
Trapping of gaseous pollutants on defective N-doped graphene†
Dibyajyoti
Ghosh
*a and
Swapan K.
Pati
*b
aChemistry and Physics of Materials Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore 560064, India. E-mail: dibyajnc@gmail.com
bTheoretical Sciences Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore 560064, India. E-mail: pati@jncasr.ac.in
First published on 22nd November 2016
Abstract
The use of pristine as well as structurally modified two-dimensional materials to efficiently trap and separate various harmful gases from the atmosphere is an intensively explored field of current research. In this paper, we have computationally demonstrated the efficient trapping of several polar poisonous and greenhouse gases on top of different kinds of pyridinic/pyrrolic nitrogen-doped graphene sheets. van der Waals corrected Born–Oppenheimer molecular dynamics (BOMD) studies evidently demonstrate the trapping of the gas molecules on top of the defect-sites of the sheets at room-temperature. Importantly, the trapping of molecules does not lead to any chemical bond formation between the adsorbates and the adsorbents. Thorough investigations evidently demonstrate the formation of hydrogen-bonds between nitrogen of the adsorbents and hydrogen of the adsorbates. Furthermore, interestingly, as most abundant atmospheric gases (i.e. N2 and O2) get repealed by the defect-sites, these sheets appear to be efficient to selectively trap the pollutants from the open atmosphere. We also suggest different ways to enhance the gas-trapping capacity of these sheets, such as increasing the defect concentration, and adsorbing pollutants on both sides of the free-standing defective sheets. Finally, thermal treatment of these sheets at high temperature is demonstrated as an efficient way to recycle the adsorbent.
1 Introduction
Emission of toxic and greenhouse gases due to various industrial and domestic usages, creates alarming air-pollution and is a major concern now-a-days.1,2 These hazardous gases affect acutely all the living species as well as the environment in various direct and/or indirect ways. A number of porous materials, such as, zeolites, metal–organic frameworks etc. have been explored for their possible promising usage as gas-sensors, adsorbents, catalytically active pollutant-degrading agents etc.3,4 However, to achieve these fascinating properties, one needs to achieve precise control over the pore-size/shape, and chemical nature of the surface in these adsorbent systems. Furthermore, to have recyclable adsorbents, chemical bond formation between adsorbates and adsorbents is undesirable, in general. And considering this point, it appears to be quite challenging to have control over these physical and chemical factors experimentally in the above mentioned adsorbent materials.Graphene, a two dimensional honeycomb lattice of sp2-hybridized carbon, is a quite promising candidate to solve these problems. It has already revolutionized the field of nanoscopic material science due to the emergence of a number of extraordinary properties.5–7 Apart from its several outstanding applications in the fields of electronics,8–10 electrocatalysis,11,12 DNA sensing,13,14 optics,15,16 and energy storage (i.e. batteries),17–19 graphene has been demonstrated as a highly efficient adsorbent for gas-storage, separation, capturing and sensing.20–24 Importantly, the theoretically measured surface area of graphene is 2630 m2 g−1, which is quite high and useful to store a large amount of gases.25 However, the low binding energies between pristine graphene and the adsorbate molecules severely affect the adsorption processes of the gas molecules in these sheets at room temperature. For example, Leenaerts et al. have demonstrated a small amount of charge transfer between different gas molecules and pristine graphene, resulting in very weak interactions between them.26 To increase the binding strength between the adsorbates and graphene, different chemical and structural modifications of the adsorbent have been proposed.27,28 Recently, experimental as well as theoretical studies have pointed out that substitutional doping in graphene can be a promising way in this regard.29,30 The inclusion of different kinds of p-type dopants, such as, boron (B), aluminium (Al) and n-type dopants, like, oxygen (O), nitrogen (N), sulphur (S) etc. enhances the adsorption capacity, and the binding strength of gases on the graphene surface by several orders of magnitude.27,31 Experimentally, Parambhath et al. have shown that pyrrolic-N dominant graphene increases H2 storage capacity by 66% compared to pristine graphene.32 Using first-principle calculations, Dai et al. have demonstrated high binding affinities of B-, Al-doped graphene towards various pollutants such as NO, NO2, SO2etc.31 Apart from these chemical doping studies, the incorporation of structural defects, such as point defects, in pristine graphene can also result in better performance towards gas adsorption due to charge accumulation at the defect sites.27,33 Combining these two potential factors, as indicated in earlier work, one can predict efficient adsorption of various gas molecules on different types of N-doped vacancies in graphene sheets.34 Interestingly, these nitrogen-doped defective sheets are already in the limelight of research because of their better activity than pristine graphene in the fields of electrocatalysis, field-effect transistors, lithium ion batteries, supercapacitors, biosensing etc.35 Moreover, recent experimental as well as theoretical studies have demonstrated strong trapping of single metal atoms in these defect sites.36,37 However, the potential application of these monolayers towards adsorptive removal of toxic and greenhouse gases remains quite unexplored. Interestingly, in their recent study, Zuo et al. have shown enhanced adsorption of different organic contaminants on N-doped multiwalled carbon nanotubes.38 This combined experimental and theoretical study have shown a 2–10 folds increase for adsorption of 2-naphthol and 1-naphthalmine in N-doped carbon nanotubes.
Considering all these factors, in the present study, we have explored the commonly found nitrogen-doped graphene systems, i.e. pyridinic graphene, for their potential use as adsorbents for adsorbing various poisonous gases, such as hydrogen fluoride (HF) and hydrogen cyanide (HCN), and greenhouse gases like, chlorodifluoromethane (CHClF2) and trifluoromethane (CHF3), which have high potential to adsorb heat. Using Born–Oppenheimer molecular dynamics (BOMD) simulations, we demonstrate that these gas molecules get adsorbed specifically on the defect-sites at room-temperature. Electrostatic interactions between the nitrogen atoms of the defect-sites and the hydrogen atoms of the adsorbent molecules results in stability of the adsorbate–adsorbent interactions. Detailed analyses show that there is no chemical bond formation/breakage in these adsorbate–absorbent systems, suggesting a strong physisorption of the gases on defects. Density functional theory (DFT) based calculations further helped us to gain an atomistic understanding about the adsorbate–adsorbent interaction in these systems. Geometric signature, considerable high binding energies and absence of effective charge transfer strongly indicate the formation of moderately strong hydrogen-bonds in these nano-systems. Further, different important practical issues such as selective adsorption, ways to increase the capacity to trap gas molecules, and recyclability of the adsorbents have also been explored thoroughly.
2 Computational details
For the present simulations, we perform BOMD calculations using the QUICKSTEP module as provided by the CP2K code.39 The CP2K code has implemented a mixed basis set where the Kohn–Sham orbitals are expanded in an atom-centered Gaussian basis set and the electronic charge density is described using an auxiliary plane wave basis set.40 We have used an energy cut-off of 280 Ry to describe the plane wave charge density. Further, analytical dual-space pseudopotential, as implemented by Goedecker, Teter, and Hutter (GTH), are used to represent the electron–ion interactions for all the elements.41 The valence electrons are modeled by double-ζ basis set with one set of polarization functions i.e. DZVP basis set.42 Generalized gradient approximation (GGA) with the form of Perdew–Burke–Ernzerhof (PBE) is used to capture exchange and correlation effects of electrons.43 Additionally, as the noncovalent interactions play an important role in our present study, we apply empirical van der Waals interactions, as prescribed by Grimme (DFT-D2) for all the BOMD calculations.44 To study the interactions between adsorbate and adsorbents in ambient condition, we perform BOMD simulation considering canonical (NVT) ensemble. We apply a Nose–Hoover thermostat to perform the constant temperature simulation.45,46 To integrate the equations of motion, we use the time step of 0.5 femtosecond (fs). To avoid the spurious interaction of nonperiodic direction, we consider the simulation box, containing 30 Å vacuum space in that direction. We generate 15 picosecond (ps) trajectories for all adsorbate–adsorbent systems. As all the systems attain the equilibrium within 10 ps, we take the trajectories of the last 5 ps run for further analysis.For electronic structure calculations, we use the accurate frozen-core full-potential projector augmented-wave (PAW) based method, as incorporated in the Vienna ab initio simulation package (VASP).47 Like BOMD simulations, here also we used GGA-PBE functionals for density functional theory based calculations. A plane-wave kinetic energy cutoff of 400 eV is used for all the DFT calculations. To obtain the ground state geometry at 0 K temperature, we relax the adsorbate–adsorbent systems using the conjugate-gradient method. We sample the full Brillouin zone by using Γ-centred Monkhorst–Pack scheme.48 For geometry optimization and accurate binding energy calculations, we use 5 × 5 × 1 and 9 × 9 × 1 k-points grids, respectively. The structural minimization of these systems are performed until the interatomic forces acting there become less than 0.02 eV Å−1. To incorporate van der Waals corrections, we use Grimme's DFT-D2 method here also. We further calculate the charge transfer between adsorbates and adsorbents using the method described by Bader.49
The binding energy of the gas molecules on the adsorbate is calculated using following formula,
| Ebind = Esheet–molecule − Esheet − Emolecule | (1) |
2.1 Structural models
To simulate the adsorbent, we consider a 7 × 7 supercell of two-atom rhombic unit cell of graphene. In pristine form, this supercell contains 98 atoms of carbon. To introduce N-doped point defects in this sheet, we modify the pristine graphene accordingly. The defective graphene-based adsorbents which we have explored majorly in our study, are shown in Fig. 1(a–d). These sheets where single (Fig. 1(a and b)) and double (Fig. 1(c and d)) carbon atoms are missing (i.e. SV and DV), are named as SV-3N and DV-4N, respectively. More details about the adsorbents are given in the ESI.† The concentrations of N-doping in the SV-3N and DV-4N appear as 3.09% and 4.17%, respectively. We also increase the doping concentration of N, as stated in the later part of the paper. As experimentally, pyrrolic and graphitic nitrogen-doped cavities also appear quite frequently, we have also performed AIMD calculations to investigate the gas-trapping on these defective sheets (see Fig. S1 in the ESI†). | ||
| Fig. 1 Snapshots of (a and c) top and (b and d) side view of equilibrated structures of SV-3N and DV-4N sheets after BOMD simulations at constant temperature (300 K). Grey and blue colored balls depicts carbon and nitrogen, respectively. | ||
3 Results and discussion
To begin with, we discuss about the stability of the single and double vacancies (SV and DV, respectively) at finite temperatures. Performing constant temperature (i.e. at 300 K) BOMD studies, defect-sites at both SV-3N and DV-4N sheets appear to be stable, without having any major structural reconstruction at equilibrium. As shown in Fig. 1(a–d), apart from the well known rippling, the equilibrated sheets are majorly planar which is in agreement with the previous experimental observation by Wang et al.50 and computational study by Yang-Xin Yu.51 Further details about the structures of these defective sheets at finite temperature are given in the ESI.†Before exploring the pollutant gas adsorption on the defective graphene sheets, we study the dynamics of these gas molecules on the pristine graphene sheet at 300 K. BOMD simulations (see Fig. S2(a–d), ESI†) evidently show that HF and CHClF2 molecules remain quite mobile inside the simulation box, without exhibiting any kind of binding interactions with the graphene π-surface. Similar kind of dynamics are also found for other two gas molecules on top of pristine graphene sheet. Fundamentally, highly stable π-electron surface prevents the binding of these polar adsorbates on the pristine graphene. Thus, other weak interactive forces (such as van der Waals interaction) play the major role in the present adsorption processes. It appears that these forces are too weak to bind the adsorbates on the graphene surface at room temperature, resulting in negligible adsorption. Interestingly, a very recent experimental study by Kulkarni et al. demonstrated the fast desorption rate of various polar molecules from pristine graphene at room temperature.52 These findings strongly support the repellent nature of pristine graphene towards adsorption of different small polar molecules at room temperature, explored in the present BOMD simulations.
Next, we investigate the pollutant gas adsorption on the defective graphene sheets by performing BOMD simulations of the adsorbate–adsorbent systems at 300 K. We begin the simulation by placing the adsorbate molecules on top of the N-doped point-defects. BOMD simulations of 15 ps for each adsorbate–adsorbent system evidently demonstrate the gas adsorption on the defective graphene-sheets. As shown in Fig. 2(a–h) and 3(a–h), both the adsorbents i.e. SV-3N and DV-4N prominently trap small polar gas molecules on their defect-sites. Importantly, within the investigated time-scale and temperature, we do not observe any event of desorption of gas molecules from these defect-sites. Further, we focus on the distance between adsorbate and adsorbent systems in equilibrium. To compute this, we calculate the distance between the centre of mass of the gas molecule and that of the defect-sites (considering the position of doped N atoms only) after attaining the equilibrium. As shown in Table 1, the adsorbed molecules remain in the range of 2.24 Å to 3.6 Å apart, from the adsorbents. These distances are quite similar to the other non-covalently bonded graphene–molecule composites as found in different studies.17,28 Note that, for all the equilibrated systems, the H-atoms of the adsorbate molecules remain close to the defect-sites. More specifically, as shown in Fig. 2(a–f) and 3(a–f), these H-atoms are quite directional towards the N atoms of the sheets. Thus, at ambient condition, these N-doped defect-sites trap both kinds of gases i.e. linear as well as tetrahedral shaped molecules.
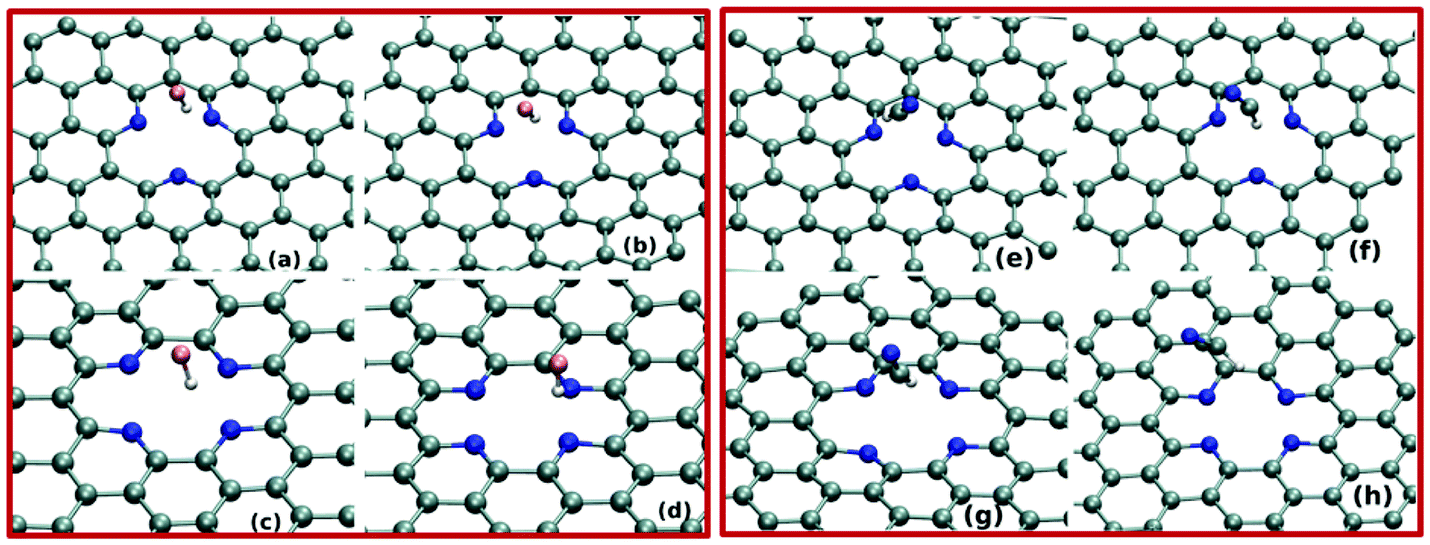 | ||
| Fig. 2 Various snapshots of configurations of HF trapped on the defect-site of SV-3N and DV-4N after (a and c) 10 ps and (b and d) 15 ps BOMD simulation. Snapshots of trapping of HCN on SV-3N and DV-4N after (e and g) 10 ps and (f and h) 15 ps are given. | ||
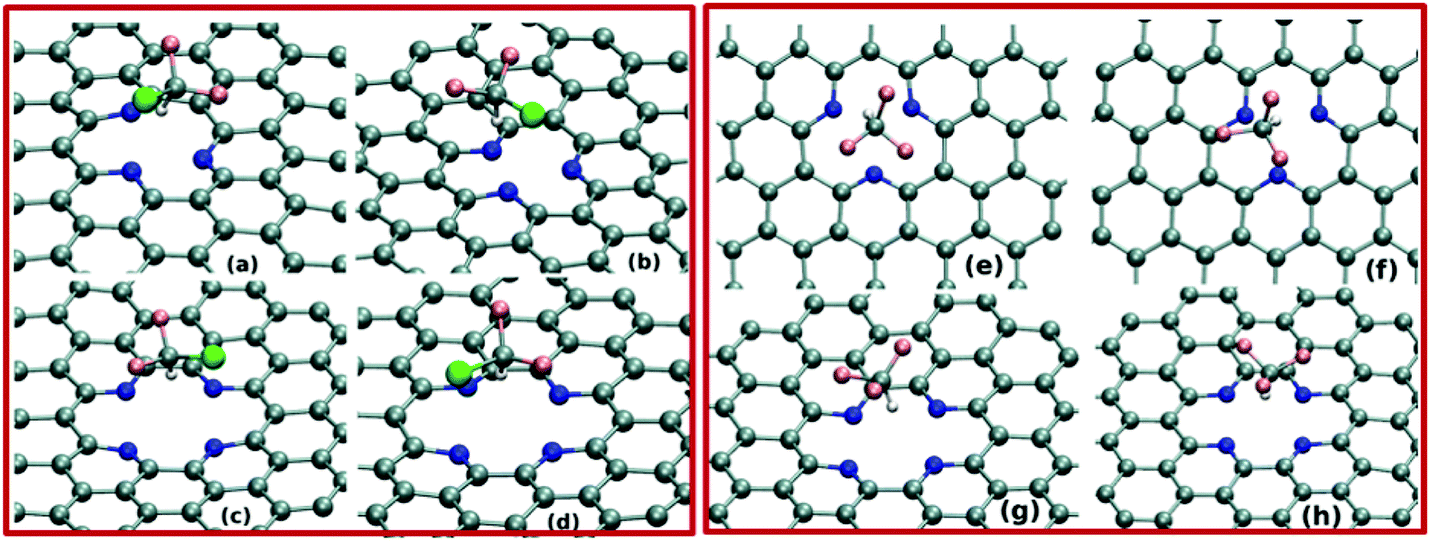 | ||
| Fig. 3 Various snapshots of configurations of CHClF2 trapped on the defect-site of SV-3N and DV-4N after (a and c) 10 ps and (b and d) 15 ps BOMD simulation. Snapshots of trapping of CHF3 on SV-3N and DV-4N after (e and g) 10 ps and (f and h) 15 ps are given. | ||
| Systems | d mol–grap(avg) | d F–H/N–H(avg) | E bind | e tran | d N–H(smallest) | d X–H,opt |
|---|---|---|---|---|---|---|
| HF@SV-3N | 2.4 | 0.97(0.92) | −55.96 | 0.07 | 1.66 | 0.99 |
| HCN@SV-3N | 3.48 | 1.10(1.06) | −51.13 | 0.05 | 2.20 | 1.11 |
| CHClF2@SV-3N | 3.6 | 1.10(1.08) | −31.84 | 0.04 | 2.16 | 1.10 |
| CHF3@SV-3N | 3.06 | 1.10(1.08) | −50.172 | 0.04 | 2.06 | 1.10 |
| HF@DV-4N | 2.24 | 1.00 | −71.40 | 0.05 | 2.00 | 0.97 |
| HCN@DV-4N | 3.15 | 1.13 | −61.35 | 0.04 | 2.31 | 1.10 |
| CHClF2@DV-4N | 3.1 | 1.13 | −63.68 | 0.04 | 2.38 | 1.10 |
| CHF3@DV-4N | 3.19 | 1.14 | −60.79 | 0.04 | 2.34 | 1.09 |
During the synthesis of N-doped graphene, other kinds of defects such as pyrrolic, graphitic N-doped structures also appear quite frequently. Thus, we briefly look into the gas trapping in these two defective adsorbent systems as well. BOMD studies at 300 K reveal that at equilibrium, single vacancy with pyrrolic N-doped defect-sites trap the gas molecules of our interest (see Fig. S3(a and b), ESI†). On the other hand, the graphitic-N sites are not efficient to trap these pollutant gas molecules at ambient condition (see Fig. S3(c and d), ESI†). Thus, it is quite clear that point defects with N-doping is necessary for trapping the polar gases on the graphene surface at ambient condition.
With these promising results, we now focus on the fundamental reasons behind these gas-trapping phenomena. To quantify the adsorbate–adsorbent interaction strength, we perform DFT-based calculations to have optimized geometries of gas-trapped SV-3N and DV-4N structures at 0 K. As shown in Table 1, calculated binding energies from these optimized structures indicate strong adsorption of the polar molecules on both kind of N-doped defective sites. Note that, here we concentrate on the origin of the static interactions, playing major role in these adsorbate–adsorbent systems, by investigating the DFT-based optimized geometries. Important structural information are given in Table 1. Further details of the optimized geometries of these defects with different pollutant gas molecules can be found in the ESI.†
Focusing on binding energies as given in Table 1, all gases bind quite strongly with both SV-3N and DV-4N defect sites with binding energies in the range of −31.84 to −71.40 kJ mol−1 (more information about the binding strength can be found in the ESI†). These strong adsorbate–adsorbent binding affinities evidently support the trapping of these gas molecules on SV/DV defect-sites at 300 K. Note that, calculated binding energies are quite high and fall in the range between weak chemisorption and strong physisorption phenomena. Thus, we look back into the dynamics of BOMD equilibrated adsorbate–adsorbent systems carefully to confirm the sorption process. The equilibrium bond lengths (i.e. for 10–15 ps simulations) of F–H for HF and C–H for other gases, as shown in Fig. 4(a–d), demonstrate little fluctuating behaviour when these molecules are trapped on the SV-3N defect-sites. A similar kind of fluctuations also appear for molecules adsorbed on DV-4N sites. These fluctuations in bond lengths are quite common due to the presence of vibration of molecules at finite temperatures. Note that, fluctuation amplitudes of the bond-length in all the systems are in the range of 0.12 to 0.34 Å which is much smaller than their corresponding F–H or C–H bond lengths. Further, as given in Table 1, time-average of F–H or C–H bond lengths in each equilibrated system do not show any major deviation from the corresponding gas-phase molecular bond length values (given in Table 1). All these phenomena strongly indicate that in the present cases, the gas molecules remain in their molecular form without undergoing any kinds of bond breaking/making in the presence of the N-doped defect-sites.
 | ||
| Fig. 4 The bond length of F–H/C–H in the adsorbate molecules, adsorbed on top of SV-3N, during the BOMD simulation for 10–15 ps time-scale. The adsorbate molecule is given in the plot. | ||
To find the microscopic origin of the strong adsorbate–adsorbent interaction in these systems, we first look into the charge distribution in the adsorbates and adsorbents, considering their corresponding isolated structures. Here, all these isolated gas molecules contain partially positively charged H atoms. Consequently, these molecules are strongly polar in nature and contain considerable permanent electric dipole moments of 1.82 D, 2.97 D, 1.38 D, 1.62 D for HF, HCN, CHClF2 and CHF3, respectively. Quite obviously, the dipole moment of all these molecules remains directed along the F–H/C–H bond, which is mostly polar in nature. The presence of strong electronegative atoms, such as N, F, Cl, in the molecules is responsible for the polar nature of these adsorbates.
Further, as shown in Fig. 5(a and b), the charge distributions in the adsorbents demonstrate accumulation of negative charges at the defect-sites. The pyridinic nitrogen atoms are sp2-hybridized in nature where two of these orbitals get involved to form C–N σ-bonds, leaving the other one as an in-plane dangling sp2-orbital. These orbitals which are in-plane of the sheet and directed towards the centre of the SV/DV, remain doubly occupied. Importantly, as each N atom has one singly occupied 2pz orbital to enable its participation in π-space aromatic structures, no charge transfer occurs from the lone pair of the sp2-orbital to the π-space. Also the electron distribution plots evidently show the strongly localized charge accumulation at the SV/DV sites of these sheets. Thus, these lone-pairs of electrons of nitrogen make the defect-sites electron-rich in nature and consequently provide suitable pockets for trapping the polar gases.
 | ||
| Fig. 5 The charge density distribution at the defect-site of (a) SV-3N and (b) DV-4N sheets. The isosurface is 0.25 e Å−3. The grey colored surface demonstrates the electron density. | ||
Now, focusing on the adsorbent–adsorbate systems, as shown in Table 1, the calculated charge transfer between the gas molecules and graphene sheets appears to be quite small for all the studied systems. It directly indicates that the partial positive charge at H-atoms of the molecules and the partial negative charge at the N-atoms of SV/DV remain strongly localized at their corresponding atomic sites. Moreover, as shown in Table 1, the optimized distance between these polarized atoms i.e. H and N, remains quite larger than their single bond length, indicating no direct overlaps of electron densities between adsorbate and adsorbent systems. Therefore, non-covalent electrostatic interactions between the gas molecules and defect-sites govern the adsorption process. Most importantly, the large partial positive charge in the H atom of the gas molecules and the accumulated negative charge at the defects illustrates the formation of moderately strong H-bonding at the adsorption-site.53 Here, F–H/C–H bonds of adsorbate and N atoms of defect-sites act as proton donor and acceptor, respectively.
To confirm the H-bond formation, we focus on various parameters, which reliably provide the signatures of the presence of H-bonding. First, we consider the sum of van der Waals radii of H and N which amounts to 2.75 Å. As the smallest distances between H and the nearest N (see Table 1) are smaller than 2.75 Å in all the adsorbate–adsorbent systems, the presence of non-covalent bonding interaction in these systems is quite evident. Further, it is quite well known that a major structural feature for H-bonded systems is the formation of angles greater than 130° between the donor and the acceptor moieties.53 As shown in optimized (see ESI†) as well as equilibrated (by BOMD simulation; Fig. 3) geometries of adsorbate–adsorbent systems, the ∠N⋯H–F/∠N⋯H–C (for other molecules apart from HF) angles remain in the range of 150°–180°. Thus, the formation of nearly linear angles between donors and acceptors strongly supports the existence of H-bonding in the presently studied systems. Moreover, we compare the bond lengths of F–H/C–H with N⋯H to find the strength of the H-bonds here. As Table 1 depicts, the smallest distance between N and H atoms in optimized geometries i.e. dsmallest are in the range of 1.66 to 2.38 Å which are quite larger than the donor i.e. F–H/C–H bond lengths i.e. dF–H/N–H,opt. Thus, according to the well accepted guidelines prescribed by Steiner,53 the H-bonds are moderately strong in these present systems. The electrostatic nature of the adsorbate–adsorbent interactions and the calculated binding energies further support the formation of a moderately strong H-bond here.
Interestingly, performing BOMD simulations of these systems at 300 K temperature, we find that gas molecules remain quite mobile on the SV and DV sites without staying to any one particular N atom. The H atom of the molecules stay nearer to any one of the three or four equivalent N atoms at a particular instant of simulation time. And during the simulations, the H atom as well as the corresponding gas molecule changes its position by changing the interacting N atom-site over time. Thus, due to thermal fluctuations at finite temperatures, the molecules remain trapped on top of the N-doped defect-sites but with local motions. Further, these motions indicate that the gas molecules are not chemically bonded to the N atoms of the defect-sites.
Selective adsorption of the pollutant gases in the open atmosphere is one of the major criteria for the adsorbents to be used in efficient practical applications. Thus, we investigate the adsorption of most abundant gases of the atmosphere i.e. N2 and O2 on the SV-3N and DV-4N sites of N-doped graphene at ambient condition. BOMD simulations at 300 K demonstrate strong repulsion between defect-sites and N2 and O2 molecules (see Fig. S4(a–d), ESI†). And consequently, these non-polar gas molecules do not get adsorbed on the SV/DV sites. Performing DFT-based calculations, we find positive binding energies of 5.61 and 6.65 kJ mol−1 for N2 and O2 adsorbed 3N-SV defect. These also evidently demonstrate the reluctance of the electron-rich defect-sites towards trapping the non-polar gases. Fundamentally, the high electron densities of the adsorbates and adsorbents results in the strong repulsion among them. Thus, the present adsorbent systems are quite selective towards polar gas molecules.
In the following section we focus on the strategy to increase the gas adsorption capacity on these sheets. Two different approaches are explored in this regard: (1) increasing the defect concentration on the adsorbents; (2) adsorbing more gas molecules on the defect sites. Concentrating on the first strategy, we stepwise increase the defect concentration in the adsorbent. Note that, several recent experiments have already demonstrated much higher N-doping concentration in graphene.54–56 Thus, this presently considered sheet is quite realistic for modeling the gas-trapping on defective graphene sheets.
BOMD simulation at 300 K evidently depicts the stability of this highly defective N-doped sheet in the equilibrium. To check the dynamics of the gas molecules on these highly defective sheets, we introduce HF atoms on top of the defect-sites. As shown in Fig. 6(a), the equilibrated structures clearly show the trapping of HF molecule on each defect-sites. Thus, trapping of gas molecules does not get affected due to the presence of other defect-sites with adsorbed molecules even if they remain quite close to each other (∼9.8 Å for present case). Further, we also perform simulation, taking more bulky tetrahedral molecules i.e. CHClF2 as the adsorbate. As shown in Fig. 6(b), a similar kind of dynamics i.e. strong trapping of gases on each defect site is found here also. Thus, as shown in these simulations, one can increase the gas-adsorption capacity of these defective sheets by increasing the N-doped defect-concentration on the adsorbents.
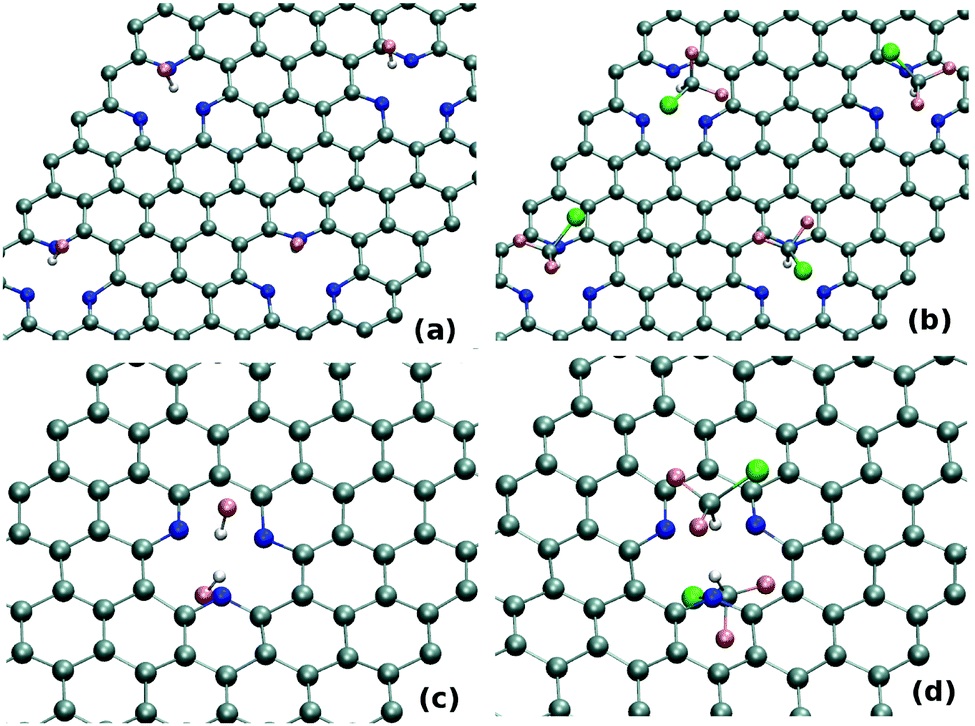 | ||
| Fig. 6 Snapshots of configurations of (a) HF and (b) CHClF2 on concentrated defective sheets with SV-3N after 10 ps BOMD simulation. Snapshots of trapping of (c) HF and (d) CHClF2 on both sides of 3N-SV after 10 ps BOMD simulation. | ||
Exploring the second approach, we perform the BOMD simulation by initially keeping two HF molecules on the same as well as different sides of 3N-SV. Same sided HF molecules does not increase the trapping capacity as only one gas molecule remains adsorbed (the other one gets desorbed due to steric hindrance). However, as shown in Fig. 6(c), we find that both the HF molecules in the opposite side of defect remain trapped at equilibrium. The same kind of BOMD study with CHClF2 also reveals the trapping of this gas molecules in both sides of 3N-SV (see Fig. 6(d)). BOMD calculations also demonstrate that the trapping of gas molecules on both side of the defect happens for other defect-sites as well as for other gas molecules. Thus, in these sheets, the presence of one trapped molecule does not affect the trapping of another molecule on the other side.
Combining these two approaches, we further study the trapping of these gas molecules on both sides of defect-densed sheets. BOMD simulations at ambient condition demonstrate the trapping of the gases on the defect-sites. Details of these have been provided in the ESI.† Thus, one can trap more hazardous gases on sheets by flowing the polluted air through the highly defective free-standing N-doped graphene sheets.
As the gas molecules get physisorbed on the adsorbents, there are many ways to desorb these adsorbates and efficiently recycle the sheets. Just to have a qualitative picture of the desorption process, we simulate the adsorbate trapped adsorbent at quite high temperature (i.e. more than 600 K). Interestingly, the BOMD study of the pollutants trapped SV-3N as well as DV-4N sheets shows that the gas molecules desorb from the sheets at the high temperatures, keeping the adsorbents quite stable structurally. A snapshot of the BOMD simulations of HF desorption can be found in the ESI.† Note that, a quantitative and thorough desorption study needs very large time-scale simulation and consideration of various other factors which are beyond the scope of our present study.
4 Conclusions
To conclude, in this study, we demonstrate the efficient trapping of different poisonous and greenhouse gases on the different pyridinic N containing graphene sheets. BOMD simulations show that polar gases, such as HF, HCN, CHClF2 and CHF3, get trapped on the defect-site at 300 K temperature. Moderately strong H-bond formation between the N atoms of the defect-sites and the H atoms of the adsorbate molecules results in this stable adsorption phenomena. Importantly, these defect-sites, which strongly repel the non-polar N2 and O2 gases, appear to be selective towards the polar gas molecules. Further, increasing the defect concentration of the N-doped defects and flowing the polluted air on both sides of the free-standing graphene sheets, we find an increase in the gas adsorption capacity of these adsorbents. With all this evidence, we strongly believe that N-doped defective graphene can emerge as an efficient adsorbent for various polar pollutant gas molecules and consequently can be quite promising to reduce air pollution.References
- D. A. Lashof and D. R. Ahuja, Nature, 1990, 344, 529–531 CrossRef CAS.
- C. Snyder, T. Bruulsema, T. Jensen and P. Fixen, Agric. Ecosyst. Environ., 2009, 133, 247–266 CrossRef CAS.
- J.-R. Li, Y. Tao, Q. Yu, X.-H. Bu, H. Sakamoto and S. Kitagawa, Chem. – Eur. J., 2008, 14, 2771–2776 CrossRef CAS PubMed.
- E. Barea, C. Montoro and J. A. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- K. Novoselov, A. K. Geim, S. Morozov, D. Jiang, M. Katsnelson, I. Grigorieva, S. Dubonos and A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J.-C. Idrobo, S. J. Pennycook and H. Dai, Nat. Nanotechnol., 2012, 7, 394–400 CrossRef CAS PubMed.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., 2009, 121, 4879–4881 CrossRef.
- S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, Adv. Funct. Mater., 2010, 20, 453–459 CrossRef CAS.
- A. Vakil and N. Engheta, Science, 2011, 332, 1291–1294 CrossRef CAS PubMed.
- A. Grigorenko, M. Polini and K. Novoselov, Nat. Photonics, 2012, 6, 749–758 CrossRef CAS.
- Y.-X. Yu, ACS Appl. Mater. Interfaces, 2014, 6, 16267–16275 CAS.
- D. Das, S. Kim, K.-R. Lee and A. K. Singh, Phys. Chem. Chem. Phys., 2013, 15, 15128–15134 RSC.
- Y.-X. Yu, J. Mater. Chem. A, 2013, 1, 13559–13566 CAS.
- F. Schedin, A. Geim, S. Morozov, E. Hill, P. Blake, M. Katsnelson and K. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- D.-E. Jiang, V. R. Cooper and S. Dai, Nano Lett., 2009, 9, 4019–4024 CrossRef CAS PubMed.
- H. W. Kim, H. W. Yoon, S.-M. Yoon, B. M. Yoo, B. K. Ahn, Y. H. Cho, H. J. Shin, H. Yang, U. Paik and S. Kwon, et al. , Science, 2013, 342, 91–95 CrossRef CAS PubMed.
- S. Blankenburg, M. Bieri, R. Fasel, K. Müllen, C. A. Pignedoli and D. Passerone, Small, 2010, 6, 2266–2271 CrossRef CAS PubMed.
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149–3171 RSC.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- O. Leenaerts, B. Partoens and F. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125416 CrossRef.
- Y.-H. Zhang, Y.-B. Chen, K.-G. Zhou, C.-H. Liu, J. Zeng, H.-L. Zhang and Y. Peng, Nanotechnology, 2009, 20, 185504 CrossRef PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- R. Gholizadeh and Y.-X. Yu, J. Phys. Chem. C, 2014, 118, 28274–28282 CAS.
- R. Gholizadeh and Y.-X. Yu, Appl. Surf. Sci., 2015, 357, 1187–1195 CrossRef CAS.
- J. Dai, J. Yuan and P. Giannozzi, Appl. Phys. Lett., 2009, 95, 232105 CrossRef.
- V. B. Parambhath, R. Nagar and S. Ramaprabhu, Langmuir, 2012, 28, 7826–7833 CrossRef CAS PubMed.
- D. Dutta, B. C. Wood, S. Y. Bhide, K. G. Ayappa and S. Narasimhan, J. Phys. Chem. C, 2014, 118, 7741–7750 CAS.
- D. Ghosh, G. Periyasamy and S. K. Pati, J. Phys. Chem. C, 2013, 117, 21700–21705 CAS.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- A. W. Robertson, B. Montanari, K. He, J. Kim, C. S. Allen, Y. A. Wu, J. Olivier, J. Neethling, N. Harrison and A. I. Kirkland, et al. , Nano Lett., 2013, 13, 1468–1475 CrossRef CAS PubMed.
- A. Krasheninnikov, P. Lehtinen, A. S. Foster, P. Pyykkö and R. M. Nieminen, Phys. Rev. Lett., 2009, 102, 126807 CrossRef CAS PubMed.
- L. Zuo, Y. Guo, X. Li, H. Fu, X. Qu, S. Zheng, C. Gu, D. Zhu and P. J. Alvarez, Environ. Sci. Technol., 2016, 50, 899–905 CrossRef CAS PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS.
- J. VandeVondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103–128 CrossRef CAS.
- S. Goedecker, M. Teter and J. Hutter, Phys. Rev. B: Condens. Matter, 1996, 54, 1703 CrossRef CAS.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef.
- W. G. Hoover, Phys. Rev. A: At., Mol., Opt. Phys., 1985, 31, 1695 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- Y. Wang, Y. Shao, D. W. Matson, J. Li and Y. Lin, ACS Nano, 2010, 4, 1790–1798 CrossRef CAS PubMed.
- Y.-X. Yu, Phys. Chem. Chem. Phys., 2013, 15, 16819–16827 RSC.
- G. S. Kulkarni, K. Reddy, W. Zang, K. Lee, X. Fan and Z. Zhong, Nano Lett., 2016, 16, 695–700 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- Z. Luo, S. Lim, Z. Tian, J. Shang, L. Lai, B. MacDonald, C. Fu, Z. Shen, T. Yu and J. Lin, J. Mater. Chem., 2011, 21, 8038–8044 RSC.
- L.-S. Zhang, X.-Q. Liang, W.-G. Song and Z.-Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055–12059 RSC.
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural details of model, BOMD-equilibrated, DFT-optimized systems, structure of pyrrolic and graphitic nitrogen containing sheets are given. Snapshots of gas molecules on top of pure graphene and on top of pyrrolic and graphitic nitrogen doped systems are shown. See DOI: 10.1039/c6cp06247d |
| This journal is © the Owner Societies 2017 |