
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePressure inverse solubility and polymorphism of an edible γ-cyclodextrin-based metal–organic framework†
Ewa
Patyk-Kaźmierczak
a,
Mark R.
Warren
b,
David R.
Allan
b and
Andrzej
Katrusiak
 *a
*a
aDepartment of Materials Chemistry, Faculty of Chemistry, Umultowska 89b, 61-614 Poznań, Poland. E-mail: katran@amu.edu.pl
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot, Oxon OX11 0DE, UK
First published on 3rd March 2017
Abstract
A very exceptional effect of pressure-induced dissolution has been revealed for an edible metal–organic framework, γ-CD-MOF-1, formed using γ-cyclodextrin and KOH base. In addition, a new polymorph of γ-CD-MOF-1 has been obtained. The trigonal structure is a symmetry sub-group modification of the cubic form. The pressure-induced dissolution of γ-CD-MOF-1 and its polymorphism are shown to be closely related and regulated by adsorption in the pores, as well as the guest framework interactions.
Introduction
Metal–organic frameworks (MOFs) have been known for decades,1 and they continuously gain new scientific interest. Owing to their large active area, as well as their structural and functional diversity, MOFs have various applications in industry and science.2–9 For example, they are promising materials for increasing the efficiency of fuel gas storage.10,11 However, most MOFs and their syntheses are toxic, which limits their applicability. Therefore safer, cheaper, easier-to-synthesize and more environment-friendly alternatives are needed. In 2010, the first edible MOFs were reported,12 which meant new possibilities for applications in pharmaceutical and nutrition technologies. These edible MOFs can be easily obtained from readily available components, e.g. γ-cyclodextrin (γ-CD) and potassium hydroxide, and only water and methanol or ethanol is needed to yield high quality crystals. The γ-CD-MOFs mark an interesting, new direction in MOF chemistry. They have already been shown to be highly selective CO2 adsorbers,13 and they are also efficient for separating mixed solvents.14In γ-CD-MOFs, the cations, preferentially of the 1st and 2nd group metals, are coordinated by the γ-cyclodextrin molecules in the presence of hydroxide counter-ions. In particular, potassium easily yields high-quality crystals with γ-cyclodextrin. The cyclodextrin cavities arrange into a three-dimensional porous structure with channels that intersect at the voids between the molecules. Owing to the 8 glucose units constituting the γ-CD molecule, it can adopt a four-fold symmetric conformation. This directly influences the structure of the γ-CD-MOF crystals, enabling their highly-symmetric space group I432. However, it is known that, in general, MOFs are highly susceptible to temperature and pressure, as well as the contents of the pores.12 Hence this high-pressure and varied-temperature study was aimed at determining the pressure stability of γ-CD-MOF-1 and its possible symmetry changes. We have found a new trigonal β form of this MOF and compared it with the previously reported cubic polymorph, now labelled with the letter α.
The effects of the external stimuli on the structure and properties of MOFs can be very different.15–19 In extreme cases, a porous structure can even be destroyed.20 The synthesis conditions of the pressure, temperature, and solvent etc. can affect the product structure. The response to the pressure stimuli and stability of MOFs depends on the framework architecture as well as its interactions with the pressure-transmitting medium. In this respect, the γ-CD linker, which is flexible and contains an open cavity, can easily deform and affect the whole MOF-crystal structure. The K+-coordination bonds are weaker compared to the heavier metal ions that are commonly used for making MOFs, which increases their sensitivity to external conditions.
Experimental
γ-Cyclodextrin (γ-CD) and potassium hydroxide (KOH) from Sigma-Aldrich were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 molar ratio and dissolved in distilled water. The flask with this solution was sealed in a container filled with methanol (in other experiments it was filled with isopropanol) and large transparent hexahedral crystals were obtained by the vapor diffusion method. The crystals, which were covered with the mother liquor (either methanol:water, isopropanol:water or methanol), were studied by X-ray diffraction inside a quartz capillary at both ambient conditions and in the 195–310 K temperature range. For high-pressure experiments, the sample was recrystallized in situ (Fig. 1 and Fig. S1, S2, ESI†) in a modified Merrill–Bassett diamond-anvil cell (DAC)21 and measured after compression in the 0.16–1.76 GPa pressure range. Beyond these pressure limits, the sample crystals dissolved, which hampered the X-ray diffraction experiments at still higher pressures (Fig. 1, 7 and Fig. S3, ESI†). The pressure inside the DAC was measured by the ruby fluorescence method22 with a Photon Control Inc. spectrometer affording an accuracy of 0.02 GPa.
:8 molar ratio and dissolved in distilled water. The flask with this solution was sealed in a container filled with methanol (in other experiments it was filled with isopropanol) and large transparent hexahedral crystals were obtained by the vapor diffusion method. The crystals, which were covered with the mother liquor (either methanol:water, isopropanol:water or methanol), were studied by X-ray diffraction inside a quartz capillary at both ambient conditions and in the 195–310 K temperature range. For high-pressure experiments, the sample was recrystallized in situ (Fig. 1 and Fig. S1, S2, ESI†) in a modified Merrill–Bassett diamond-anvil cell (DAC)21 and measured after compression in the 0.16–1.76 GPa pressure range. Beyond these pressure limits, the sample crystals dissolved, which hampered the X-ray diffraction experiments at still higher pressures (Fig. 1, 7 and Fig. S3, ESI†). The pressure inside the DAC was measured by the ruby fluorescence method22 with a Photon Control Inc. spectrometer affording an accuracy of 0.02 GPa.
 | ||
| Fig. 1 Dissolution of γ-CD-MOF-1-α powder (a) and the single crystal growth (b–d) from the water:methanol mixture inside the DAC: (a) polycrystalline sample at 0.16 GPa/295 K; (b) one small crystal seed by the left edge of the gasket; (c) crystal growth at 345 K; (d) the single crystal at 295 K and 0.16 GPa. A small ruby sphere lies by the left edge of the chamber. cf. Fig. S2 in the ESI.† | ||
The in situ microscopic observation of the pressure-induced dissolution of the sample crystals was continued to over 2.0 GPa (Fig. 2). The compression of the sample crystal eventually led to its complete dissolution at 2.1 GPa. The attempted recrystallization by further increasing the pressure resulted most likely in the water of mother liquor frozen in the form of ice VI. Upon decrease of the pressure, small hexahedral crystals appeared at 0.53 GPa (Fig. 2 and Fig. S3, ESI†). The γ-CD-MOF-1 crystals grown under ambient conditions, and compressed in the DAC in fluorinert, methanol or ethanol, showed very weak or even hardly detectable diffraction, and gave no satisfactory crystallographic information.
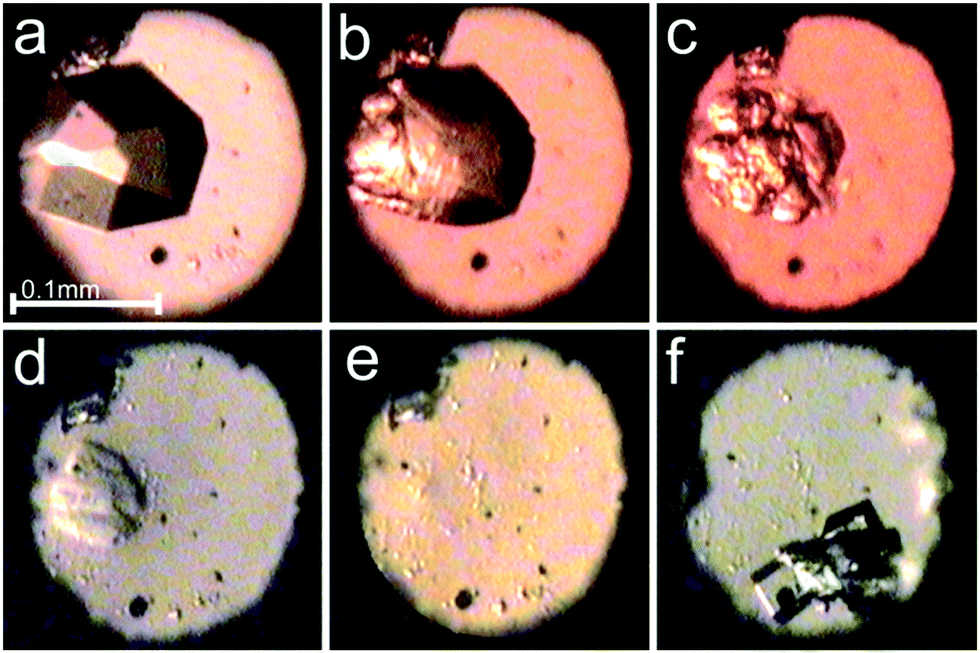 | ||
| Fig. 2 Selected photographs of the pressure dissolution of a γ-CD-MOF-1-α crystal in the water:methanol mixture inside the DAC (a–e) and a few single crystals subsequently grown upon lowering the pressure (f); (a) a single crystal at 0.19 GPa/295 K; (b–e) the crystal dissolving at 0.67, 2.09, 2.15 and 2.10 GPa; (f) single crystals at 295 K and 0.53 GPa. A small ruby chip lies by the left edge of the chamber. cf. Fig. S3 in the ESI.† | ||
The X-Ray diffraction experiments were performed either with synchrotron radiation (λ = 0.6889 Å) using a 4-circle Newport diffractometer equipped with a Pilatus 300K detector at beamline I19 of Diamond Light Source or with MoKα graphite monochromated radiation and a 4-circle Xcalibur diffractometer equipped with an EOS CCD detector and a gaseous-nitrogen open-flow Oxford Cryosystems attachment. For the synchrotron measurements, GDA software was used for collecting the data and converting them for the CrysAlisPro software, which was used for determining the UB-matrix, absorption corrections and data reduction for all collected data. Destruction of the sample (Fig. 3 and 7) affected the data sets and precision of the refinements at 0.41 and 0.49 GPa, as well as hindered the crystal structure solution and refinement at 0.26 and 1.76 GPa. The temperature measurements were aimed at obtaining the temperature dependence of the unit-cell parameters and volume only, and due to the low resolution of collected data no structure refinement was performed for the crystals, except for those in the isopropanol:water mixture. The difference in the diffracting properties of the crystals plausibly originates from the local differences in the crystal structure distortions due to the different packing and interactions of the small molecules of methanol.
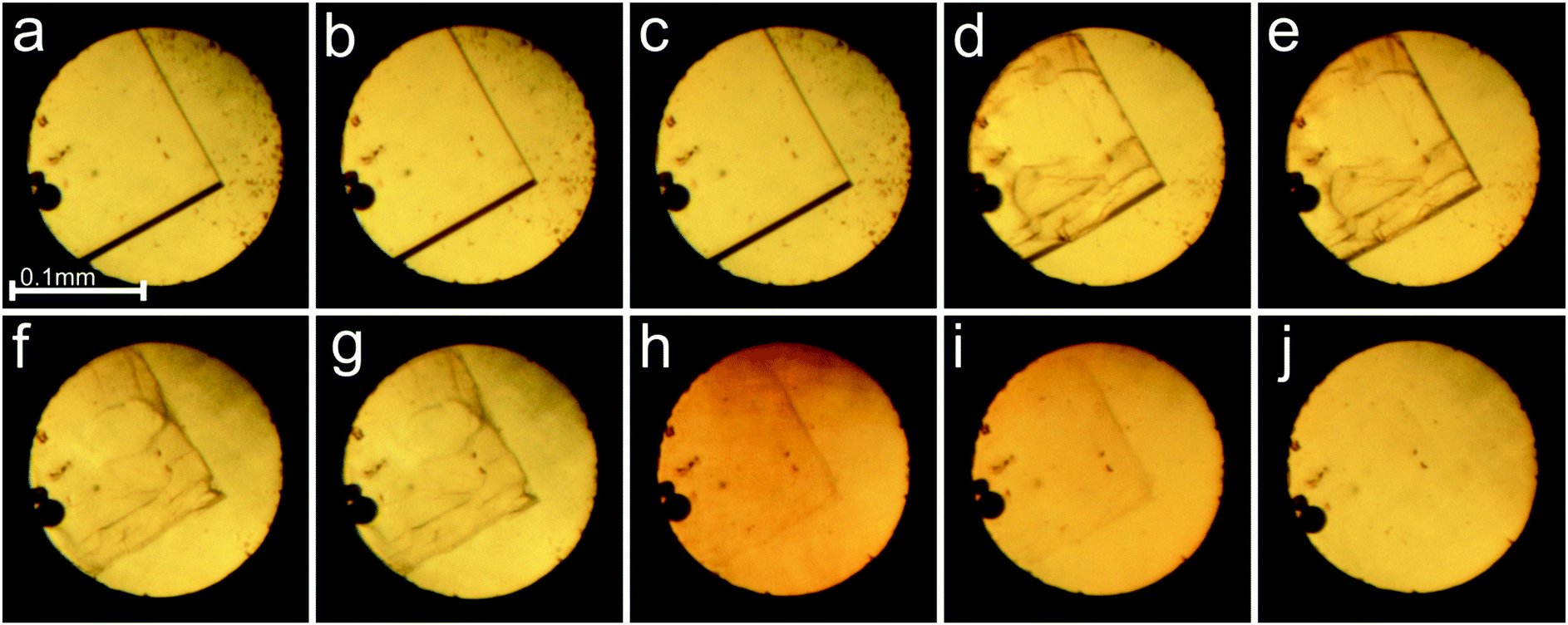 | ||
| Fig. 3 Pressure dissolution and radiation damage of the γ-CD-MOF-1-α crystal; the pictures were taken before and after the measurements at 0.16 GPa (a and b); 0.25 GPa (c and d); 0.33 GPa (e and f); 0.41 GPa (g and h); 0.49 GPa (i and j). | ||
The upper limit of the temperature study resulted from the significant experimental difficulties related to the low quality of the crystals, the concomitant mixed phases I and II, their twinning and the disordering in their structures. Due to these features, the resolution of diffraction data was low when measured with the laboratory diffractometers equipped with sealed X-ray tubes, and we had to resort to synchrotron radiation. Even for the single-crystal diffraction data measured with a state of the art synchrotron beam, the structural refinements required constraints on the molecular dimensions and temperature factors. These sample-quality related constraints confined the diffraction experiments to low-temperature and high-pressure measurements, and no meaningful high-temperature diffraction data above 310 K could be recorded.
The crystal structures were solved either by direct methods using the program Shelxs,23 or intrinsic phasing using Shelxt,23followed by refining using least-squares in the Shelxl program.23 The hydrogen atoms were located using the molecular geometry with Uiso equal to 1.2 Ueq for the C-carriers and 1.5 Ueq for the O-carriers. The O–H and C–H bond lengths were fixed to the distances of 0.82 Å for the oxygen atoms, and 0.98 or 0.97 Å for the tertiary and secondary carbon atoms, respectively. The crystal structures of γ-CD-MOF-1 have been deposited with the Cambridge Crystallographic Data Centre (CCDC 1529141–1529165) and with the Crystal Open Database.
The single-crystal X-ray diffraction experiments on γ-CD-MOF-1 revealed, apart from the known cubic form (hereafter labelled as phase α), a new trigonal form of the space group R32 (labelled as phase β), similar to the γ-CD-MOF-1 crystal with adsorbed benzoate ions.12 The crystallographic data of the γ-CD-MOF-1 phases are listed in Tables S1 and S2 in ESI,† and summarized in Table 1.
| Phase | γ-CD-MOF-1-α | γ-CD-MOF-1-β | |
|---|---|---|---|
| Pressure (GPa) | 0.16 | 1.36 | 0.0001 |
| Temperature (K) | 295 | 295 | 295 |
| Crystal system | Cubic | Cubic | Trigonal |
| Space group | I432 | I432 | R32 |
| a (Å) | 31.347(1) | 30.898(3) | 43.676(1) |
| c (Å) | 28.186(1) | ||
| Volume (Å3) | 30802(3) |
29499(9) |
46565(3) |
| Z | 12 | 12 | 18 |
| Final R1 (I > 4σI) | 0.0745 | 0.0907 | 0.1354 |
| R unit cell | |||
| a (Å) | 27.156(4) | 26.756(2) | 26.928(1) |
| α (°) | 109.47(2) | 109.40(1) | 108.47(<1) |
| V (Å3) | 15419.00(2) | 14783(2) |
15563.9(8) |
We have observed the symmetry change of the sample crystal, from trigonal to cubic, when kept in an isopropanol:water mixture and monitored by X-ray diffraction at 295 K/0.1 MPa over 12 days. The crystal transforms from phase α to phase β after lowering the temperature to 245 K. Moreover, crystals of mixed cubic and trigonal phases were grown as the pressure was increased to a few MPa (Fig. S4, ESI†), however only crystals of the pure phase α were obtained after recrystallization at 0.25 GPa. The performed experiments are described in detail in the ESI.†
Results and discussion
We have established that γ-CD-MOF-1 can crystallize into two similar crystal forms, either the cubic space group I432 (phase α) or trigonal space group R32 (phase β). The γ-CD-MOF-1 structures appear very similar when viewed down the directions of the channel pores: in the trigonal phase, β, along the crystal directions [21![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) ], [
], [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1] and [] and in the cubic phase, α, along the directions [100], [010] and [001] (Fig. 4). However, in γ-CD-MOF-1-β, the glucose units are distorted and the four-fold symmetry of the channels is broken. The overall symmetry of the γ-CD-MOF-1-β crystal is lowered, and the channel voids are not ideally perpendicular (like in phase α), but at 87.96°.
1] and [] and in the cubic phase, α, along the directions [100], [010] and [001] (Fig. 4). However, in γ-CD-MOF-1-β, the glucose units are distorted and the four-fold symmetry of the channels is broken. The overall symmetry of the γ-CD-MOF-1-β crystal is lowered, and the channel voids are not ideally perpendicular (like in phase α), but at 87.96°.
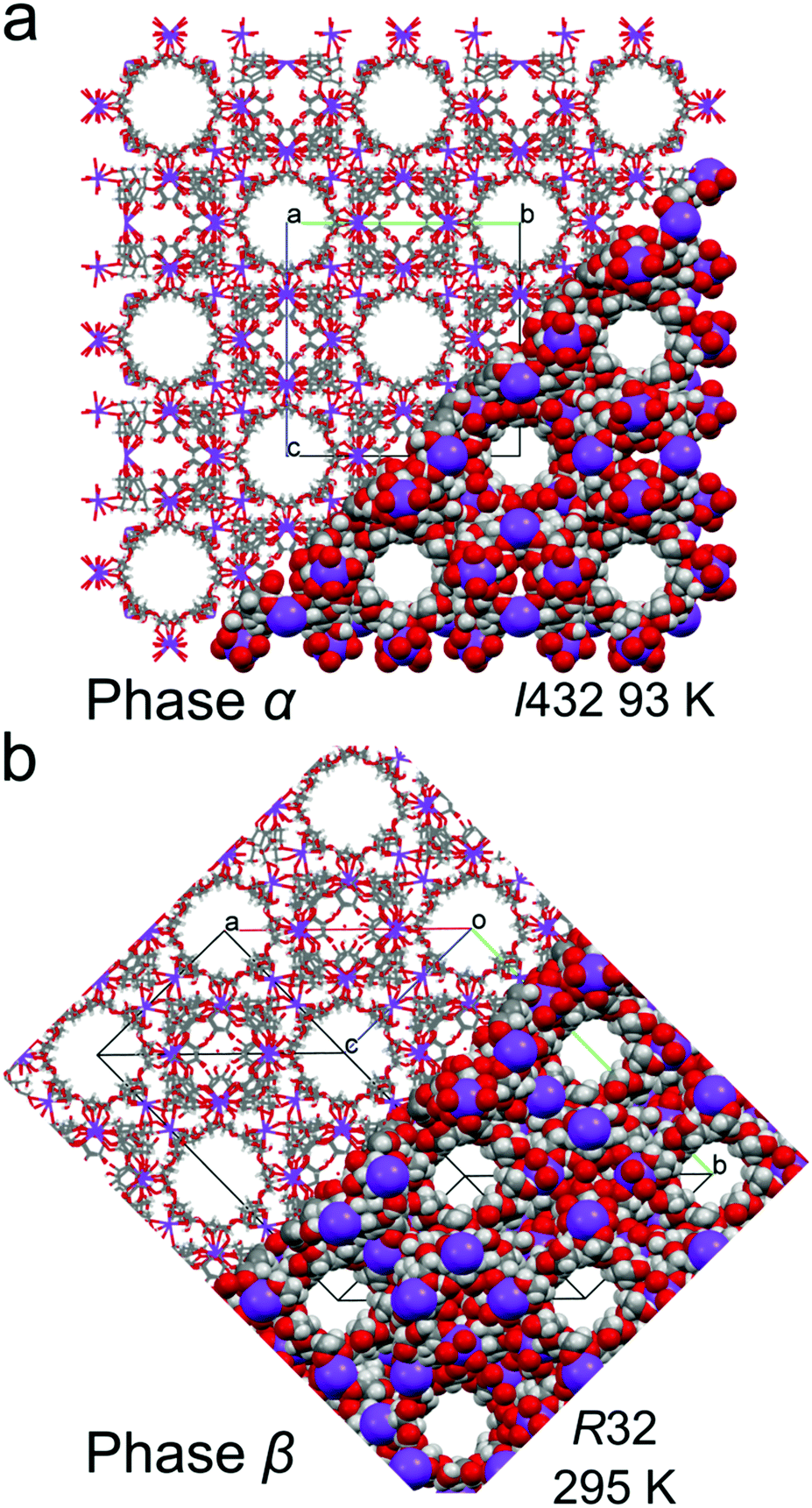 | ||
| Fig. 4 Crystal structures of (a) cubic γ-CD-MOF-1-α and (b) trigonal γ-CD-MOF-1-β, presented in part as the capped-stick and space-filling models, viewed along the channel voids. | ||
Each γ-CD molecule, both in phases α and β, is bonded by 8 potassium cations, and each cation is 8-fold coordinated by 4 γ-CD molecules. In γ-CD-MOF-1-α, only one fourth of the γ-CD molecule is symmetry independent (Fig. S5, ESI†). Consequently, the γ-CD ring consists of 4 identical maltose units. In γ-CD-MOF-1-β, small displacements of the atoms and their thermal vibrations distort the individual glucose units, which differentiates their positions and breaks the 4-fold symmetry. As shown in Fig. 5, the γ-CD rings and coordinated K+ cations of the α and β phases superimpose nearly exactly, with the exception of small distortions.
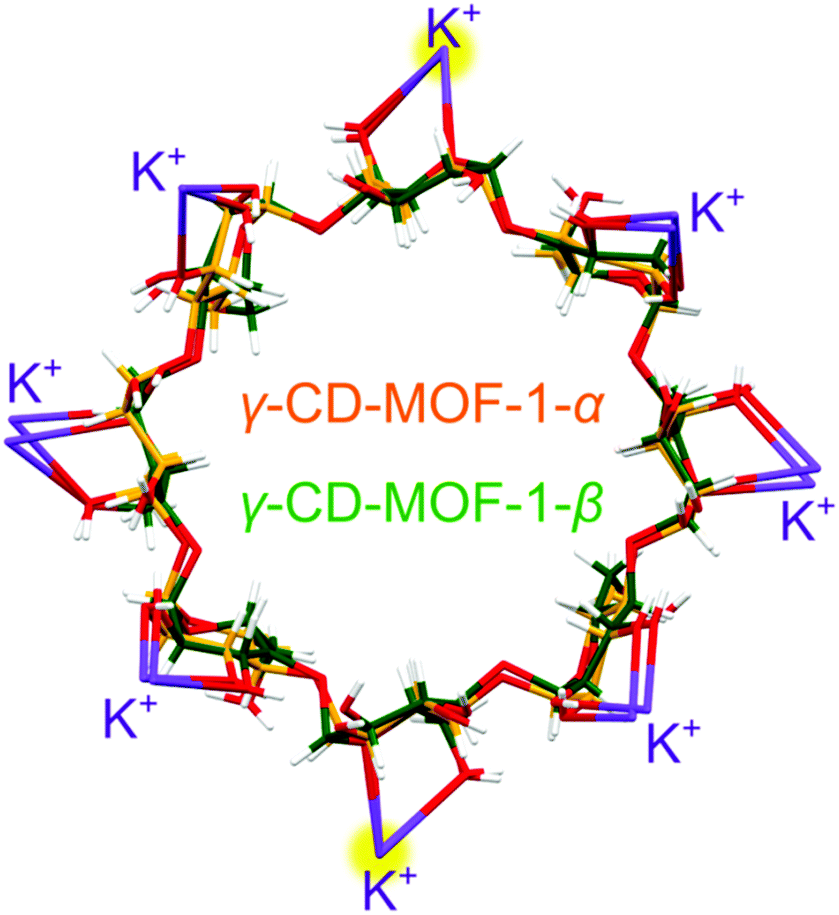 | ||
| Fig. 5 The γ-cyclodextrin molecules of the γ-CD-MOF-1 phases α (orange) and β (green) at 295 K/0.1 MPa, which are overlaid in one drawing, together with their 8 coordinated potassium cations shown in purple; two of the K+ cations are exactly superimposed and highlighted in yellow. | ||
It is well known that the cubic body-centered Bravais lattice I can be transformed to the equivalent rhombohedral R lattice (Fig. S12, ESI†). Consequently, the R lattice of phase β can be considered as a distorted version of the cubic phase α. The strain in the lattice of phase β can be expressed by the distortion of the angle αR of the rhombohedral unit cell derived from the hexagonal unit-cell parameters aH and cH by equation:
| α = 2sin−1{1.5·[3 + (cH/aH)2]−0.5}. |
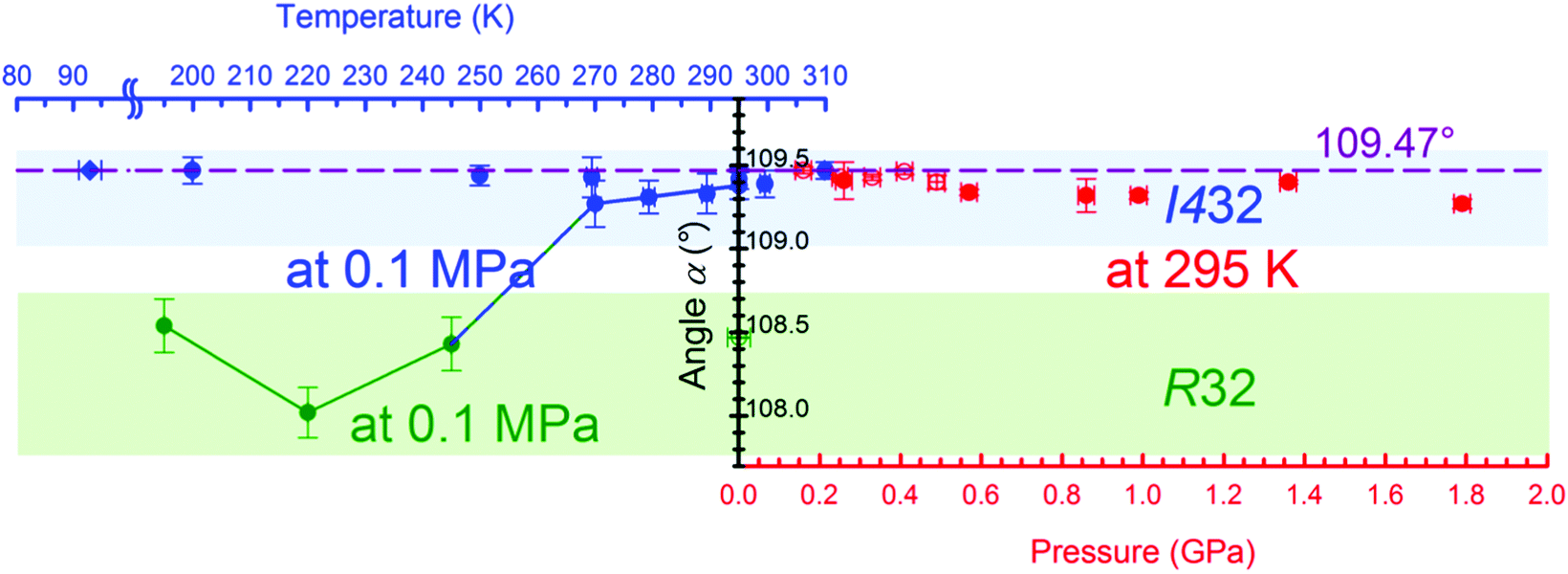 | ||
| Fig. 6 The pressure (red) and temperature (blue and green) dependence of angle α in the rhombohedral lattice assigned for the phases α and β of γ-CD-MOF-1. The ideal value of the α angle for the cubic I lattice is marked with the purple dashed line. The solid blue and green lines guide the eye for the sample transforming from cubic phase α (highlighted in blue) to trigonal phase β (highlighted in green). | ||
The intriguing temperature behavior of the γ-CD-MOF-1 crystals, which are exhibiting different symmetries at the same temperature and pressure conditions (Fig. S8 and S9, ESI†), can be related to the inclusion of solvent molecules in the channel pores. It appears that the solvent adsorbed in the pores supports the 4-fold symmetry of the cyclodextrin molecules and the cubic symmetry of the crystal in phase α. The adsorption is a kinetic process, and the trigonal crystals of phase β in the mother liquor acquire the cubic symmetry of phase α after some time. On lowering the temperature to 245 K the pores decrease their volume by forcing the guest molecules out, however the mother liquor, which partly consists of water, starts to freeze and prevents any transport to and from the pores. The differences in the unit-cell parameters of the crystal samples immersed in different solvents and their mixtures (Fig. S8–S10, ESI†) originate from the adsorption of the molecules in the porous channels. Depending on the type and ratio of the components of the mother liquor, their different interactions with the framework affect the γ-CD-MOF structure and the crystal symmetry. Similarly, when γ-CD-MOF is recrystallized under high pressure, the solvent is forced into the porous channels and enforces higher symmetry of the crystal. Thus it is very likely that phases α and β differ in their composition (i.e. type and amount of solvent in the channel pores), which in strict terms excludes their classification as polymorphs and also as one compound of defined composition undergoing a phase transition in the strictly physical sense. However, it is characteristic of transforming MOFs that some transport of guest molecules will be involved, and such different MOF structures are treated as polymorphs.24,25
The cubic form α of the space group I432 is retained during the compression up to at least 1.76 GPa, when the crystal dissolves (Fig. 7). It is noteworthy that the molecular volume (Vm = V/Z) of γ-CD-MOF-1-β at 295 K/0.1 MPa is higher than that of the ambient and high-pressure structures of γ-CD-MOF-1-α (Fig. 7). This, along with the filling effect of the solvent molecules forced into the porous channels, may be responsible for the preferential crystallization of phase α at high pressure.
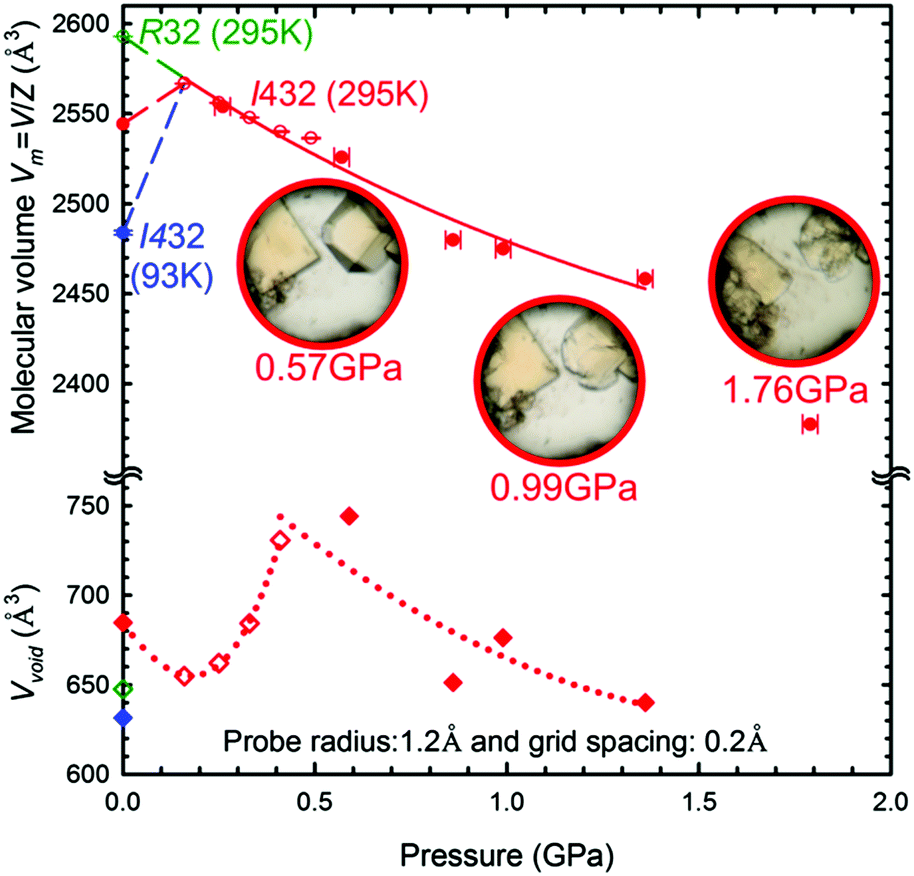 | ||
| Fig. 7 The pressure dependence of the molecular volume (Vm = V/Z; circles) and solvent-accessible volume of the pores (Vvoid; diamonds) in γ-CD-MOF-1 phases α (red and blue) and β (green). The open and filled symbols indicate the diffraction data collected with the synchrotron and MoKα radiation, respectively. The insets show the pressure-induced dissolution and radiation damage in the sample crystals in the 0.57–1.76 GPa range. The solid, dashed and doted lines are for guiding the eye only. | ||
We have established that the γ-CD-MOF-1-α crystals resemble those of pristine α-cyclodextrin26 in this respect that they both exhibit the effect of pressure-induced dissolution. This behavior is the opposite to that observed for most of substances. Thus the γ-CD-MOF-1 crystals can be grown in solution on releasing the pressure (Fig. S3, ESI†). This extremely unusual behavior is analogous to that of pressure-induced melting of ice Ih, which is connected to the lower specific density of ice compared to that of water, i.e. V(Ih) > V(water). It is plausible that the pressure-induced dissolution of γ-CD-MOF-1-α is connected to an analogous reversed volume relation between the crystal and solution. Such a reversal could be caused by the better access of the solvent to the γ-CD cavities in the isolated molecules at high pressure than in the compressed γ-CD-MOF-1-α structure. It complies with the pressure-promoted solvation observed in MOFs and molecular crystals.27–30 It allows more compact packing of the molecules in the limited space, enabling the formation of solvates with possible technological applications.31 The molecular volume of the γ-CD-MOF-1 phase α crystal at 0.16 GPa is higher than that at ambient conditions. This pressure-induced increase can be explained only by the solvent-intake stretching the framework (Fig. 7). When the γ-CD-MOF-1-α crystal is gradually pressurized, additional solvent molecules are pushed into the channel pores, which increases the solvent accessible volume (Fig. 7). The solvent-accessible volume within the trigonal γ-CD-MOF-1-β crystal structure is about 25% of the total unit-cell volume and it is lower than that in the cubic α form in the presence of isopropanol under the same conditions (295 K/0.01 MPa). At 0.16 GPa the volume in phase α is 25.5%, which is 1.5% lower than that at ambient conditions, probably due to the presence of methanol instead of isopropanol in the mother liquor, and it increases up to 0.5 GPa where it saturates around 30% (Fig. 7 and Fig. S11, ESI†). The increased volume within the channel voids, involving the γ-cyclodextrin cavities, contrasts with the decreasing volume of the unit-cell, so it appears that the expansion of the cyclodextrin cavities is compensated by changing the conformation and distances of the cyclodextrin molecules, and regulated by the coordination bonds between the oxygen atoms of the glucose units and potassium ions (Fig. S6, ESI†). Thus, the strained cohesion forces counteract the increased intake of the solvent, which at some pressures destabilizes the structure and leads to its dissolution.
Attempts to recrystallize γ-CD-MOF-1, after it completely dissolved when increasing the pressure further to above 2 GPa, resulted in the formation of crystals that were probably ice VI. Thus at this pressure, the solution components separated, which can lead to a new form of γ-CD-MOF-1 crystal, or its amorphous state.
Conclusions
Crystals of γ-CD-MOF-1 are susceptible to the environmental conditions. Two crystalline forms of this compound, cubic γ-CD-MOF-1-α and trigonal γ-CD-MOF-1-β, exist at ambient and low temperature conditions. Despite the symmetry difference, both of these forms are highly similar in their lattices, the positions of the molecules and ions, and the architecture of the porous channels. At high pressure, the α form is more favorable than β, which is not obtained in pure form by the high-pressure crystallization at all. The formation of the β phase has been connected to the kinetics of the adsorption of the solvent in the pores, which is quicker at high-pressure and slows down at low temperature. Above 1.7 GPa, no transitions of the α phase were detected, however a very unusual dissolution of the sample was observed with increasing pressure. This exceptional feature can be employed technologically for exchanging the guest molecules in the γ-CD cavities for pharmacological purposes. The pressure-induced dissolution has been explained by the volume changes relating to the interactions of the solvent molecules, and their better access to the γ-CD cavities that are partly blocked in the crystal. In addition, the swelling of γ-CD-MOF-1-α in the range up to 0.5 GPa implies increased adsorption from the surrounding fluid, and also that pressure can be used for modifying the composition of this promising non-toxic porous material.Acknowledgements
We are grateful to Mrs Michalina Anioła from the Chemistry Faculty of the Adam Mickiewicz University, the Wielkopolskie Centrum Zaawansowanych Technologii for the experimental support, and to Dr Stephen Moggach from the University of Edinburgh for his useful discussion. We gratefully acknowledge the Diamond Light Source Ltd, where part of the experimental work was conducted, for hosting Dr Ewa Patyk-Kaźmierczak as a part of the Erasmus+ program. This study was founded by the Preludium program no. 2015/17/N/ST5/01927 of the National Science Center, Poland. Dr Ewa Patyk-Kaźmierczak is a laureate of the START program of the Foundation for Polish Science and Poznań City Hall stipend for young researchers.Notes and references
- J. H. Rayner and H. M. Powell, J. Chem. Soc., 1952, 319–328 RSC.
- S.-M. Xie, M. Zhang, Z.-X. Fei and L.-M. Yuan, J. Chromatogr. A, 2014, 1363, 137–143 Search PubMed.
- H.-Y. Huang, C.-L. Lin, C.-Y. Wu, Y.-J. Cheng and C.-H. Lin, Anal. Chim. Acta, 2013, 779, 96–103 CrossRef CAS PubMed.
- R. Plessius, R. Kromhout, A. L. D. Ramos, M. Ferbinteanu, M. C. Mittelmeijer-Hazeleger, R. Krishna, G. Rothenberg and S. Tanase, Chem. – Eur. J., 2014, 20, 7922–7925 CrossRef CAS PubMed.
- F.-X. Coudert, A. U. Ortiz, V. Haigis, D. Bousquet, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, N. Geoffroy, J.-P. Bellat, G. Ortiz, G. Chaplais, J. Patarin and A. Boutin, J. Phys. Chem. C, 2014, 118, 5397–5405 CAS.
- J. Zhuang, C.-H. Kuo, L.-Y. Chou, D.-Y. Liu, E. Weerapana and C.-K. Tsung, ACS Nano, 2014, 8, 2812–2819 CrossRef CAS PubMed.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- L. Ma and W. Lin, in Functional Metal–Organic Frameworks: Gas Storage, Separation and Catalysis, ed. M. Schröder, Springer Berlin Heidelberg, 2009, pp. 175–205 Search PubMed.
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44–53 RSC.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2013, 5, 32–51 RSC.
- R. A. Smaldone, R. S. Forgan, H. Furukawa, J. J. Gassensmith, A. M. Z. Slawin, O. M. Yaghi and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8630–8634 CrossRef CAS PubMed.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS PubMed.
- K. J. Hartlieb, J. M. Holcroft, P. Z. Moghadam, N. A. Vermeulen, M. M. Algaradah, M. S. Nassar, Y. Y. Botros, R. Q. Snurr and J. F. Stoddart, J. Am. Chem. Soc., 2016, 138, 2292–2301 CrossRef CAS PubMed.
- A. J. Graham, D. R. Allan, A. Muszkiewicz, C. A. Morrison and S. A. Moggach, Angew. Chem., Int. Ed., 2011, 50, 11138–11141 CrossRef CAS PubMed.
- W. Cai, A. Gładysiak, M. Anioła, V. J. Smith, L. J. Barbour and A. Katrusiak, J. Am. Chem. Soc., 2015, 137, 9296–9301 CrossRef CAS PubMed.
- W. Cai and A. Katrusiak, Nat. Commun., 2014, 5, 4337 CAS.
- K. J. Gagnon, C. M. Beavers and A. Clearfield, J. Am. Chem. Soc., 2013, 135, 1252–1255 CrossRef CAS PubMed.
- M. Andrzejewski and A. Katrusiak, J. Phys. Chem. Lett., 2017, 8, 279–284 CrossRef CAS PubMed.
- S. C. McKellar, A. J. Graham, D. R. Allan, M. I. H. Mohideen, R. E. Morris and S. A. Moggach, Nanoscale, 2014, 6, 4163–4173 RSC.
- L. Merrill and W. A. Bassett, Rev. Sci. Instrum., 1974, 45, 290–294 CrossRef.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- D. Aulakh, J. R. Varghese and M. Wriedt, Inorg. Chem., 2015, 54, 8679–8684 CrossRef CAS PubMed.
- M. Wriedt, A. A. Yakovenko, G. J. Halder, A. V. Prosvirin, K. R. Dunbar and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 4040–4050 CrossRef CAS PubMed.
- R. Granero-García, F. J. Lahoz, C. Paulmann, S. Saouane and F. P. A. Fabbiani, CrystEngComm, 2012, 14, 8664–8670 RSC.
- H. Tomkowiak, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2013, 13, 121–125 CAS.
- M. Anioła, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2014, 14, 2187–2191 Search PubMed.
- A. Olejniczak, M. Podsiadło and A. Katrusiak, New J. Chem., 2016, 40, 2014–2020 RSC.
- E. V. Boldyreva, Z. Kristallogr., 2013, 229, 236–245 Search PubMed.
- F. P. A. Fabbiani, G. Buth, D. C. Levendis and A. J. Cruz-Cabeza, Chem. Commun., 2014, 50, 1817–1819 RSC.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Extended experimental; detailed crystallographic information. CCDC 1529141–1529165. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cp00593h |
| This journal is © the Owner Societies 2017 |