
Investigating the behavior of various cocatalysts on LaTaON2 photoanode for visible light water splitting†
Wenping
Si
 a,
Daniele
Pergolesi
a,
Fatima
Haydous
a,
Aline
Fluri
a,
Alexander
Wokaun
a and
Thomas
Lippert
*ab
a,
Daniele
Pergolesi
a,
Fatima
Haydous
a,
Aline
Fluri
a,
Alexander
Wokaun
a and
Thomas
Lippert
*ab
aThin Films and Interfaces, Research with Neutrons and Muons Department, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland. E-mail: thomas.lippert@psi.ch
bLaboratory of Inorganic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland
First published on 30th November 2016
Abstract
We performed a comparative study on the photoelectrochemical performance of LaTaON2 loaded with NiOx, Ni0.7Fe0.3Ox, CoOx and IrOx as cocatalysts. Ni-based oxides lead to the highest improvement on the photoelectrochemical performance, while CoOx and IrOx also enhance the performance though to a lower extent, but they simultaneously introduce more pseudocapacitive current thus resulting in an inefficient utilization of the photo-generated holes. Repetitive voltage cycling between 1.0 VRHE and 1.6 VRHE transforms the NiOx and Ni0.7Fe0.3Ox into oxyhydroxides known to possess higher catalytic activities. However, these oxyhydroxides lead to lower photoelectrochemical performance compared to the as-loaded oxides, most probably due to the decay of the passivation centers at the photoelectrode–cocatalyst interface. High catalytic activities cannot be achieved without sufficient passivation of surface recombination states. Despite that the photoelectrochemical performance of LaTaON2 can be improved by cocatalysts, the maximum achievable photocurrent density is still not comparable to that reported for other oxynitride compounds. Our study suggests that poor electronic conductivity or severe bulk recombination of the photo-generated electron–hole pairs are the main limiting factors for the photon-to-current conversion efficiency in LaTaON2 photoanodes.
Introduction
The conversion of solar energy into chemical energy as a clean fuel, thus making solar energy available for use at night and meeting peak power demands, promises great potential for the development of a renewable and sustainable integrated power generation system. This could be realized by solar light driven photoelectrochemical (PEC) water splitting to produce oxygen and hydrogen. In such a system, a photoanode (n-type conductivity) drives the oxidation of water at the electrode–electrolyte interface, while a photocathode (p-type conductivity) drives the reduction of water. Oxynitrides of transition metals and rare earth metals are one class of promising semiconductor materials for solar water splitting. The width and energy positions of the band gap of some oxynitrides make it theoretically possible that both reactions of water oxidation and reduction occur within one material, resulting in the so-called overall water splitting. Since the efficiency of water splitting is mainly limited by the oxygen evolution reaction (OER),1,2 many oxynitrides are particularly investigated as photoanodes for the water oxidation half reaction.The PEC performance of a photoanode is usually probed using a three-electrode configuration where the immobilized photoanode is used as the working electrode, Pt as the counter electrode, and the potential of the photoanode is monitored by a reference electrode. The measured photocurrent reflects the photon-to-current conversion efficiency of the photoanode. Photoanodes, deposited by electrophoretic method (EPD) on fluorine-doped tin oxide (FTO), such as TaON,3–5 Ta3N5,3 and BaTaO2N,6,7 show photocurrent densities for water oxidation in the range of several mA cm−2 at 1.23 VRHE (the water oxidation potential) under an irridation by a 300 W Xe lamp fitted with a 400 nm or 420 nm cut-off filter. Materials that are used for OER electrocatalysis such as IrOx,3,6 CoOx,4,6 Co–Pi (inorganic cobalt phosphate)8 and NiOx9,10 are often also employed as cocatalysts for photoanodes to promote the kinetics of water oxidation and suppress the recombination of photo-generated electrons and holes by passivating the surface recombination centers.11 It has been reported that loading CoOx or IrOx cocatalysts on TaON, Ta3N5, BaTaO2N, SrNbO2N and LaTiO2N usually result in 50–400% higher currents and more negative onset potentials for water splitting.3,6,12–14 However, no obvious improvement in photocurrent was observed after loading cocatalysts Co3O4, Co(OH)2 or Co–Pi on LaTaON2 photoanode.15 Here we would like to take LaTaON2 as an example and investigate whether and how proper cocatalysts can be found to further improve the PEC performance of this oxynitride.
Recently, in the research of water electrolysis, NiyFe1−yOx has been reported to exhibit a higher catalytic capability than IrOx in basic media for the OER,16,17 which is attributed to the in situ formation of layered NiyFe1−yOOH oxyhydroxide species upon voltage cycling with nearly every Ni atom being electrochemically active.16 It has also been suggested that the Fe ion might be the real catalytic active site in nickel iron oxides/oxyhydroxides.1 The use of Ni(Fe)OOH cocatalyst indeed remarkably enhanced the PEC performances of hematite,9 BiVO4,18 TiO2,19etc. Depending on the ion permeability, cocatalysts have been demonstrated to form different interfaces with the photoelectrode.19,20 Ion-permeable Ni(OH)2 or NiOOH form an adaptive junction with the photoelectrode, where the effective Schottky barrier height changes with the oxidation level of the cocatalyst,19 while ion-impermeable cocatalysts such as dense IrOx form a constant-barrier-height ‘buried’ junction with the photoelectrode.19 However, it has also been reported that the PEC performance of the photoelectrode–cocatalyst system is nearly independent on the activity of ion-permeable cocatalysts, while strongly influenced by the activity of ion-impermeable cocatalysts.20,21
The comprehensive understanding of the photoelectrode–cocatalyst interface is further complicated by the existence of various chemical species of the cocatalysts characterized by different coordination environments and/or valence states. As an example, researchers have categorized NiOx on BiVO4 photoanode into three types: OER catalytic centers, recombination centers, and passivation centers.10 By selective removal of the first two centers while keeping the NiOx passivation centers at the surface, a significantly increased photocurrent was obtained.10
These considerations lead to the following questions. What is the most important asset of a cocatalyst for determining a high PEC performance? Should we introduce a material with higher catalytic activity or better passivation and minimal recombination effects? This study aims at gaining insights into the roles of the cocatalysts in affecting the photoanode performance. To achieve this goal, various metal oxides cocatalysts were loaded on LaTaON2 photoanodes and the PEC performance were measured and compared. Specifically, in order to enlarge the effects of cocatalysts on a LaTaON2 photoanode, a very small area was illuminated in comparison to the whole size of the photoanode in this work. The dark electrocatalytic behaviors of the cocatalysts were therefore “exaggerated” and the differences between them could be easily distinguished.
Experimental
Synthesis of LaTaON2
The oxide precursor LaTaO4 was first prepared by a solid state reaction of La2O3 (Alfa Aesar puratronic, 99.993%) and Ta2O5 (Alfa Aesar puratronic, 99.993%) in the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, with calcination at 1000 °C for 2 h, and then at 1400 °C for 10 h in air. Three cycles of heating were conducted with intermediate grinding until the reaction was fully completed. Afterwards, 0.5 g LaTaO4 was mixed with a mineralizer of 0.5 g NaCl and put in an alumina boat for ammonolysis at 950 °C under an ammonia flow of 200 mL min−1 for 30 h to prepare LaTaON2.
:1, with calcination at 1000 °C for 2 h, and then at 1400 °C for 10 h in air. Three cycles of heating were conducted with intermediate grinding until the reaction was fully completed. Afterwards, 0.5 g LaTaO4 was mixed with a mineralizer of 0.5 g NaCl and put in an alumina boat for ammonolysis at 950 °C under an ammonia flow of 200 mL min−1 for 30 h to prepare LaTaON2.
Materials characterization
The oxide precursor LaTaO4 and the nominal ammonolysis product LaTaON2 were characterized by powder X-ray diffraction (XRD) on a Bruker–Siemens D500 X-ray Diffractometer. The optical band gap of LaTaON2 was determined by measuring the diffuse reflectance spectrum in the wavelength range between 200 nm and 1000 nm with a Cary 500 Scan UV-Vis-NIR spectrophotometer using an integrating sphere. Scanning electron microscopy (SEM) images were obtained with a Zeiss Supra VP55 Scanning Electron Microscope. N2 adsorption–desorption analysis was conducted on a BEL-Mini device, and the specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) theory.Preparation of LaTaON2 photoanodes
LaTaON2 photoanodes were prepared via the electrophoretic deposition method (EPD) followed by a necking treatment.3 In detail, 120 mg LaTaON2 powders were dispersed in 50 mL acetone with 10 mg iodine and sonicated for 60 min. Iodine reacts with acetone releasing H+, thus the LaTaON2 powders were positively charged. Two parallel FTO substrates (1 × 2 cm, ∼7 Ω sq−1, Sigma Aldrich) were then placed in a LaTaON2 dispersion with a distance of 1 cm under a bias of 20 V for 5 min. LaTaON2 powders were deposited on the negative electrode with a geometric area of ∼1 × 1 cm2.The so-called necking treatment was applied to enhance the contacts between LaTaON2 particles. 25 μL of 10 mM TaCl5 methanol solution was dropped on LaTaON2 photoanodes and dried in air. This procedure was repeated for three times, followed by heating at 350 °C in air for 30 min. This temperature was chosen to avoid oxidation of the oxynitrides. For a comparison to the post-treatment in air, the as-prepared LaTaON2 photoanodes and the samples with pre-loaded cocatalysts were also treated for 30 min under NH3 flow at 450 °C and 400 °C, respectively. The use of FTO substrates determines an upper limit of 500 °C for the heat treatment in ammonia.3 A relatively lower temperature was used to treat cocatalysts-loaded samples under ammonia since we tried to avoid a reduction of the cocatalysts.
It is worth noting that all the cocatalysts used in this paper were supposed to be dense cocatalysts due to thermal treatments. NiOx, Ni0.7Fe0.3Ox, and CoOx cocatalysts (2 wt%, calculated as metal) were pre-loaded on LaTaON2 powders by impregnation from their aqueous nitrate solutions, followed by drying and heating at 150 °C for 30 min in air. A mixture of Ni(NO3)2 and Fe(NO3)3 solution was used to load Ni0.7Fe0.3Ox. IrOx was post-loaded on the as-prepared LaTaON2 photoanodes by impregnation from a colloidal IrO2 aqueous solution, which was prepared by hydrolysis of Na2IrCl6.22 The colloidal IrO2 was prepared as follows: 0.008 g Na2IrCl6·6H2O was dissolved in 50 mL H2O, and the pH was adjusted to 11–12 with 1 M NaOH. The solution was heated at 80 °C for 30 min and cooled to room temperature by immersion in an ice water bath. The pH of the cooled solution was adjusted to 10 with HNO3. Subsequent heating at 80 °C for 30 min resulted in a blue solution containing colloidal IrO2. The photoanodes were immersed in the IrO2 colloid for 30 min, heated at 300 °C for 30 min and then washed with distilled water before PEC measurements.
Photoelectrochemical (PEC) measurements
PEC measurements were performed in a three electrode configuration in 0.5 M NaOH (pH = 13.0) aqueous solution. The LaTaON2 photoelectrode was used as the working electrode, while a coiled Pt wire and Ag/AgCl were used as the counter and the reference electrodes, respectively. The light source was a 405 nm laser diode (Laser 2000) with 5 mW power output and a spot size of 3 mm2. PEC measurements were carried out on a Solartron 1286 electrochemical interface. Potentiodynamic measurements with a scan rate of 10 mV s−1 in the potential window of 0–1.8 VRHE were performed to investigate the PEC performance of LaTaON2. The chopped dark-light current densities were normalized according to the illuminated area of 3 mm2. Cyclic voltammetry with a scan rate of 10 mV s−1 in the potential window of 1.0–1.6 VRHE was used to study the voltage cycling effect on Ni-based cocatalysts.Results and discussion
Characterization
After the solid state reaction at 1400 °C, the La2O3 and Ta2O5 powder mixture was completely converted to white LaTaO4 powder, which corresponds to a mixture of orthorhombic and monoclinic polymorphs as shown in Fig. 1a.23 The XRD pattern of the ammonolysis product corresponds to LaTaON2 with an orthorhombic perovskite structure24 with a trace of Ta3N5 indicated by an arrow at 28.06°. The LaTaON2 powders have a dark red color and a specific surface area of ∼5 m2 g−1 as measured by the BET method.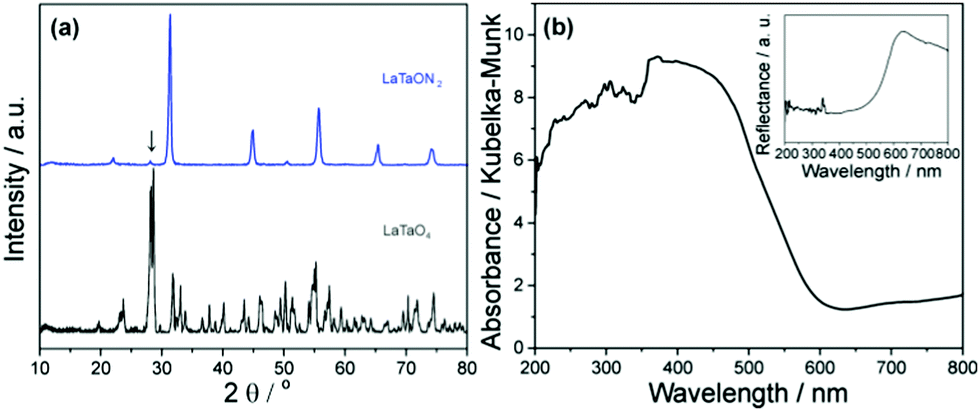 | ||
| Fig. 1 (a) XRD patterns for LaTaO4 and LaTaON2. (b) Kubelka–Munk absorbance for LaTaON2 powder. Inset is the UV-Vis reflectance spectrum. | ||
The Kubelka–Munk absorbance in Fig. 1b derived from a UV-Vis diffuse reflectance spectrum (inset) shows an absorption edge at around 600 nm. Thus the optical bandgap of LaTaON2 is around 2.1 eV. The Kubelka–Munk absorbances for cocatalyst-loaded LaTaON2 are also shown in Fig. S1 (ESI†). In comparison with the bare LaTaON2, only slight changes occur in the absorbance at 405 nm (light source for PEC measurement) for the cocatalysts loaded LaTaON2.
An electrophoretic deposition method was utilized to immobilize the LaTaON2 powders from the suspension onto FTO substrates, followed by a necking treatment using TaCl5 methanol solution and a heat treatment to improve the particle-to-particle and particle-to-substrate contacts by the formation of Ta2O5 thin layers, as reported previously.3 Four materials NiOx, Ni0.7Fe0.3Ox, CoOx and IrOx were used as cocatalysts on the oxynitrides to investigate their effects on the PEC performance.
Under visible light illumination, Ta2O5 does not contribute to the photocurrent due to its large band gap, which was also confirmed in our experiment. Thus, we assume that Ta2O5 only helps the charge transport between the LaTaON2 particles. Fig. 2a shows a schematic illustration of the cross-section for the cocatalyst-loaded LaTaON2 photoanode.
 | ||
| Fig. 2 (a) Schematic illustration of the cross-section for the cocatalyst-coated LaTaON2 photoanode. (b–f) SEM images of bare LaTaON2, NiOx–LaTaON2, Ni0.7Fe0.3Ox–LaTaON2, CoOx–LaTaON2, and IrOx–LaTaON2. | ||
The SEM image in Fig. 2b shows that the bare LaTaON2 particles with diameters in a range of 50–200 nm, have smooth surfaces. The preloading process generated highly dispersed cocatalyst nanoparticles (NiOx, Ni0.7Fe0.3Ox, and CoOx in Fig. 2c–e) with diameters less than 10 nm on the LaTaON2 surface. However, the post-loaded IrOx nanoparticles (see Fig. 2f) were poorly dispersed on the LaTaON2 surface, along with a large portion of uncovered surfaces, similar to the post-loaded CoOx and IrOx on TaON electrode reported previously.4
PEC measurement
Fig. 3 shows the PEC performances of LaTaON2 photoelectrodes which were treated with TaCl5 followed by heat treatment in air or NH3 to form tantalum oxide or oxynitride to connect LaTaON2 particles. In comparison to the air-treatment, NH3-treated photoanodes showed an evident increase of the photocurrent due to the improved electronic conductivity after the nitridation of Ta2O5 layer. At 1.23 VRHE, air-treated LaTaON2 exhibited a photocurrent density of 18.2 μA cm−2, while a value of 54.4 μA cm−2 was measured for NH3-treated LaTaON2. At potentials higher than 1.6 VRHE, the voltage-driven OER (dark OER) dominated in the water oxidation, which makes the light-driven photocurrent undetectable. | ||
| Fig. 3 Potentiodynamic measurements under chopped light in 0.5 M NaOH (pH = 13.0) for as-prepared LaTaON2 after TaCl5 necking treatment and a heat treatment in air at 350 °C (black), under NH3 flow at 450 °C (red), pre-loaded NiOx–LaTaON2 (blue), Ni0.7Fe0.3Ox–LaTaON2 (green), CoOx–LaTaON2 (olive) and post-loaded IrOx–LaTaON2 (wine) followed by TaCl5 necking treatment in air at 350 °C. Photocurrent densities were normalized based on the illuminated area of 3 mm2. | ||
Since photocurrents on the scale of mA cm−2 have been reported for TaON,3–5 Ta3N5,3 and BaTaO2N6,7 after the decoration with cocatalysts, we expect that higher photocurrent density could be achieved if appropriate cocatalysts are loaded on LaTaON2. As mentioned above, we loaded NiOx, Ni0.7Fe0.3Ox, CoOx, and IrOx cocatalysts on LaTaON2 by the impregnation method. In order to best represent the buried junction scenario,20 only the first scans of all the samples are shown in Fig. 3 unless otherwise noted.
Although literature has reported the use of NH3 post-treatment for the cocatalysts loaded photoelectrodes,3,6 we observed an adverse effect of ammonia on the PEC performance of the system (see Fig. S2, ESI†). All the samples loaded with cocatalysts were therefore only annealed in air.
The pre-loaded NiOx–LaTaON2 and Ni0.7Fe0.3Ox–LaTaON2 (see Fig. 3) after necking in air exhibited very similar performances, both higher than the NH3-treated LaTaON2. At low bias, NiOx–LaTaON2 exhibited a slightly smaller photocurrent than Ni0.7Fe0.3Ox–LaTaON2, while at potentials higher than 1.0 VRHE, NiOx–LaTaON2 showed a gradually larger photocurrent. At 1.23 VRHE, the photocurrent densities of NiOx–LaTaON2 and Ni0.7Fe0.3Ox–LaTaON2 were 140.8 μA cm−2 and 105.3 μA cm−2, respectively. In addition, the loading of NiOx and Ni0.7Fe0.3Ox both negatively shifted the onset of the dark OER current to ∼1.4 VRHE in comparison to ∼1.6 VRHE for as-prepared LaTaON2, indicating that they both were well-loaded cocatalysts, which is also confirmed by the SEM images in Fig. 2. Although it has been reported that Fe incorporation increases the electrocatalytic activity of NiOx,1,9 our results show that the Ni0.7Fe0.3Ox does not result in higher PEC performance than NiOx at 1.23 VRHE. One possible explanation for this discrepancy is that the electrolysis of water is mainly limited by the OER kinetics, but other factors related to the charge separation efficiency play more important roles for OER in photoelectrochemical water splitting, such as the surface recombinations of photo-generated charge carriers.
CoOx and IrOx both negatively shifted the onset potential of the dark OER to ∼1.45 VRHE, indicating that they are active OER cocatalysts (Fig. 3). At 1.23 VRHE, the photocurrent densities of CoOx–LaTaON2 and IrOx–LaTaON2 were 80.8 μA cm−2 and 76.6 μA cm−2, respectively, larger than NH3-treated LaTaON2 but still smaller than NiOx and Ni0.7Fe0.3Ox loaded LaTaON2.
The chopped dark-light current densities were normalized according to the illuminated area. It is important to mention that in this work the light source is a laser with a spot size of 3 mm2. On the other hand, not only the illuminated area contribute to the dark current, but the whole area of the photoanode in contact with the electrolyte, which is in our case 1 cm2. Taking this into account, it is possible to readily distinguish the different dark behaviors from the various cocatalysts. Obviously, CoOx and IrOx both experienced dark oxidation of metal ions at voltages below 1.23 VRHE, while the Ni-based oxides did not.
According to the Pourbaix diagram, Co2+ is oxidized to Co3+ between 0.6–1.4 VRHE,25 which will be further oxidized to Co4+ by photo-generated holes as evidenced by the anodic transients of CoOx–LaTaON2 in Fig. 3.27,28 Hamann et al. have reported that water oxidation occurs only when a sufficient concentration of Co4+ species is present.27 While Ir is oxidized to Ir3+ hydrous oxide between 0.4–0.8 VRHE and further to Ir4+ hydrous oxide between 0.8–1.2 VRHE.26 Because of the oxidations of surface metal ions, large redox capacitances are observed for these samples. In other words, before the oxidation of water, part of photo-generated holes were “wasted” to oxidize surface Co or Ir species, and less holes participated in PEC water splitting, resulting in lower PEC currents.
As further validations of the experimental results, we also used sputtered metallic Ni and non-catalytic CuOx as cocatalysts for LaTaON2 (Fig. S3, ESI†). As expected, metallic Ni also introduced a large pseudocapacitive current, due to the oxidation of Ni to Ni2+, while the non-catalytic CuOx had no detectable impact on the photocurrent.
As mentioned before, the enhancement in PEC performance can be attributed to both the passivation of recombination centers and catalytic improvement of the water oxidation from the cocatalysts. The fact that Ni-based oxides outperform CoOx and IrOx can be explained by the higher redox capacitance of the CoOx and IrOx, which results in an extra consumption of photo-generated holes. What is not yet clear is whether the catalytic or passivating effect plays a more important role.
H2O2 is considered as an efficient hole scavenger in electrolytes to suppress surface recombinations.29 The addition of H2O2 into the electrolyte could therefore help to maximize the photocurrent. Thus we also performed potentiodynamic measurements of cocatalysts loaded LaTaON2 in the presence of H2O2, as shown in Fig. S4 (ESI†). Unfortunately with Ni-based oxides, we did not observe any significant improvement in photocurrents comparing measurements with and without H2O2 in the electrolyte, indicating that the amount of holes reaching the surface is probably the rate determining factor. This implies that pronounced recombination of photo-generated charge carriers or the low electronic conductivity of the LaTaON2 bulk could be the bottleneck for limited PEC performances. It is worthwhile developing perovskite oxynitrides with a low number of crystalline defects, e.g., single crystalline layer, to eliminate bulk recombination and get the highest possible PEC performance. In addition, introducing Mo6+ or W6+ as donors to the Ta5+ site can increase the charge carrier concentration and enhance the electronic conductivity to further improve the PEC performance.7 With Co- and Ir-oxides, however, the light-driven photocurrent was undetectable due to the very high voltage-driven catalytic activity (dark reaction).29–32
Voltage cycling effect
Repetitive voltage cycling transforms NiOx and Ni0.7Fe0.3Ox into layered oxyhydroxides, which usually have higher OER catalytic activities than oxides.1,33 Since NiOx and Ni0.7Fe0.3Ox exhibit very similar behaviors, here we only show the results for Ni0.7Fe0.3Ox–LaTaON2. The transformation is confirmed by the anodic peak at ∼1.44 VRHE corresponding to the oxidation of α-Ni(OH)2 to γ-NiOOH, as shown in Fig. 4a.33 Prior to PEC tests, 50 scans of cyclic voltammetry (CV) were performed between 1.0 VRHE and 1.6 VRHE to convert the oxides into oxyhydroxides. A drastic change of the CV behavior for Ni0.7Fe0.3Ox–LaTaON2 occurred during the first 10 scans, but only a slight difference was observed between the 30th and 40th scan, and the 50th scan was basically identical to the 40th, indicating that the oxides have been completely converted into oxyhydroxides.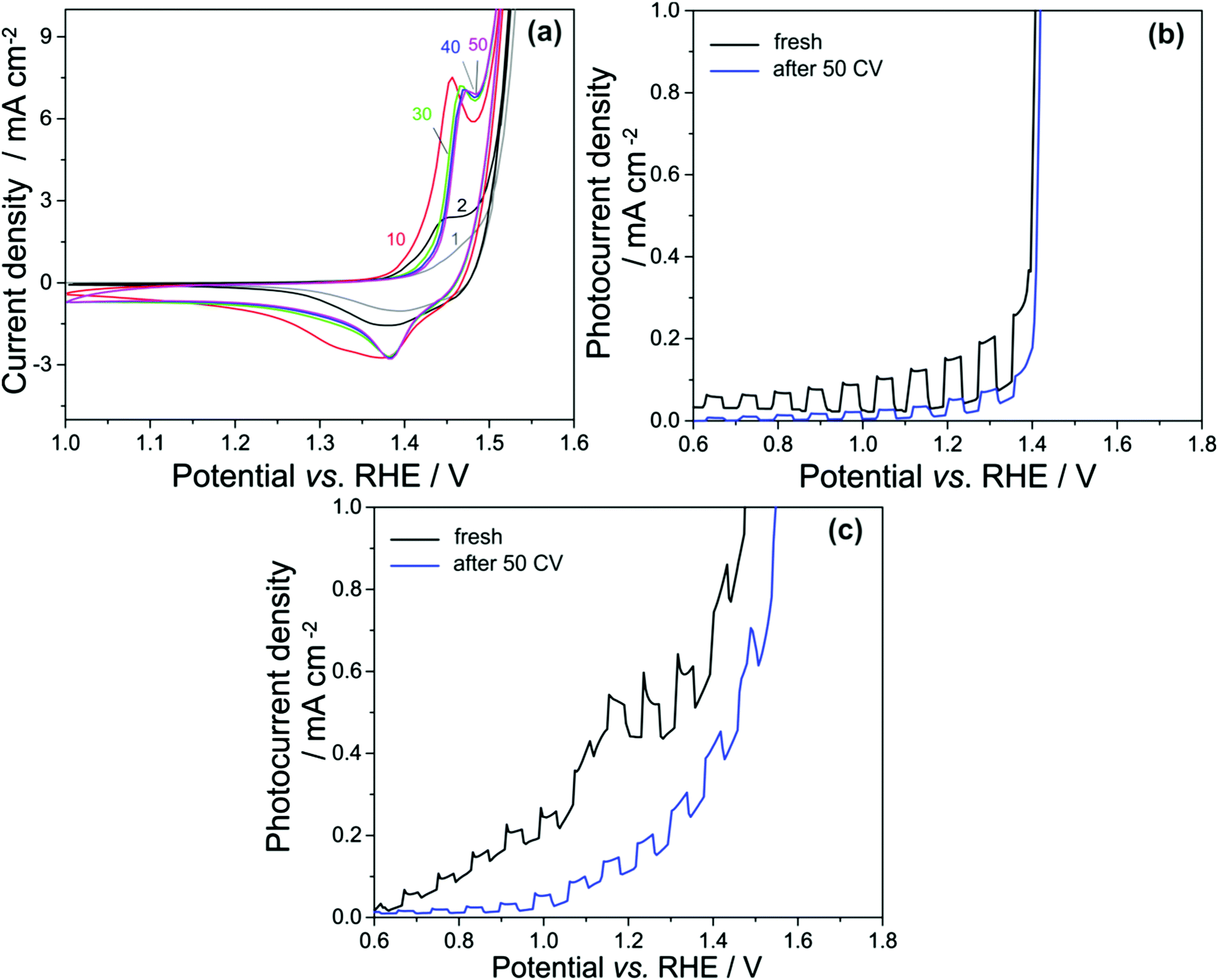 | ||
| Fig. 4 (a) Cyclic voltammetry of Ni0.7Fe0.3Ox–LaTaON2 in 0.5 M NaOH (pH = 13.0); comparison of potentiodynamic measurements before and after CV for (b) Ni0.7Fe0.3Ox–LaTaON2 and (c) CoOx–LaTaON2. Both dark current and photocurrent densities were normalized based on the illuminated area of 3 mm2. | ||
After the CV measurements, however, the system showed a decreased PEC performance, as shown in Fig. 4b. Assuming that the catalytic activity should be higher for oxyhydroxides, a possible explanation could be the change in the surface passivation centers of LaTaON2. NiOx and Ni0.7Fe0.3Ox at passivation centers have been transformed into oxyhydroxides, destroying the passivation and resulting therefore in a lower PEC performance. We can now conclude that high performance cocatalysts will not work without a good passivation as a prerequisite, i.e., the interface between the photoelectrode and cocatalysts should be stable. It should be noted that although in literature Ni-based oxides have been often used as highly catalytic cocatalysts, the passivation properties and the resulting stability were rarely discussed. Our study shows that it is actually not appropriate to directly contact Ni-based oxides with both the photoelectrode and electrolyte at the same time. An alternative way would be inserting a robust layer with suitable band alignments in-between the photoelectrode and cocatalysts, for instance, a thin layer of TiO2 or Al2O3 deposited by atomic layer deposition, to ensure a stable passivation.34,35
The voltage cycling effect of CoOx is shown in Fig. 4c. After the CV measurements, the dark pseudocapacitive current is largely reduced, implying that the oxidation states of surface Co ions have been probably saturated. Compared to the first scan, we also observed a slight decrease in the photocurrent of CoOx–LaTaON2, but not as pronounced as for Ni0.7Fe0.3Ox–LaTaON2. In this sense, ion-impermeable CoOx is a more stable cocatalyst for the passivation of the photoelectrode–cocatalyst interface.
In summary, a good surface passivation is required for photoelectrodes to exhibit high PEC performance. The high catalytic activity of cocatalysts does not improve the PEC performance unless the surface recombination centers of the photoelectrodes are passivated. It is therefore recommended to first passivate the surface with an inert thin layer of e.g. TiO2 or Al2O3 and then load the cocatalysts to catalyze water oxidation.
Conclusions
We have investigated the different behaviors of various cocatalysts on LaTaON2 photoelectrodes. NiOx and Ni0.7Fe0.3Ox result in higher photocurrents than CoOx and IrOx, due to the fact that the latter consume more photo-generated holes for the oxidation of metal ions. Repetitive voltage cycling transforms the NiOx and Ni0.7Fe0.3Ox into oxyhydroxides with higher catalytic activities, however, resulting in lower photocurrents. This is ascribed to the degradation of the passivation centers at LaTaON2–cocatalyst interface during the formation of oxyhydroxides. A robust and stable passivation layer should therefore be inserted between the photoelectrode and cocatalysts especially when Ni-based oxides are used. The limited improvement on the PEC performance observed using various cocatalysts suggests that the most important overall rate determining process in LaTaON2 are a strong bulk recombination and low electronic conductivity. To further improve the PEC performance of perovskite oxynitrides, perovskite structure with a low number of defects and high electronic conductivity should be selected.Acknowledgements
W. Si would like to thank colleague Markus Pichler and Christof Schneider for discussions and technical help. This research was supported by the Paul Scherrer Institute and the NCCR MARVEL, funded by the Swiss National Science Foundation.References
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, Energy Environ. Sci., 2011, 4, 4138–4147 CAS.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
- E. S. Kim, N. Nishimura, G. Magesh, J. Y. Kim, J.-W. Jang, H. Jun, J. Kubota, K. Domen and J. S. Lee, J. Am. Chem. Soc., 2013, 135, 5375–5383 CrossRef CAS PubMed.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2013, 135, 10238–10241 CrossRef CAS PubMed.
- M. Higashi, Y. Yamanaka, O. Tomita and R. Abe, APL Mater., 2015, 3, 104418 CrossRef.
- C. Wang, T. Hisatomi, T. Minegishi, Q. Wang, M. Zhong, M. Katayama, J. Kubota and K. Domen, J. Phys. Chem. C, 2016, 120, 15758–15764 CAS.
- C. G. Morales-Guio, M. T. Mayer, A. Yella, S. D. Tilley, M. Grätzel and X. Hu, J. Am. Chem. Soc., 2015, 137, 9927–9936 CrossRef CAS PubMed.
- Y. Liang and J. Messinger, Phys. Chem. Chem. Phys., 2014, 16, 12014–12020 RSC.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2012, 46, 1900–1909 CrossRef PubMed.
- R. Abe, M. Higashi and K. Domen, J. Am. Chem. Soc., 2010, 132, 11828–11829 CrossRef CAS PubMed.
- K. Maeda, M. Higashi, B. Siritanaratkul, R. Abe and K. Domen, J. Am. Chem. Soc., 2011, 133, 12334–12337 CrossRef CAS PubMed.
- S. Landsmann, A. E. Maegli, M. Trottmann, C. Battaglia, A. Weidenkaff and S. Pokrant, ChemSusChem, 2015, 8, 3451–3458 CrossRef CAS PubMed.
- L. Zhang, Y. Song, J. Feng, T. Fang, Y. Zhong, Z. Li and Z. Zou, Int. J. Hydrogen Energy, 2014, 39, 7697–7704 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- F. Lin and S. W. Boettcher, Nat. Mater., 2014, 13, 81–86 CrossRef CAS PubMed.
- F. Lin, B. F. Bachman and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 2427–2433 CrossRef CAS PubMed.
- T. J. Mills, F. Lin and S. W. Boettcher, Phys. Rev. Lett., 2014, 112, 148304 CrossRef PubMed.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- N.-Y. Park and Y.-I. Kim, J. Mater. Sci., 2012, 47, 5333–5340 CrossRef CAS.
- S. H. Porter, Z. Huang and P. M. Woodward, Cryst. Growth Des., 2014, 14, 117–125 CAS.
- E. M. Garcia, J. S. Santos, E. C. Pereira and M. B. J. G. Freitas, J. Power Sources, 2008, 185, 549–553 CrossRef CAS.
- J. Juodkazytė, B. Šebeka, I. Valsiunas and K. Juodkazis, Electroanalysis, 2005, 17, 947–952 CrossRef.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, J. Am. Chem. Soc., 2012, 134, 16693–16700 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, J. Bisquert and T. W. Hamann, Energy Environ. Sci., 2012, 5, 7626–7636 CAS.
- H. Dotan, K. Sivula, M. Gratzel, A. Rothschild and S. C. Warren, Energy Environ. Sci., 2011, 4, 958–964 CAS.
- B. P. Minks, G. Oskam, D. Vanmaekelbergh and J. J. Kelly, J. Electroanal. Chem. Interfacial Electrochem., 1989, 273, 119–131 CrossRef CAS.
- A. Theuwis, I. E. Vermeir and W. P. Gomes, J. Electroanal. Chem., 1996, 410, 31–42 CrossRef.
- F. F. Abdi and R. van de Krol, J. Phys. Chem. C, 2012, 116, 9398–9404 CAS.
- L.-A. Stern and X. Hu, Faraday Discuss., 2014, 176, 363–379 RSC.
- F. Le Formal, N. Tetreault, M. Cornuz, T. Moehl, M. Gratzel and K. Sivula, Chem. Sci., 2011, 2, 737–743 RSC.
- T. Wang, Z. Luo, C. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7469–7484 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Potentiodynamic measurements for NiOx–LaTaON2 after treatment in air and under NH3, potentiodynamic measurements for sputtered Ni–LaTaON2 and CuOx–LaTaON2, PEC measurements for all samples in 0.5 M NaOH with the presence of 0.1 M H2O2. See DOI: 10.1039/c6cp07253d |
| This journal is © the Owner Societies 2017 |