
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater in cellulose: evidence and identification of immobile and mobile adsorbed phases by 2H MAS NMR†
E. L.
Lindh‡
abc,
C.
Terenzi‡§
 *ab,
L.
Salmén
bc and
I.
Furó
a
*ab,
L.
Salmén
bc and
I.
Furó
a
aDivision of Applied Physical Chemistry, Department of Chemistry, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden. E-mail: ct499@cam.ac.uk
bWallenberg Wood Science Center, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden
cInnventia AB, Box 5604, SE-114 86 Stockholm, Sweden
First published on 18th January 2017
Abstract
The organization of water molecules adsorbed onto cellulose and the supramolecular hydrated structure of microfibril aggregates represents, still today, one of the open and complex questions in the physical chemistry of natural polymers. Here, we investigate by 2H MAS NMR the mobility of water molecules in carefully 2H-exchanged, and thereafter re-dried, microcrystalline cellulose. By subtracting the spectral contribution of deuteroxyls from the spectrum of hydrated cellulose, we demonstrate the existence of two distinct 2H2O spectral populations associated with mobile and immobile water environments, between which the water molecules do not exchange at the NMR observation time scale. We conclude that those two water phases are located at differently-accessible adsorption sites, here assigned to the cellulose surfaces between and within the microfibril aggregates, respectively. The superior performance of 2H MAS NMR encourages further applications of the same method to other complex systems that expose heterogeneous hygroscopic surfaces, like wood cell walls.
Introduction
One of the leading challenges in the study of the ever-increasing family of new materials derived from cellulose – the most available and useful natural polymer – is to fully understand the way in which water molecules are organized upon adsorption onto the cellulose microfibrils.1–7 There are many entangled reasons why a definite answer to this apparently simple question, pertinent also to other systems,8 still seems to elude us. Cellulose has a strong affinity for other hydroxyl-bearing systems, specifically for small molecules like water. In addition, it has a complex supramolecular organization with anisotropic microfibrils – stabilized by van der Waals interactions and by a network of intra- and inter-molecular hydrogen bonds – joined into larger microfibril aggregates. As the detailed physico-chemical structure of cellulose remains itself a matter of lively debate, inhomogeneities such as (i) the site-specific reactivity of surface hydroxyls and (ii) the existence of crystalline and amorphous domains, differently accessible to adsorbed water, inherently add further complexities.Besides being of fundamental interest, the interaction of cellulose with water carries a lot of practical importance, too. As one well-known example,9,10 mechanical properties, structural stability and hydration state are inherently connected in cellulose. As another example, the processing of cellulose by enzymatic degradation also exhibits a water content dependence.11 Likewise, other biomolecules depend on water in order to function: in particular, the enzymatic activity of proteins typically ceases under dry conditions. Besides adjacent to protein surfaces, water in proteins may also exist as deeply-buried and strongly-hindered molecules, which may undergo very slow exchange with the surrounding mobile water phase, and thus can be seen as a structural element of the protein.12–18 Hence, understanding the state of immobile water molecules in hydrated natural polymers has a high biological relevance.
Here, we investigate the state of the water adsorbed in cellulose (hereby referred to as WAC), i.e. of water molecules that exist in cellulose at moisture contents far below the fiber saturation point1 or, with alternative formulation, far below capillary condensation.2 This fraction is generally called “bound water”, typically further sub-classified as “strongly bound”, “moderately bound” or “weakly bound”, a nomenclature that is rather qualitative, prone to controversy and, thus, far from strict.3 Clearly, an unambiguous and quantitative classification of this water phase is not an easy task3,19 and we find that, in cellulose, there has been given little19 evidence that such phases exist as discrete populations.
As we are going to demonstrate below by 2H magic-angle-spinning (MAS) NMR spectroscopy20 measurements, two distinct WAC phases do exist in cellulose fibers as concerning mobility and resulting 2H NMR behavior, with the minor population exhibiting molecular dynamics that is significantly slower than that shown by the other, more mobile, water population.
2H NMR studies for the quantitative investigation of WAC must involve 2H2O molecules added to the system under highly controlled conditions during the sample preparation (to this point, essential in the present study, we will return later). Yet, the advantages of 2H NMR – over the more frequently chosen 1H NMR methods – are several. First, 2H NMR is more selective, because the largest and unreactive part of cellulose – including non-exchangeable hydroxyl groups – does not contribute to the recorded 2H spectrum.21,22 Second, the dominant spectral broadening mechanism for cellulose-water systems with high 1H-density is the multi-spin dipole–dipole interaction,20,23,24 of both intra- and inter-molecular character. In 2H NMR the dominant spin interaction is instead the quadrupole coupling, of intra-molecular and single-spin character. As a consequence, 2H nuclei are more sensitive probes of the intra-molecular dynamics of WAC than 1H nuclei, and this has been recently proven to be of practical advantage when studying water mobility in cellulose nanocomposites.25,26 In general, broad 2H NMR spectra of “Pake pattern” (or, “powder pattern”) character20,23,24,27 are expected for immobile molecules, as opposed to the narrow Lorentzian peaks typical of mobile domains, with characteristic motionally averaged spectral shapes for molecules experiencing intermediate dynamical regimes.26,28,29
The most direct precursors and motivators to our present work are both 2H and 1H spectroscopic studies of WAC.30–32 Regarding 2H NMR spectroscopy directed toward detecting line shapes, there are two main approaches. In conventional 2H single-pulse experiments, broad spectral components (say, 10–20 kHz wide or wider) are not detected for instrumental reasons. Using such conventional measurements, Vittadini et al.32 investigated cellulose samples with different water contents. At low water content, the authors reported no detectable 2H NMR signal, evidence which led those authors to the hypothesis that some water molecules may be rather immobile at those hydration levels. Earlier than the work of Vittadini et al.,32 Radloff et al.31 used quadrupole echo (QE) as detection method on cellulose systems. This pulse sequence,33 combined with the use of short (few μs) 90° pulses, refocuses the large quadrupolar broadening and, as a result, both broad and narrow spectral components can be detected. Radloff et al.31 detected water molecules in sparsely hydrated cellulose samples that, at low temperature, exhibited dominantly broad 2H spectra and, thereby, very slow molecular dynamics. More recently, Garvey et al.30 used a more complex experimental scheme and detected, by 1H NMR, magnetization exchange between water and cellulose in paper. The magnitude of the observed exchange rate led the latter authors to the conclusion that some water molecules could be in a rather immobile state. Among the experimental schemes mentioned above, the most informative method is the one based on 2H QE NMR spectroscopy.31,34,35 Yet, a major shortcoming of that method is that the relevant 2H NMR intensity is spread over a large spectral range and, therefore, the signal-to-noise ratio (SNR) is inherently low. This makes it difficult to achieve reliable spectral subtraction or decomposition that is necessary for the identification and quantitation of distinct molecular populations,22,36 in our present case different water molecules with fast or, if any, slow molecular dynamics.
Hence, here we rely mainly on a different approach: 2H MAS NMR. While the corresponding high-resolution solid-state method of 13C MAS NMR is a frequent tool for investigating cellulosic materials,37–402H MAS NMR has been used only sparingly for investigating cellulose systems41 (though, it is more popular in other bio-fibrous materials42) and has not been previously applied for detailed investigation of WAC. In general, 2H NMR relaxation experiments26,32,43 have been used in cellulose and related materials far less than corresponding 1H ones, even though the interpretation of the latter is complicated by various factors, some of which were mentioned above. Recently, 2H line-width data in hydrated hemicellulose/cellulose system reported by Terenzi et al.26 provided information about fast exchange of water molecules between environments characterized by high molecular mobility.
In 13C MAS NMR studies of cellulose, the main use of the method was to recover the chemical sensitivity by removing spectral broadening that obscures the isotropic chemical shift. For that purpose, it suffices to investigate the so-called central band. In the 2H MAS experiment presented below, we shall mainly rely on the analysis of the spinning side bands20 (SSBs). The SSBs are located at integer multiples of the spinning frequency, their line width is not influenced by the static quadrupole coupling, and their intensities follow roughly the spectral shape in the QE-detected NMR spectra, but the SNR is higher for the individual narrow SSBs. As has been demonstrated previously in dry 2H-exchanged cellulose,442H MAS NMR provides useful information about the state of hydroxyls and the site-specific 1H–2H exchange mechanisms. Here we propose the use of 2H MAS NMR in cellulose containing adsorbed water in it.
Experimental
Materials and sample preparation
The cellulose samples consisted of microcrystalline cellulose (MCC) from cotton linters provided by Sigma-Aldrich, with a crystallinity 58 ± 3% as estimated by 13C CP MAS NMR45 (triplicate samples). The N2-gas used was from Strandmøllen (purity approximately 99.995%). The 2H2O used was from Sigma-Aldrich (99.9 atom% 2H). Information regarding manufacturer and purity of the inorganic salts used are collected in Table S1 of ESI.†Since we intended to carry out hydrogen–deuterium exchange at maximum efficiency and reproducibility, we initially dried (using the same setup as presented below) all our cellulose samples so that residual 1H2O was efficiently removed. Indeed, never-dried and dried cellulose have slightly different structures (due to hornification46,47) and, thus, it was important for reasons of consistency to start from a controlled dry condition. The initial drying step lasted for a week, after which we observed no detectable decrease in the sample weight.
After having been dried, each MCC sample was placed in a separate vessel containing saturated 2H2O solution of a particular inorganic salt. The used salts and the achieved RH levels are described in Table S1 of ESI.† The RH measurements were performed using suitable data loggers (HOBO UX100-011 and Testo 174H) placed inside the individual vessels. This equilibration step in controlled 2H2O atmosphere lasted for 17 days. The average weight increase shown in Fig. 1 in the 33–93% RH range arises mostly from the increase in the water content while, as observed by 2H NMR, a small fraction of it is caused by 1H–2H exchange in hydroxyl groups (see below). 2H MAS NMR measurements were performed on single replica samples within each series prepared at a given RH. The experimental trend in Fig. 1 seems to follow well the expected shape of an adsorption isotherm of type II.2
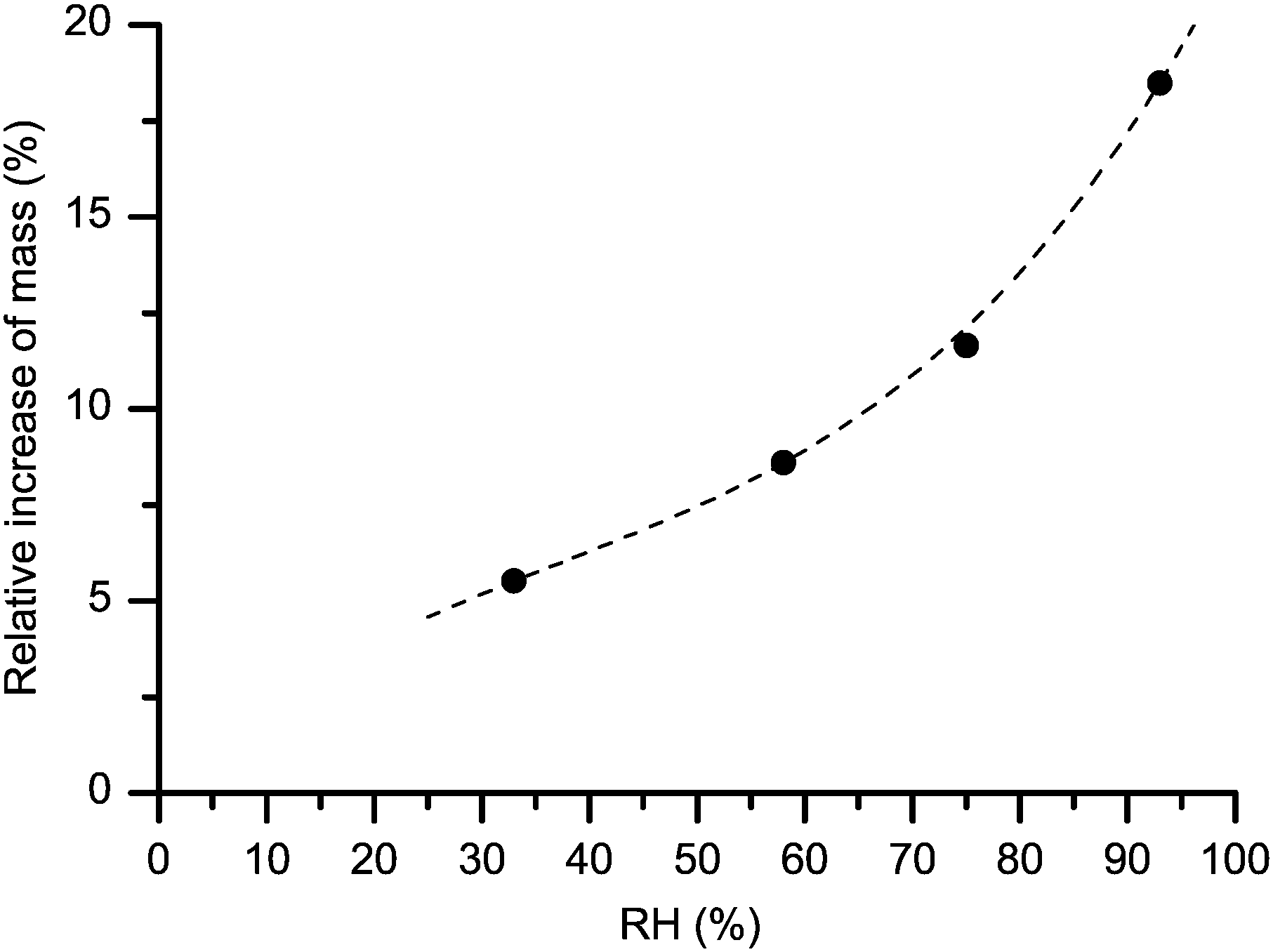 | ||
| Fig. 1 Mass gain upon hydration, expressed as weight fraction of the initially dry cellulose (MCC) samples kept at selected relative humidities. The dashed line is a polynomial fit serving as a guide to the eye. The estimated standard deviation of ±0.5% is represented by the symbol size. | ||
Re-dried (partly-deuterated) MCC samples were then prepared by drying each sample, which was initially hydrated at a specific 2H2O atmosphere, directly in its own MAS rotor (or vial for some gravimetric measurements). The drying was carried out in a heated vacuum chamber (set to 50 °C) connected to a vacuum pump (Alcatel, 2012AC), which generated a pressure of maximum 0.3 mbar inside the chamber. The vacuum-oven and the scale (Mettler-Toledo ML104) for gravimetric measurements were placed inside a glove-box: hence, it was essential to constantly purge the glove-box with N2 gas in order to keep the relative-humidity (RH) level uniformly low (namely, below the detection limit of 2.5% for the used RH loggers, see above). This arrangement was necessary in order to minimize contact with atmospheric 1H2O vapor when handling the dry deuterated cellulose samples inside the glove-box (e.g., when closing the cellulose-containing vials and MAS NMR rotors in an air-tight manner). In our experience, an exposure to environmental moisture of few tens of seconds produces appreciable effects on the 1H–2H exchange. We note that even at 0.3 mbar some 1H2O vapor molecules could still remain in the vacuum-chamber and, in turn, unavoidably lead to a small re-exchange of deuteroxyls in cellulose. This exchange was negligible over 24 hours of re-drying, a period that was long enough to allow a close to complete removal of the excess adsorbed water. It is important to stress here that for the MAS experiments the samples were dried while being within the MAS sample-holding rotor. 2H MAS NMR measurements were performed again on those individual samples that were first investigated in their respective hydrated states.
The mass difference for each MCC sample between its initial dry and re-dried states was on average of ca. 1.4 wt%. Previous findings of Taniguchi et al.48 point to a weight difference of only about 0.3–0.4 wt% due to hydrogen–deuteron exchange in similar situations. From the 2H MAS NMR intensities in dry deuterated cellulose (dominated by deuteroxyl groups, discussed in more detail below) we can estimate (after comparing the intensities to that of suitable reference material,36 in this case d5-PMMA) that ca. 0.4–0.5 wt% of the weight difference is due to hydroxyl protons exchanging to heavier deuterons and, therefore, the samples may contain about 1 wt% water after the re-drying procedure used here. We note that the samples investigated previously by Vittadini et al.32 were initially dried by freeze-drying. As it seems to be rather well established,49 such procedure may leave a few wt% of water adsorbed on cellulose, and thus the actual moisture contents in the samples of that work could not be reliably considered as reference for our study.
2H NMR measurements
The 2H QE NMR spectra were recorded on a Bruker Avance II 300 spectrometer equipped with a high-power static solenoid NMR probe. The samples were closed in an air-tight manner in short (5 mm) glass NMR tubes. The spectra were recorded at room temperature and at a resonance frequency of 46.05 MHz. The length of the 90° pulse was calibrated to 2.3 μs. The spectra presented in Fig. 2 arise from QE-detected inversion recovery experiments performed by the 180°–τinv–90°–τE–90°–acq pulse sequence. The echo delay was set to τE = 10 μs and the inversion times, τinv, were increased roughly in geometric progression (see Table S2 and phase cycling scheme in ESI†). The recycle delay was set to 4 s, and the individual spectra were acquired with 2k scans. The spectra were obtained by Fourier transforming the time-dependent signal following the actual echo top (where this last procedure was implemented to avoid spectral distortions50). Whereas the pulse lengths were rather short, at short inversion times unavoidable imperfections in the 180° pulses caused signal distortion effects, mostly visible as slightly reduced intensities at the edges of the spectra in Fig. 2.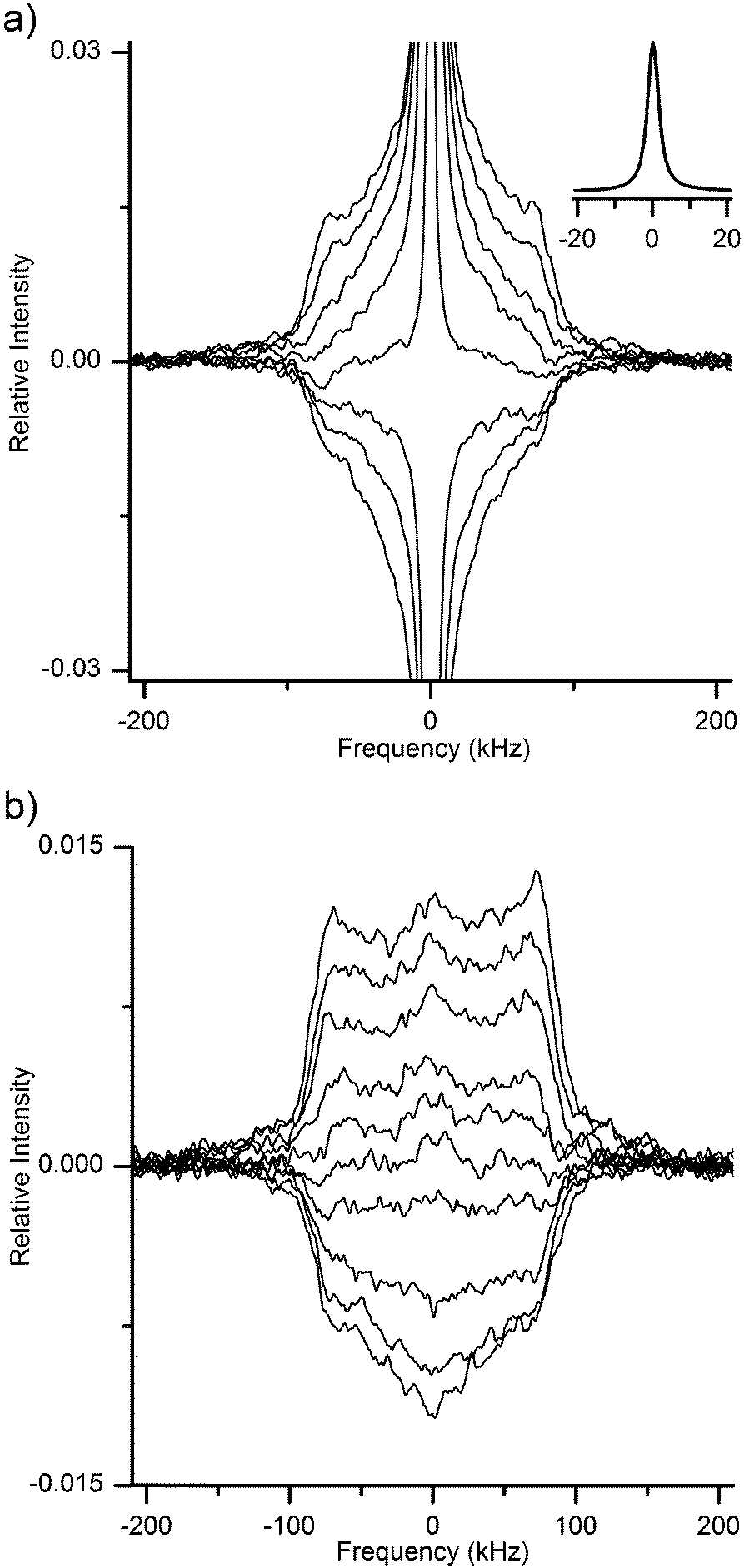 | ||
| Fig. 2 Time-evolution of the 2H QE NMR spectra of MCC hydrated at 93% RH of 2H2O (a) and then re-dried (b). Spectra were recorded at set inversion times (see list of delay times in ESI†) after an 180° inversion pulse, thus reflecting the longitudinal relaxation of the nuclear magnetization. The inset in (a) shows the central peak for the fully-relaxed spectrum of hydrated MCC. Spectral intensities are all normalized to the maximum intensity of the equilibrium spectrum in (a). | ||
The 2H MAS NMR spectra were recorded on a Bruker Avance HD 500 spectrometer equipped with a high-power 4 mm MAS probe. The samples were conventional NMR rotors into which ca. 40 mg of MCC (depending on the actual moisture content) was tightly packed. The rotor was air-tight as tested by observing a constant weight for dry samples before and after the NMR measurements. The spectra were recorded by single-pulse experiments at room temperature and at a resonance frequency of 76.1 MHz, using a sample spinning speed of 10 kHz. The length of the 90° pulse was calibrated to 3.2 μs. Signal acquisition started after the customary delay of 100 μs (the inverse of the spinning frequency) at the first rotational echo top. Each spectrum was collected with a minimum of 2k scans with a recycle delay of at least 4 s. As further underlined below, all paired spectra (for hydrated and re-dried states) were acquired under identical experimental conditions. When comparing the 2H MAS NMR signal intensity of the different samples, equilibrated at different RH values, the peaks (central peak and SSB manifold) in the spectra were integrated between −180 kHz and 180 kHz.
Results and discussion
In the first two sub-sections below, we illustrate and compare the different performances of QE and MAS solid-state 2H NMR spectra acquired in MCC samples that were previously hydrated over heavy-water solutions and subsequently re-dried (thus, respectively, with and without WAC). Additionally, we perform a critical review of the limitations and sensitivity of the QE-based 2H NMR approach, already adopted a few decades ago for a similar purpose by Radloff et al.31 We then proceed to demonstrate the superior capability of 2H MAS detection in aiding, under its much improved SNR conditions as compared to 2H QE NMR, the quantitative separation of signals arising from distinct 2H populations, namely deuteroxyls and adsorbed heavy-water molecules. Finally, in the last sub-section, we identify different phases of adsorbed water with strikingly distinct degrees of mobility, as testified by their 2H NMR spectral shapes and line-widths. We put particular emphasis here on presenting the spectral quantification of the relatively small fraction of immobile WAC.Individual components within 2H QE NMR spectra of hydrated and re-dried cellulose
The 2H QE NMR spectrum of cellulose hydrated in heavy water, acquired using short high-power r.f. pulses, is necessarily a composite spectrum with at least two distinct populations.26,31 As heavy-water molecules enter the cellulose system, they (i) undergo exchange with some of the accessible44 surface hydroxyl groups and (ii) are adsorbed onto the cellulose microfibrils. Due to their slow (with correlation times of microseconds or longer) and anisotropic molecular dynamics deuteroxyls are detected as a very broad (∼200 kHz wide) 2H QE signal.22,44 Contrarily, more mobile adsorbed water molecules exhibit faster and isotropic molecular motions and thereby contribute to an intense narrow central peak in the 2H QE spectrum. Yet, water molecules with more restricted dynamics may give rise to a sub-population that does not exhibit the same narrow spectral feature as that of mobile WAC. Addressing this latter issue in a quantitative way is the main point of the present paper.We found that, although both the spectral assignment and the molecular dynamics assessment are undoubtedly more straightforward in 2H than in 1H NMR measurements, properly analyzing the composite 2H NMR spectrum of hydrated cellulose in such a way that the different 2H contributions can be reliably identified and extracted is far from being a trivial task.
Radloff et al.31 based the separation of their QE-detected broad and narrow signals in cellulose on the different longitudinal relaxation times experienced by those individual signal components. One of the 2H populations was identified as water and the related spectrum could be recorded after suppression (by pre-saturation) of the signal of the other slow-relaxing component. For completeness, we report here in Fig. 2 our own 2H QE spectra detected in hydrated (2a) and re-dried (2b) MCC samples during an NMR inversion-recovery experiment. The narrow central line in Fig. 2a obviously arises from the mobile water molecules. The identification of the remaining spectral component(s) is less straightforward, given the low SNR of the measurements. Qualitatively, it is rather clear that there are more than one longitudinal relaxation time present even in the case of re-dried cellulose (see Fig. 2b).
Yet, a more detailed analysis of those data, left alone a quantitative assignment of each distinct 2H T1 population based on spectral deconvolution, remains prohibitively difficult for several reasons. First, the spectral shapes in cellulose re-dried after hydration are not31 the familiar Pake pattern,27 probably because there exists in the cellulose system a distribution of the magnitude of quadrupole coupling and/or of restricted molecular motions. Second, in such an anisotropic material the 2H relaxation is also unavoidably anisotropic,51,52 resulting in different 2H T1 values at different spectral frequencies. Third, the SNR is low due to the broad distribution of 2H frequencies in the QE spectrum. The latter issue presents a major limitation in quantitative studies of 2H–1H exchange kinetics in cellulose, as lengthening the experiment time (in order to average the signal over more acquisitions) far beyond a day as used here is both impractical for kinetics studies and may also result in the contamination of the deuterated sample by atmospheric moisture during measurements.
Regarding the main outcome of the results reported by Radloff et al.,31 at 300 K the spectral component that they assign to water seems to contain both a narrow peak and a broad peak. The latter spectral component was recorded by suppressing (with the use of a short recycle delay) any signal exhibiting long longitudinal relaxation times.
However, in dry deuterated MCC the 2H MAS signal from 2H-exchanged hydroxyl groups exhibits a rather fast-relaxing (with longitudinal relaxation time T1 in the order of 40–60 milliseconds) component.44 Assuming similar relaxation behavior also in hydrated cellulose one is led to conclude that the method used by Radloff et al.,31 while yielding an intensity reduction by a factor of ca. 4–6, does not provide complete suppression of the deuteroxyl signal. Hence, the broad spectral component reported by those authors, and attributed merely to water molecules, instead likely contained significant contribution from deuteroxyls at room temperature. The water contribution to the broad spectral component appeared to be dominant only upon lowering the temperature down to 190 K, which was a natural consequence of slowing down the molecular motions of adsorbed water molecules. Recently, the same experimental strategy (2H QE with and without saturation filter) was also applied to probe the state of water in hydrated chitosan.53 Assumedly, some of the limitations discussed above may apply in that case, too.
We note here that an additional problem in the data of Radloff et al.31 is that, at their alleged water content of only 1–2 wt%, they detected a relatively intense narrow peak in the 2H spectrum. As reported by Vittadini et al.,32 and further verified by us on our MCC samples (data not shown), at such low nominal water contents a narrow peak is definitely missing in the conventional 2H spectrum of cellulose. This seems to point to the possibility of very inhomogeneously dried samples or of some other imperfections in the sample preparation process used by Radloff et al.31
Individual components within 2H MAS NMR spectra of hydrated and re-dried cellulose
For reasons illustrated in the previous section, the 2H NMR approach pursued here relies not on QE, but on MAS detection. Fig. 3a and b show the spectra obtained, respectively for samples after being hydrated at 33% and 93% RH of 2H2O (spectra in magenta) and after subsequent re-drying (spectra in black). The envelope of the SSB peaks in MAS spectra roughly follows the spectral shape observed in the static QE spectra of Fig. 2. Hence, in Fig. 3a one can distinguish a dominant narrow central peak for the MCC sample hydrated at 33% RH, which is entirely missing in the spectrum of the same sample after re-drying. We note here that a slightly elevated central intensity could be expected in the 2H MAS spectral manifold even if the respective static spectrum lacks a central line – this is due to more intricate details of rotational averaging.54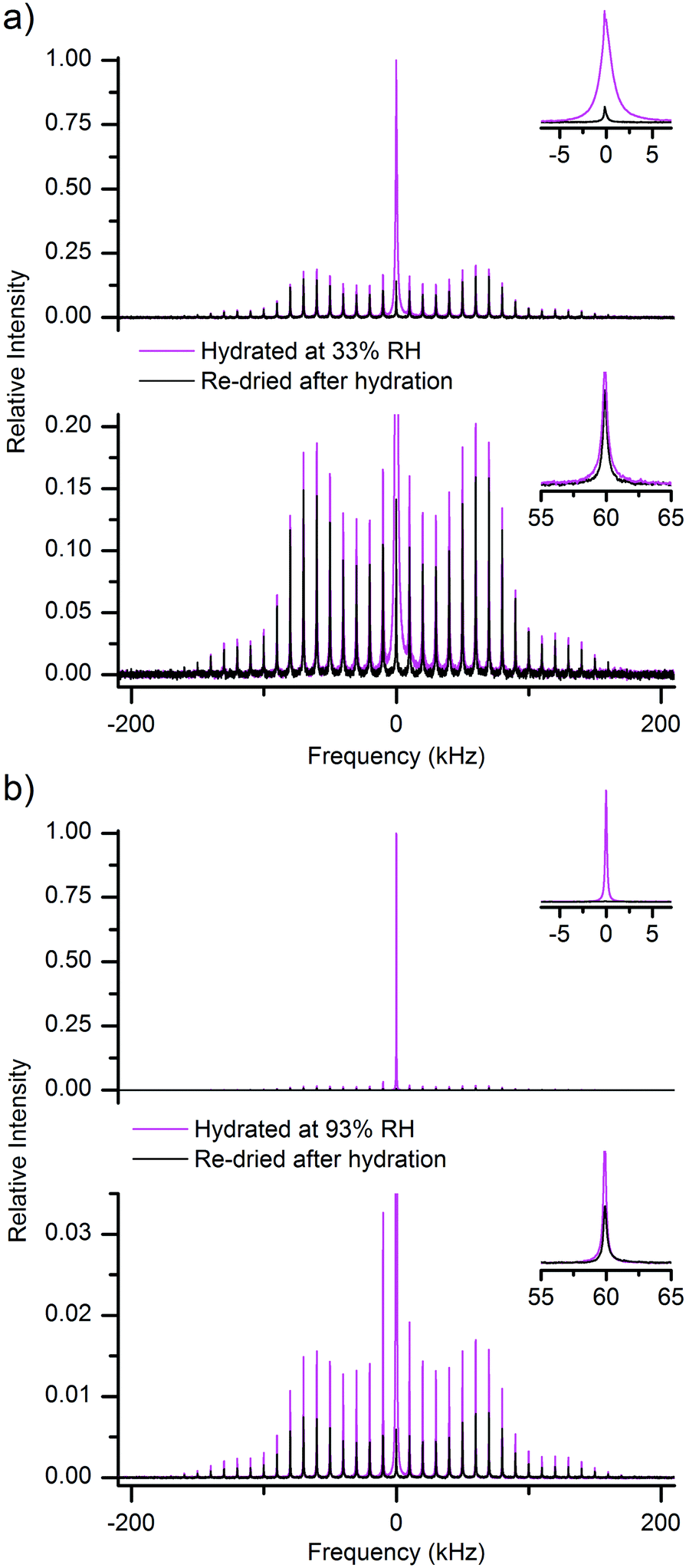 | ||
| Fig. 3 (a) 2H MAS NMR spectra of a hydrated (at 33% RH of 2H2O, magenta) and then re-dried (black) MCC sample, both shown in full scale (top) and magnified (bottom) to illustrate the signals arising respectively from mobile water (intense central peak) and from the other deuterium-containing fractions of the sample (the SSBs spread over a broad frequency range). The spectra are normalized relative to the maximum amplitude in the hydrated spectrum. The insets show the central line (top) and a selected SSB (bottom). (b) Same as in (a), but for a MCC sample hydrated at 93% RH of 2H2O. | ||
The spectra in Fig. 3 are very broad and exhibit SSBs at integer multiples of the spinning frequency offset from the central frequency. Clearly, they arise from 2H nuclei residing within molecules that undergo little re-orientational dynamics and the observed spectral broadening is here due to static quadrupole coupling. We note that the paired spectra in Fig. 3a and b were recorded under identical conditions, including the same sample filling factor, and that the samples were all dried within their MAS rotors.
The SNR in the MAS spectra is clearly much higher than that in the QE spectra of Fig. 2. Indeed, the SNR for a given narrow SSB is roughly νrot/Δ1/2, where νrot is the sample spinning frequency and Δ1/2 is the line width of the individual SSB. Hence, upon MAS at 10 kHz and with Δ1/2 ∼ 400–500 Hz, the SNR of individual SSBs is more than one order of magnitude higher than that obtained, under the same experimental conditions, for the QE spectrum.
The other point regarding data in Fig. 3 is that the dry, partly-deuterated, state was obtained by re-drying the same sample from its respective hydrated state, whose characteristic spectrum is also reported within the same figure. This is important, because the amount of hydroxyl groups exchanged in a cellulose sample depends on the actual amount of water that has been adsorbed in it.48 A careful re-drying procedure is expected to leave unaltered the deuterated hydroxyl groups, which thus are the sole contributors to the broad SSB manifold in the dry spectra of Fig. 3.
Since drying does not remove 2H or 1H from hydroxyls, but only those in water, the difference between each pair of spectra in Fig. 3 must provide the spectrum that arises merely from the heavy-water molecules in the respective hydrated system. The spectral difference for each pair of spectra in Fig. 3, along with the additional difference spectrum obtained for another MCC sample prepared at intermediate hydration level (namely, 58% RH of 2H2O), are reported in Fig. 4.
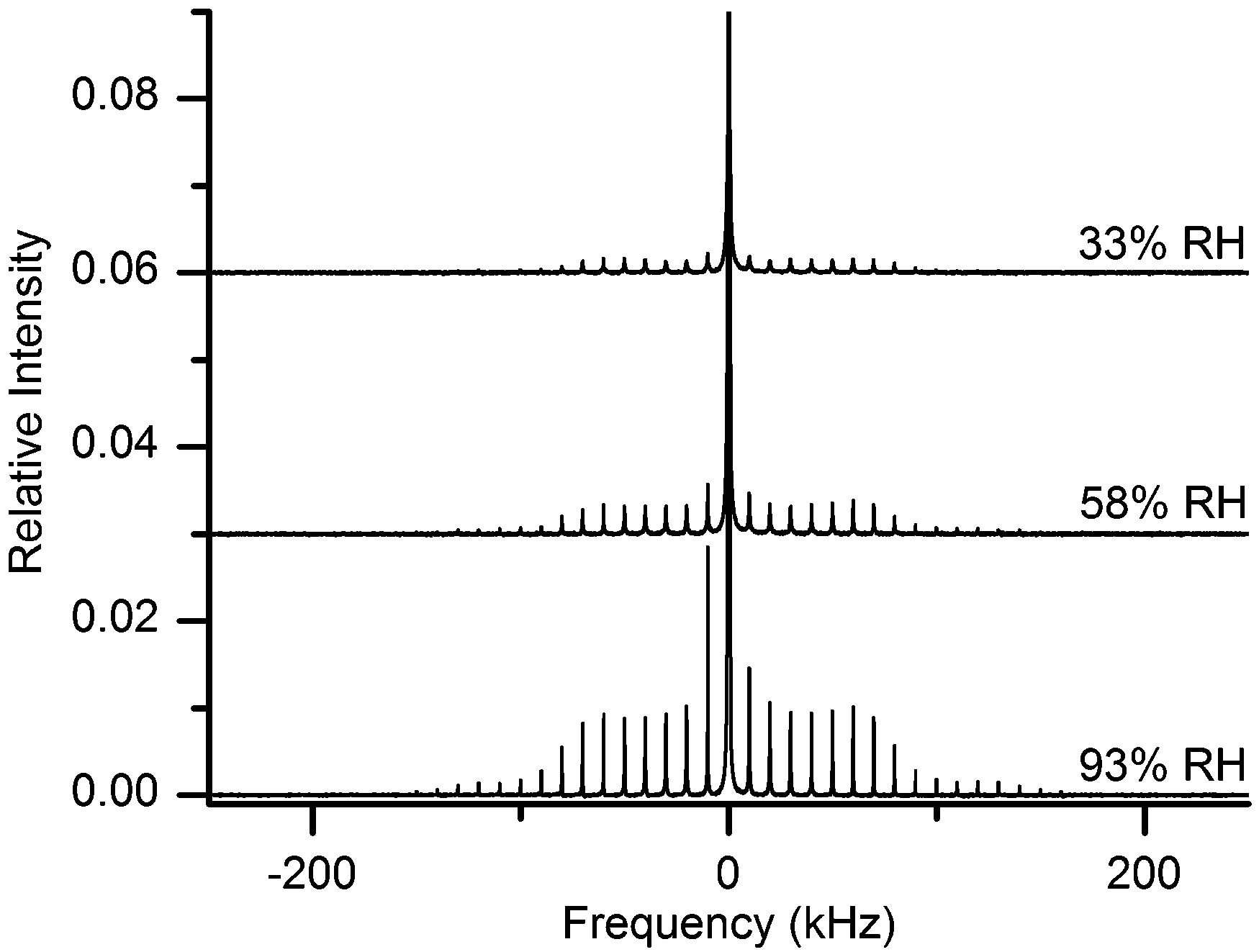 | ||
| Fig. 4 2H MAS NMR spectra of water in MCC samples hydrated at different relative humidities of 2H2O atmosphere. The central lines are truncated at different thresholds; all spectra are relative to the maximum intensity of the spectrum at 93% RH (bottom), while the spectra at RH of 58% (middle) and 33% (top) are shifted vertically for better visibility. Magnified spectra for better visibility are shown in ESI.† | ||
The sole assumption behind the adopted subtraction procedure as method yielding results significant for the state of water is that the spectral shape of the macromolecular deuteroxyl 2H signals in each sample, as measured after hydration and after subsequent re-drying, remains the same.
If anything, one could expect to observe a possible spectral narrowing for the deuteroxyl signals upon hydration, if the presence of more water permits faster dynamics of surface hydroxyls. In the latter case, the difference spectra in Fig. 4 should as a consequence exhibit negative peaks at the spectral edges of each SSB (while closer to the centre the peaks should be positively contributed by deuteroxyl deuterons). This is not observed (see also magnified spectra in ESI†), thus showing that the spectra arising from the 2HO groups in cellulose are rather insensitive (within our experimental uncertainty) to the removal of adsorbed water.
In addition, if our statement that the spectra in Fig. 4 arise solely from water is true, their total (central line + SSBs) integral intensity must be roughly proportional to the mass difference between the hydrated/re-dried paired states (although this mass difference includes also a <0.5 wt% contribution from the proton-deuteron exchange48 in some hydroxyl groups, see below). As shown in Fig. 5 this is, indeed, the case. We can thus safely conclude that the spectra in Fig. 4 are actually representative of the sole contribution arising from adsorbed heavy-water molecules.
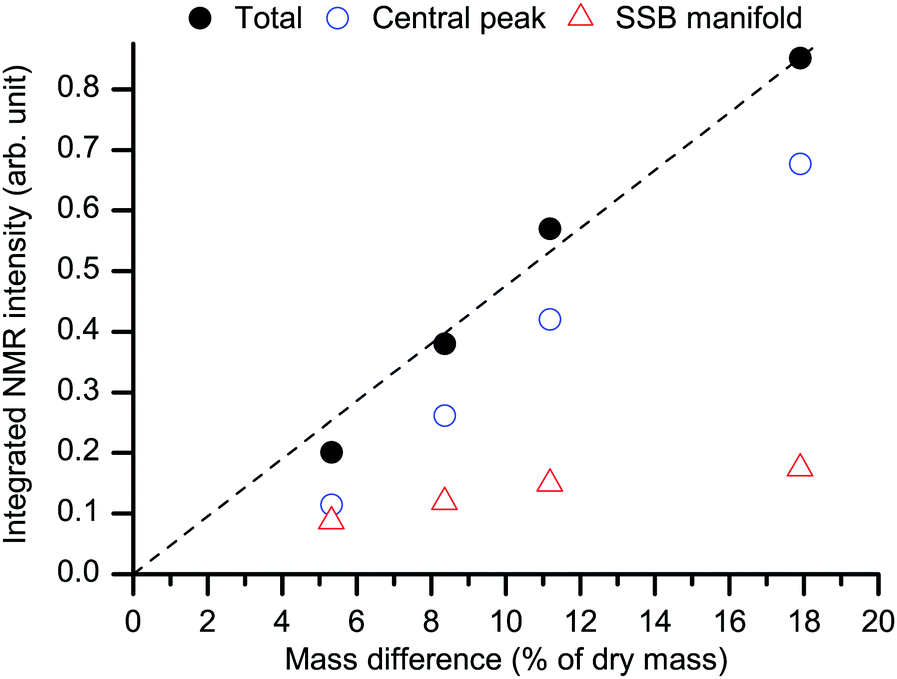 | ||
| Fig. 5 Behavior of the total integral NMR intensity of the water spectra as in Fig. 4 (black filled circles) as a function of the mass difference upon re-drying (see RH levels in Fig. 1). The dashed black line is a linear fit with zero-intercept. The separate integral intensities for the central peak (blue empty circles) and for the SSB manifold (red empty triangles) are also shown. The average standard deviation for the total intensity points is ±1%. | ||
When considering the mass-signal relation one must also recall that, upon putting initially dry cellulose in a heavy-water atmosphere, the resulting increase of the mass, Δm, is contributed by (i) the adsorption of a number n(2H2O) of heavy-water molecules and (ii) the 1HO–2HO exchange occurring at a number n(2HO) of selected44 hydroxyl sites. As shown below in eqn (1a), the gravimetric water contribution exceeds that of 2HO groups by a factor of 20.
On the other hand, eqn (1b) shows that in the corresponding increase of the 2H NMR signal (in arbitrary units), ΔI, the water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :deuteroxyls ratio is only 2, meaning that the relative contribution of deuteroxyls to ΔI is 10 times larger than that to Δm, as described in the following:
:deuteroxyls ratio is only 2, meaning that the relative contribution of deuteroxyls to ΔI is 10 times larger than that to Δm, as described in the following:
| Δm = [n(2H2O)·20 + n(2HO)·1] g mole−1 | (1a) |
| ΔI ∝ n(2H2O)·2 + n(2HO)·1. | (1b) |
These equations formalize the reason why it is necessary, when obtaining the spectra in Fig. 4, to subtract the signal for the dry state that was prepared from the respective hydrated state of the same cellulose sample. As is clear from previous studies,48n(2HO) varies between samples prepared at different RH values: higher RH leads to larger n(2HO). The corresponding increase (from 33% to 93% RH) in Δm is deceptively small,48 only 0.2–0.3% of the total cellulose mass. Yet, when normalized to the mass of the adsorbed water (recall Fig. 1), such contribution is ca. one order of magnitude higher, a few %. As is shown in eqn (1a) and (b), the gravimetric contribution of a proton-deuteron exchange is 20 times smaller than that due to the adsorption of a water molecule. Yet, the same difference in terms of 2H NMR intensity growth is only a factor 2. This clarifies the observation in Fig. 3a, where the deuteroxyl signal dominates the broad spectral component despite its <0.5 wt% gravimetric contribution.48 Conversely, subtracting from the spectrum of cellulose hydrated at a low RH that of a re-dried sample prepared at a high RH may even yield negative SSBs.
The behavior of the immobile water component
The water spectra in Fig. 4 present two distinct contributions, one narrow (represented by the central peak) and one broad (the SSB manifold at offset frequencies at multiples of the spinning speed). Since the two spectral contributions are distinct, the first statement is that the water populations yielding them do not exchange with each other on a time scale (∼10 μs) in the order of the inverse width of the broad spectral component. The shape of the SSB manifold in Fig. 4 (see also the magnified spectra in ESI†) barely changes upon increasing hydration even though molecular dynamics is strongly dependent on the water content, see Fig. 6. In case of conformational exchange,55–57 the rate of exchange dynamics on the intermediate time scale was shown to have a very strong influence on the line shape of the SSB manifold. Hence, the constancy of the spectral shape upon increasing hydration implies that intermediate exchange has no major effect on our observations. Methods58–60 that are more sensitive to this dynamical mode may be useful to investigate further if there is any minor influence.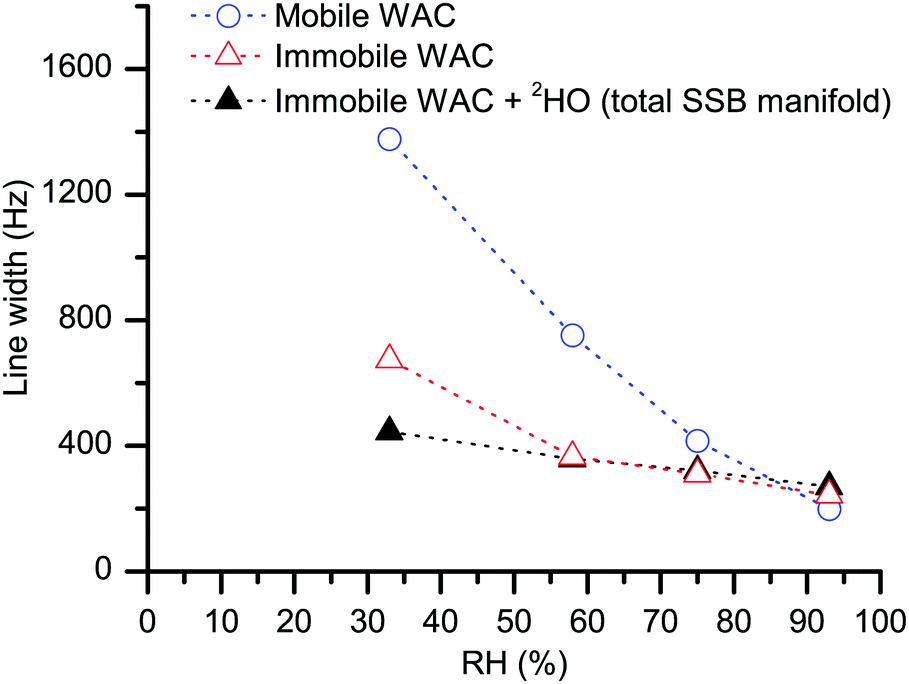 | ||
| Fig. 6 The line width of the central peak (blue empty circles) and the average line width of the SSB manifold (black filled and red empty triangles) in, respectively, the measured 2H MAS spectra (as in Fig. 3) and the difference 2H MAS spectra (as in Fig. 4). The dotted lines serve as a guide to the eye, the standard deviation for the individual points is below symbol size. | ||
It is difficult to imagine that water molecules adjacent to each other can refrain from exchanging their positions over such a long time. Hence, the most plausible explanation of the lack of exchange between the two water populations is that, within the cellulose system, the molecules in the two inferred water phases have completely different locations, separated by multiples of water molecular dimensions. Below, we label the WAC subpopulation that yields the narrow peak as “mobile WAC” and the one that yields the SSB manifold outside the central peak as “immobile WAC”. Within these two broad populations, there might exist respective distributions of molecular properties; this latter point is not addressed here in any detail. As illustrated in Fig. 6, the line width of the central peak (blue empty circles) decreases strongly with increasing RH. Hence, we can conclude that the molecules in the mobile WAC phase become more mobile upon increasing hydration. Consequently, the regions within the cellulose system where the mobile WAC phase resides must permit extensive water–water contact, in turn further promoting faster molecular motions upon increasing water content.
On the other hand, the line width of the immobile WAC phase changes much less (less than two-fold) in the 33–93% RH range and, as is shown in Fig. 4, also the shape of the water SSB manifold remains roughly the same upon increasing hydration. Moreover, the residence time for the immobile WAC phase remains longer than 10 μs over the full range of hydration. From these observations we infer that there is only limited water–water contact at the sites where the molecules of the immobile WAC phase reside. The line width of the deuteroxyl peaks in the hydrated samples remains roughly the same, as is witnessed by the line width data for the total SSB manifold (Fig. 6, at 33% RH the total SSB line width is dominated by that of deuteroxyls).
The spectral shape of the signal from immobile WAC is clearly not that of a Pake pattern27 (see Fig. 4). Yet, although it lacks the characteristic “90°-peak” of the Pake pattern, it presents a shoulder at ca. 70 kHz. For rigid water molecules in ice,61 the location of the 90°-peak is at ca.
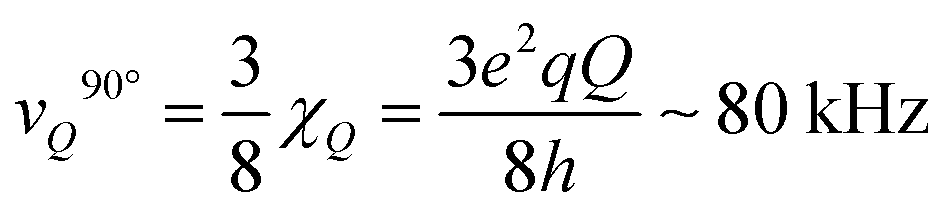 | (2) |
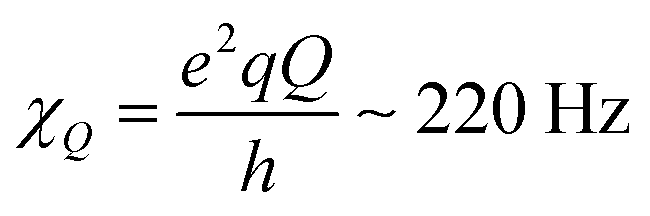 is the quadrupole coupling constant of 2H in water molecules.23,24,61 Hence, the water molecules in the immobile WAC phase exhibit some limited motion, but are clearly strongly hindered and cannot isotropically re-orient. It also seems probable that there is a distribution of molecular properties within this phase.
is the quadrupole coupling constant of 2H in water molecules.23,24,61 Hence, the water molecules in the immobile WAC phase exhibit some limited motion, but are clearly strongly hindered and cannot isotropically re-orient. It also seems probable that there is a distribution of molecular properties within this phase.
In clear contrast, the central peak from the mobile WAC phase presents no splitting. While a splitting smaller than the spinning speed does not manifest itself in the MAS spectrum, the central line in our static QE spectra (see inset in Fig. 2a) also lacks splitting. In some studies and at certain degrees of hydration, small (few kHz) splitting of the corresponding water signal in QE spectra was observed.32,34,35
Hence, we can conclude that the water molecules in the mobile WAC phase exhibit fast and essentially isotropic re-orientational dynamics. The line width for the mobile WAC being larger than that for the immobile WAC (up to 75% RH) is a consequence of complete averaging23,62 of the quadrupole coupling to a negligible residual value in the mobile WAC signal. In the immobile WAC, anisotropically restricted molecular motions retain a large part of the quadrupole coupling un-averaged62 (hence the broad SSB manifold) and thereby without contribution to the MAS line width (that is, transverse relaxation).
If one assumes that (i) the re-orientational motional averaging in the mobile WAC phase is characterized by a single correlation time τC and that (ii) the only coupling to be averaged is the total quadrupole coupling of 2H nuclei in water molecules, the value of τC can be estimated from the line width Δ1/2 as23,24
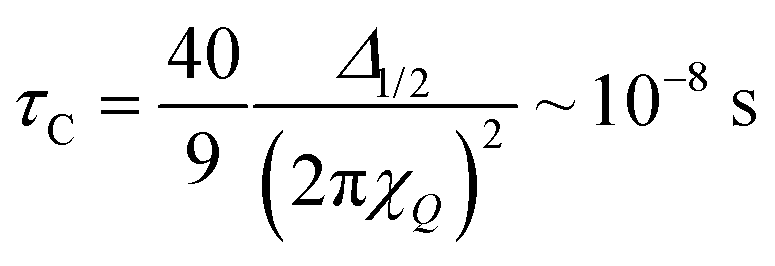 | (3) |
Of course, the motional averaging can be a multi-step process, in which case distinct characteristic correlation times will be distributed around the mean estimate given by eqn (3). In the immobile WAC, the motionally averaged fraction of the quadrupole coupling is almost an order of magnitude less than that in the mobile WAC. Hence (consider eqn (3)), the data in Fig. 6 indicate that the mean motional correlation time in the immobile WAC is always (even at 33% RH) much longer than that in the mobile WAC.
Regarding the specific location of the water molecules in the mobile and immobile WAC phases, it is well established that the adsorption sites in cellulose are hydroxyl groups to which water molecules may hydrogen-bond.63,64 The question that remains to be addressed is: what is the distinct location of the hydroxyl sites in cellulose that are responsible for the distinct behaviors of the mobile and immobile WAC phases? Clearly, what locations may be considered as available in cellulose depends on which structural model one accepts. In our explanation below, we rely on the well-established model65,66 where the basic cellulose unit, the microfibril, is associated into microfibril aggregates. In this model, there appear to be two distinct types of cellulose surfaces that are both able to expose hydroxyl groups. One is the outer surface of the aggregates, sometimes also termed as the external surface,67 which does not necessarily face air: indeed, the microfibril aggregates are largely in contact with each other or with hemicellulose. This contact may involve hydrogen bonds between microfibrils which face each other while belonging to different microfibril aggregates, and/or hydroxyls belonging to hemicellulose. The other surface is instead at the interface between microfibrils within the same aggregate. Across this surface, often termed the internal surface,67 numerous hydrogen bonds join the microfibrils that belong to the same aggregate.68 Despite earlier suggestions that cellulose surfaces within aggregates are inaccessible to solvents,40 it has been shown that moisture is able to penetrate between microfibrils to some extent.45 Hence, we propose here that the mobile WAC consists of inter-aggregate water molecules adsorbed at the external surfaces, while water molecules responsible for the observed immobile WAC phase reside at intra-aggregate microfibril interfaces.
Assumedly, the microfibril-microfibril contact within the aggregates is more regular and stronger than the contact between different aggregates.68,69 If so, swelling of the aggregates and thereby the resulting microfibril–microfibril separation is limited since it proceeds against strong forces between the microfibrils within the aggregate. That limitation must be weaker at the external surfaces. Thereby, the mobile WAC phase can increase more upon increasing hydration, while the immobile water phase should exhibit limited growth and/or saturation. This is indeed one feature that is clear in Fig. 5.
The arrangement of hydroxyl groups along the internal surfaces is plausibly regular. If, in addition, the regularity is such that binding sites appear with favorable geometry for coordinating water molecules from several directions,70,71 water molecules at those sites will be strongly and rather rigidly bound. It is for that reason that they exhibit slow and anisotropic re-orientational dynamics and yield the spectral signature of immobile WAC. Strong binding also favors adsorption at those sites, until they become saturated at higher RH. Hence, one expects that at low hydration levels the immobile WAC phase is dominant. Indeed, as shown in Fig. 5, the amount of the mobile and immobile WAC is roughly the same at our lowest explored hydration level. The mobile WAC phase, not affected by saturation, becomes dominant at higher RH.
The supposed organization of surface hydroxyls along/across the internal surfaces in such way that they may coordinate water molecules well is, of course, speculative. However, based on what we know of organic (and, inorganic) hydrates,13,72 immobile water molecules in them require exactly that type of arrangement. Comparison with crystalline hydrates with water molecules coordinated within the crystalline lattice13,72 may even tempt one to explore less conventional models. Hence, by analogy, another possible speculative location for the immobile water molecules would then be in-between the cellulose chains in the “paracrystalline”73,74 outer region of the microfibrils. What we currently know about cellulose does not support an interpretation of the presently reported and other findings75 in terms of water molecules penetrating all the way into the microfibril core.
Conclusions
In the present work, we report a direct experimental proof of the existence of an immobile adsorbed water phase in hydrated cellulose. In addition, we describe and validate a robust experimental approach that allows to separate and quantitatively measure the mobile and immobile water populations in cellulose. By directly probing the 2H NMR MAS spectral response in 2H-exchanged cellulose, the organization of water in distinct environments is manifested in the form of two separate, narrow and broad, spectral components undergoing slow molecular exchange. Such unambiguous distinction of water sub-populations in a complex material like cellulose has not been experimentally accessible via previously explored 2H QE NMR measurements, due their prohibitively low SNR. Also, reproducible and quantitative (gravimetric and NMR) data require that lengthy drying/hydration steps and highly controlled sample handling protocol during and after the 2H–1H exchange process are followed.Looking at the present results and at the existing models for the cellulose structure, we propose that the here quantified mobile water fraction adsorbs at the external surface of microfibril aggregates, i.e. at aggregate–aggregate interfaces, while the newly-detected immobile water fraction adsorbs at the interfaces between microfibrils belonging to the same aggregates. We envisage future experiments (including the characterization of the re-drying kinetics) to validate or further refine the model preliminarily suggested here. Similar measurements in other and more complex systems that expose different hydrated interfaces, such as chitin, chitosan53 or native wood cell walls,76 are both feasible and desirable.
Acknowledgements
The authors gratefully acknowledge the Wallenberg Wood Science Center for having financially supported this work. IF also acknowledges the Swedish Research Council (VR) for funding.Notes and references
- S. L. Berry and M. L. Roderick, New Phytol., 2005, 168, 25–37 CrossRef CAS PubMed.
- E. T. Engelund, L. G. Thygesen, S. Svensson and C. A. S. Hill, Wood Sci. Technol., 2013, 47, 141–161 CrossRef CAS.
- V. J. McBrierty, S. J. Martin and F. E. Karasz, J. Mol. Liq., 1999, 80, 179–205 CrossRef CAS.
- A. N. Prusov, S. M. Prusova and A. G. Zakharov, Russ. Chem. Bull., 2014, 63, 1926–1945 CrossRef CAS.
- C. Skaar, Wood-Water Relations, Springer, Berlin, 1988 Search PubMed.
- A. Venkateswaran, Chem. Rev., 1970, 70, 619–637 CrossRef CAS.
- G. Zografi, M. J. Kontny, A. Y. S. Yang and G. S. Brenner, Int. J. Pharm., 1984, 18, 99–116 CrossRef CAS.
- A. Striolo, Adsorpt. Sci. Technol., 2011, 29, 211–258 CrossRef CAS.
- M. Åkerholm and L. Salmén, J. Appl. Polym. Sci., 2004, 94, 2032–2040 CrossRef.
- K. Nakamura, T. Hatakeyama and H. Hatakeyama, Text. Res. J., 1983, 53, 682–688 CrossRef CAS.
- M. J. Selig, L. G. Thygesen and C. Felby, Biotechnol. Biofuels, 2014, 7, 10 CrossRef PubMed.
- M. C. Bellissent-Funel, A. Hassanali, M. Havenith, R. Henchman, P. Pohl, F. Sterpone, D. van der Spoel, Y. Xu and A. E. Garcia, Chem. Rev., 2016, 116, 7673–7697 CrossRef CAS PubMed.
- A. J. Benesi, M. W. Grutzeck, B. O'Hare and J. W. Phair, J. Phys. Chem. B, 2004, 108, 17783–17790 CrossRef CAS.
- O. Carugo, Curr. Protein Pept. Sci., 2015, 16, 259–265 CrossRef CAS PubMed.
- A. W. Friedline, M. M. Zachariah, K. Johnson, K. J. Thomas, A. N. Middaugh, R. Garimella, D. R. Powell, P. A. Vaishampayan and C. V. Rice, J. Phys. Chem. B, 2014, 118, 8945–8955 CrossRef CAS PubMed.
- A. W. Friedline, M. M. Zachariah, A. N. Middaugh, R. Garimella, P. A. Vaishampayan and C. V. Rice, J. Phys. Chem. B, 2015, 119, 14033–14044 CrossRef CAS PubMed.
- B. Halle, Philos. Trans. R. Soc., B, 2004, 359, 1207–1223 CrossRef CAS PubMed.
- S. Kaieda, B. Setlow, P. Setlow and B. Halle, Biophys. J., 2013, 105, 2016–2023 CrossRef CAS PubMed.
- A. M. Olsson and L. Salmén, Carbohydr. Res., 2004, 339, 813–818 CrossRef CAS PubMed.
- M. J. Duer, Solid-State NMR Spectroscopy, Blackwell, Oxford, 2004 Search PubMed.
- V. J. Frilette, J. Hanle and H. Mark, J. Am. Chem. Soc., 1948, 70, 1107–1113 CrossRef CAS.
- Y. Horikawa and J. Sugiyama, Cellulose, 2008, 15, 419–424 CrossRef CAS.
- A. Abragam, The Principles of Nuclear Magnetism, Clarendon, Oxford, 1961 Search PubMed.
- B. Cowan, Nuclear Magnetic Resonance and Relaxation, Cambrigde University Press, Cambridge, 1997 Search PubMed.
- K. Prakobna, C. Terenzi, Q. Zhou, I. Furó and L. A. Berglund, Carbohydr. Polym., 2015, 125, 92–102 CrossRef CAS PubMed.
- C. Terenzi, K. Prakobna, L. A. Berglund and I. Furó, Biomacromolecules, 2015, 16, 1506–1515 CrossRef CAS PubMed.
- G. E. Pake, J. Chem. Phys., 1948, 16, 327–336 CrossRef CAS.
- H. W. Spiess, Colloid Polym. Sci., 1983, 261, 193–209 CAS.
- R. J. Wittebort, E. T. Olejniczak and R. G. Griffin, J. Chem. Phys., 1987, 86, 5411–5420 CrossRef CAS.
- C. J. Garvey, I. H. Parker, G. P. Simon and A. K. Whittaker, Holzforschung, 2006, 60, 409–416 CrossRef CAS.
- D. Radloff, C. Boeffel and H. W. Spiess, Macromolecules, 1996, 29, 1528–1534 CrossRef CAS.
- E. Vittadini, L. C. Dickinson and P. Chinachoti, Carbohydr. Polym., 2001, 46, 49–57 CrossRef CAS.
- J. H. Davis, K. R. Jeffrey, M. Bloom, M. I. Valic and T. P. Higgs, Chem. Phys. Lett., 1976, 42, 390–394 CrossRef CAS.
- T. C. Wong, Mol. Phys., 1983, 48, 495–502 CrossRef.
- T. C. Wong and T. T. Ang, J. Phys. Chem., 1985, 89, 4047–4051 CrossRef CAS.
- E. L. Lindh, P. Stilbs and I. Furó, J. Magn. Reson., 2016, 268, 18–24 CrossRef CAS PubMed.
- M. Foston, Curr. Opin. Biotechnol., 2014, 27, 176–184 CrossRef CAS PubMed.
- K. Wickholm, P. T. Larsson and T. Iversen, Carbohydr. Res., 1998, 312, 123–129 CrossRef CAS.
- A. Palme, A. Idström, L. Nordstierna and H. Brelid, Cellulose, 2014, 21, 4681–4691 CrossRef CAS.
- P. T. Larsson, K. Wickholm and T. Iversen, Carbohydr. Res., 1997, 302, 19–25 CrossRef CAS.
- F. H. Larsen, M. Schobitz and J. Schaller, Carbohydr. Polym., 2012, 89, 640–647 CrossRef CAS PubMed.
- X. Y. Shi, G. P. Holland and J. L. Yarger, Biomacromolecules, 2015, 16, 852–859 CrossRef CAS PubMed.
- E. Vittadini, L. C. Dickinson, J. P. Lavoie, X. Pham and P. Chinachoti, J. Agric. Food Chem., 2003, 51, 1647–1652 CrossRef CAS PubMed.
- E. L. Lindh, M. Bergenstråhle-Wohlert, C. Terenzi, L. Salmén and I. Furó, Carbohydr. Res., 2016, 434, 136–142 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- R. Pönni, L. Rautkari, C. A. S. Hill and T. Vuorinen, Cellulose, 2014, 21, 1217–1226 CrossRef.
- R. Pönni, T. Vuorinen and E. Kontturi, BioResources, 2012, 7, 6077–6108 CrossRef.
- T. Taniguchi, H. Harada and K. Nakato, Nature, 1978, 272, 230–231 CrossRef CAS.
- K. Y. Lee and A. Bismarck, Cellulose, 2012, 19, 891–900 CrossRef CAS.
- M. Bloom, J. H. Davis and M. I. Valic, Can. J. Phys., 1980, 58, 1510–1517 CrossRef CAS.
- B. Halle, P. O. Quist and I. Furó, Liq. Cryst., 1993, 14, 227–263 CrossRef CAS.
- R. Y. Dong, Prog. Nucl. Magn. Reson. Spectrosc., 2002, 41, 115–151 CrossRef CAS.
- F. F. Wang, R. C. Zhang, T. H. Chen and P. C. Sun, Polymers, 2016, 8, 11 CrossRef.
- J. H. Kristensen, H. Bildsoe, H. J. Jakobsen and N. C. Nilesen, J. Magn. Reson., 1991, 92, 443–453 CAS.
- M. Hologne and J. Hirschinger, Solid State Nucl. Magn. Reson., 2004, 26, 1–10 CrossRef CAS PubMed.
- J. H. Kristensen, H. Bildsoe, H. J. Jakobsen and N. C. Nielsen, J. Magn. Reson., 1992, 100, 437–443 CAS.
- O. Weintraub and S. Vega, Solid State Nucl. Magn. Reson., 1995, 4, 341–351 CrossRef CAS PubMed.
- M. Cutajar, S. E. Ashbrook and S. Wimperis, Chem. Phys. Lett., 2006, 423, 276–281 CrossRef CAS.
- N. H. M. Hogg, P. J. T. Boulton, V. E. Zorin, R. K. Harris and P. Hodgkinson, Chem. Phys. Lett., 2009, 475, 58–63 CrossRef CAS.
- F. H. Larsen, H. J. Jakobsen, P. D. Ellis and N. C. Nielsen, Chem. Phys. Lett., 1998, 292, 467–473 CrossRef CAS.
- R. J. Wittebort, M. G. Usha, D. J. Ruben, D. E. Wemmer and A. Pines, J. Am. Chem. Soc., 1988, 110, 5668–5671 CrossRef CAS.
- B. Halle and H. Wennerström, J. Chem. Phys., 1981, 75, 1928–1943 CrossRef CAS.
- B. Lindman, G. Karlström and L. Stigsson, J. Mol. Liq., 2010, 156, 76–81 CrossRef CAS.
- J. F. Matthews, C. E. Skopec, P. E. Mason, P. Zuccato, R. W. Torget, J. Sugiyama, M. E. Himmel and J. W. Brady, Carbohydr. Res., 2006, 341, 138–152 CrossRef CAS PubMed.
- A. N. Fernandes, L. H. Thomas, C. M. Altaner, P. Callow, V. T. Forsyth, D. C. Apperley, C. J. Kennedy and M. C. Jarvis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1195–1203 CrossRef PubMed.
- M. Martinez-Sanz, M. J. Gidley and E. P. Gilbert, Soft Matter, 2016, 12, 1534–1549 RSC.
- S. Okubayashi, U. J. Griesser and T. Bechtold, Carbohydr. Polym., 2004, 58, 293–299 CrossRef CAS.
- R. L. Silveira, S. R. Stoyanov, A. Kovalenko and M. S. Skaf, Biomacromolecules, 2016, 17, 2582–2590 CrossRef CAS PubMed.
- T. Kuribayashi, Y. Ogawa, C. Rochas, Y. Matsumoto, L. Heux and Y. Nishiyama, ACS Macro Lett., 2016, 5, 730–734 CrossRef CAS.
- C. Joly, R. Gauthier and M. Escoubes, J. Appl. Polym. Sci., 1996, 61, 57–69 CrossRef CAS.
- V. Kocherbitov, S. Ulvenlund, M. Kober, K. Jarring and T. Arnebrant, J. Phys. Chem. B, 2008, 112, 3728–3734 CrossRef CAS PubMed.
- L. W. Reeves, Prog. Nucl. Magn. Reson. Spectrosc., 1969, 4, 193–233 CrossRef CAS.
- K. Kulasinski, S. Keten, S. V. Churakov, D. Derome and J. Carmeliet, Cellulose, 2014, 21, 1103–1116 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074–9082 CrossRef CAS PubMed.
- S. Hediger, A. Lesage and L. Emsley, Macromolecules, 2002, 35, 5078–5084 CrossRef CAS.
- P. B. White, T. Wang, Y. B. Park, D. J. Cosgrove and M. Hong, J. Am. Chem. Soc., 2014, 136, 10399–10409 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Table listing manufacturer and purity of the used inorganic salts; inversion-recovery delays of Fig. 2. See DOI: 10.1039/c6cp08219j. |
| ‡ These authors contributed equally. |
| § Present address: Magnetic Resonance Research Centre, Department of Chemical Engineering and Biotechnology, University of Cambridge, Cambridge CB3 0HE, UK. |
| This journal is © the Owner Societies 2017 |