
An efficient 2D 11B–11B solid-state NMR spectroscopy strategy for monitoring covalent self-assembly of boronic acid-derived compounds: the transformation and unique architecture of bortezomib molecules in the solid state†
J.
Brus
*a,
J.
Czernek
a,
M.
Urbanova
a,
L.
Kobera
a and
A.
Jegorov
b
aInstitute of Macromolecular Chemistry, Academy of Sciences of the Czech Republic, Heyrovsky sq. 2, 162 06 Prague 6, Czech Republic. E-mail: brus@imc.cas.cz
bTeva Czech Industries s.r.o., Branisovska 31, 370 05 Ceske Budejovice, Czech Republic
First published on 22nd November 2016
Abstract
The difficulty in the prediction of the complicated solid-state structure of boronic acid derivatives, resulting from the complex pathway of reversible covalent interactions, represents a significant obstacle to the development of a new generation of advanced supramolecular systems such as covalent organic frameworks of efficient anticancer drugs. In this contribution, various 2D 11B–11B solid-state NMR correlation techniques supported by DFT calculations were explored to formulate a reliable tool for monitoring the covalent assembly of boronic acid residues in the solid state. This way, the self-condensation of bortezomib molecules was investigated, different local constitutions of boroxine motifs were unveiled, and the previously unreported boroxine structures of bortezomib polymorphs exhibiting secondary coordination were discovered and described in detail. The recorded 11B NMR parameters responded sensitively to subtle changes in the local geometries, which were reliably interpreted and directly visualized by the DFT calculations. A uniform 2.6 Å distance in bortezomib 11B–11B spin pairs was conclusively identified by the through-space 11B–11B double-quantum (DQ) coherence build-up curves, whereas distinct 2D 11B–11B DQ correlation patterns revealed unique boroxine structures existing in the crystalline as well as amorphous state. The boroxine rings were found to be internally stabilized through the transformation of the trigonal boron sites toward tetrahedral geometry, as the secondary five-membered rings were formed. This way, the nature of bortezomib polymorphism is disclosed, and an efficient strategy for exploring the assembly of boronic acid derivatives in the solid state, for which no crystallographic data are available, is thus demonstrated.
Introduction
Boron-containing compounds have long been recognized as potentially active pharmaceutical ingredients.1–3 Recent investigations have resulted in the discovery of many promising highly efficient pharmaceuticals exhibiting anticancer and antibacterial activity such as bortezomib, MLN978, CEP-18770, GSK2251052 etc. Consequently, research in peptidic boronic acid derivatives in pharmaceutical laboratories is rapidly gaining in intensity.4Boronic acid derivatives have also played a key role in many fields of organic, bioorganic and macromolecular chemistry, and interest in the dynamic covalent assembly of boronic acid derivatives has increased in the field of supramolecular chemistry.5,6 The reversible self-condensation of boronic acids to generate boroxine anhydrides as well as the assembly of boronic acids with organic diols to form boronate esters have opened new routes to the synthesis of complex supramolecular systems,7,8 polymers,9 hydrogels10,11 and covalent organic frameworks.12,13 Full exploitation of such boron-based systems, however, requires their precise structural characterization. This requirement is particularly urgent for amorphous systems where no diffraction structures are available.
Crystalline bortezomib (Scheme 1), a proteasome inhibitor approved for the treatment of multiple myeloma,14 represents a unique combination of a complex supramolecular solid-state structure with an extremely high multilateral pharmaceutical activity.15 Bortezomib crystallizes from dry ethyl acetate as Form I, from wet ethyl acetate as Form II, or can exist in the amorphous state. As described in the registration dossier and patent literature,16,17 solid-state bortezomib presumably exists in the form of boroxine anhydride (Scheme 1), although proof of its boroxine structure has apparently never been published. Consequently, a thorough understanding of the solid-state structure of bortezomib, a peptidic derivative of boronic acid, would further not only stimulate the development of new pharmaceuticals, but also provide a clue to the way in which self-assembly of organo-boron compounds can be controlled toward the synthesis of well-defined supramolecular systems.
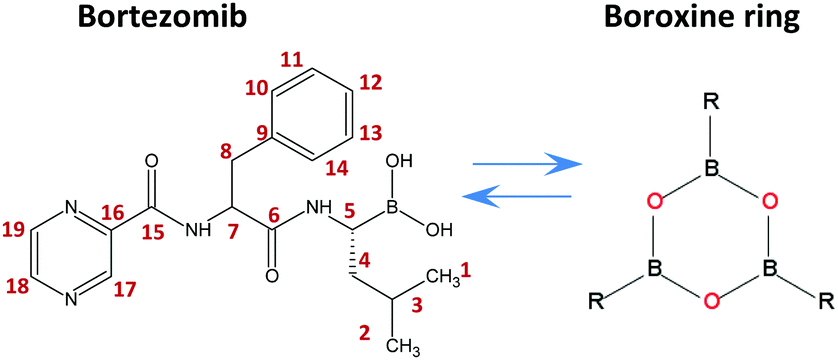 | ||
| Scheme 1 Chemical structure of bortezomib (C19H25BN4O4), boroxine ring and the labeling scheme used herein. | ||
11B solid-state nuclear magnetic resonance (ssNMR) spectroscopy has evolved into an important tool for structural studies in a variety of inorganic,18 metalorganic,19 and macromolecular20 systems. Recently, advanced 11B ssNMR techniques (see ref. 21 and 22 for a survey) have been successfully applied to a number of materials including glasses,23 pharmaceutical systems,24,25 resins,26 zeolites,27,28 and even supramolecular hydrogels.10,11 However, despite this effort, a comprehensive methodology suitable for the clear identification of boroxine motifs and exploration of assembly of boronic acid building blocks at an atomic resolution in the solid state is still missing. This fact thus represents a significant obstacle for the further development of covalently assembled boron-based materials, and for the advancement of the second generation of proteasome inhibitors built on boron-containing peptides.
Experimental
Materials
The bortezomib polymorphs, Forms I and II, and the amorphous form were obtained from TEVA Pharmaceutics (C19H25BN4O4; Czech Republic). Model compounds such as cesium [3-cobalt(III) bis(1,2-dicarbollide)](-1) (CsCoD; Katchem Ltd, Prague, Czech Republic.); borax (disodium tetraborate, Na2[B4O5(OH)4]·8H2O, Sigma-Aldrich); boric acid (H3BO3, Sigma-Aldrich); 2-methyl-propyl-boronic acid ((CH3)2CHCH2B(OH)2, Sigma-Aldrich); and [(E)-4-(2-cyano-2-ethoxycarbonylvinyl)-phenyl] boronic acid pinacol ester (C18H22BNO4, Sigma-Aldrich) were used as purchased.Solid-state NMR spectroscopy
Solid-state NMR spectra were measured at 11.7 T on a Bruker Avance III HD 500 US/WB NMR spectrometer (Karlsruhe, Germany, 2013). The following techniques were applied: (i) one-dimensional (1D) 11B and 13C MAS and CP/MAS NMR experiments; (ii) two-dimensional (2D) 1H-13C FSLG HETCOR experiments;29 (iii) NOESY-type, three-pulse 11B–11B proton-driven spin-diffusion experiments30 (2D 11B–11B PDSD MAS NMR); (iv) modified triple-quantum (TQ) experiments31 with an incorporated 11B–11B spin-exchange period (2D 11B–11B TQ/MAS PDSD NMR); and (v) 11B–11B double-quantum (DQ) experiments32,33 with the BR212 recoupling sequence34,35 (2D 11B–11B DQ/SQ BR212 MAS NMR). The frictional heating36,37 of the spinning samples was compensated by active cooling, and the temperature calibrations were performed with Pb(NO3)2. (For all experimental parameters, see the ESI,† Section S1.)Computational methods
Numerous initial structures of bortezomib in monomeric, dimeric, trimeric and cyclic trimeric boroxine forms in a vacuum were prepared using interactive computer graphics (program Insight II (2000), Accelrys Inc., San Diego, California). In all initial models boron atoms were in trigonal coordination. Considering the relatively large size of the system (150 atoms as boroxine), the structures were first preoptimized using the Hartree–Fock (HF) method and the STO-3G basis set. The structures that turned out to be minima of the potential-energy surface (PES) of bortezomib at this level of quantum chemical theory, namely, those whose harmonic vibrational frequencies were real, were considered further. Thus, the HF/STO-3G estimates of the PES served as the input for the geometrical optimization using the density functional theory (DFT)-based B3LYP/6-311G** method. The stationary points of the PES located in this way were subjected to the B3LYP/6-311G** calculations of the harmonic vibrational frequencies. The structures identified as minima were used for the predictions of the NMR parameters. The B3LYP/6-311G** method, which successfully approximates the NMR chemical shifts in boron-containing systems,26,38–40 was used for the electric field gradient (EFG) and chemical shielding calculations. In the latter, the GIAO41,42 strategy to overcome the gauge problem was adopted. The Gaussian 09 suite of computer codes was used throughout.43Results and discussion
11B–11B correlations in typical assemblies of boron-containing compounds
For monitoring the covalent assembly of boronic acid residues in the solid state, we explored an experimental strategy based on the analysis of 11B–11B solid-state NMR correlation spectra complemented by the DFT optimization of local structures and computations of 11B NMR shielding parameters. In order to measure the B⋯B interatomic distances and to inspect the changes in local structures in the vicinity of boron sites, we optimized three complementary 11B–11B MAS NMR correlation techniques. First we applied the 2D 11B–11B PDSD MAS NMR experiment based on the detection of SQ coherences in both frequency dimensions, which utilizes the exchange of the longitudinal 11B magnetization. Second, we tested the modified 2D 11B–11B TQ/MAS PDSD NMR experiment employing the same polarization transfer detecting the TQ coherences in the indirect dimension, and third, we optimized the 2D 11B–11B DQ/SQ BR212 MAS NMR experiment, which indirectly detects 11B DQ coherences (DQC) generated by manipulating the transverse 11B polarization during the rotor-synchronized, rf-field-optimized recoupling period.The structure-related outputs from these techniques were validated using crystalline compounds representing typical molecular assemblies with different boron–boron interatomic distances. One-bond, 11B–11B spin pairs separated by a distance of 1.77 Å dominate in the CsCoD system,44 whereas medium-range B–O–B distances of ca. 2.50 Å prevail in borax.45 In contrast, hydrogen-bonded boric acid molecules represent long-range B⋯B spin pairs separated by a distance of ca. 3.60 Å.46 Somewhat larger B⋯B distances (>4 Å) are expected in 2-methyl-propyl-boronic acid, whereas crystalline CEPBA-PE represents the system expected to contain boron atoms that are essentially isolated and separated by distances greater than 5 Å. Due to the well-defined crystal structure and the presence of two chemically distinct boron sites (BIII and BIV) that are covalently bound through an oxygen atom, crystalline borax represents the most suitable model for searching for the boroxine motifs. In particular, the bi-cyclic borax molecule, in which boron atoms adopting different local geometries are separated by 2.5 Å (BIII⋯BIV) and 3.6 Å (BIII⋯BIII), offers an ideal system to probe the evolution of 11B–11B spin-exchange NMR correlation signals that can be expected in the boroxine units. (For borax structure, see the ESI,† Fig. S5.)
The obtained results that are discussed in detail in the ESI† (Section S2) show that the 2D 11B–11B PDSD MAS NMR experiment is a robust technique that facilitates the probing of 11B–11B dipolar contacts over a range of interatomic distances from ca. 1.8 to 3.6 Å. As a result of the combination of several anisotropy-driven recoupling mechanisms,47 particularly due to the non-parallel relative orientations of quadrupolar tensor Vzz components48 (Fig. 1a), the 11B–11B polarization transfer can also be observed between the chemically equivalent sites.
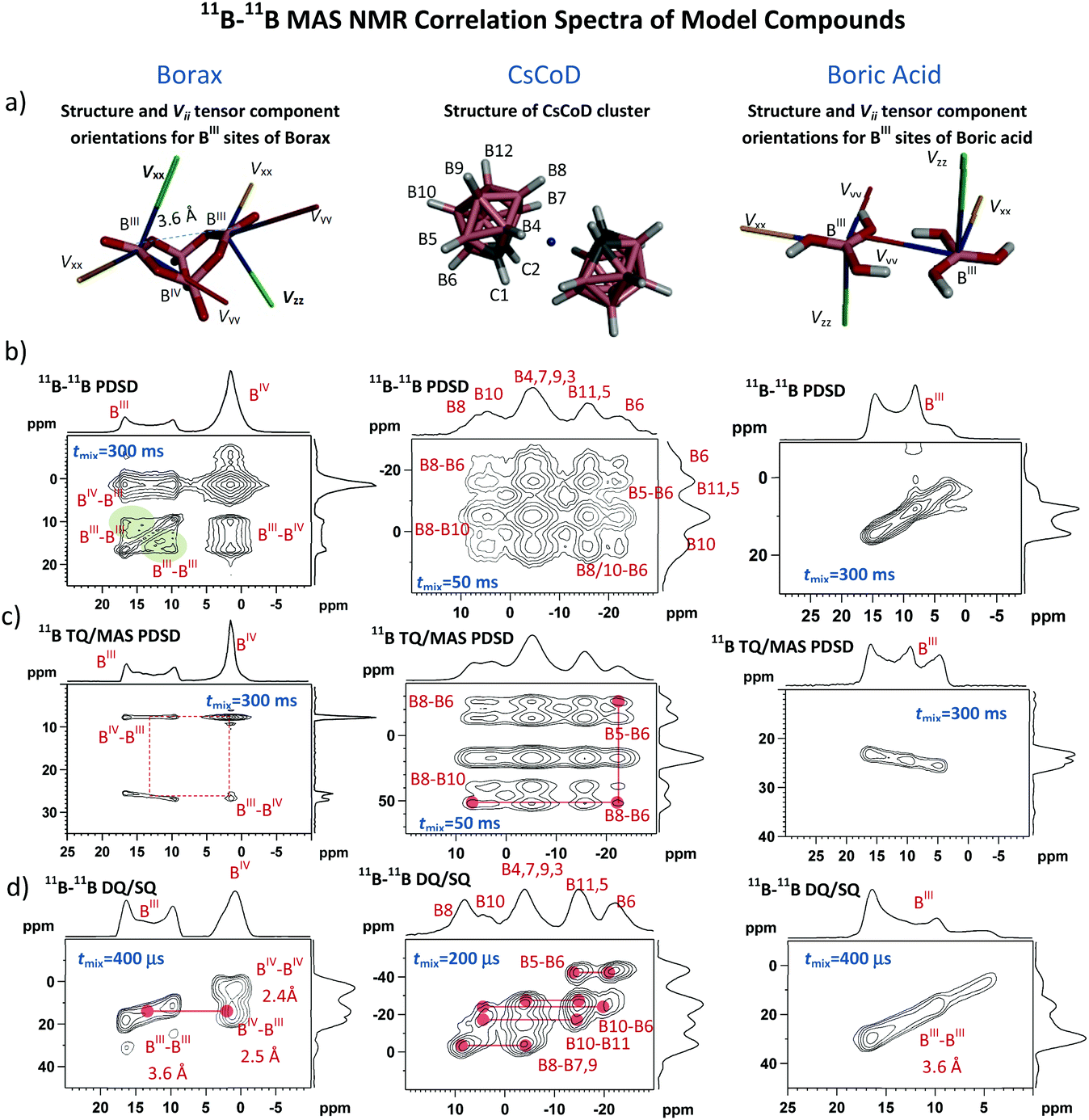 | ||
| Fig. 1 Structures of selected boron-containing model compounds: borax, CsCoD and H3BO3 (a), and the corresponding 2D 11B–11B correlation NMR spectra measured using the 2D 11B–11B PDSD MAS NMR experiment (b); 2D 11B–11B TQ/MAS PDSD NMR technique (c); and 2D 11B–11B DQ/SQ BR212 MAS NMR pulse sequence (d). The green areas in the 2D 11B–11B PDSD MAS NMR spectrum of borax (b) highlight autocorrelation signals arising from the non-parallel orientation of the quadrupolar tensor principal components Vzz. Orientations of principal components Vii for borax and boric acid are demonstrated in the corresponding structures (a). Typical experimental time for each of the 2D NMR experiments did not exceed 12–24 hours. | ||
This is demonstrated by autocorrelation signals observed for trigonal BIII sites of borax (Fig. 1b, the green areas). The same autocorrelation signals also evolve for BIV sites. However, due to the small quadrupolar splitting (Qcc = 0.9 MHz) these signals are less apparent. (For the expanded 2D 11B–11B PDSD MAS NMR spectra of borax BIV sites, see the ESI,† Fig. S6.)
In contrast, if the tensor Vzz components of the interacting sites are in a parallel orientation, as in crystalline boric acid (Fig. 1a), the magnetic inequivalence of these sites disappears and the autocorrelation signals do not evolve (Fig. 1b), although the interatomic distance between the BIII atoms (3.6 Å) is comparable with that in borax (3.6 Å). Information about the relative orientation of neighbouring sites thus can be, in principle, extracted from the 11B–11B PDSD MAS NMR correlation patterns. As indicated previously38 such an analysis requires extensive simulation of 2D spectra including variation of the sign and size of quadrupolar coupling constants, Qcc. These simulations, which are beyond the scope of this contribution, will be the subject of our future studies.
In some cases, the evolution of autocorrelation signals reduces spectral resolution and thus creating obstacles in the interpretation of NMR data. This is particularly apparent for the spectra overcrowded by a range of different resonances (e.g. CsCoD, Fig. 1b; see also the ESI,† Fig. S12). Therefore, if the spectral resolution is the major limitation to finding the required interatomic contacts, then the application of the 2D 11B–11B TQ/MAS PDSD NMR technique is more suitable. Here, the formation of the autocorrelation signals is suppressed, and the spectral resolution is enhanced by the detection of TQ coherences in the isotropic F1 dimension (Fig. 1c; see also the ESI,† Fig. S8 and S13).
In general, however, 2D 11B–11B PDSD MAS NMR correlation spectroscopy loses its efficiency when probing long-range internuclear contacts, particularly between magnetically equivalent sites. In contrast, the 2D 11B–11B DQ/SQ BR212 MAS NMR technique is perfectly suited to overcome this insufficiency. The DQ coherence can be generated for spin pairs in which the internuclear distance far exceeds 5–6 Å49,50 (see also the ESI,† Fig. S27 and S28). Moreover, the build-up curves of 11B–11B DQ coherence sensitively reflect the changes in internuclear distances as demonstrated in Fig. 2a, and the obtained maxima of DQ build-up curves follow an inverse third power dependence upon increasing the B⋯B interatomic separation (Fig. 2b). Consequently it is clear that structural information that is vital for identifying the medium-range spin-pairs expected in boroxine rings can be derived from these build-up curves and 2D 11B–11B DQ/SQ MAS NMR spectra measured with a short mixing period (ca. 200–400 μs).
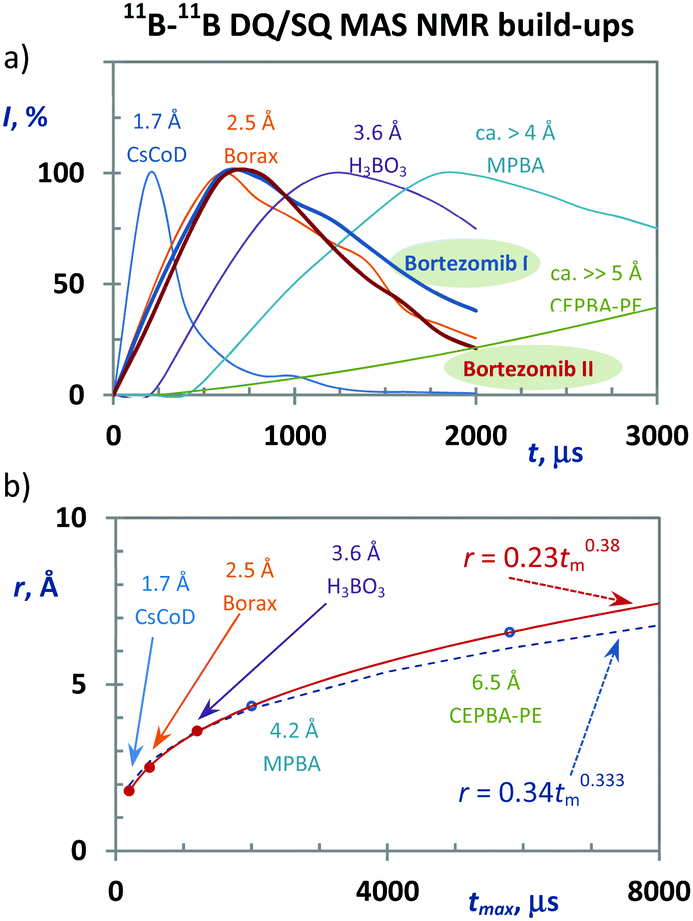 | ||
| Fig. 2 11B–11B DQC build-up curves recorded for model compounds, and bortezomib Forms I and II (a). Dependence between the recoupling time in which the maximum intensities of the DQ coherences are reached and the interatomic 11B⋯11B distances (b). The red line represents the best fit of the experimental data. The dashed line represents the pure third-power function. | ||
2D 11B–11B solid-state NMR of bortezomib polymorphs
By employing the obtained methodological findings, and in the absence of X-ray diffraction data, we analyzed the assembly of bortezomib molecules in the polymorphic Forms I and II. The 13C CP/MAS and 11B TQ/MAS NMR spectra, in which three sets of carbon and boron signals always appeared (Fig. 3a and b), revealed the existence of three bortezomib fragments, which exhibit an unexpectedly large inequivalence. Whereas the boron site B1 resonates at ca. 10 ppm, the boron site B3 exhibits a resonance frequency at ca. 30 ppm. These differences in resonance frequencies indicate considerable differences in local structures that are seemingly inconsistent with the expected formation of boroxine rings (Scheme 1). However, when measuring the 11B–11B PDSD MAS NMR spectra (Fig. 3c), the formation of broad correlation signals involving all resonance frequencies indicates the spatial proximity of all these three distinct boron species. This finding is supported by the 11B–11B TQ/MAS PDSD NMR spectra (ESI,† Fig. S29). However, the resolution of these PDSD NMR spectra is insufficient for in-depth structural insights.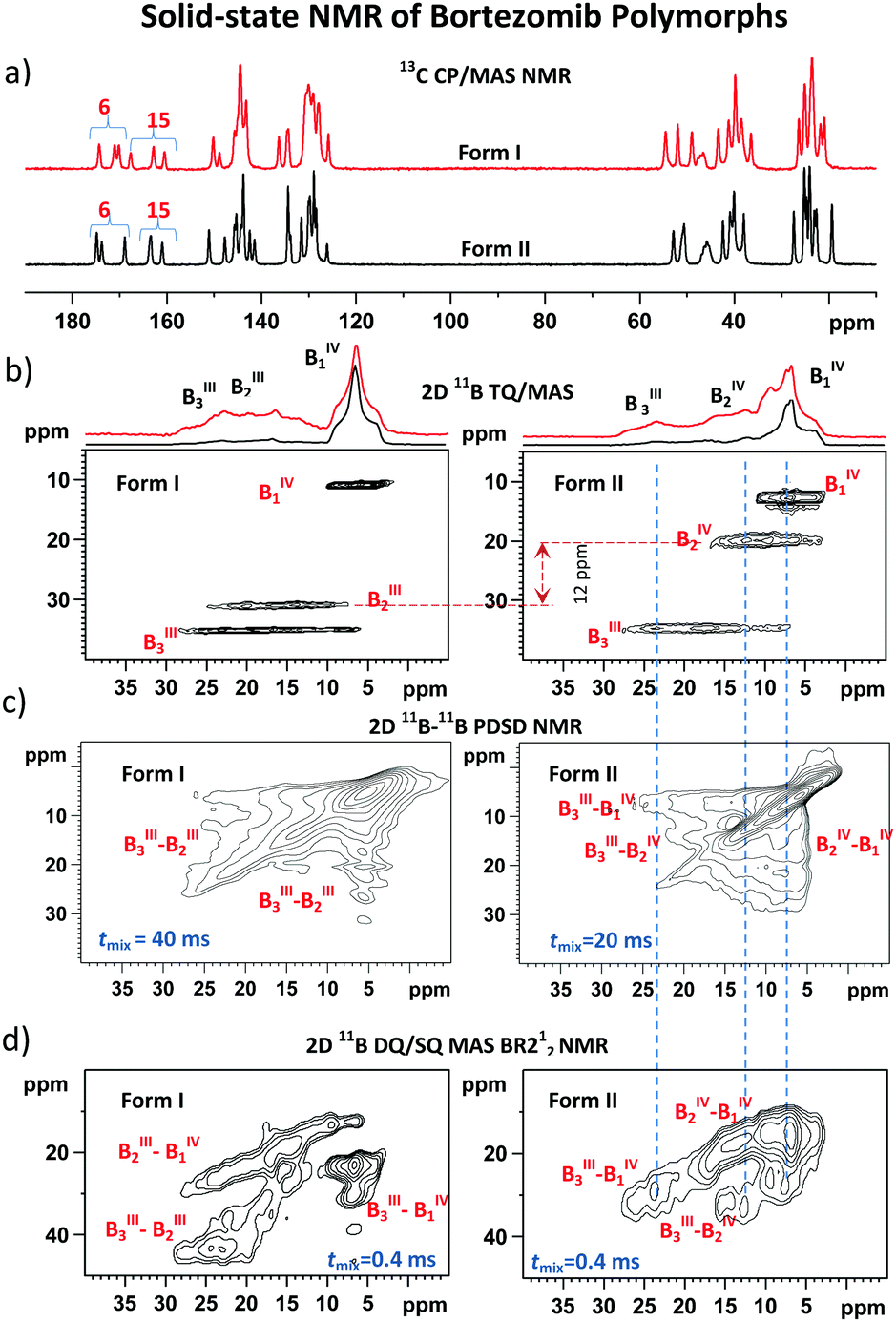 | ||
| Fig. 3 13C CP/MAS NMR (a), 11B TQ/MAS NMR (b), 11B–11B PDSD MAS NMR (c), and 11B–11B DQ/SQ BR212 MAS NMR spectra (d) of bortezomib Forms I and II. 11B MAS NMR spectra are presented as red lines in panel (b). | ||
Therefore, to search for precise structural data the 11B–11B DQ/SQ BR212 MAS NMR data were analyzed. At first, the DQ coherence build-up curves detected for both bortezomib polymorphs (Fig. 2a) exhibit maxima at ca. 600 μs, indicating that typical 11B–11B interatomic distances in the bortezomib assemblies lie at approximately 2.6–2.7 Å, which are comparable with the structural motifs of crystalline borax. Second, focusing on the medium-range boron–boron pairs, the interaction networks showing the individual 11B–11B spin pairs and their connectivity were traced in the 2D 11B–11B DQ/SQ BR212 MAS NMR spectra measured for short recoupling times of 200–600 μs. For both polymorphic forms, the interaction network showing the interconnected pairs B1–B2, B1–B3 and B2–B3 was fully developed within 600 μs (Fig. 3d). Moreover, the same DQ correlation signals were detected in the 2D spectra measured with the shortest recoupling period (200 μs; ESI,† Fig. S31 and S32), thus supporting the existence of compact structural motifs in which all three boron sites mutually interact.
Considering that the interatomic distances between these boron sites are not greater than 2.6–2.7 Å, the obtained data suggest the covalent assembly of bortezomib molecules into the boroxine cycles in both polymorphs. On the other hand, the distributions of the DQ coherences in the recorded 2D 11B–11B DQ/SQ BR212 NMR spectra differ substantially (Fig. 3d). Specifically, for the polymorphic Form I, the B2–B3 correlation signal resonates in a high DQ frequency region (ca. 45 ppm), whereas the polymorphic Form II shows the resonance frequency of the corresponding B2–B3 correlation signal as strongly shifted toward lower frequencies. This observation thus clearly reveals the existence of two distinct boroxine structures that differ considerably in their local architectures.
In this regard we considered the application of 2D J-resolved 11B MAS NMR spectroscopy51 because the measurement of through-bond (B–O–B) J-couplings would provide a straightforward support for the existence of boroxine rings. However, our preliminary calculations as well as literature data52,53 predicted extremely low two-bond 2J(11B,11B) coupling constants in boroxine rings, ca. 2–5 Hz, the detection of which is beyond the current experimental capabilities.
Local architecture of bortezomib boroxine assemblies
The differences in the local structures are also found to be encoded in the isotropic NMR chemical shifts and quadrupolar parameters. As extracted from the 1H–13C FSLG HETCOR spectra (Fig. 4, ESI,† Fig. S33 and S34) and the skyline projections of 11B TQ/MAS NMR signals, changes in shielding parameters reach ca. 5 ppm for C6 carbonyl atoms, 2 ppm for H7 protons and 10 ppm for B2 boron sites. In an attempt to understand the observed dispersion of the 11B NMR parameters and explain the above-demonstrated large inequivalence of the boron sites in bortezomib boroxine units, DFT-based B3LYP/6-311G** geometry optimization of a set of potential structures was carried out, followed by GIAO-B3LYP/6-311G** calculations of the 11B NMR parameters (the computed isotropic chemical shieldings were converted to the estimates of the chemical shifts using the procedure described in ref. 26).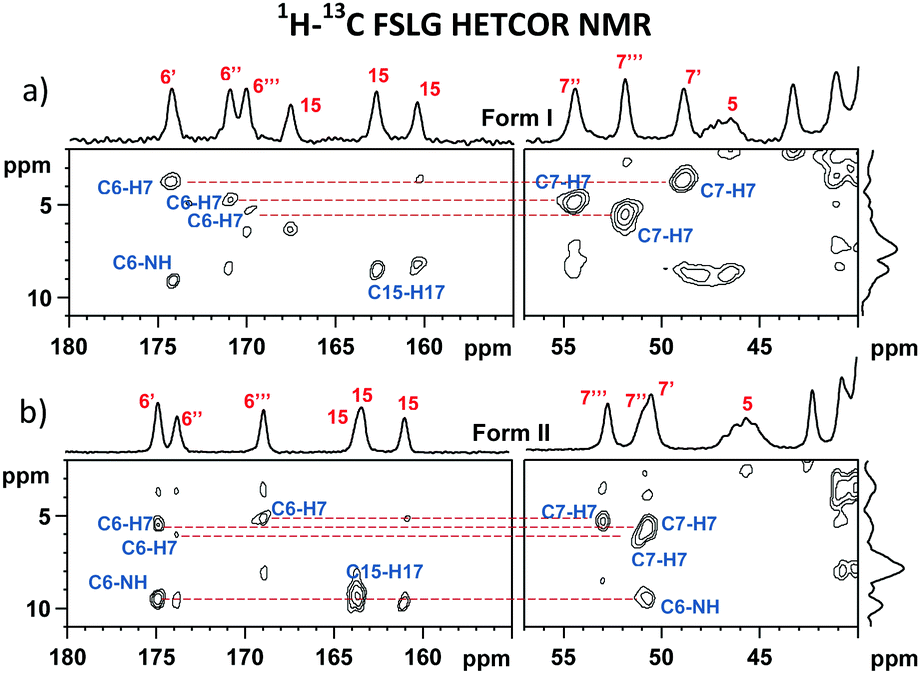 | ||
| Fig. 4 Expanded regions of the 1H–13C FSLG HETCOR NMR spectra of bortezomib Forms I (a) and II (b) measured at 0.3 ms CP mixing time. | ||
The performed geometrical optimizations revealed two NMR-consistent candidates for which the calculated 11B NMR parameters agreed with the experimental data (Tables 1 and 2). These two structures represent the boroxine motifs with alternating coordination configurations of the boron sites (Fig. 5). Contrary to the generally accepted planar structure of the boroxine moiety caused by the expected trigonal coordination of boron atoms, the boroxine rings in the predicted DFT-optimized structures consist of either two boron sites in trigonal configuration BIII and one in tetrahedral coordination BIV and/or two tetrahedral sites accompanied by one BIII atom with trigonal geometry. The trigonal coordination is characterized by the large quadrupolar constants Qcc > 3 MHz and high-frequency isotropic shifts δ(11B) > 25 ppm, whereas the increasing symmetry around the BIV sites reduces these parameters to ca. 1.5–2.1 MHz and 7–15 ppm, respectively.
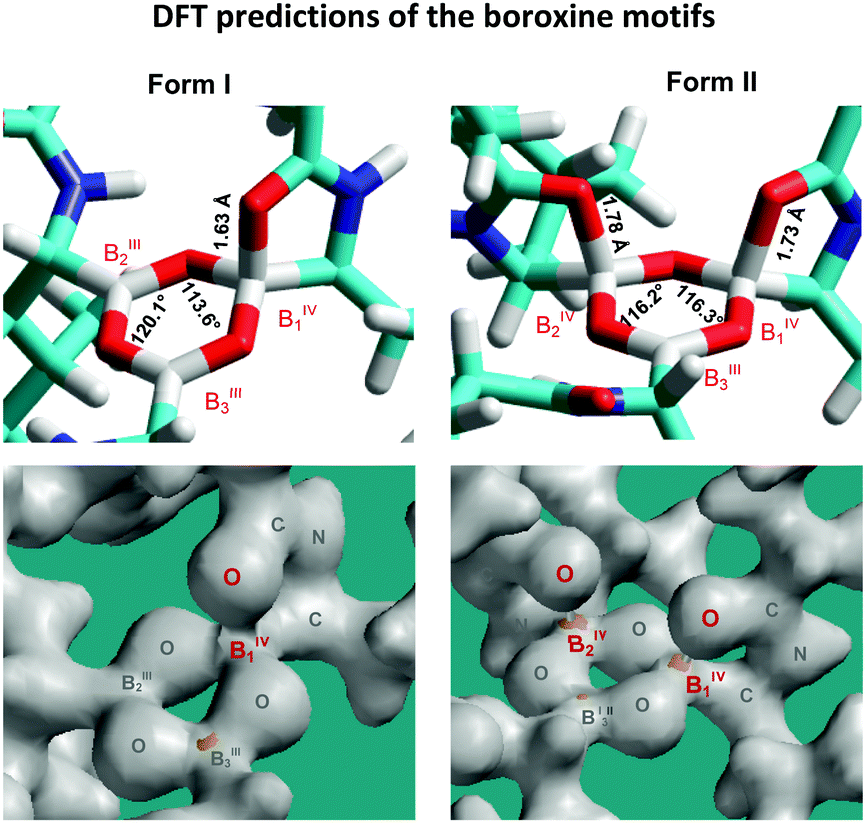 | ||
| Fig. 5 Predicted B3LYP/6-311G** structures of the bortezomib boroxine motifs representing Forms I and II, with the corresponding bond-density plots shown below. | ||
| Isotropic chemical shifts δ(11B), ppm | ||||||
|---|---|---|---|---|---|---|
| Form I | Form II | |||||
| BIV1 | BIII2 | BIII3 | BIV1 | BIV2 | BIII3 | |
| *Estimated experimental error is ±0.2 ppm. | ||||||
| δ(11B)exp | 9.5 | 24.9 | 28.9 | 10.7 | 16.5 | 27.5 |
| δ(11B)cal | 8.0 | 30.0 | 30.6 | 11.5 | 12.9 | 29.0 |
| Quadrupolar coupling constants Qcc, MHz | ||||||
|---|---|---|---|---|---|---|
| Form I | Form II | |||||
| BIV1 | BIII2 | BIII3 | BIV1 | BIV2 | BIII3 | |
| *Estimated experimental error is ±0.3 MHz. | ||||||
| Q cc(exp) | 1.7 | 2.9 | 3.0 | 1.5 | 2.1 | 3.0 |
| Q cc(cal) | 1.8 | 3.4 | 3.4 | 2.1 | 2.4 | 3.3 |
It should be noted that for both the models representing bortezomib polymorphs Form I and Form II the experimental and calculated 11B NMR parameters are in good agreement for BIV1 and BIII3 sites. Specifically, the differences between the calculated and experimental isotropic chemical shifts δ(11B) lie in the 1.0–1.5 ppm range, and the differences in quadrupolar coupling constants Qcc are smaller than 0.5 MHz. This finding thus indicates that the predicted geometries of tetra-coordinated BIV1 sites and tri-coordinated BIII3 sites precisely describe the corresponding local structures of boroxine rings in the investigated systems. These sites also represent two distinctly different, well-defined and relatively stable local geometries of boroxine rings. In contrast, a worse agreement between the calculated and experimental isotropic chemical shifts observed for B2 sites (in particular, the chemical shift differences in the 3.6–5.1 ppm range) indicates the possible disorder around those sites. Nevertheless, the GIAO-B3LYP calculations correctly reproduced the trend in chemical shift changes between the BIII and BIV moieties.
In the absence of crystallographic data, and due to many degrees of freedom within the organic residue of bortezomib, the DFT-calculated 1H and 13C isotropic chemical shifts were not analysed in detail. In addition, the complete, explicit signal assignment is unachievable for the investigated systems at the natural isotopic abundance. Nevertheless, we performed an analysis of 13C NMR parameters and compared the experimentally determined and calculated 13C NMR isotropic chemical shifts for carbonyl units C6 that can be involved in the coordination with boron atoms (–B⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–). Specifically, we compared 13C NMR isotropic chemical shifts (experimentally determined) to their corresponding isotropic chemical shieldings as calculated by the GIAO-B3LYP/6-311G** approach. As demonstrated graphically (see the ESI,† Fig. S35 and S36) there is a clear agreement between experimental and calculated 13C NMR parameters. Carbonyl carbon atoms C6 involved in the secondary coordination with boron atoms always exhibit considerably smaller shielding (higher chemical shifts) as compared to the uncoordinated carbonyl carbons. Moreover, in agreement with the experimental data the performed calculations correctly predicted the increase in isotropic shielding (the decrease of the isotropic chemical shift) for the carbonyl carbons C15 in the distal segments of bortezomib molecules.
C–). Specifically, we compared 13C NMR isotropic chemical shifts (experimentally determined) to their corresponding isotropic chemical shieldings as calculated by the GIAO-B3LYP/6-311G** approach. As demonstrated graphically (see the ESI,† Fig. S35 and S36) there is a clear agreement between experimental and calculated 13C NMR parameters. Carbonyl carbon atoms C6 involved in the secondary coordination with boron atoms always exhibit considerably smaller shielding (higher chemical shifts) as compared to the uncoordinated carbonyl carbons. Moreover, in agreement with the experimental data the performed calculations correctly predicted the increase in isotropic shielding (the decrease of the isotropic chemical shift) for the carbonyl carbons C15 in the distal segments of bortezomib molecules.
In accord with the observed changes in isotropic chemical shifts, the C6 carbonyl groups play a key role in the structural diversity of bortezomib boroxine moieties (Fig. 5). When placed above the boroxine ring, the C6 carbonyl oxygen interacts with the boron site BIII, whose geometry then becomes tetrahedral. Consequently, a secondary five-membered [–B⋯OC–N(H)–C–] ring perpendicular to the boroxine plane is formed. The corresponding O–B–O valence angle decreases from the ideal value of 120° to ca. 113°, and the planarity of the boroxine ring is effectively removed, adopting a dihedral angle of 4° for the BIII3–O–BIII2–O fragment in Form I and a BIV1–O–BIV2–O dihedral angle of 23° in Form II. The predicted small torsion involving the trigonal BIII3 and BIII2 sites corresponds to the weak BIII3–BIII2 correlation signal detected in the 11B–11B PDSD NMR spectrum (Fig. 3c), reflecting the nearly parallel orientation of the Vzz components of the quadrupolar tensors of the BIII3 and BIII2 sites (Fig. 6).
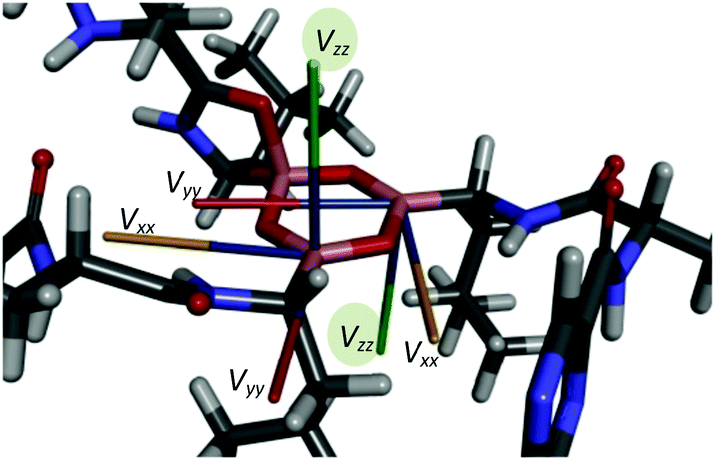 | ||
| Fig. 6 Orientations of the principal axes of the quadrupolar tensors in the boroxine cycle of bortezomib Form I. | ||
As reflected by the B⋯O distances and bond-density plots (Fig. 5) the strength of the B⋯O(C) coordination interactions depends on the local architecture of the boroxine moieties. If the tetrahedrally coordinated boron atom is surrounded by two trigonal BIII sites, the B⋯O distance predicted by the B3LYP/6-311G** method is 1.63 Å, whereas if two BIV sites adjoin each other, the bond strength is weakened and the predicted distances increase to 1.73 and 1.78 Å. This finding agrees well with the increase in isotropic 11B chemical shifts experimentally observed for the BIV1 and BIV2 sites of Form II and suggests some degree of variability and receptivity in these boroxine moieties.
An example of such receptivity is shown in the 11B TQ/MAS NMR spectrum of bortezomib Form II treated at 160 °C for 2 hours (Fig. 7a), in which a new signal appears at 15.0 ppm. The narrow isotropic projection of this resonance observed in the TQ dimension F1 (Fig. 7b) indicates a uniform and well-defined geometry around this thermally converted BIV1(T) site. Moreover, the secondary set of narrow, low-intensity, 13C CP/MAS NMR signals (Fig. 7c) reflects the formation of a new, structurally distinct arrangement of bortezomib molecules.
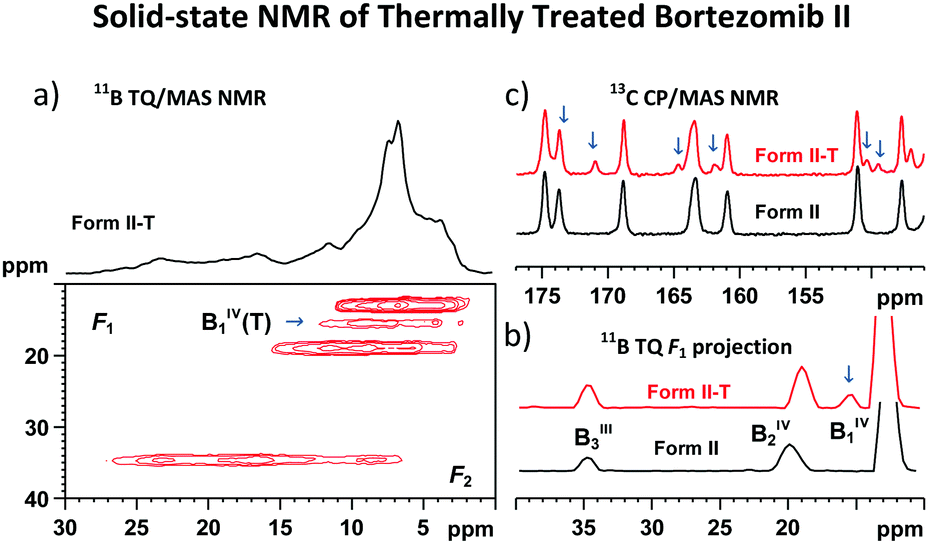 | ||
| Fig. 7 11B TQ/MAS NMR spectrum of the thermally treated bortezomib Form II-T (a); comparison of the 11B TQ isotropic projections (b); and comparison of the expanded parts of the 13C CP/MAS MMR spectra of the thermally treated and untreated bortezomib Form II, respectively (c). | ||
To verify the general applicability of the proposed experimental approach an amorphous form of bortezomib was also investigated. Although the 13C CP/MAS NMR and 11B MAS NMR spectra are significantly broadened due to the loss of uniformity in the molecular arrangement, the 2D 11B TQ/MAS NMR and 11B–11B DQ/SQ BR212 NMR spectra still allow the identification of the preferred structural motifs. Interestingly, the recorded 2D 11B–11B DQ/SQ NMR correlation spectra of the amorphous system are almost identical to the spectra measured for the crystalline Form II (ESI,† Fig. S37). This finding thus reveals not only the existence of boroxine rings in the amorphous form of bortezomib but also suggests their nonrandom structure. The bortezomib boroxine rings in the amorphous phase rather adopt conformations existing in the crystalline Form II.
Conclusions
In summary, we formulated a reliable experimental approach for monitoring the covalent assembly of boronic acid residues in the solid state. This strategy is based on 11B–11B MAS NMR correlation spectroscopy, supported by DFT structural searches, followed by predictions of the 11B NMR parameters. These 11B NMR parameters (isotropic chemical shifts and quadrupolar constants) respond sensitively to subtle changes in the local geometries. The 11B–11B DQ coherence build-up curves then accurately reflect the changes in the 11B–11B spin-pair distances, whereas the correlation patterns detected in the 2D 11B–11B DQ/SQ BR212 MAS NMR spectra are sufficiently distinctive to trace the interaction networks of the 11B–11B spin pairs in different assemblies. When complemented by 11B–11B PDSD MAS NMR spectra containing orientation information, these correlation data combined with DFT searches can be used to reconstruct the local structure around the boron sites in a variety of boronic acid-derived materials with high precision. The proposed strategy works well for the systems in crystalline, partially disordered as well as amorphous states.Using this approach, we successfully described the covalent assembly of structurally complex bortezomib molecules existing exclusively in the solid state. We discovered and described in detail the unique, previously unreported boroxine structures in bortezomib polymorphs. We identified the formation of boroxine motifs in crystalline as well as amorphous forms, although the local geometries around these boroxine rings differed considerably due to intramolecular interactions with carbonyl groups. This contribution thus describes the previously unknown boroxine structures with secondary coordination and discloses the nature of polymorphism of bortezomib. In future studies we hope to extend the proposed methodological approach by complete simulation of 2D 11B–11B PDSD NMR spectra, and by applying the entire concept of powder NMR crystallography54–56 to predict the complete crystal structures of all polymorphic forms of bortezomib. The agreement between our preliminary analysis of electron powder diffraction data and crystal structure predictions has already confirmed the feasibility of this approach (ESI,† Fig. S38). However, further investigation and structural searches are needed.
Acknowledgements
The authors thank the Czech Science Foundation (grant no. GA14-03636S and GA16-04109S) and the Ministry of Education, Youth and Sports of the CR within the National Sustainability Program I (NPU I), Project LO1507 POLYMAT, for their financial support. Computational resources were partially provided under program LM2010005, and in the Centre CERIT Scientific Cloud, part of the Operational Program Research and Development for Innovations, reg. no. CZ.1.05/3.2.00/08.0144.Notes and references
- S. J. Baker, C. Z. Ding, T. Akama, Y. K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed.
- R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS PubMed.
- S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC.
- L. R. Dick and P. E. Fleming, Drug Discovery Today, 2010, 15, 243–249 CrossRef CAS PubMed.
- N. Iwasawa and H. Takahagi, J. Am. Chem. Soc., 2007, 129, 7754–7755 CrossRef CAS PubMed.
- R. Nishiyabu, Y. Kubo, T. D. James and J. S. Fossey, Chem. Commun., 2011, 47, 1124–1150 RSC.
- N. Nishimura, K. Yoza and K. Kobayashi, J. Am. Chem. Soc., 2010, 132, 777–790 CrossRef CAS PubMed.
- K. Ono, K. Johmoto, N. Yasuda, H. Uekusa, S. Fujii, M. Kiguchi and N. Iwasawa, J. Am. Chem. Soc., 2015, 137, 7015–7018 CrossRef CAS PubMed.
- W. J. Niu, M. D. Smith and J. J. Lavigne, J. Am. Chem. Soc., 2006, 128, 16466–16467 CrossRef CAS PubMed.
- G. M. Peters, L. P. Skala, T. N. Plank, B. J. Hyman, G. N. M. Reddy, A. Marsh, S. P. Brown and J. T. Davis, J. Am. Chem. Soc., 2014, 136, 12596–12599 CrossRef CAS PubMed.
- G. M. Peters, L. P. Skala, T. N. Plank, H. Oh, G. N. M. Reddy, A. Marsh, S. P. Brown, S. R. Raghavan and J. T. Davis, J. Am. Chem. Soc., 2015, 137, 5819–5827 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- M. K. Smith and B. H. Northrop, Chem. Mater., 2014, 26, 3781–3795 CrossRef CAS.
- J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304–311 CrossRef CAS PubMed.
- B. Stadelmann, D. Aeschbacher, C. Huber, M. Spiliotis, J. Muller and A. Hemphill, PLoS Neglected Trop. Dis., 2014, 8, e3352 CrossRef PubMed.
- R. V. Palle, R. Kadaboina, V. Murki, A. Manda, N. Gunda, R. R. Pulla, M. Hanmanthu, N. N. Mopidevi and S. K. Ramdoss, US20100226597A1, 2010.
- M. Janca and P. Dobrovolny, WO2009004350A1, 2009.
- N. D. Feng, Q. Wang, A. M. Zheng, Z. F. Zhang, J. Fan, S. B. Liu, J. P. Amoureux and F. Deng, J. Am. Chem. Soc., 2013, 135, 1607–1616 CrossRef CAS PubMed.
- P. Matejicek, J. Brus, A. Jigounov, J. Plestil, M. Uchman, K. Prochazka and M. Gradzielski, Macromolecules, 2011, 44, 3847–3855 CrossRef CAS.
- J. Brus, A. Zhigunov, J. Czernek, L. Kobera, M. Uchman and P. Matejicek, Macromolecules, 2014, 47, 6343–6354 CrossRef CAS.
- F. A. Perras and D. L. Bryce, J. Chem. Phys., 2013, 138, 174202 CrossRef PubMed.
- A. Brinkmann and M. Eden, Phys. Chem. Chem. Phys., 2014, 16, 7037–7050 RSC.
- F. Angeli, O. Villain, S. Schuller, T. Charpentier, D. de Ligny, L. Bressel and L. Wondraczek, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 054110 CrossRef.
- F. G. Vogt, G. R. Williams and R. C. B. Copley, J. Pharm. Sci., 2013, 102, 3705–3716 CrossRef CAS PubMed.
- S. Sene, S. Begu, C. Gervais, G. Renaudin, A. Mesbah, M. E. Smith, P. H. Mutin, A. van der Lee, J. M. Nedelec, C. Bonhomme and D. Laurencin, Chem. Mater., 2015, 27, 1242–1254 CrossRef CAS.
- L. Kobera, J. Czernek, M. Streckova, M. Urbanova, S. Abbrent and J. Brus, Macromolecules, 2015, 48, 4874–4881 CrossRef CAS.
- P. V. Wiper, J. Amelse and L. Mafra, J. Catal., 2014, 316, 240–250 CrossRef CAS.
- A. Hough, A. F. Routh, S. M. Clarke, P. V. Wiper, J. A. Amelse and L. Mafra, J. Catal., 2016, 334, 14–22 CrossRef CAS.
- B. J. vanRossum, H. Forster and H. J. M. deGroot, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- N. M. Szeverenyi, M. J. Sullivan and G. E. Maciel, J. Magn. Reson., 1982, 47, 462–475 CAS.
- J. P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116–118 CrossRef CAS PubMed.
- I. Schnell, S. P. Brown, H. Y. Low, H. Ishida and H. W. Spiess, J. Am. Chem. Soc., 1998, 120, 11784–11795 CrossRef CAS.
- S. P. Brown and H. W. Spiess, Chem. Rev., 2001, 101, 4125–4155 CrossRef CAS PubMed.
- M. Eden, D. Zhou and J. H. Yu, Chem. Phys. Lett., 2006, 431, 397–403 CrossRef CAS.
- Q. Wang, B. Hu, O. Lafon, J. Trebosc, F. Deng and J. P. Amoureux, J. Magn. Reson., 2009, 200, 251–260 CrossRef CAS PubMed.
- B. Langer, L. Schnell, H. W. Spiess and A. R. Grimmer, J. Magn. Reson., 1999, 138, 182–186 CrossRef CAS PubMed.
- J. Brus, Solid State Nucl. Magn. Reson., 2000, 16, 151–160 CrossRef CAS PubMed.
- M. R. Hansen, G. K. H. Madsen, H. J. Jakobsen and J. Skibsted, J. Phys. Chem. A, 2005, 109, 1989–1997 CrossRef CAS PubMed.
- J. W. E. Weiss and D. L. Bryce, J. Phys. Chem. A, 2010, 114, 5119–5131 CrossRef CAS PubMed.
- S. W. Oh, J. W. E. Weiss, P. A. Kerneghan, I. Korobkov, K. E. Maly and D. L. Bryce, Magn. Reson. Chem., 2012, 50, 388–401 CrossRef CAS PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- A. Zalkin, T. E. Hopkins and D. H. Templeton, Inorg. Chem., 1967, 6, 1911–1915 CrossRef CAS.
- G. J. Gainsford, T. Kemmitt and C. Higham, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, I24–I25 CrossRef CAS PubMed.
- M. Gajhede, S. Larsen and S. Rettrup, Acta Crystallogr., Sect. B: Struct. Sci., 1986, 42, 545–552 CrossRef.
- M. Eden and L. Frydman, J. Phys. Chem. B, 2003, 107, 14598–14611 CrossRef CAS.
- P. Hartmann, C. Jager and J. W. Zwanziger, Solid State Nucl. Magn. Reson., 1999, 13, 245–254 CrossRef CAS PubMed.
- G. Mali, V. Kaucic and F. Taulelle, J. Chem. Phys., 2008, 128., 204503 CrossRef PubMed.
- G. Mali and F. Taulelle, Chem. Commun., 2004, 868–869 RSC.
- F. A. Perras and D. L. Bryce, J. Am. Chem. Soc., 2013, 135, 12596–12599 CrossRef CAS PubMed.
- F. A. Perras and D. L. Bryce, Chem. Sci., 2014, 5, 2428–2437 RSC.
- N. S. Barrow, J. R. Yates, S. A. Feller, D. Holland, S. E. Ashbrook, P. Hodgkinson and S. P. Brown, Phys. Chem. Chem. Phys., 2011, 13, 5778–5789 RSC.
- E. Salager, G. M. Day, R. S. Stein, C. J. Pickard, B. Elena and L. Emsley, J. Am. Chem. Soc., 2010, 132, 2564–2566 CrossRef CAS PubMed.
- M. Baias, C. M. Widdifield, J. N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein, A. Lesage, G. M. Day and L. Emsley, Phys. Chem. Chem. Phys., 2013, 15, 8069–8080 RSC.
- J. Leclaire, G. Poisson, F. Ziarelli, G. Pepe, F. Fotiadu, F. M. Paruzzo, A. J. Rossini, J. N. Dumez, B. Elena-Herrmann and L. Emsley, Chem. Sci., 2016, 7, 4379–4390 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Parameters of solid-state NMR experiments; validation of 2D 11B–11B correlation experiments; 2D 11B–11B correlation spectra of bortezomib; 1H–13C FSLG HETCOR NMR spectra of bortezomib; spectroscopic data for amorphous bortezomib; and preliminary electron powder diffraction data. See DOI: 10.1039/c6cp06555d |
| This journal is © the Owner Societies 2017 |