
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic and microscopic investigations of tautomerization in porphycenes: condensed phases, supersonic jets, and single molecule studies
P.
Fita
 *a,
L.
Grill
*b,
A.
Listkowski
c,
H.
Piwoński
d,
S.
Gawinkowski
d,
M.
Pszona
d,
J.
Sepioł
d,
E.
Mengesha
d,
T.
Kumagai
*e and
J.
Waluk
*cd
*a,
L.
Grill
*b,
A.
Listkowski
c,
H.
Piwoński
d,
S.
Gawinkowski
d,
M.
Pszona
d,
J.
Sepioł
d,
E.
Mengesha
d,
T.
Kumagai
*e and
J.
Waluk
*cd
aInstitute of Experimental Physics, Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland. E-mail: Piotr.Fita@fuw.edu.pl
bDepartment of Physical Chemistry, University of Graz, Heinrichstrasse 28, 8010 Graz, Austria. E-mail: leonhard.grill@uni-graz.at
cFaculty of Mathematics and Natural Sciences, College of Science, Cardinal Stefan Wyszyński University, Dewajtis 5, 01-815 Warsaw, Poland
dInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224, Warsaw, Poland. E-mail: waluk@ichf.edu.pl
eDepartment of Physical Chemistry, Fritz-Haber Institute of the Max-Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: kuma@fhi-berlin.mpg.de
First published on 16th January 2017
Abstract
We describe various experimental approaches that have been used to obtain a detailed understanding of double hydrogen transfer in porphycene, a model system for intramolecular hydrogen bonding and tautomerism. The emerging picture is that of a multidimensional tautomerization coordinate, with several vibrational modes acting as reaction-promoters or inhibitors through anharmonic intermode coupling. Tunnelling processes, coherent in the case of isolated molecules and incoherent in condensed phases, are found to play a major role even at elevated temperatures. Single-molecule spectroscopy studies reveal large fluctuations in hydrogen transfer rates observed over time for the same chromophore. Scanning probe microscopy is employed to directly observe the structure and tautomerization dynamics of single molecules adsorbed on metal surfaces and demonstrates how the interactions of the molecules with atoms of the supporting surface affect their static and dynamic properties: different tautomeric forms are stabilized for molecules depending on the surface structure and the reaction mechanism can also change, from a concerted to a stepwise transfer. The scanning probe microscopy studies prove that tautomerization in single molecules can be induced by different stimuli: heat, electron attachment, light, and force exerted by the microscope's tip. Possible applications utilizing tautomerism are discussed in combination with molecular architectures on surfaces, which could pave the way for the development of single-molecule electronics.
 P. Fita | Piotr Fita obtained his PhD at the Faculty of Physics, University of Warsaw in 2008. During his graduate studies he carried out research on ultrafast processes, in particular proton transfer, in organic molecules. Afterwards he spent two years in the group of Prof. Eric Vauthey at the University of Geneva where he learned to use surface second harmonic generation for the investigation of liquid/liquid interfaces. Currently he is an assistant professor at the Faculty of Physics, University of Warsaw, where he continues to apply time-resolved spectroscopy for studies of proton transfer and other ultrafast processes in organic compounds. |
 L. Grill | Leonhard Grill did his PhD in experimental physics in Trieste, and then joined the Rieder group (FU Berlin) as a postdoc before he became a group leader at the Fritz-Haber-Institute Berlin in 2009. His research focusses on the manipulation of single functional molecules at surfaces and the characterization of specific molecular functions by scanning probe microscopy. The electronic, electrical or mechanical properties of individual molecules are studied and controlled with the goal to obtain the fundamental understanding of physical and chemical processes. He received the Feynman Prize in Nanotechnology (2011) and has been a professor of Physical Chemistry at the University of Graz since 2013 (http://www.nanograz.com). |
 Top, left to right: Dr Arkadiusz Listkowski, Dr Hubert Piwoński, Dr Sylwester Gawinkowski and Maria Pszona. Bottom: Prof. Jerzy Sepioł, Dr Ephriem T. Mengesha and Prof. Jacek Waluk | Warsaw team: top, left to right: Dr Arkadiusz Listkowski, organic chemist interested in syntheses of porphycenes; Dr Hubert Piwoński, specialist in single molecule fluorescence. He was a postdoc with Prof. Gilad Haran at the Weizmann Institute of Sciences (Rehovot, Israel), focusing on single-molecule enzymatic activity. Currently he is working at King Abdullah University of Sciences and Technology, Thuwal, Saudi Arabia, developing near-IR-emitting conjugated polymer nanoparticles; Dr Sylwester Gawinkowski, PhD in the vibrational spectroscopy and single molecule SERS detection of porphycenes. Since 2015, a postdoctoral researcher and Marie Curie Sklodowska Actions European Fellowship recipient at the Institute of Photonic Sciences, Castelldefels-Barcelona. His research interests include femtosecond nonlinear Raman spectroscopy, molecular nanophotonics and single molecule SERS; Maria Pszona, finishing her PhD thesis on single molecule SERRS. Bottom: Prof. Jerzy Sepioł, whose scientific interests encompass single molecule and supersonic jet spectroscopy; Dr Ephriem T. Mengesha, from Ethiopia, PhD in proton tunneling in porphycenes isolated in supersonic jets; currently a postdoc (Eurotalents Fellow) in the Laser Interactions and Dynamics Laboratory at CEA-Saclay, working on conformation and dynamics of floppy molecular/biomolecular complexes in helium nanodroplets; Prof. Jacek Waluk, Head of Photochemistry and Spectroscopy Department, Institute of Physical Chemistry of the Polish Academy of Sciences. |
 T. Kumagai | Takashi Kumagai finished his PhD program at Kyoto University, Japan, in March 2011, with the theme of Visualization of Hydrogen-Bond Dynamics. In April 2011, he joined the Fritz-Haber Institute (FHI) as a research fellow of the Japan Society for the Promotion of Science. Since April 2013 he has been a research group leader of the Nanoscale Surface Chemistry Group at FHI and studies single molecule chemistry on surfaces using low-temperature scanning probe microscopy. He was awarded several prizes for early career scientists, including the Inoue Research Award for Young Scientists, the Morino Award for Molecular Science, and the Gerhard Ertl Young Investigator Award. |
Introduction
Proton or hydrogen transfer reactions look simple on paper, but, because of nuclear quantum effects of the proton, the precise description of the transfer dynamics may be extremely complicated and still remains a challenging research topic. The understanding of these fundamental processes is crucial not only for chemists: such reactions are ubiquitous in nature, often involving multiple proton transfer events. In many cases hydrogen/proton transfer reactions occur via a hydrogen bond and the hydrogen-bonding geometry largely affects the potential landscape, i.e. the reaction dynamics. Therefore, it is of fundamental importance to elucidate the correlation between them. The hydrogen-bonding geometry may be susceptive to the surrounding of a molecule in condensed phases and the transfer rate can be significantly changed under different conditions. However, it is difficult to obtain molecular-level understanding about such environmental impacts and single-molecule experiments are desirable to achieve this purpose.Porphycenes, structural isomers of porphyrins,1,2 emerge as good models for studying intramolecular single or double hydrogen transfer processes.3–7 Tautomerization, occurring in an inner molecular cavity consisting of four nitrogen atoms, is observed in both ground and excited electronic states. Three doubly degenerate tautomeric forms are possible (Fig. 1). The dimensions of the cavity, which determine the strength of the intramolecular hydrogen bonds, can be modified in a controlled fashion by appropriate substitution of peripheral groups, providing a series of compounds useful to examine correlations between the hydrogen bonding geometry and tautomerization dynamics.8 A variety of electronic and vibrational spectroscopy techniques have been applied to investigate the molecular structure and tautomerization dynamics, including supersonic jet isolation and single molecule studies using fluorescence imaging and surface-enhanced resonance Raman scattering.9–13 The direct observation of single-molecule tautomerization has also been performed using low-temperature scanning probe microscopy.14–18
 | ||
| Fig. 1 Tautomeric forms of porphycene and their estimated relative energies. The arrows represent the S0–S1 electronic transition dipole moment. | ||
In this perspective article we describe emerging methodologies and recent progress in tautomerization studies of porphycenes using various and different experimental techniques. Spectroscopic studies on molecules isolated in supersonic jets are discussed first. Their electronic spectra exhibit characteristic tunnelling splitting of vibronic levels, a clear signature of coherent tunnelling of the inner protons. The splitting values are found to be susceptive to excitation of a specific vibrational mode of a molecule, which demonstrates that the tautomerization coordinate has a multidimensional character and proton delocalization is significantly affected by the modulation of the potential landscape.19
Next, the results obtained for porphycenes in condensed phases are described. An important finding is that the main mechanism of tautomerization involves tunnelling, which is dominant even at room temperature in solutions.20 A spectacular confirmation of tunnelling is the variation of the tautomerization rate – exceeding three orders of magnitude – with the distance between nitrogen atoms bridging the intramolecular hydrogen bonds.8
In the third part, the main results obtained from single molecule studies are presented. Three complementary techniques have been used: (a) fluorescence imaging; (b) surface-enhanced resonance Raman scattering; (c) low-temperature scanning tunnelling microscopy. Investigations of tautomerization in single molecules allowed observations that would have been impossible to obtain in bulk studies, such as temporal variations of hydrogen transfer rates by many orders of magnitude, the detection of rare cis isomers, or the local environmental impact on the tautomerization behaviour.
We conclude by summarizing the current state of knowledge on tautomerism obtained from porphycene studies and by discussing future challenges aiming at elucidating the complex multidimensional nature of the tautomerization dynamics and possible applications of single-molecule tautomerization as a molecular switch in combination with molecular architecture on surfaces.
Results and discussion
Molecules isolated in supersonic jets
Fluorescence spectra measured in supersonic jets usually consist of spectrally narrow lines. Moreover, it is possible to observe non-relaxed emission occurring from the initially excited vibronic level (single vibronic level fluorescence, SVLF). Therefore, such measurements provide detailed information about the vibrational structure, not only in the ground electronic state, but also in S1. The latter is obtained by recording laser-induced fluorescence excitation (LIF) spectra.Both LIF and SVLF spectra obtained for porphycene isolated in supersonic jets reveal the splitting of vibronic levels into doublets (Fig. 2).19,21–24 The splitting disappears when the inner protons are replaced by deuterons, or when porphycene forms a complex with water or alcohol in the jet,21 suggesting that its origin can be attributed to coherent tunnelling of the inner protons occurring in a symmetric double minimum potential of the lowest energy trans tautomers. The spacing between doublet components is constant in LIF, but significantly varies for vibrational levels in the ground state probed by SVLF.19 These results indicate that the tunnelling splitting in S1 is negligible compared to S0, and the constant value of separation between the doublet components observed in LIF, 4.4 cm−1, results from the splitting in the vibrational levels in S0. This interpretation has been confirmed by measurements for porphycene embedded in an ultra-cold environment of helium nanodroplets (0.37 K).25 Under these conditions, the upper level of the doublet is not populated in S0, and the splitting is not observed in fluorescence excitation spectra. However, it does appear in the emission, because the splitting of the lowest vibrational level in S1 is very small, so that both energy levels can be populated.
 | ||
Fig. 2 Spectra of porphycene isolated in a supersonic jet. Bottom, fluorescence excitation (LIF) and top, fluorescence recorded for excitation into the 0–0 origin band (16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 163 cm−1). Tunnelling splitting values are presented for selected vibronic levels. Frequencies are given relative to the 0–0 excitation energy. 163 cm−1). Tunnelling splitting values are presented for selected vibronic levels. Frequencies are given relative to the 0–0 excitation energy. | ||
The observation of different splitting for different vibrational modes indicates a multidimensional character of the tautomerization coordinate. Depending on the vibrational state, porphycene experiences a different barrier to double hydrogen transfer. Both tautomerization-promoting and inhibiting modes are observed, which make tunnelling splitting larger or smaller than the value for the ground vibrational state, respectively.19 For the majority of vibronic bands, the splitting is similar to that observed for the transition between the lowest vibrational levels in S0 and S1; therefore, the corresponding modes are described as “neutral”. However, a neutral mode can become promoting in the porphycene in which all twelve peripheral hydrogen atoms were replaced by deuterium, because the displacement vector of the mode is different from that of the non-deuterated molecule.24
Simulations of tautomerization in porphycene using Car–Parrinello molecular dynamics26 confirmed the model of promoting and inhibiting vibrational modes. An interesting case arises for simultaneous excitation of two vibrations of opposite nature. Calculations predict that the enhancing character dominates. Indeed, experimentally observed splitting in such cases shows values between those observed for neutral and promoting modes.
Molecules in condensed phases
Due to the inhomogeneous and/or fluctuating environment of individual molecules, tautomerization of porphycenes in condensed phases, such as solutions, glasses, polymer matrices or crystals, becomes an (incoherent) rate process.8,20,27–32 It seems natural to assume that due to perturbations of solute molecules by the environment, coherence is quickly lost and the superposition state of protons is quenched. On the other hand, thermal fluctuations might deliver energy enabling the protons to move over the potential barrier, enhancing a classical thermally activated mechanism. Nevertheless, an incoherent tunnelling process was observed for porphycene in condensed phases even at room temperature.20In order to elucidate the tautomerization mechanism in porphycene and its derivatives, the reaction rates have been determined for a number of differently substituted derivatives under various conditions.8,20,28–32 It is not a trivial task to measure the rate, since the initial and final tautomeric forms are identical. Therefore, the underlying principle of the measurements is based on the fact that the directions of electronic transition dipole moments rotate significantly (e.g. by α = 72° for the S1 ← S0 transition in bare porphycene, see Fig. 1) when a molecule undergoes trans–trans tautomerization. Therefore, fluorescence emitted by an ensemble of immobilized molecules which are excited with pulses of linearly polarized light becomes less polarized with time elapsed after excitation, as more and more molecules undergo tautomerization. This gradual depolarization of the emitted light can be described in terms of fluorescence anisotropy r defined as33
 | (1) |
| r(t) = (r0 − r∞)e−2kt + r∞, | (2) |
| r∞ = ½(r0 + rα), | (3) |
 | (4) |
 | ||
| Fig. 3 (a) Anisotropy kinetics recorded for porphycene in paraffin oil with S1 ← S0 excitation at 635 nm and probe wavelengths of 550 or 635 nm (when the S2 ← S0 transition is probed at 550 nm, the initial anisotropy is close to −0.2 because the S2 ← S0 and S1 ← S0 transition moments are almost perpendicular to each other). (b) Tautomerization rates recorded for a series of porphycene derivatives in the ground (k0) and first singlet excited state (k1) plotted as a function of the calculated N–N donor–acceptor distance. (c) The same data as in panel b, but for a subset of porphycene derivatives with the N–N distance close to 2.65 Å. | ||
Eqn (2) describing anisotropy kinetics does not take into account its decay due to rotational diffusion. Even though it can be modified to include this effect, accurate determination of tautomerization rates requires timescales of tautomerization and molecular rotations to be well separated. This condition can be best realized by embedding the target molecules in polymer matrices where the rotations are hindered on the timescales of anisotropy decays. However it turned out to be sufficient to dissolve them in highly viscous solvents such as paraffin oil or ethylene glycol.8,28 Transient absorption anisotropy decays are also affected by the deactivation of the excited state, which cannot be neglected if decay times of the excited state population and anisotropy become comparable. The exact formula describing transient absorption anisotropy kinetics is derived in ref. 8.
The choice of solvents raised a question concerning their influence on the reaction rates and the mechanism. Measurements carried out at room temperature did not indicate any significant differences between various solvents and PMMA matrices. This observation leads to a conclusion that tautomerization is insensitive to the environment of the molecule and the geometry of the molecular cavity consisting of four nitrogen atoms is not affected by the surroundings. Nevertheless, recent experiments suggest that the environment may affect the tautomerization in polymer matrices at cryogenic temperatures (below 150 K). If such effects are confirmed, they could explain a large discrepancy between the results obtained by transient absorption20 and low-temperature steady-state fluorescence anisotropy34 measurements.
In contrast to the lack of the influence from the surroundings of a molecule, room temperature tautomerization rates of porphycenes are strongly modified by substitution at the molecular periphery, either at the pyrrole rings or at the meso positions.8 Such a substitution changes the geometry of a molecule, in particular the donor–acceptor N–N distance in the cavity. Thus, the effect of the substitution on tautomerization rates can be explained in terms of the potential landscape modification, i.e. the variation of the hydrogen bond strength. This effect is drastic, as the decrease of the N–N distance by merely 10% leads to the increase of the tautomerization rate by three orders of magnitude (Fig. 3b). Such a strong dependence of the reaction rate on the barrier shape is a strong indication that tunnelling may be involved in the process, because the tunnelling probability depends (in WKB approximation) exponentially on the square root of the barrier height and width. Nevertheless, the donor–acceptor N–N distance is not the only factor affecting tautomerization rates, which becomes apparent from a closer look at molecules with the distances close to 2.65 Å (Fig. 3c). Even though the geometry of the substituted molecules is very similar, hydrogen transfer rates can differ significantly, with the bare porphycene tautomerizing at the slowest rate (for this subset of molecules the tautomerization rate seems to weakly increase with the N–N distance). This observation can be explained by the influence of additional, low frequency vibrational modes introduced by the substituting groups, which are thermally populated. These modes can couple to the tautomerization path as it was revealed in high resolution spectroscopic studies discussed in the preceding section. The larger number of vibrational states available for thermal excitation at room temperature should facilitate hydrogen transfer in such a case.
When the contribution of quantum tunnelling in hydrogen transfer reactions is suspected, the rates should be examined upon isotope substitution of the transferred hydrogen by deuterium and/or across a range of temperatures in order to confirm it. According to a commonly applied Bell model35 which explicitly takes tunnelling into account, H/D substitution should lead to at least 7-fold reduction of the reaction rate at 300 K if tunnelling is involved. Experiments with deuterated porphycene and its derivative, 2,7,12,17-tetra-t-butyl porphycene in solution (mixtures of ethanol-d1 with methanol-d1 or methanol-d1 with ethylene glycol-d2), revealed the room temperature isotope effect within this limit for the parent molecule (kH/kD = 4.3 ± 0.3 in S0 and 5.0 ± 0.4 in S1) and slightly exceeding it (kH/kD = 8.0 ± 0.5 in S0 and 7.7 ± 0.4 in S1) for the tert-butyl derivative.29 Here, values close to the theoretical limit are inconclusive because the Bell model does not take into account the multidimensionality of the potential surface and coupling of hydrogen movements to vibrational degrees of freedom (e.g. skeletal modes). It may also not take into account the zero-point energy correction in the transition state. Therefore, further insight into the mechanism of porphycene tautomerization in the condensed phase could be obtained only by temperature-dependent measurements. For the parent porphycene and its 2,7,12,17-tetra-t-butyl derivative, anisotropy kinetics in glass forming mixtures of ethanol and methanol were measured down to 60 and 30 K, respectively. Additionally, measurements at elevated temperatures in ethylene glycol were carried out up to 400 K. As expected, anisotropy decays slowed down as temperature decreased. However, this was not the only effect seen in anisotropy decays: at the lowest temperatures (below approx. 150 K) asymptotic anisotropy values increase when temperature decreases (Fig. 4a). It is very unlikely that the geometry of porphycene changes, and therefore this effect should be rather interpreted as due to an increased number of molecules which do not undergo tautomerization and maintain the initial anisotropy (r0 = 0.4). This explanation is particularly plausible in the context of single-molecule studies described below, which show that tautomerization at room temperature can be stopped in certain molecules. Such an effect is even more likely at low temperatures, when molecules are “frozen” into a rigid environment. Depending on the local structure of this environment, tautomerization may be blocked or not.
 | ||
| Fig. 4 (a) Anisotropy kinetics recorded for porphycene (PC) in the ethanol/methanol mixture at low temperatures. (b and c) Arrhenius plots for PC and 2,7,12,17-tetra-t-butyl porphycene (TTPC). (d) Contribution of tunnelling and other channels to the overall tautomerization rate in PC (solid line) and TTPC (dashed line). (e) Isotope effect in tautomerization of PC and TTPC. | ||
The temperature dependence of tautomerization rates clearly exhibits non-classical behaviour: deviations from straight lines in Arrhenius plots of lnk as a function of 1/T (Fig. 4b and c). In particular, plateaus reached at the lowest temperatures correspond to a region of temperature-independent pure tunnelling in the vibrational ground state of a molecule. When temperature increases, thermally activated channels contribute to the process. Taking into account various vibrational modes that can couple to the hydrogen transfer we should model the temperature dependence of tautomerization rates by the function:
 | (5) |
It is instructive to compare incoherent tunnelling rates in the condensed phase with tunnelling splitting Δ measured in isolated molecules. In the latter, coherent tunnelling in a symmetric double-well potential leads to periodic Rabi oscillations of a particle wavefunction between the two wells with a frequency Δ/h, where h is the Planck constant. Twice of this frequency can be interpreted as a rate of particle transfer between the two positions. The measured value of Δ = 4.4 cm−1 in the vibrational ground state of parent porphycene in a supersonic jet and helium droplets is equal to 2.6 × 1011 s−1. The tunnelling rate obtained for the lowest vibrational level in S0 in solutions, k0 = (0.60 ± 0.02) × 1011 s−1, is only 4 times lower than the coherent limit. This means that the perturbation of the symmetric double-well potential by interactions with the environment only moderately hinders tunnelling in the studied example. The solvent exerts even a weaker effect on tunnelling in 2,7,12,17-tetra-t-butyl porphycene, for which Δ = 9 cm−1 which gives the upper tunnelling rate limit at approximately 5.4 × 1011 s−1. This value is only 40% higher than the incoherent tunnelling rate measured to be k0 = (3.8 ± 0.2) × 1011 s−1.
Inspection of tunnelling splitting values also allows the verification of the proposed mechanism of tunnelling enhancement through vibrational excitation. The incoherent tunnelling rate should be proportional to the square of the tunnelling splitting.37 The latter was measured to be 12 ± 1 cm−1 at the v = 1 level of the 180 cm−1 (2Ag) mode of the parent porphycene. The tunnelling rate in this vibrational state, k1 in eqn (5), was determined to be (5.4 ± 0.2) × 1011 s−1. The ratio k1/k0 = 9.0 ± 0.5 (incorrectly reported to be 7.0 in ref. 20) is in acceptable agreement with the square of the tunnelling splitting ratio, (12 cm−1/4.4 cm−1)2 = 7.4 ± 1.3.
Analogous temperature-dependent measurements of tautomerization rates in molecules with two inner protons substituted by deuterons surprisingly revealed a very weak (by less than a factor of 3) enhancement of tunnelling by the 180 cm−1 mode. In WKB approximation, the probability of tunnelling is proportional to the exponential function of the square root of barrier dimensions multiplied by the particle mass. Thus, it seems that any effects related to the modulation of the tautomerization barrier should be stronger for a more massive particle. This does not agree with the observations. Apparently, a static model cannot be applied here and the theory of the so-called vibrationally assisted tunnelling in a double-well potential is more appropriate.38–40 According to this model, periodic modulations of the potential that are much faster than the tunnelling rate increase the tautomerization rate. The degree of this enhancement is much lower if the mass of a tunnelling particle is doubled, in agreement with our observations. The isotope effect should be understood here as the ratio of vibrational enhancements (k1/k0) for both isotopologues. Its value could not be reliably established, because the k1 values for deuterated molecules were very close to 0 and determined with relative uncertainties exceeding 100%. Nevertheless, the isotope effect is expressed here as a qualitative change in the dynamics of the studied molecules, as one of the tautomerization channels is practically blocked upon H/D exchange. This finding can also explain why the room temperature isotope effect on total tautomerization rates is rather low (Fig. 4e) and not significantly exceeding the Bell model limit, even though, as shown earlier, tunnelling effects are crucial for non-deuterated molecules.20 First, in deuterated molecules one of the tunnelling channels is inactive and classical processes can dominate at room temperature. Second, neither vibrationally enhanced tunnelling nor dynamic effects of periodic potential modulation via anharmonic intermode coupling, apparently very important in porphycenes, are taken into account in this model.
Summing up, tautomerization studies of porphycenes in the condensed phase have revealed two tunnelling channels: one occurs in the vibrational ground state and the other, significantly accelerated, in the vibrationally excited state. These two quantum channels dominate the overall reaction rate at low temperatures and their contribution may exceed 50% even above room temperature. The influence of decoherence due to interactions with a surrounding environment seems to be rather weak concerning the transfer rates because those in the condensed phases are not much lower than the upper limit of coherent tunnelling. The tunnelling rate enhancement by vibrational excitation appears to be an effect of quantum dynamics in a periodically modulated double-well potential, which results in a lack of this effect for deuterated molecules. Measurements at cryogenic temperatures (in particular below 150 K) indicated that the tautomerization properties of porphycenes may be affected by the local environment, in contrast to the regime of room temperature, where tautomerization rates seem to be independent of the medium. Bulk measurements, obviously, cannot investigate the differences between individual molecules surrounded by various local environments. This problem will be addressed in the following section.
A general remark seems to be appropriate regarding the methodology of tautomerization rate measurements based on the rotation of transition moments upon trans–trans conversion. While this effect is obvious when the two tautomers are chemically equivalent (self-exchange reaction), in an unsymmetrically substituted compound the transition moment direction may become “locked”. So far, such behavior has been detected for only one derivative, 9-amino-2,7,12,17-tetraphenylporphycene,30,41 but it can be expected also for other, strongly asymmetric porphycenes. Since the transition moment directions in the two trans forms should not be exactly parallel, the determination of tautomerization rates is still possible, albeit experimentally more challenging, because rather small anisotropy changes have to be analysed.
Tautomerization in single molecules
The observation of tautomerization in single molecules enabled us to reveal an effect that would have been inaccessible in bulk studies. Analysis of nearly 900 molecules of 2,7,12,17-tetraphenylporphycene, one by one, revealed the existence of a small subpopulation in which the double hydrogen transfer could be temporarily frozen for periods of seconds to minutes.11 This was demonstrated by visualizing the spatial fluorescence intensity patterns recorded in consecutive scans (Fig. 5). Their shape changes from a double lobe to doughnut, indicating the localization of the inner hydrogens on the timescale of the image recording, or a rapid tautomerization, respectively. The image evolution indicates that after (or before) the molecule is in the regime of rapid tautomerization, the protons become localized. This is a spectacular phenomenon, given that the average reaction time determined in ensemble studies was of the order of single picoseconds. The effect was explained by the deviation of the molecular geometry and/or the environment from a symmetric double minimum potential. For the molecule under study, the torsional motion of the phenyl substituents is a good candidate for the distortion coordinate.
 | ||
| Fig. 5 Changes in the tautomerization rate illustrated by fluorescence imaging of single molecules of 2,7,12,17-tetraphenylporphycene. Three consecutively taken images are shown. The molecule indicated by a square switches from a regime of localized protons to fast reaction, and back to the localized state, but now with the protons located on other nitrogens. | ||
 | ||
| Fig. 6 Time evolution in a series of single-molecule SERRS spectra of porphycene (Pc-d0) and its isotopologue deuterated at the peripheral carbon atoms (Pc-d12). Porphycenes were placed on gold nanostructures and excited at 633 nm. The bands that appear and disappear over time, indicating transitions between the trans and cis1 tautomers, are marked by asterisks. At the bottom, calculated Raman spectra of both forms. Adapted from ref. 12. Copyright 2016 The Royal Society of Chemistry. | ||
In order to study the static and dynamic properties at the single-molecule level, porphycene molecules were deposited on a single-crystalline metal surface under ultra-high vacuum conditions (10−10 mbar) where an atomically well-defined environment can be obtained. Fig. 7 shows the topographic STM images of a single porphycene molecule adsorbed on a Cu(110) surface at 5 K. The sample was prepared in situ and the molecules were thermally evaporated from a Knudsen cell at 460–500 K onto an atomically clean surface. The relative stability of the tautomeric form is determined by the interaction between the molecule and the surface. It was found that the cis1 configuration is more favourable than the trans configuration on Cu(110),15 in contrast to isolated molecules in the gas phase. This also implies that the presence of the surface could have a significant impact on the tautomerization mechanism. As shown in the previous sections, tautomerization of porphycene in the gas phase or in condensed phases occurs very rapidly via quantum tunnelling either in a coherent (isolated molecules) or incoherent (condensed phases) manner.19–21,23–25 However, spontaneous tautomerization of porphycene on Cu(110) via tunnelling was not observed at 5 K. The absence of the tunnelling process could be attributed to the reduced symmetry of the molecule caused by the adsorption.
 | ||
| Fig. 7 Porphycene on a Cu(110) surface. (a) Schematic of the atomic arrangement of Cu(110). (b) Topographic STM image of a single porphycene molecule at 5 K (Vbias = 0.1 V and It = 10 nA). Reversible cis ↔ cis tautomerization can be induced by various external stimuli (i.e. heat, electron, and force). (c) Trace of tunnelling current recorded over a molecule while fixing the STM tip at the position indicated by the star in (b). (d) Schematic of “High” and “Low” current states where the inner H atoms are located under and away from the tip, respectively (the surface is omitted). (e) Heat-induced cis ↔ cis tautomerization (Arrhenius plot). The inset shows the STM images at elevated temperatures. The solid line represents the best fitted result of the experimental data to the Arrhenius equation and a barrier of 168(±12) meV was obtained. | ||
DFT calculations have predicted that the molecular frame of porphycene on Cu(110), which is fully planar in the gas phase, is distorted due to the relatively strong interaction of free (amine) N atoms of pyrrole rings with the surface Cu atoms.15 Additionally, the adsorption position of porphycene slightly deviates with respect to the high symmetry axis of the surface lattice underneath. The thermally induced cis1 ↔ cis1 tautomerization was observed above ∼78 K and its rate rapidly increases upon increasing the temperature (see the inset STM images of Fig. 7e).14 The tautomerization was directly monitored by recording the tunnelling current while fixing the STM tip position over a molecule (a feedback loop of the STM was disabled). In this method the time-resolution, determined by the bandwidth of the amplifier, is approximately 1 ms in the setup used. Tautomerization results in a random telegraph noise in the current trace (Fig. 7c). The tautomerization rate was obtained by a statistical analysis of time intervals between the tautomerization events. Fig. 7e shows the temperature dependence of the tautomerization rate (i.e. Arrhenius plot) and the activation barrier was estimated to be 168(±12) meV by fitting the experimental data to the Arrhenius equation.
Low energy electrons from STM can be used to control adsorbate reactions, as has, for a pioneering example, been demonstrated for the dissociation of oxygen molecules44 and the rotation of acetylene45 that can be induced by vibrational excitation through inelastic electron tunnelling processes. This capability makes STM a powerful tool to investigate single molecule chemistry on surfaces.46 We found that the cis1 ↔ cis1 tautomerization of porphycene can also be triggered by tunnelling electrons, even at 5 K if the bias voltage (i.e. energy of a tunnelling electron) is sufficiently high, exceeding 150 meV (Fig. 8a–c). For tunnelling electron-induced reactions, investigating the voltage and current dependence of the reaction rate (or yield) can provide deep insights into the excitation mechanism (so-called STM action spectroscopy).47 Additionally, substitution of transferred hydrogen atoms with deuterium is a very common and effective approach to elucidate the mechanism behind hydrogen transfer reactions, as also described in previous sections. Fig. 8d shows the voltage dependence of the tautomerization yield (per electron) measured for h- and d2-porphycene. A threshold voltage is found around 150 mV, similar at opposite bias polarity (see the inset of Fig. 8), thus revealing the threshold gap of 300 mV, which is too small to be rationalized by the electronic transition of porphycene since the S1 state is located at 2.0 eV.21 More importantly, a pronounced isotope effect was observed at higher voltages, indicating that the reaction is triggered by vibrational excitation via an inelastic electron tunnelling process. The threshold voltage around 150 mV that reveals a weak isotope shift could be assigned to excitation of a skeletal mode(s) of porphycene. For d2-porphycene a steep increase of the yield appears around 270 mV that nicely matches the N–D stretching energy. However, the second onset that should be associated with the N–H stretching mode is not clear for h-porphycene and a moderate increase was observed from 250 to 380 mV. The rising gradient of the yield was found to be related to the distribution of the vibrational density of states48 and the widespread feature of the N–H stretch could be attributed to noticeable anharmonic nature.49
 | ||
| Fig. 8 Electron-induced cis ↔ cis tautomerization of porphycene on a Cu(110) surface. (a–c), STM images at 5 K obtained at different bias voltages. (d) Bias voltage dependence of the tautomerization yield (STM action spectra) measured for h- and d2-porphycene. The inset displays the yields at both bias polarities in the regime of the threshold voltage. | ||
As discussed in precedent sections, tautomerization may be susceptible to a local environment in condensed phases, e.g. an electrostatic field from neighbouring molecules or solvation. Investigation of such a local impact on tautomerization dynamics is extremely difficult using conventional spectroscopic methods that detect spatially averaged signals over large areas and therefore for many molecules at the same time. The local probing capability of STM instead allows directly addressing the dynamics of individual molecules in different local environments. Fig. 9a shows an STM image of a porphycene dimer connected with a single Cu atom. Interestingly, tautomerization was not observed in the dimer, suggesting that the potential landscape that is symmetric for an isolated molecule is considerably distorted, i.e. asymmetric (as shown in Fig. 9b), in the dimer. Note that, for simplicity, we show a double-well potential in Fig. 9, but the theoretical simulation predicts the stepwise reaction mechanism in which the meta-stable trans configuration exists.17 However, tautomerization in the central molecule of a trimer (Fig. 9c) indicates that the potential landscape is restored to be symmetric, due to the local symmetry of the central molecule (Fig. 9d). Additionally, tautomerization was also observed for inner molecules in larger clusters (Fig. 9e and f), indicating a rather weak influence from the second neighbouring molecule.
 | ||
| Fig. 9 One-dimensional porphycene assemblies on a Cu(110) surface at 5 K. (a) Dimer. (b) Schematic of the dimer and the tautomerization potential. (c) Trimer. (d) Schematic of the trimer and the tautomerization potential. (e) Tetramer. (f) Pentamer. All STM images were obtained at Vbias of 300 mV which is sufficiently higher than the threshold required to induce the cis ↔ cis tautomerization. | ||
As mentioned above, the stable tautomeric form is sensitive to the surface underneath. On a Cu(111) surface (Fig. 10a) porphycene prefers to adsorb in the trans configuration (Fig. 10b). However, the cis1 configuration (Fig. 10c) also exists as a meta-stable form and the unidirectional conversion from the trans to cis1 configuration can be induced by tunnelling electrons or photo-irradiation. Fig. 10d illustrates the schematic potential diagram of the porphycene on Cu(111). Fig. 10e displays the STM images before and after illumination at 405 nm and the switched molecules are marked by white circles. When the surface was exposed to light, all the molecules were converted to the cis1 configuration, indicating that no backward reaction occurred. It should be noted that the reversible cis1 ↔ cis1 tautomerization also occurs under illumination. The illumination time-evolution of the trans → cis1 conversion was found to follow a first-order process.18 On the other hand, the backward cis1 → trans conversion can be induced by heating the substrate up to ∼30 K and the temperature dependence measurement revealed the activation barrier of ∼42 meV.
 | ||
| Fig. 10 Porphycene on a Cu(111) surface. (a) Atomic arrangement of Cu(111). (b and c) STM image of trans and cis tautomers, respectively. The former is the thermodynamically stable form. (d) Chemical structure in the trans and cis configuration and schematic potential. (e) Large-scale STM images (size: 25 × 25 nm2, Vbias = 0.05 V, and It = 0.05 nA) before and after irradiation with a 405 nm diode laser (CW, 2.7 mW) for 2 min. The switched molecules are marked by white circles. The STM tip was retracted by ∼2 μm from the surface during irradiation. | ||
Photochemical processes on a metal surface differ substantially from those in the gas phase or in solution.50 In the former case, the excitation mechanism can be divided into a direct and indirect process. In the direct process, a photon is absorbed by molecules and the process resembles the conventional one, i.e. electronic transition of the molecule followed by a chemical reaction. However, a spectral response is expected to be blurred because discrete molecular states are considerably broadened through the interaction with the surface (mixing of molecular orbitals with continuous bands of a metal) as illustrated in Fig. 11a. This direct process is not usually a dominant channel, due to a relatively small absorption cross-section of adsorbates at low molecular coverages (e.g. below 1 monolayer). In the indirect process, photons are absorbed by the substrate and, as a consequence, hot carriers (excited electrons and holes) are generated near the surface. They are subsequently transferred to the adsorbed molecule, resulting in the reaction. In this case, the spectral response reflects the electronic structure of the substrate. Fig. 11b shows the wavelength dependence of the tautomerization cross-section measured with p- and s-polarized light. The cross-section rapidly increases at around 2 eV and the p-polarized light is more efficient than the s-polarized one at all wavelengths. No obvious molecular resonance appears and the onset at ∼2 eV matches the Cu d-band edge below the Fermi level.51 Thus, it can be concluded that the indirect mechanism is the dominant excitation channel in this case. It should be mentioned that the observed cross-section of ∼10−19 cm2 in the visible-ultraviolet region is much higher than that of previously studied molecular switches on a metal surface, for example, azobenzene derivatives (10−23–10−22 cm2). Tautomerization of porphycene could therefore be a promising candidate as a photoactive molecular switch working directly on metal electrodes.
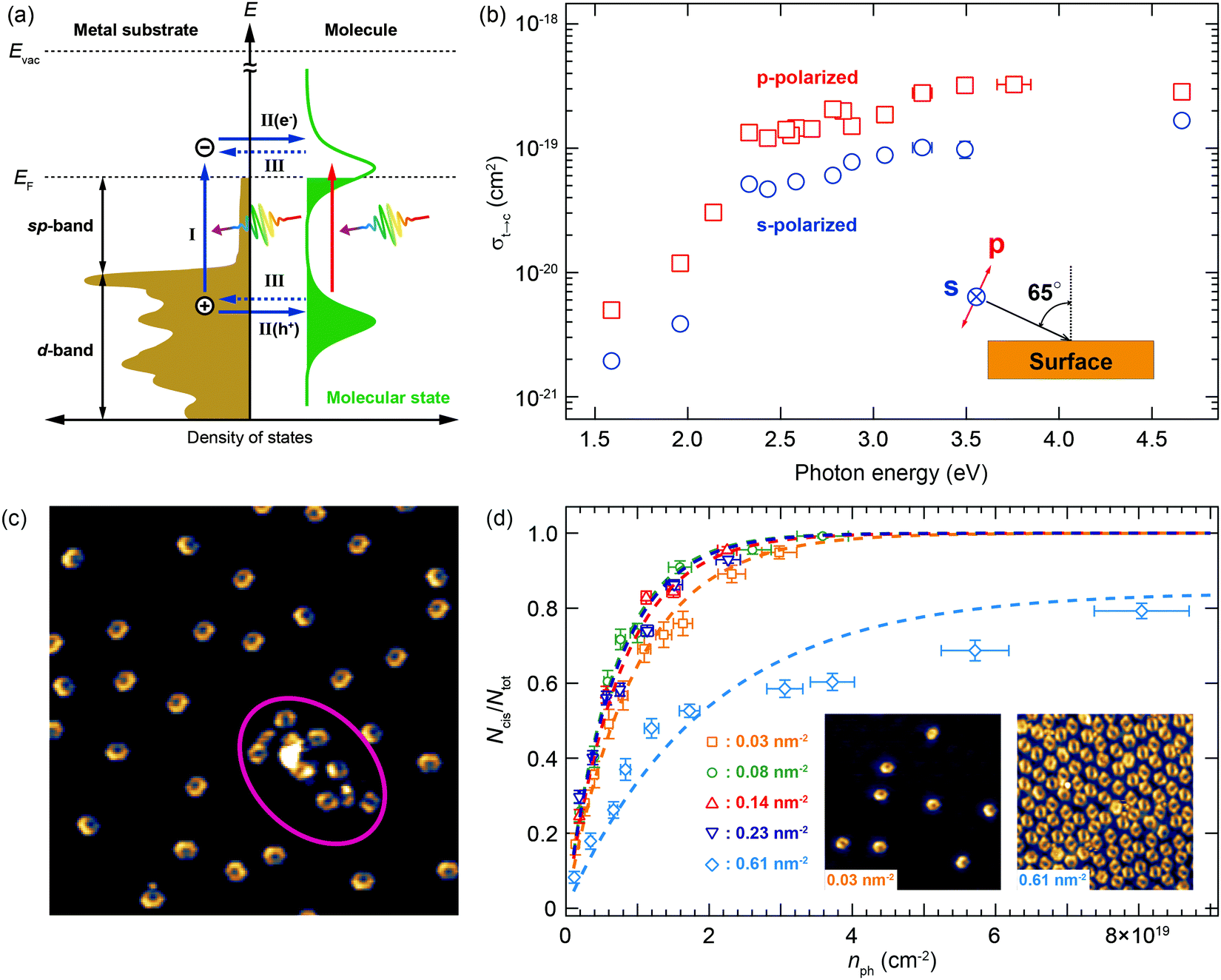 | ||
| Fig. 11 Light-induced tautomerization of porphycene on a Cu(111) surface. (a) Schematic energy diagram of a metal–molecule interface. Upon illumination photons are absorbed either by the molecule (red arrow, direct mechanism), or by the substrate (blue arrows, indirect mechanism). In the latter process, the photon absorption creates hot carriers in the substrate, which is followed by carrier attachment (either hot electron or hole) the molecular state, leading to a reaction. (b) Wavelength dependence of the trans → cis1 tautomerization cross-section measured with p- and s-polarized beam (linearly polarized perpendicular and parallel to the surface, respectively, as shown in the inset). (c) STM image after a long illumination (sufficient to switch all trans molecules on the terrace). The molecules nearby a surface defect are not converted (indicated by the red circle). (d) Coverage dependence of the trans → cis1 tautomerization efficiency. The dashed lines represent the best fitted curves by the first-order rate equation. Typical STM images at a low and high coverage are shown in the inset. | ||
As discussed above, LT-STM allows directly addressing the role of the local environment in surface reactions. Fig. 11c shows an STM image of porphycene on Cu(111), where intrinsic surface defects are involved (highlighted by the red ellipse). The molecules near the defect did not switch from trans to cis1, even after very long irradiation times during which all the other molecules on the clean terrace have reacted. In order to examine such an effect of the local surroundings in a systematic manner, tautomerization has been investigated at varying molecular densities (Fig. 11d). At low densities (0.03–0.23 nm−2), the molecules are well-separated from each other, and the reaction follows a first-order process. The cross-section was determined to be 9.8(±0.2) × 10−20, 1.34(±0.04) × 10−19, 1.37(±0.05) × 10−19, and 1.22(±0.04) × 10−19 at 0.03, 0.08, 0.14, and 0.23 nm−2, respectively, for 532 nm excitation wavelength. The cross-section appears to be slightly lower at 0.03 nm−2, while a negligible difference for molecular densities of 0.08–0.23 nm−2 suggests that a long-range intermolecular interaction (e.g. dipole–dipole interaction) does not affect the tautomerization process in these regimes. However, the surface state of Cu(111) is expected to be influenced by molecular adsorption, in particular above a molecular density of ∼0.08 nm−2.16 This may cause changes in the optical absorption of the substrate and/or hot carrier dynamics and, consequently, the subtle difference in the tautomerization cross-section between 0.03 and 0.08–0.23 nm−2. On the other hand, the cross-section substantially decreased at a higher molecular density (0.61 nm−2), and the experimental data do not perfectly follow a first-order process. Additionally, not all of the molecules were converted to the cis1 configuration, no matter how long they were exposed to light. At this density, the intermolecular distances become much smaller and some of the molecules are in contact with each other, as can be seen in the inset STM image of Fig. 11d. In this situation, molecule–molecule interactions would play a critical role. Furthermore, the lack of a highly ordered molecular arrangement may cause subtly different local interactions between molecules and, consequently, different tautomerization behaviour from molecule to molecule. This could be the reason why the evolution deviates from a first-order process at a molecular density of 0.61 nm−2. The presence of unreacted molecules indicates quenching of tautomerization due to the intermolecular interaction.
As discussed above, porphycene represents a molecular switch responsive to various external stimuli. An interesting perspective for such functional molecules is the linking of different molecular building blocks to combine the various functions in a controlled fashion. It is a central objective in the field of monomolecular electronics52,53 to form well-defined arrays of functional molecules (as, for instance, molecular switches, rectifiers or wires). Such an assembly might represent a real nano-circuit with electronic behaviour, by analogy with classical semiconductor electronics. Self-assembly of molecules in supramolecular nanostructures by weak non-covalent interactions is a very common process in nature.54,55 These structures, however, lack high stability and thus durability under various environmental conditions. Furthermore, they typically do not exhibit efficient charge transport across the intermolecular bonds, which is a fundamental prerequisite in electronic applications. Hence, covalent linking of molecules appears to be much more promising in view of monomolecular electronics, as such bonds are characterized by their strength and the possibility of electron delocalization across it, capable of electrical transport.
Various groups have attempted to covalently link molecular building blocks on flat surfaces in the last few years and this so-called on-surface synthesis (for oligomers of limited length) or on surface polymerization (for extended chains) in a bottom-up fashion became a rapidly growing field.56–60 Most studies are carried out under ultra-high vacuum conditions to provide well-defined conditions and to exclude side reactions with the atoms/molecules from the surrounding liquid or gaseous environment. While covalent linking is a standard procedure in conventional synthetic chemistry with isotropic solution or gas phase environments, the polymerization on flat crystalline surfaces offers specific advantages: (i) two-dimensional confinement for molecular diffusion and polymerization,61 (2) an atomically defined environment that enables us to understand the influence of the surface on the elementary chemical processes (as for instance in catalytic reactions62), and (3) the possibility for characterization of the molecules and molecular nanostructures in real space by scanning probe microscopy.
On-surface synthesis is based on specially designed monomers, which react at specific pre-defined connection sites during the polymerization step. These monomers should be chemically inert and become reactive only after an activation step to prevent uncontrolled growth. One particularly popular approach is equipping the molecular building blocks with halogen substituents.63 The concept (described in Fig. 12a) is that molecular building blocks are equipped with halogen substituents and the carbon–halogen bonds are the weakest in the entire molecule. Hence, after deposition of the molecules (but potentially already in the evaporator63) these bonds break first if the sample is heated. Accordingly, reactive sites are created at well-defined positions in the molecular skeleton, at which the molecules can form covalent bonds upon surface diffusion (particularly stimulated by the heating step) and molecular oligomers are formed.
 | ||
| Fig. 12 (a) Scheme of on-surface polymerization. After deposition of the molecular building blocks onto a surface, they are activated and connected to covalently bound molecular nanostructures. The creation of linear (b) and two-dimensional (c) molecular assemblies composed of porphyrin derivatives depends on the number and location of halogen substituents in the initial building blocks. From ref. 62. | ||
Two examples are given in Fig. 12b and c where porphyrin building blocks are equipped with either two or four Br substituents. In the former case, the two halogen atoms are placed in a trans configuration which is important for the growth process. According to these different initial chemical structures, either one-dimensional chains (Fig. 12b) or two-dimensional networks (c) are formed. The one-dimensional structures received particular interest in the last few years, resulting in molecular wires of different shapes and compositions.64–68 Very recently, the construction of an asymmetric molecular node, which represents a key component in view of future nano-circuits in molecular electronics, has been achieved by combining different molecular building blocks under suitable conditions.69
The power of on-surface synthesis is evident in Fig. 12b and c as the structure of the final polymer on the surface is precisely determined by the chemical structure of the initial building blocks, in particular their symmetry and the position of the halogen substituent groups, which encode their connectivity. This inherent chemical precision has been used for the development of a hierarchical growth scheme where different potentially reactive sites are sequentially activated.70 Accordingly, linear chains are formed in a first step, which are then linked sideways in the second step – each step of this “programmed reactivity” involves different halogen substituents that are specifically addressed and activated by heat. Moreover, it has been demonstrated in the synthesis of graphene nanoribbons with precise control over the edge-structure, ranging from armchair65 to zig-zag edges71 that exhibit very particular chemical structures that are associated with different molecular properties.
If tautomerization could be controlled in molecular structures, the covalent assembly of the involved molecules offers high potential for future research. Specifically, there are several advantages in view of functional assemblies of molecules and monomolecular electronics.
(1) Tautomerization in molecules can be recognized as a switching process because various properties are modified in a controlled way, in particular the conductance of the molecules if located between two electrodes.72 Hence, these molecules are of interest as potential molecular switches in miniaturized electronic circuits where they control the electric current by the tautomeric state. An important property in this regard is the molecular conformation during the process which changes strongly in the case of the prototypical azobenzene isomerization.73–75 Tautomerization is a very attractive alternative because the proton transfer is typically not linked to any strong conformational changes. Accordingly, it should be possible to maintain a contact of the molecule with other molecules or electrodes during the switching process.
(2) The switching state of the molecules can still be identified by scanning probe microscopy, due to their rather flat structure that is maintained in a two-dimensional nano-circuit on a surface. In the same regard, it should also still be possible to access the individual molecules in such an arrangement, thus being able to change their state in a controlled fashion. Note that there are different possible stimuli how the tautomerization can be induced (as discussed above and sketched for instance in Fig. 10d).
(3) The process of covalent linking is very versatile, because (as discussed above) the final architectures can be designed precisely via the chemical structure of the initial building blocks.70 In the case of porphyrin or porphycene, each molecule offers (at least) four potential connection sites in the two dimensions that could in principle be linked to other molecules in a sequential fashion, thus allowing additional control of the final structures.
Conclusions
Systematic studies of tautomerization of porphycenes by various spectroscopic and microscopic methods and under various conditions provide detailed information about the tautomerization mechanisms, which can readily be extended to other proton or hydrogen transferring systems. First of all, the reaction is dominated by tunnelling, even under conditions usually considered typical for “above the barrier” processes, i.e. room temperature solutions. In isolated molecules, coherent tunnelling is significantly affected by vibrational excitation. Thus, tautomerization is a multidimensional process, with contributions from both high (NH stretching) and lower frequency (skeletal) modes. Moreover, the impact of a specific vibrational mode on tautomerization can change upon isotope substitution of peripheral protons by deuterons, which are apparently far from the reaction centre (molecular cavity). This makes a proper description of tautomerism even more challenging.Both experiment and theory suggest cooperativity between the two moving protons, resulting in a synchronous trans–trans conversion. However, the picture becomes much more complicated for a molecule adsorbed on a metal surface and specifically interacting with the surface atoms. Depending on which surface is used, both trans and cis forms can be detected. Moreover, both single and double hydrogen transfer reactions have been observed.
The knowledge about the vibrational modes involved in tautomerization can be useful for quantum control experiments based on selective vibrational excitation. It seems that such control is possible even for room temperature solutions, since for several porphycenes the reaction time is shorter than vibrational decoherence.
The use of three different techniques to monitor tautomerization in single molecules resulted in significant extension of the previous knowledge. Especially attractive in this regard were the results obtained by scanning probe microscopy. Tautomerization, induced by different external stimuli, i.e. heat, electron attachment, light, and force, could be directly visualized. Moreover, the reaction could be controlled by manipulating single atoms and molecules nearby the target molecule, revealing a local environmental impact and also providing precise control of a single-molecule switch. In this respect, it is worth mentioning that, in jet-isolated porphycenes, one observes a superposition of states, which is a realization of a qubit. A significant challenge for the future is a reliable control of such quantum states in condensed phases for the time periods suitable for practical use. Naturally, tautomerization of porphycenes can also be exploited in a classic fashion, e.g. in the design of rapid optical switches or logic gates. A clear benefit of the use of organic molecules is their capacity to self-assemble into ordered supramolecular architectures, enabling functionalized molecules to arrange in a programmed manner. A promising strategy for the use of porphycenes as molecular switches in the future is their linking in nano-circuits of single-molecule electronics by on-surface synthesis. The strong, covalent, intermolecular interactions should not hinder tautomerization in the individual molecules, but at the same time ensure high stability and charge transport from one molecule to another.
Acknowledgements
This work was partially supported by the Polish National Science Centre (NCN) through Grants DEC-2011/02/A/ST5/00443, DEC-2013/10/M/ST4/00069, and DEC-2011/01/B/ST2/02053, and by the European Project ACRITAS. T. K. acknowledges the financial support from Morino Foundation for Molecular Science. Open Access funding provided by the Max Planck Society.References
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed., 1986, 25, 257–259 CrossRef.
- D. Sánchez-Garcia and J. L. Sessler, Chem. Soc. Rev., 2008, 37, 215–232 RSC.
- J. Waluk, Chem. Rev., 2017 DOI:10.1021/acs.chemrev.1026b00328.
- J. Waluk, Acc. Chem. Res., 2006, 39, 945–952 CrossRef CAS PubMed.
- J. Waluk, in Hydrogen-Transfer Reactions, ed. J. T. Hynes, J. P. Klinman, H. H. Limbach and R. L. Schowen, Wiley-VCH, Weinheim, 2007, vol. 1, pp. 245–271 Search PubMed.
- J. Waluk, in Handbook of Porphyrin Science, ed. K. Smith, K. Kadish and R. Guilard, World Scientific, Singapore, 2010, vol. 7, p. 359 Search PubMed.
- J. Waluk, in CRC Handbook of Organic Photochemistry and Photobiology, ed. M. Oelgemöller, A. Griesbeck and F. Ghetti, CRC Press, 2012, pp. 809–829 Search PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, M. Kijak, S. Nonell, D. Kuzuhara, H. Yamada, C. Radzewicz and J. Waluk, J. Phys. Chem. B, 2015, 119, 2292–2301 CrossRef PubMed.
- H. Piwoński, C. Stupperich, A. Hartschuh, J. Sepioł, A. Meixner and J. Waluk, J. Am. Chem. Soc., 2005, 127, 5302–5303 CrossRef PubMed.
- H. Piwoński, A. Hartschuh, N. Urbańska, M. Pietraszkiewicz, J. Sepioł, A. Meixner and J. Waluk, J. Phys. Chem. C, 2009, 113, 11514–11519 Search PubMed.
- H. Piwoński, A. Sokołowski, M. Kijak, S. Nonell and J. Waluk, J. Phys. Chem. Lett., 2013, 4, 3967–3971 CrossRef.
- S. Gawinkowski, M. Pszona, A. Gorski, J. Niedziółka-Jönsson, I. Kamińska, W. Nogala and J. Waluk, Nanoscale, 2016, 8, 3337–3349 RSC.
- W. Nogala, P. Kannan, S. Gawinkowski, M. Jönsson-Niedziolka, M. Kominiak, J. Waluk and M. Opallo, Nanoscale, 2015, 7, 10767–10774 RSC.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Phys. Rev. Lett., 2013, 111, 246101 CrossRef PubMed.
- T. Kumagai, F. Hanke, S. Gawinkowski, J. Sharp, K. Kotsis, J. Waluk, M. Persson and L. Grill, Nat. Chem., 2014, 6, 41–46 CrossRef CAS PubMed.
- J. N. Ladenthin, L. Grill, S. Gawinkowski, S. Liu, J. Waluk and T. Kumagai, ACS Nano, 2015, 9, 7287–7295 CrossRef CAS PubMed.
- J. Ladenthin, T. Frederiksen, M. Persson, J. Sharp, S. Gawinkowski, J. Waluk and T. Kumagai, Nat. Chem., 2016, 8, 935–940 CrossRef CAS PubMed.
- H. Böckmann, S. Liu, J. Mielke, S. Gawinkowski, J. Waluk, L. Grill, M. Wolf and T. Kumagai, Nano Lett., 2016, 16, 1034–1041 CrossRef PubMed.
- E. T. Mengesha, J. Sepioł, P. Borowicz and J. Waluk, J. Chem. Phys., 2013, 138, 174201 CrossRef PubMed.
- P. Ciąćka, P. Fita, A. Listkowski, C. Radzewicz and J. Waluk, J. Phys. Chem. Lett., 2016, 7, 283–288 CrossRef PubMed.
- J. Sepioł, Y. Stepanenko, A. Vdovin, A. Mordziński, E. Vogel and J. Waluk, Chem. Phys. Lett., 1998, 296, 549–556 CrossRef.
- Y. Nosenko, J. Jasny, M. Pietraszkiewicz and A. Mordziński, Chem. Phys. Lett., 2004, 399, 331–336 CrossRef CAS.
- A. Vdovin, J. Sepioł, N. Urbańska, M. Pietraszkiewicz, A. Mordziński and J. Waluk, J. Am. Chem. Soc., 2006, 128, 2577–2586 CrossRef CAS PubMed.
- E. T. Mengesha, A. Zehnacker-Rentien, J. Sepioł, M. Kijak and J. Waluk, J. Phys. Chem. B, 2015, 119, 2193–2203 CrossRef CAS PubMed.
- A. Vdovin, J. Waluk, B. Dick and A. Slenczka, ChemPhysChem, 2009, 10, 761–765 CrossRef CAS PubMed.
- Ł. Walewski, J. Waluk and B. Lesyng, J. Phys. Chem. A, 2010, 114, 2313–2318 CrossRef PubMed.
- J. Waluk and E. Vogel, J. Phys. Chem., 1994, 98, 4530–4535 CrossRef CAS.
- P. Fita, N. Urbańska, C. Radzewicz and J. Waluk, Chem. – Eur. J., 2009, 15, 4851–4856 CrossRef CAS PubMed.
- P. Fita, N. Urbańska, C. Radzewicz and J. Waluk, Z. Phys. Chem., 2008, 222, 1165–1173 CrossRef CAS.
- P. Fita, M. Pszona, G. Orzanowska, D. Sánchez-García, S. Nonell, E. Vauthey and J. Waluk, Chem. – Eur. J., 2012, 18, 13160–13167 CrossRef CAS PubMed.
- P. Fita, P. Garbacz, M. Nejbauer, C. Radzewicz and J. Waluk, Chem. – Eur. J., 2011, 17, 3672–3678 CrossRef CAS PubMed.
- P. Fita, P. Ciąćka, I. Czerski, M. Pietraszkiewicz, C. Radzewicz and J. Waluk, Z. Phys. Chem., 2013, 227, 1009–1020 CrossRef CAS.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Kluwer Academic, New York, 2nd edn, 1999, pp. 291–318 Search PubMed.
- M. Gil and J. Waluk, J. Am. Chem. Soc., 2007, 129, 1335–1341 CrossRef CAS PubMed.
- R. P. Bell, Tunnel Effect in Chemistry, Springer US, Boston, MA, 1980 Search PubMed.
- M. F. Shibl, M. Pietrzak, H. H. Limbach and O. Kühn, ChemPhysChem, 2007, 8, 315–321 CrossRef CAS PubMed.
- J. H. Weiner, J. Chem. Phys., 1978, 69, 4743–4749 CrossRef CAS.
- F. Grossmann, P. Jung, T. Dittrich and P. Hänggi, Z. Phys. B: Condens. Matter., 1991, 84, 315–325 CrossRef.
- J. M. G. Llorente and J. Plata, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 45, R6958–R6961 CrossRef; Erratum Phys. Rev. E, 1994, 49, 3547 Search PubMed.
- M. Grifoni and P. Hänggi, Phys. Rep., 1998, 304, 229–354 CrossRef CAS.
- M. Duran-Frigola, R. Tejedor-Estrada, D. Sánchez-García and S. Nonell, Phys. Chem. Chem. Phys., 2011, 13, 10326–10332 RSC.
- A. M. Chizhik, R. Jäger, A. I. Chizhik, S. Bär, H. G. Mack, M. Sackrow, C. Stanciu, A. Lyubimtsev, M. Hanack and A. J. Meixner, Phys. Chem. Chem. Phys., 2011, 13, 1722–1733 RSC.
- S. Gawinkowski, Ł. Walewski, A. Vdovin, A. Slenczka, S. Rols, M. R. Johnson, B. Lesyng and J. Waluk, Phys. Chem. Chem. Phys., 2012, 14, 5489–5503 RSC.
- B. C. Stipe, M. A. Rezaei, W. Ho, S. Gao, M. Persson and B. I. Lundqvist, Phys. Rev. Lett., 1997, 78, 4410 CrossRef CAS.
- B. C. Stipe, M. A. Rezaei and W. Ho, Phys. Rev. Lett., 1998, 81, 1263 CrossRef CAS.
- W. Ho, J. Chem. Phys., 2002, 117, 11033 CrossRef CAS.
- Y. Kim, K. Motobayashi, T. Frederiksen, H. Ueba and M. Kawai, Prog. Surf. Sci., 2015, 90, 85–143 CrossRef CAS.
- T. Kumagai, A. Shiotari, H. Okuyama, S. Hatta, T. Aruga, I. Hamada, T. Frederiksen and H. Ueba, Nat. Mater., 2012, 11, 167–172 CrossRef CAS PubMed.
- S. Liu, D. Baugh, K. Motobayashi, X. Zhao, S. Levchenko, S. Gawinkowski, J. Waluk, L. Grill, M. Persson and T. Kumagai, to be published.
- X.-L. Zhou, X.-Y. Zhu and J. M. White, Surf. Sci. Rep., 1991, 13, 73–220 CrossRef CAS.
- N. V. Smith, R. L. Benbow and Z. Hurych, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 21, 4331 CrossRef CAS.
- J. R. Heath and M. A. Ratner, Phys. Today, 2003, 56, 43 CrossRef CAS.
- C. Joachim, J. K. Gimzewski and A. Aviram, Nature, 2000, 408, 541 CrossRef CAS PubMed.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Top. Curr. Chem., 2009, 287, 1 CrossRef CAS PubMed.
- J. A. A. W. Elemans, S. Lei and S. DeFeyter, Angew. Chem., Int. Ed., 2009, 48, 7298 CrossRef CAS PubMed.
- A. Gourdon, Angew. Chem., Int. Ed., 2008, 47, 6950 CrossRef CAS PubMed.
- D. F. Perepichka and F. Rosei, Science, 2009, 323, 216 CrossRef CAS PubMed.
- L. Bartels, Nat. Chem., 2010, 2, 87 CrossRef CAS PubMed.
- M. Lackinger and W. M. Heckl, J. Phys. D: Appl. Phys., 2011, 44, 464011 CrossRef.
- J. Mendez, M. F. Lopez and J. A. Martin-Gago, Chem. Soc. Rev., 2011, 40, 4578 RSC.
- Polymerization in other confined geometries as for instance zeolite channels has been reported: K. Kageyama, J. Tamazawa and T. Aida, Science, 1999, 285, 2113 CrossRef CAS PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 1990, 29, 1219 CrossRef.
- L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed.
- L. Lafferentz, F. Ample, H. Yu, S. Hecht, C. Joachim and L. Grill, Science, 2009, 323, 1193 CrossRef CAS PubMed.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- J. Cai, C. A. Pignedoli, L. Talirz, P. Ruffieux, H. Söde, L. Liang, V. Meunier, R. Berger, R. Li, X. Feng, K. Müllen and R. Fasel, Nat. Nanotechnol., 2014, 9, 89 CrossRef PubMed.
- G. Reecht, F. Scheurer, V. Speisser, Y. J. Dappe, F. Mathevet and G. Schull, Phys. Rev. Lett., 2014, 112, 047403 CrossRef PubMed.
- C. Nacci, F. Ample, D. Bleger, S. Hecht, C. Joachim and L. Grill, Nat. Commun., 2015, 6, 7397 CrossRef PubMed.
- C. Nacci, A. Viertel, S. Hecht and L. Grill, Angew. Chem., Int. Ed., 2016, 55, 13724–13728 CrossRef CAS PubMed.
- L. Lafferentz, V. Eberhardt, C. Dri, C. Africh, G. Comelli, F. Esch, S. Hecht and L. Grill, Nat. Chem., 2012, 4, 215 CrossRef CAS PubMed.
- P. Ruffieux, S. Wang, B. Yang, C. Sanchez-Sanchez, J. Liu, T. Dienel, L. Talirz, P. Shinde, C. A. Pignedoli, D. Passerone, T. Dumslaff, X. Feng, K. Müllen and R. Fasel, Nature, 2016, 531, 489 CrossRef CAS PubMed.
- S. J. v. d. Molen and P. Liljeroth, J. Phys.: Condens. Matter, 2010, 22, 133001 CrossRef PubMed.
- M. Alemani, M. V. Peters, S. Hecht, K.-H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446 CrossRef CAS PubMed.
- N.-J. Choi, S.-J. Kahng, S. Kim, H. Kim, H. W. Kim, Y. J. Song, J. Ihm and Y. Kuk, Phys. Rev. Lett., 2006, 96, 156106 CrossRef PubMed.
- J. Henzl, M. Mehlhorn, H. Gawronski, K.-H. Rieder and K. Morgenstern, Angew. Chem., Int. Ed., 2006, 45, 603 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2017 |