
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pressure-driven phase transition mechanisms revealed by quantum chemistry: L-serine polymorphs†
Denis A.
Rychkov
 *ab,
Jernej
Stare
*c and
Elena V.
Boldyreva
*a
*ab,
Jernej
Stare
*c and
Elena V.
Boldyreva
*a
aInstitute of Solid State Chemistry and Mechanochemistry, Siberian Branch of Russian Academy of Sciences, ul. Kutateladze, 18, Novosibirsk 630128, Russian Federation. E-mail: rychkov.dennis@gmail.com; eboldyreva@yahoo.com
bNovosibirsk State University, ul. Pirogova, 2, Novosibirsk 630090, Russian Federation
cNational Institute of Chemistry, 19 Hajdrihova str., SI-1000, Ljubljana, Slovenia. E-mail: jernej.stare@ki.si
First published on 2nd February 2017
Abstract
The present study delivers a computational approach for the understanding of the mechanism of phase transitions between polymorphs of small organic molecules. By using state of the art periodic DFT calculations augmented with dispersion corrections and an external stress tensor together with gas-phase cluster calculations, we thoroughly explained the reversible phase transitions of three polymorphs of the model system, namely crystalline L-serine in the pressure range up to 8 GPa. This study has shown that at the macroscopic level the main driving force of the phase transitions is the decrease in the volume of the crystal unit cell, which contributes to the enthalpy difference between the two forms, but not to the difference in their internal crystal energies. At the microscopic level we suggest that hydrogen bond overstrain leads to a martensitic-like, cooperative, displacive phase transition with substantial experimental hysteresis, while no such overstrain was found for the “normal type”, atom per atom, reconstructive phase transition. The predicted pressures for the phase transitions deducted by the minimum enthalpy criterion are in reasonable agreement with the observed ones. By delivering unambiguous explanations not provided by previous studies and probably not accessible to experiment, this work demonstrates the predictive and explanatory power of quantum chemistry, confirming its indispensable role in structural studies.
The control of polymorphism is becoming increasingly important to understand the basics of crystal structure formation, but also to ensure control over the properties of various molecular materials, including non-linear optics, explosives, hybrid materials, etc. This is a crucial point especially in the pharmaceutical industry.1–4 The progress in lattice energy calculations achieved in recent years makes it possible to explain, and in some cases – even predict the existence of polymorphs and their crystal structures.5–11 However, it remains a challenge to find the experimental conditions to obtain a predicted polymorph in the laboratory.12 The main routes for obtaining a new polymorph include crystallization (under very different conditions) as well as solid-state phase transitions or solid-state chemical reactions. The mechanisms of these processes and the role of thermodynamic and kinetic factors in the formation of a given polymorph often remain unclear. In particular, though many pressure-induced phase transitions in molecular crystals have been documented, it remains difficult to rationalize a priori if and when a solid-state transition may occur for any selected compound.13–19 In the present contribution we demonstrate the capability of density functional theory (DFT) calculations to elucidate the mechanisms of pressure-induced phase transitions and greatly improve our understanding of this compelling topic. In particular, the present work focuses on polymorphs of molecular compounds with conformationally flexible molecules that are linked in a hydrogen bond (H-bond) network.
L-Serine polymorphs have been chosen as a case study. The behavior of this crystalline amino acid with increasing pressure has been extensively studied experimentally by optical microscopy, Raman spectroscopy, powder and single-crystal X-ray diffraction, and neutron powder diffraction.20–26 Two reversible isosymmetric (space group P212121) phase transitions have been reported, giving polymorph II at 5.3 GPa, and polymorph III at 7.8 GPa.‡ The transition to phase II is of a martensitic type with a pronounced hysteresis.20,24 In a single crystal, as soon as the phase transition starts, it propagates rapidly through the crystal until the transformation is complete, and the single crystal is preserved. At the same time, in a polycrystalline sample two phases – polymorphs I and II – can co-exist in a wide pressure range.24,26 Apparently, nucleation is the rate-limiting step of this transformation. The phase transition to phase III also preserves a single crystal intact; no visible changes occur, whereas Raman spectra and X-ray diffraction patterns change. Structural data (including atomic coordinates) at multiple pressure points were obtained from X-ray diffraction and later from neutron powder diffraction enabling us to follow also the changes in molecular conformations and H-bonding.22,24,25 With increasing pressure, hydrogen bonds were supposed to act as “springs” and –CH2OH side chains – as “joints”: the two pressure-induced phase transitions are accompanied by distortion, switching over of existing hydrogen bonds (from the OH⋯OH type in L-serine I to more preferable OH⋯OOC bonds in L-serine II), as well as by formation of new hydrogen bonds (additional NH⋯O hydrogen bonds in L-serine III). These changes in the hydrogen bond network were supposed to be the driving force of the phase transitions. However, there was no proof that optimizing the H-bonds (all or some of them), rather than molecular packing and non-directional van der Waals interactions, is the main driving force of the phase transitions.
Periodic DFT calculations enhanced by dispersion corrections and an external stress tensor were performed to model L-serine solid state behavior at different pressures. The models for all calculations were built on the basis of the experimental X-ray crystal and neutron powder diffraction data collected at different pressures. All solid state calculations were performed by the program package VASP27–30 using the functional of Perdew, Bruke and Ernzerhof (PBE),31 a plane-wave basis set with a kinetic energy cutoff of 500 eV and the Projector Augmented Wave atomic pseudopotentials.32,33 The integrals in the reciprocal space were calculated on a Monkhorst–Pack mesh of 8 × 8 × 4 k-points.34 The effects of external pressure were enforced by the stress tensor (PSTRESS keyword) corresponding to a selected pressure value in the range between 0.0001 GPa and 8.1 GPa.
Gas phase cluster models were used to study intermolecular interactions between pairs of molecules. All molecular pairs were extracted from crystal structures optimized at corresponding pressure and the distance between these molecules was changed while the geometry remained fixed. The Gaussian 09 program35 was used to study the intermolecular interactions between pairs of molecules. The calculations were performed using the M06-2X/6-311++G(2d,2p)36 level of theory. Technical details of the performed calculations, accuracy validation and additional data are described in the ESI.†
Crystal energies and enthalpies of the three polymorphs
The values of crystal energy and enthalpy calculated for L-serine crystal structures (optimized experimental structures documented to dominate at these pressures) are plotted in Fig. 1.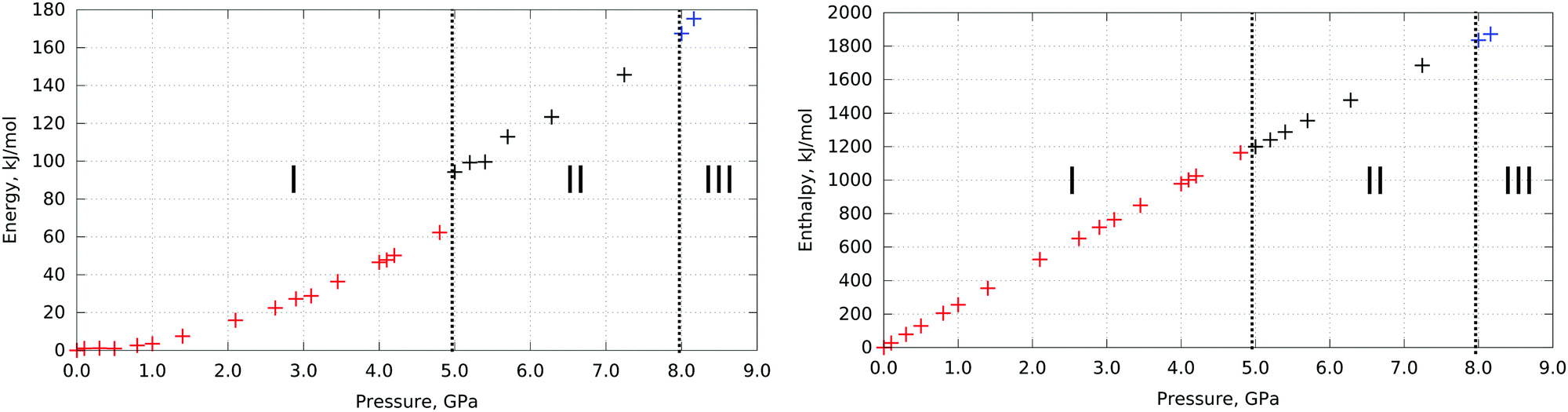 | ||
| Fig. 1 Energy (left) and enthalpy (right) differences in L-serine structure vs. pressure. Polymorphs experimentally observed at corresponding pressures are highlighted with color: L-serine I – red, L-serine II – black, L-serine III – blue. The phase transitions near 5 GPa and 8 GPa are shown with a dashed line. Internal crystal energies and enthalpies of all three polymorphs were calculated at multiple pressure points starting from the experimental data with relaxed atomic positions and cell parameters during optimization. Relative energies and enthalpies are plotted taking the minimal value in the set of calculations as zero. Energy and enthalpy differences in the L-serine structure vs. volume are plotted in Fig. S1 (ESI†). | ||
The I → II phase transition near 5 GPa manifests itself in the changes in the calculated internal crystal energy, which increases jump-wise at the transition point. The II → III phase transition is not evident from the same plot (Fig. 1).§ This type of calculation is usually performed in studies devoted to polymorphism or high-pressure phase transitions. However, this technique cannot help to explain why the phase transitions in fact occur, since we cannot compare the enthalpies and crystal energies of different phases at the same pressure. Therefore, as the next step we have calculated the energy characteristics for all polymorphs in the same pressure range, beyond the range in which they have been observed experimentally. Even if a polymorph could never be obtained at a selected pressure in reality, its structure and energy can be still simulated by DFT calculations by optimizing it to the selected pressure conditions. To predict a hypothetical structure out of the region of structural stability, it was possible to take the structure at experimental pressure and provide optimization with programmed pressure (e.g., taking initial experimental atom coordinates and cell parameters for ambient pressure and running optimization at 5.0 GPa). This technique definitely does not consider phase transitions. Instead, it allows for prediction of which hypothetical structure the same polymorph would adapt at any pressure if no phase transition took place. Such an approach is beyond experimental assessment but can reveal very important information about phase transitions. Unit cell parameters of the optimized structures can be found in Table S1 (ESI†).
Laborious verification of correctness of the employed computational approach was done (see Accuracy check, ESI†). After the calculations proved to reproduce correctly the structures in the range where they have been observed experimentally, the same methodology was used to predict hypothetical structures of all three polymorphs out of the range of their existence.
A comparison of the changes in lattice energies and enthalpies of the three L-serine polymorphs in a wide pressure range 0–9 GPa (neglecting the entropic term) shows that the decrease in volume gives a major contribution to the total decrease in free energy according to these calculations. For I → II phase transition the volume decrease can be considered as the main driving force. As for II → III transition, the same conclusion cannot be made unambiguously because of the lower accuracy of the computational technique with respect to the prediction of volume changes over pressure for polymorphs II and III. These results could be adjusted with better accuracy if the structures of polymorph II and especially polymorph III could be refined at more pressure points experimentally. Up to now the contributions of energy and PV terms to the enthalpy change during II to III transition seem to be at least comparable and in agreement with previous experimental and theoretical work.22,24,25
The phase transitions from I to II and from II to III can be expected when the free energies (in the approximation of the calculations used here – the enthalpies) of the corresponding phases become equal. These calculations predict this to happen at 3.7 GPa and 5.3 GPa, respectively (Fig. 2).
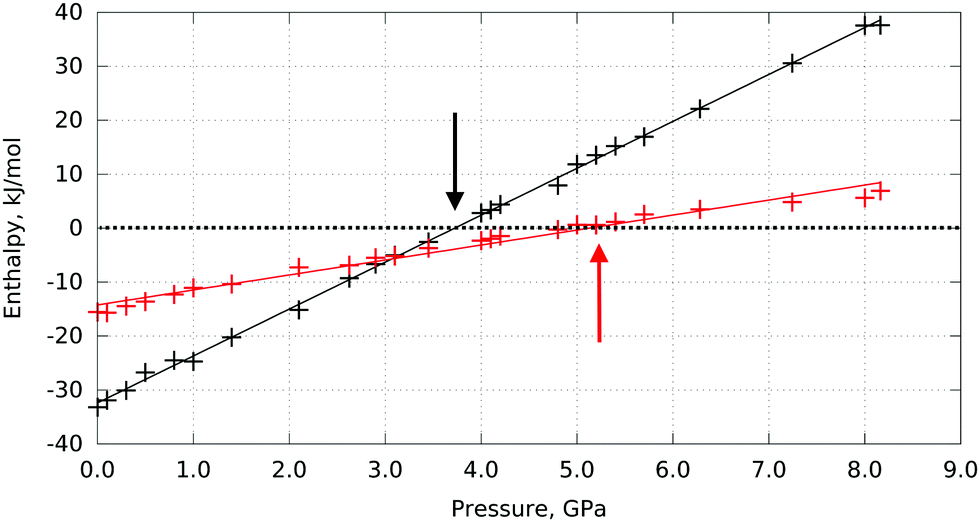 | ||
| Fig. 2 Computed enthalpy differences between L-serine polymorph I and II (black), and between polymorph II and II (red) as a function of pressure (i.e. ΔHI–II = HI − HII and ΔHII–III = HII − HIII). Phase transition between the involved polymorphs can be expected when their enthalpy difference is equal to zero, marked with black (I → II) and red (II →III) arrows. | ||
For both phase transitions the calculated transition pressures are smaller than the experimental values. This can be a consequence of the limitations of the model, but can also reflect the kinetic control over the phase transitions, so that the transition is observed at pressures higher than the equilibrium value. It is known from experiments that L-serine I, II, III can co-exist in a wide pressure range, and the transitions could be triggered at different pressure values, depending on the experimental conditions.24,26
Energy-volume profiles were calculated for all three polymorphs of L-serine, to estimate the zero-pressure bulk modulus (Fig. S3, ESI†). L-Serine I showed the highest bulk modulus value of 23.4 GPa, whilst those for L-serine II and III appear to be significantly less and very similar to each other − 14.7 GPa and 13.9 GPa respectively (Table S2, ESI†). This reflects similarity in the H-bond network in polymorphs II and III and a significant difference in comparison to L-serine I.
Modeling effect of pressure on individual H-bonds
Changes in the phase transitions of L-serine are accompanied by significant changes in the H-bond networks: some H-bonds are broken, whereas new ones are formed (Fig. 3).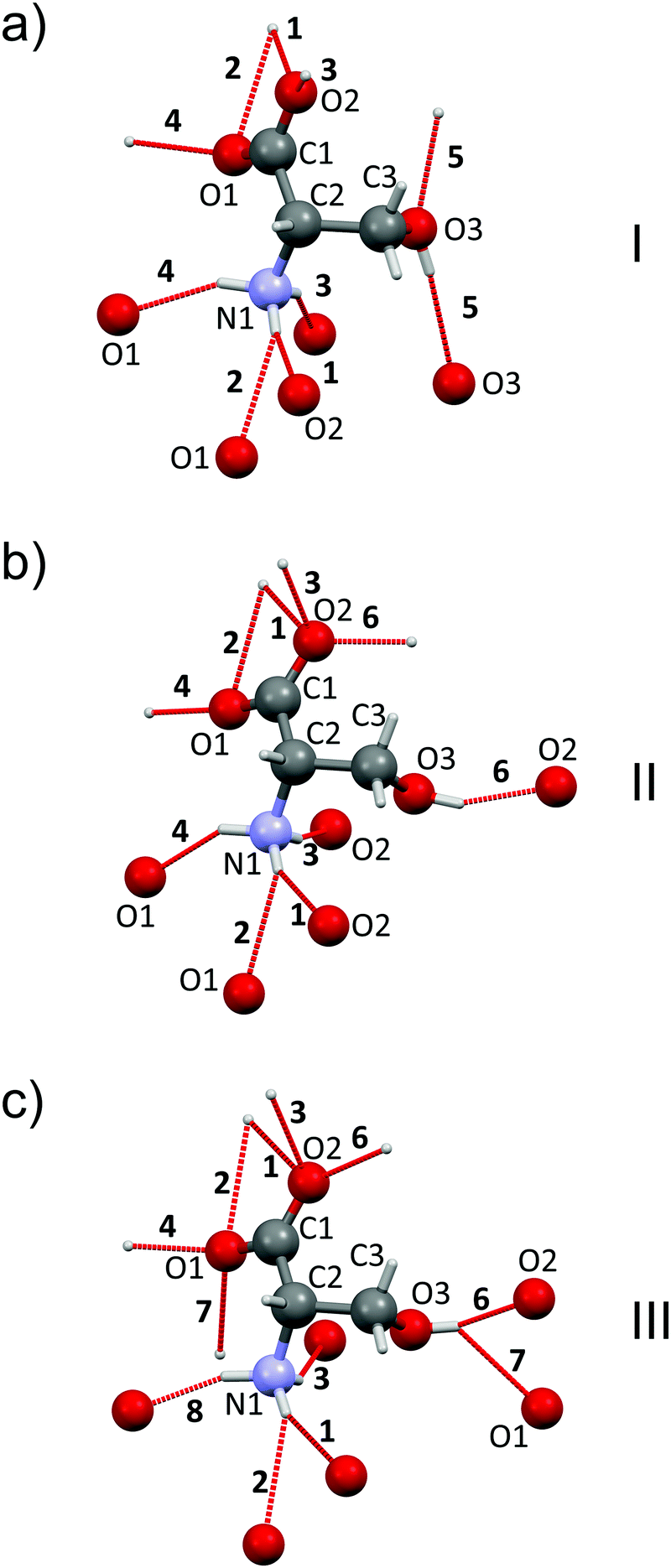 | ||
| Fig. 3 Zwitter-ions of L-serine in forms I (a), II (b), III (c). Hydrogen bonds to the neighbors are labeled by numbers and shown by dashed red lines. Numeration of atoms and H-bonds is as in Table S3, ESI.† Based on ref. 24. | ||
Such phenomena have been documented for many pressure-induced phase transitions in molecular crystals. However, it always remains an open question whether the observed change in an H-bond is directly caused by increasing pressure to optimize its energy, or if it is an indirect consequence of some other structural changes, for example of increasing the packing density.37
In the present work we have modeled the effect of pressure on individual H-bonds in the approximation of pair-wise interactions (see Techniques in the ESI†). The changes in the interaction energy vs. the distance between the proton donor and the proton acceptor for all H-bonds were compared for dimers extracted from the three polymorphs (if the selected type of bond was present for the given polymorph) (Fig. 4). The donor–acceptor distances in experimental structures for all H-bonds are summarized in Table S2 in the ESI.†
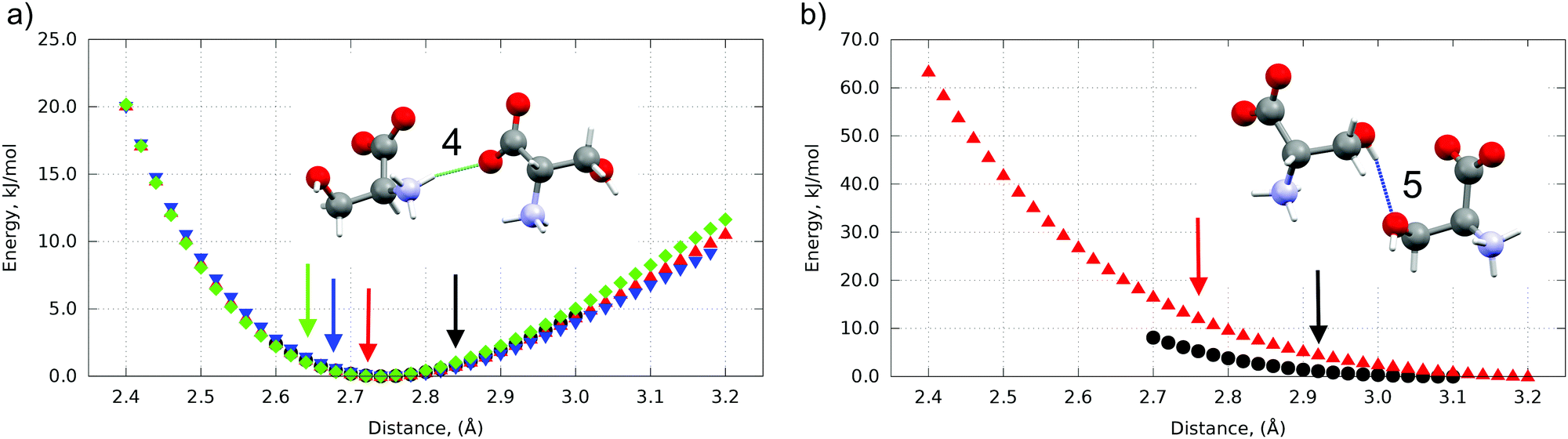 | ||
| Fig. 4 Energy wells of pair-wise intermolecular interactions along H-bond #4 (a) and H-bond #5 (b) at different pressures. Ambient pressure – black, 4.8 GPa – red, 5.4 GPa – blue, 8.0 GPa – green Colored arrows mark D⋯A distance between highlighted dimers at corresponding pressure. An overstrain (red, blue, green arrows (a), red and black arrows (b)) at corresponding pressures is clearly seen. H-bond #5 disappears after 4.8 GPa. H-bond numeration according to Fig. 3. | ||
The bottom of the energy well for a selected hydrogen bond corresponds to the optimum distance between the donor and the acceptor, while any change results in the weakening of this interaction. The bond can still be compressed with gain in energy if the donor–acceptor distance is longer than the minimum value (the right side of the well), whereas the distances shorter than the optimum distance (the left side of the well) correspond to over-straining of the bond (Table S3 in the ESI†), which can be expected to trigger a structural transition, in order to release the stress. For all three polymorphs additional energy (in comparison to Emin for the donor–acceptor distance different from optimum) for every H-bond was calculated at pressures of 0.0001, 4.8, 5.4 and 8.0 GPa if this H-bond exists at this pressure. The H-bond vector connecting the donor and acceptor atom can be mapped to x, y and z directions coinciding with unit cell vectors a, b and c, respectively. The energy contribution along each direction was calculated proportionally to the values of vector components. The highest overstrain for all three phases was found along the x vector (see Table S3 in the ESI†).
The profiles of some hydrogen bonds (#4 and #6) were almost identical for all three polymorphs (see Fig. 4(a) for H-bond #4). One can suppose that these hydrogen bonds can hardly have any significant effect on the pressure-induced transitions. In contrast some hydrogen bonds (#1 and #2, #3, #8 and #5) displayed significantly different energy wells, with notable differences in the minimum for different polymorphs, suggesting that it is the overstrain of these bonds that drives the microscopic mechanism of the structural transition resulting from the increasing pressure (Fig. 4(b) for H-bond #5). All H-bond profiles are plotted in Fig. S4 in the ESI.† The two phase transitions in L-serine are significantly different in terms of hydrogen bond overstrain. If L-serine I is compressed, the OH⋯O hydrogen bond (#5) is overstrained along the a vector up to 11 kJ mol−1. This is a large value, especially remembering that the energy difference between the polymorphs is several kJ mol−1 (ca.10 kJ mol−1 as calculated in this work). The chains of zwitter-ions linked by OH⋯O H-bonds are directed along the a axis – the same direction as the direction in which the overstrain of this bond is observed. The overstrain along this hydrogen bond upon compression seems to be the reason for the I → II phase transition at the microscopic level. This bond acts as the “spring” which cannot be compressed further and therefore stretches back to the initial length at a sudden, triggering the phase transition. Such a model agrees well with the observed changes in cell parameters during I → II phase transition: the cell parameter a decreases with increasing pressure till the transition point, at which it instantly restores its initial value (Fig. S5 in the ESI†). At the same time the jump-wise change of the parameter c axis must be secondary. This also correlates well with the higher value of the bulk modulus of L-serine I in comparison to L-serine II, showing polymorph I being more stiff at the macroscopic level which can be considered as an indirect proof of its H-bond overstrain (Fig. S3 and Table S2, ESI†).
The large hysteresis observed experimentally for I → II phase transition can also be interpreted at the microscopic level by the overstrain of the OH⋯O H-bond upon increasing the pressure – one can hardly expect that the overstrained H-bond will be formed also during the pressure decrease. If we start from L-serine II and stretch the system (modeling the pressure decrease), it would be very hard to rearrange the system in such a way, so that one “spring” (the OH⋯O hydrogen bond) is pressed to the limit.
The situation with the phase transition from L-serine II to L-serine III is different. No significant overstrain could be observed for any of the pair-wise interactions (Table S3 in the ESI†). This phase transition is a result of the co-operative effect when many types of hydrogen bonds are being changed to a similar extent, the bifurcated O–H⋯O bond giving the maximum contribution to the energy gain. No change in the c parameter is observed upon II → III transition (Fig. S5 in the ESI†). The compression of the structure of L-serine II along the a axis still resembles a compression of a spring (but to a smaller extent, as compared to the I → II phase transition), until this spring relaxes at a sudden to restore its original length (Fig. S5 in the ESI†). However, in this case the limit of compression is reached not because of the overstrain of a selected individual H-bond, but because the neighboring molecules come so closely to each other that many contacts become unfavorable. This can explain the relatively small difference between the crystal structures of L-serine II and L-serine III and a smaller hysteresis of the phase transition between these two phases. The molecules approach each other as pressure is increased, so that at a certain point the formation of new H-bond becomes possible in the absence of any significant overstrain. Overall similarity of these structures, both showing no significant overstrain of H-bonds, was also showed by very close values for the bulk modulus, minimal volumes and energies in energy-volume profiles (Fig. S3 and Table S2, ESI†). This model also agrees with the very small changes in the values of b and c cell parameters, as well as those of the volume accompanying the phase transition from polymorph II to III. From aforementioned calculations one can expect that the nucleation barrier for the phase transition from I to II must be significantly higher than that for the transition from II to III. One can also expect that the rate of increasing pressure must have an effect on the phase transition from I to II. Another high-pressure phase can be formed before the overstrain of the OH⋯O hydrogen bond #5 (necessary to trigger the transformation into L-serine II) is achieved if pressure is increased slowly. It is also clear that polymorph III can be formed easily from polymorph II, but is not likely to form equally easily from polymorph I directly, even if pressure is increased very sharply immediately to the point of the second phase transition. These predictions of the modeling have been confirmed experimentally in a recent independent study.26
Summing up, in this work a new approach to improving our understanding of the reasons and mechanisms of pressure-induced phase transitions was suggested and tested using L-serine. The structures were successfully optimized also out of the range of their stability. This enabled us to explore the difference between the hypothetical structures of different polymorphs at the same pressure. Despite the entropy effects were not considered the occurrence of the two phase transitions was predicted in the correct order; though the computed transition points were lower than the experimentally observed values, the difference between the points of the two phase transitions was close to that observed in experiments. It was shown that the PV term plays the decisive role at least in the first, I → II phase transition. The modeling of the effect of pressure on individual hydrogen bonds made it possible to rationalize a radical difference between the I → II and II → III phase transitions. The first one is triggered by a large overstrain of a selected intermolecular hydrogen bond OH⋯O, which can explain the experimentally observed changes in cell parameters, and the large hysteresis. The second one seems to result from multiple small changes in many interactions without a significant overstrain of any selected hydrogen bond. The extension of this work using these results and the suggested computational approach in conjunction with new experimental data for the L-serine system will provide more understanding of phase transition L-serine I → L-serine IV and its co-existence with phases II and III.
The present work illustrates that relatively simple calculations complementing detailed experimental data can in fact give an insight into the macro- and micro- driving forces of pressure-induced phase transitions in hydrogen-bonded molecular crystals. The approaches used for model calculations are quite general and can be applied to pressure-induced transitions in other organic molecular crystals with different types of intermolecular hydrogen bonds.
Acknowledgements
This work was supported by Russian Science Foundation (project 14-13-00834) (DR, EB) and the Slovenian Research agency (program funding P1-0012) (JS). The Siberian Branch of the Russian Academy of Sciences (SB RAS) Siberian Supercomputer Center is gratefully acknowledged for providing supercomputer facilities (http://www2.sscc.ru/).References
- S. Price, Adv. Drug Delivery Rev., 2004, 56, 301–319 CrossRef CAS PubMed.
- F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- J. Bernstein, Cryst. Growth Des., 2011, 11, 632–650 CAS.
- D.-K. Bučar, R. W. Lancaster and J. Bernstein, Angew. Chem., Int. Ed., 2015, 54, 6972–6993 CrossRef PubMed.
- S. L. Price, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 313–328 CrossRef CAS PubMed.
- S. L. Price, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- J. Nyman, O. S. Pundyke and G. M. Day, Phys. Chem. Chem. Phys., 2016, 18, 15828–15837 RSC.
- J. C. Cole, C. R. Groom, M. G. Read, I. Giangreco, P. McCabe, A. M. Reilly and G. P. Shields, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 1–12 Search PubMed.
- E. O. Pyzer-Knapp, H. P. G. Thompson and G. M. Day, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 1–11 Search PubMed.
- Y. N. Heit and G. J. O. Beran, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 1–16 Search PubMed.
- A. J. Cruz-Cabeza, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 437–438 CrossRef CAS PubMed.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, A. J. Davidson, A. R. Lennie, S. Parsons, C. R. Pulham and J. E. Warren, Cryst. Growth Des., 2007, 7, 1115–1124 CAS.
- E. Boldyreva, Cryst. Growth Des., 2007, 7, 1662–1668 CAS.
- B. A. Zakharov, N. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2015, 17, 2074–2079 RSC.
- A. Olejniczak, A. Katrusiak and M. Szafrański, Cryst. Growth Des., 2010, 10, 3537–3546 CAS.
- E. Patyk and A. Katrusiak, Chem. Sci., 2015, 6, 1991–1995 RSC.
- X. Tan, K. Wang, T. Yan, X. Li, J. Liu, K. Yang, B. Liu, G. Zou and B. Zou, J. Phys. Chem. C, 2015, 119, 10178–10188 CAS.
- B. A. Zakharov, S. V. Goryainov and E. V. Boldyreva, CrystEngComm, 2016, 18, 5423–5428 RSC.
- E. N. Kolesnik, S. V. Goryainov and E. V. Boldyreva, Dokl. Phys. Chem., 2005, 404, 169–172 CrossRef CAS.
- S. A. Moggach, D. R. Allan, C. A. Morrison, S. Parsons and L. Sawyer, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 58–68 Search PubMed.
- T. N. Drebushchak, H. Sowa, Y. V. Seryotkin, E. V. Boldyreva and H. Ahsbahs, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4052–o4054 CAS.
- S. A. Moggach, W. G. Marshall and S. Parsons, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 815–825 Search PubMed.
- E. V. Boldyreva, H. Sowa, Y. V. Seryotkin, T. N. Drebushchak, H. Ahsbahs, V. Chernyshev and V. Dmitriev, Chem. Phys. Lett., 2006, 429, 474–478 CrossRef CAS.
- P. A. Wood, D. Francis, W. G. Marshall, S. A. Moggach, S. Parsons, E. Pidcock and A. L. Rohl, CrystEngComm, 2008, 10, 1154 RSC.
- M. Fisch, A. Lanza, E. Boldyreva, P. Macchi and N. Casati, J. Phys. Chem. C, 2015, 119, 18611–18617 CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5, Semichem Inc, Shawnee Mission KS, 2009 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- S. A. Moggach, W. G. Marshall, D. M. Rogers and S. Parsons, CrystEngComm, 2015, 17, 5315–5328 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp07721h |
| ‡ Recently, a new high-pressure polymorph, L-serine IV, has been discovered, which up to now could be obtained only from powder samples of L-serine I under special conditions.26 Analysis of this fourth polymorph and related phase transitions remains beyond the scope of this study, since the limited experimental data is not sufficient for proper simulation of a structure response to increasing or decreasing pressure (see Techniques, Solid State Calculations). Nevertheless conclusions of the present study match well with new experimental studies about the L-serine system. |
| § Phase transitions are not evident from the enthalpy plot because the PV term has more impact (hundreds of kJ mol−1) on the enthalpy in comparison to the crystal energy (tens of kJ mol−1), so the PV term masks crystal energy changes; the enthalpy change versus pressure looks therefore nearly linear (R2 = 0.9863) in the whole range. This result is similar to the one reported previously.19 |
| This journal is © the Owner Societies 2017 |