 2011, 13, 7821–7833 RSC.
- L. Albrecht and R. J. Boyd, J. Phys. Chem. A, 2012, 116, 3946–3951 CrossRef CAS PubMed.
- S. Eigler, C. Dotzer, F. Hof, W. Bauer and A. Hirsch, Chem. – Eur. J., 2013, 19, 9490–9496 CrossRef CAS PubMed.
- S. J. Grabowski, J. Mol. Struct., 2001, 565, 137–143 CrossRef.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with data of topological and structural parameters of hydrogen bonds. See DOI: 10.1039/c7cp02303k |
| This journal is © the Owner Societies 2017 |