
The effect of gme topology on multicomponent adsorption in zeolitic imidazolate frameworks†
Anastasios
Gotzias
 *
*
Institute of Nanoscience and Nanotechnology, NCSR Demokritos, Athens, Greece. E-mail: a.gotzias@inn.demokritos.gr; Tel: +30 2106503408
First published on 29th November 2016
Abstract
We employ a simulation approach to study the adsorption of single, binary and ternary mixtures on eight gme Zeolitic Imidazolate Frameworks (ZIFs) at 298 K. Four adsorbate fluids were considered; carbon dioxide, methane, nitrogen and water. We compute the high pressure adsorption density profiles inside the micropore channels of each crystal. The profiles are compared directly for the different structures and adsorbate components and used to highlight the influence of the imidazolate ligands on pure and competitive adsorption. ZIFs with long ligands reveal an additional, accessible to the fluid space detected for the first time. This is a wedged volume on one direction of the pore walls, shaped thus because the long ligands tilt in order to be connected. We estimate the pressure required for water to become the dominating competing adsorbate within different crystal cavities. The simulated data for CO2 adsorption in ZIF69 strongly correlate with a set of Raman spectroscopy intensity values that correspond to the same adsorbate–adsorbent system.
1 Introduction
Zeolitic Imidazolate Frameworks (ZIFs) are synthesized microporous crystals, composed of tetrahedral fragments with metal and functional imidazolate units. The synthesis of such crystals can be achieved by making suitable substitutions or combinations of imidazolate units to produce materials with different chemical compositions, physical properties and porosities.1 This kind of synthesis control makes ZIFs an interesting class of porous crystallite as it provides a significant opportunity to create “designer adsorbents”, whose properties are optimized for specific industrial separation processes,2 like carbon dioxide capture and sequestration, natural gas purification, hydrogen storage, water desalination and separation of liquid petroleum gases.3–8 A critical advantage of ZIFs over other organic–inorganic frameworks (i.e. MOFs) is that, both in powder and in membrane forms, they exhibit excellent chemical and thermal stability and resistance in humid environments.ZIFs are classified into isoreticular groups comprising frameworks that inherit a particular topology. Almost all metal–imidazolate networks have topologies that have already occurred in natural zeolites and are named after them. gme ZIFs are a class of ZIF materials having the topology of gmelinite zeolite.9 This class lists 8 samples that are synthesized with Zn metal atoms and an equimolar mixture of 2-nitroimidazole (nIm) and another functionalized imidazole linker. The second linker may be either an imidazole (Im) or a benzo-imidazole (bIm) molecule, having different functionalities which affect the chemistry of the crystal surface.10 The molecular lengths of the mixing linkers control the spatial arrangement of metal nodes inside the framework. gme ZIFs have nanocavities with similar sizes and apertures, because of the comparable lengths of the imidazolate ligands used in crystallization. This particular feature of gme ZIF porosity makes these crystals exhibit adsorption and selectivity properties exceeding those of other crystallized adsorbents.11,12
Probably the most representative gme ZIF crystal is ZIF69. It is crystallized using chlorobenzoimidazole (cbIm) as a second linker. In a recent study, Kontos et al. detected the CO2–cbIm coupling using Raman spectroscopy on ZIF69 with different loadings of CO2.13 They performed Raman spectroscopy in a cell chamber that contained the sample under CO2 pressure and gave experimental evidence of fluid–ligand interactions at intermediate bulk pressures. An important effort of molecular simulation is that it contributes in the interpretation of the relevant results and exemplifies many physical and chemical properties of such materials. The adsorption in gme ZIF structures has been extensively studied using modelling methods at various scales.14–24 Babarao et al. studied the adsorption behaviour of CO2, N2 and CH4 in ZIF68 and ZIF69, by blocking groups of pore channels of the model frameworks in order to reproduce adsorption experiments.25 Fluid–ligand interactions have been computed with density functional theory and presented with probability densities, for many probe fluids in most ZIFs having gme, rho and other topologies.26–31 In most cases it is essential to concern high pressure systems and include crowded pores where packing effects are important specifically when competitive adsorbate components are simultaneously present.32
In nearly all industrial applications of interest the feed stream contains water vapor, which interferes with the mechanisms involved in the adsorption of CO2 and other gases. Water adsorption proceeds with the formation of clusters of water particles which eventually block the entrance of the remaining adsorbate components of the mixture.33,34 The desorption of water has sufficient energy cost and therefore it is crucial to concern water behaviour specifically in order to design regeneration strategies which can cope with water.35
In this work we use the grand canonical (constant volume, chemical potential and temperature) Monte Carlo method to study the adsorption of single and mixture components in eight gme isoreticular ZIFs. We use binary mixtures containing equimolar pair components of CO2, N2 and CH4. We also use ternary adsorbate mixtures, which contain water in trace levels additional to the previous components. One feature of the gme topology is the formation of three types of nano-channels that constitute the porous network. We present high pressure density profiles to describe specifically the adsorption within these channels. The profiles for the eight structures and for the different fluid components are compared directly and the discrepancies are attributed to the different functionalized imidazole molecules used in each framework. The use of density profiles enables us to quantify the adsorption isotherms within the nanocavities associated with the particular topology. Following this approach we display, in a simple way, the pore filling of the water adsorbate component inside the gme ZIF pores.
2 Materials and methods
gme ZIFs are a particular class of synthesized ZIF materials named after the gme topology which was first found in the tiling of the zeolite gmelinite.11 As seen in Fig. 1, they are hexagonal crystal systems, that feature hpr-, gme- and kno-type channels. They are crystallized using an equimolar mixture of nIm and functionalized Im or bIm linkers (linker substitutes). The nIm linkers and the linker substitutes are connected to Zn atoms in a tetrahedral arrangement. The nIm linkers occupy the shared edges between the hexagonal and rectangular rings of the topology network. The linker substitutes occupy the remaining edges. The linker substitutes are either aligned along the directional axis of the channels or directed radially inside the kno cage with their functional groups pointing to the void of the cavity.10 The eight gme ZIF structures are shown in Fig. 2, where the formation of the gme topology can be easily recognised. In Fig. 2 we display the framework carbon atoms and only the atoms on the functionalities inside the kno cavity. The remaining non-carbon atoms are hidden for clarity. It is interesting to note that the lengthier bIm ligands like nbIm of ZIF78 or mbIm of ZIF79, when connected to the backbone of the structure, attain a slightly tilted orientation. This is shown in Fig. 2, where the projected image of the ligand substitutes, on the walls of gme or hpr fragments of ZIF78 and ZIF79, is thicker than that of the respective ligands of the remaining crystals. | ||
| Fig. 1 gme ZIF tiles, structured by hpr, gme and kno fragments. Black edges denote nIm ligands, the remaining edges denote the ligand substituent. | ||
 | ||
| Fig. 2 The structure of 8 gme ZIF crystals, showing the carbon atoms (black) of a framework and the atoms on the functional groups within a kno cavity; Cl (green), O (red), N (blue) and Br (brown). | ||
The different linker substitutes are tabulated in Table 1. In the same table, we list various structural characteristics of gme ZIFs which were computed using the poreblazer code of Sarkisov and Harrison.36 In this case, we used universal force fields (uff) for all atoms. In the surface area computations, we used N2 as a van der Waals sphere with diameter σ = 3.14 Å. The effective diameter listed in Table 1 refers to the largest sphere that can fit inside the particular cavity without overlapping the van der Waals sphere of any atom in the framework. In order to compute the effective diameter of the kno (or gme) cavity we manually inserted massless spheres with σ = 10 Å along the central axis of gme (or kno) cavities. Within this concept we blocked artificially the cavities of a specific type and explored the rest of the pore volume. In the simulations, we treated gme and hpr channels as identical, because in practice the free space confined by the rectangular rings of gme fragments, is insufficient to accommodate adsorbate particles. Therefore the total pore volume of ZIF crystals consists solely of the volumes provided by gme- and kno-type confinements.
| Linker | nIm + | bIm | cbIm | Im | nbIm | mbIm | dclIm | brbIm | cnIm |
|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
|
|
|
| Pore volumeb (cm3 g−1) | 0.484 | 0.391 | 0.756 | 0.303 | 0.363 | 0.465 | 0.334 | 0.568 | |
| Surface areab (m2 g−1) | 930 | 793 | 1730 | 450 | 597 | 991 | 658 | 1308 | |
| kno diameterb (Å) | 10.18 | 7.69 | 14.58 | 6.78 | 8.81 | 12.05 | 7.15 | 11.47 | |
| gme (or hpr) diameterb (Å) | 8.38 | 8.34 | 9.14 | 6.62 | 6 | 9.04 | 8.25 | 8.69 | |





The Lennard-Jones (LJ) parameters for ZIF atoms and adsorbate particles used in the following simulations are reported in the ESI.† We used the refined potentials for the atoms on ZIFs, obtained by Liu and Smit17 and Li et al.28 We used the uff potential parameters for Br atoms on ZIF81. CH4 adsorbate was treated as a single-center LJ molecule. We used TraPPE models for CO2, N2 and O2,37–39 and we used the Jorgensen's TIP3P popular model for the definition of water, due to the low computational cost.40 The partial charge computations were carried out using the Gaussian program, using the CHELPG scheme, the basis set of 6-311G(d,p) and the wB97XD functional. The atomic charges of ZIFs along with the eight Im–Zn–nIm fragments are tabulated in the ESI.† The model structures are given in the MOFomics database and were used without any further geometry optimization.41
Grand Canonical Monte Carlo (GCMC) simulations for single and multicomponent gas adsorption were performed using the multipurpose simulation code (MUSIC).42 All GCMC simulations included 107 steps. The first 40% of the data were skipped to ensure thermal equilibration of the resulting configurations. We implemented random trial moves of adsorbate particles, namely translation, insertion and deletion. We performed rotation for the nonspherical particles and exchanging of particles for binary or ternary mixture adsorption simulations. All types of moves for each adsorbate occurred with equal probability. The atoms on the solid framework were immobile. The simulation cell contained 2 × 2 × 2 unit cells with periodic boundary conditions applied in all three dimensions. We used the minimum image and a spherical cut-off radius of 16 Å for all interacting atoms. We used Ewald summations for the Coulombic interactions, with wide (w = 6.7 Å) Gaussians, and low end cutoffs at 10−10. All intermolecular distances were measured from the centre of their masses (com). We used pre-tabulated potential energy maps to calculate solid–fluid interaction energies. The energy maps were set on a (1 × 1 × 2) unit cell over a grid with an interval of 0.2 Å, probed with a unit charge or the appropriate set of LJ parameters. Each isotherm contained 60 fugacity steps. The simulations for different adsorbate(s)–adsorbent systems were submitted as job arrays in a multi-CPU workstation where each job was executed serially.
The bulk experimental pressure was related to the vapour fugacity required in GCMC simulations, using the Peng–Robinson equation of state (EoS). The excess adsorbed amount, Nex, was calculated from the absolute adsorbed amount Nabs obtained directly from the single component simulations according to: Nex = Nabs − ρbulkVp, where ρbulk is the EoS density of the bulk gas phase and Vp is the pore volume of the ZIF taken from Table 1. The isotherms of excess densities were fitted using the Langmuir adsorption model; nads = ninf × KP/(1 + KP), where nads is the adsorption density, ninf is the isotherm's asymptote, P is the pressure and K is the Langmuir (or the Henry) constant. The isosteric heats of adsorption were calculated using the fluctuations of adsorbate particles during a GCMC run.43 If U is the intermolecular energy and N the number of adsorbed molecules in a configuration, the isosteric heat is given by: 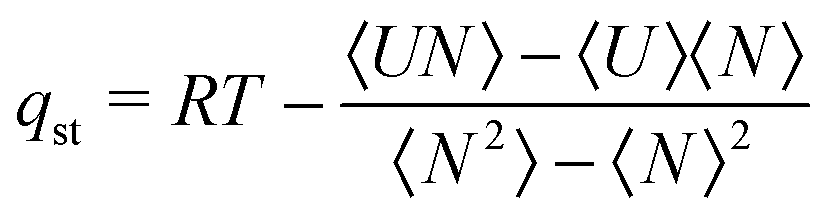 . The angle brackets denote averages in the grand canonical ensemble. In the following section we note the solid–fluid (sf) noncoulombic (nc) energy contribution to the isosteric heat. This value is computed from the expression of the isosteric heat, taking the averages of Uncsf instead of the total energy U.
. The angle brackets denote averages in the grand canonical ensemble. In the following section we note the solid–fluid (sf) noncoulombic (nc) energy contribution to the isosteric heat. This value is computed from the expression of the isosteric heat, taking the averages of Uncsf instead of the total energy U.
To compute the adsorbate density profiles in the channels of ZIFs, we used the density maps resulting from the simulations in the following procedure.44 We divided the ZIF channels into concentric regions of equal thickness δr and recorded the number of particles δN(r), located inside the regions [r + δr]. The density distributions result from the fraction p(r) = δN(r)/(2πrδr). We set the radius of kno channels rkno = 11 Å, while for hpr or gme channels, we set rgme = 7 Å.
3 Results and discussion
3.1 Single component adsorption
The pure component adsorption isotherms of CO2, CH4 and N2 of gme ZIFs at 298 K are shown in Fig. 3. ZIFs derived diverse capacities of CO2, some of which were adequately high for carbon capture. This was attributed to specific CO2 interactions with the ligands and ligand substitutes, especially with those with cyano- and oxygen-containing functional groups.45 CH4 adsorbed densities were low compared to CO2, because of the exclusion of the electrostatic contributions in the computation of CH4 adsorption energy. N2 adsorbed density was essentially smaller than CO2 or CH4 at 298 K due to the weak LJ interactions of N2. As it can be seen in the ESI,† our calculations agree with several sets of previously reported, experimental and simulated adsorption data at low pressures.10,13,16,17,25,26,28 | ||
| Fig. 3 Isotherms of CO2, CH4 and N2 adsorption in gme ZIFs at 298 K. | ||
The Langmuir parameters for the single component simulated isotherms of gme ZIFs are listed in Table 2. Low coverage calculated isosteric heats of CO2 adsorption are given in Table 3. In most cases the calculations agree with values reported in the literature.
| ZIF68 | ZIF69 | ZIF70 | ZIF78 | ZIF79 | ZIF80 | ZIF81 | ZIF82 | ||
|---|---|---|---|---|---|---|---|---|---|
| CO2 | K | 0.129 | 0.18 | 0.049 | 0.289 | 0.194 | 0.066 | 0.241 | 0.11 |
| n inf | 313 | 277 | 372 | 216 | 145 | 413 | 169 | 390 | |
| CH4 | K | 0.045 | 0.053 | 0.021 | 0.085 | 0.11 | 0.037 | 0.113 | 0.019 |
| n inf | 268 | 237 | 556 | 180 | 202 | 305 | 206 | 474 | |
| N2 | K | 0.038 | 0.033 | 0.0173 | 0.029 | 0.053 | 0.017 | 0.038 | 0.013 |
| n inf | 76 | 91 | 165 | 133 | 71 | 202 | 103 | 301 | |
| ZIF68 | ZIF69 | ZIF70 | ZIF78 | ZIF79 | ZIF80 | ZIF81 | ZIF82 | |
|---|---|---|---|---|---|---|---|---|
| a Liu & Smit (sim.).17 b Liu et al. (sim.).26 c Kontos et al. (exp.).13 d Rankin et al. (sim.).12 e Li et al. (sim.).28 f Ding & Yazaydin (sim.).22 | ||||||||
| q ncsf non coul, solid–fluid | 16.9 | 17.41 | 17.46 | 21.23 | 20.62 | 15.92 | 20.22 | 19.17 |
| q allst total isosteric | 22.9 | 22.19 | 20.77 | 26.2 | 23.46 | 20.97 | 22.94 | 21.11 |
| Reference values (total) | 23,a 23b | 26.7,a 17,c 25.9f | 21d | 25.7e | 26.1e | |||
The dissimilarities between the isotherm curves can be better interpreted with the correlation set values displayed in Fig. 4. CO2 Langmuir constants of adsorption (Table 2) and the noncoulombic CO2–ZIF term of isosteric heat (Table 3) were associated. ZIFs with an aromatic functionality on the substituted imidazolate obtained ≃2 times steeper CO2 isotherms at low pressures. Presumably the aromatic rings provide additional sites for strong interactions with CO2. CH4 adsorption was correlated with the surface area of the structure. Being supercritical at 298 K, CH4 can be adsorbed mainly in a monolayer formation, and therefore the coverage of the specific adsorbate is proportional to the accessible surface area of the solid. ZIFs with short ligands achieve enhanced adsorption capacities of CH4.
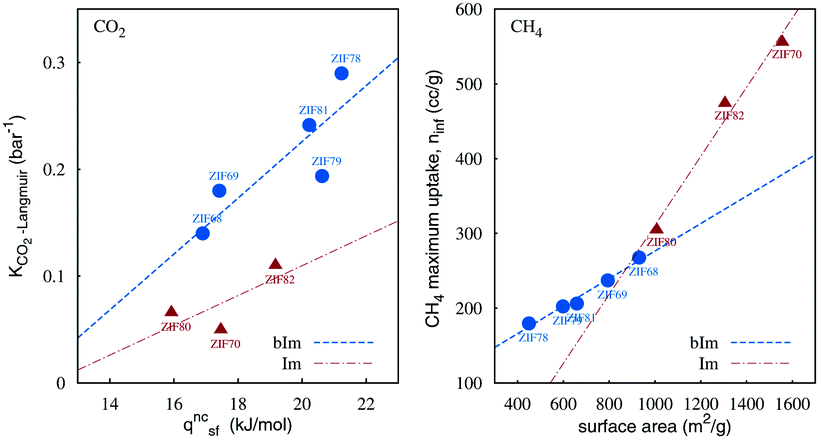 | ||
| Fig. 4 Gas adsorption related to the structural properties of gme ZIFs, with (bIm) and without (Im) benzene rings on ligand substituents. Trend-lines can be used as guidelines. | ||
3.2 Multicomponent adsorption
Fig. 5 shows the CO2 adsorption distributions within the kno cavities. In the kno channels of ZIF68, there is a sharp CO2 adsorption layer at the pore center, and a broader one at the distance of the effective radius. In ZIF69, the second layer is observed close to the aromatic rings of the Cl-containing ligands. The adsorption in ZIF70 takes place totally within the effective pore radius, as beyond this line the remaining space is narrow. In ZIF78, CO2 particles were more concentrated between the functionalized ligands than in the center of kno. This space, due to O2N-electrostatics, is also highly hydrophilic as the water content results in a great reduction of CO2 densities. The layer of CO2 particles in ZIF79 is observed in front of the methyl groups and not at the pore center due to its narrow confinement. In ZIF80 we observe a broad adsorption layer which traverses the borderline of chlorine atoms up to the walls of the cavity. The adsorption in ZIF81 takes place almost exclusively behind Br atoms, in the aperture direction of the cage. In ZIF82, the enhanced interaction energy of –CN groups with CO2, along with the large effective diameter of the particular cavity, result in high densities and broad CO2 adsorption layers. However the presence of water in the pore interior of ZIF82, decreases the CO2 adsorption capacity essentially.
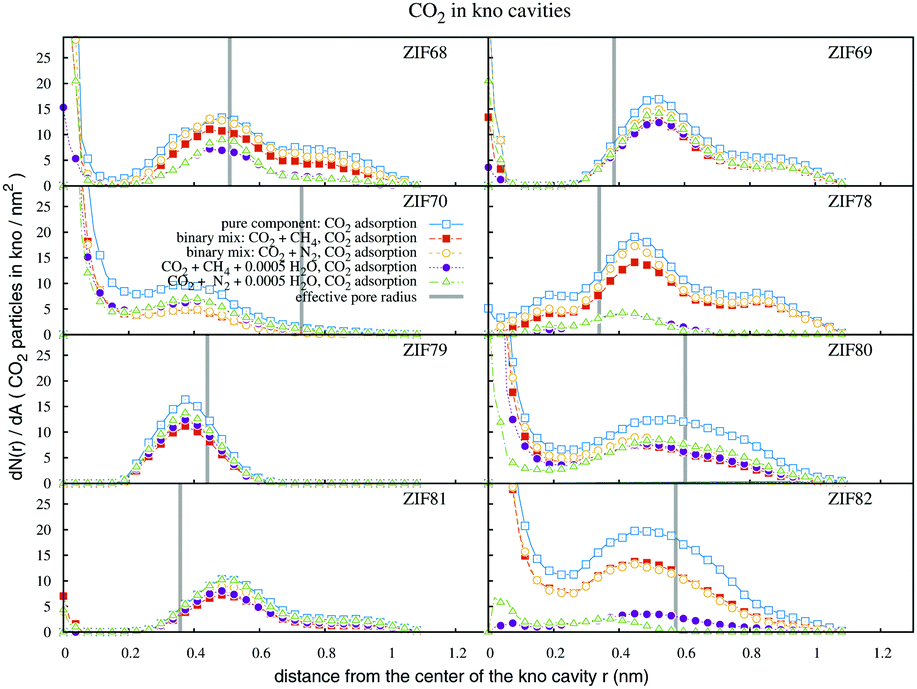 | ||
| Fig. 5 Profiles of CO2 density within the kno channels, for single and multicomponent adsorption simulations. The bulk pressure is 20 bar. The effective pore radii are given in Table 1. | ||
In Fig. 6 we present the CO2 adsorption density distributions within the gme channels. ZIFs with groups of benzoimidazolate type (like ZIF69, ZIF-78, ZIF-79 and ZIF81) gave sharp CO2 profiles in the center of the gme cavities. Apart from the center, the CO2 profiles are flat indicating that the adsorption is not ordered there. The profiles inside the gme cage of Im ZIFs are single layered, they peak at the center and cover the entire size of the cavity. It is interesting to note that for ZIF78 and ZIF79, an extra space is revealed around ≃0.6 nm from their gme pore center. This space is formed because the large bIm groups, when connected to the framework backbone, are slightly tilted, shaping small wedges along the aperture direction of gme channels.
 | ||
| Fig. 6 Profiles of CO2 density within the gme and hpr channels, for single and multicomponent simulations. The bulk pressure is 20 bar. The effective pore radii are given in Table 1. | ||
The CH4 and N2 adsorption profiles within the kno channels of ZIFs are presented in Fig. 7. We note low N2 adsorption densities in all samples. In general all profiles peak at the same position inside the pores. However in ZIF78, because of the strong electrostatic contribution of O2N-functionality, N2 adsorbate particles move closer to the aromatic ring of the ligand as compared to CH4. The opposite is observed for the corresponding profiles of ZIF79, where the ligands repulse N2 more than CH4, towards the center of the cavity. In most cases N2 and CH4 densities almost vanish in water containing adsorbate mixtures at 20 bars, indicating that water, even in minute amounts, is the dominating adsorbate for such systems.
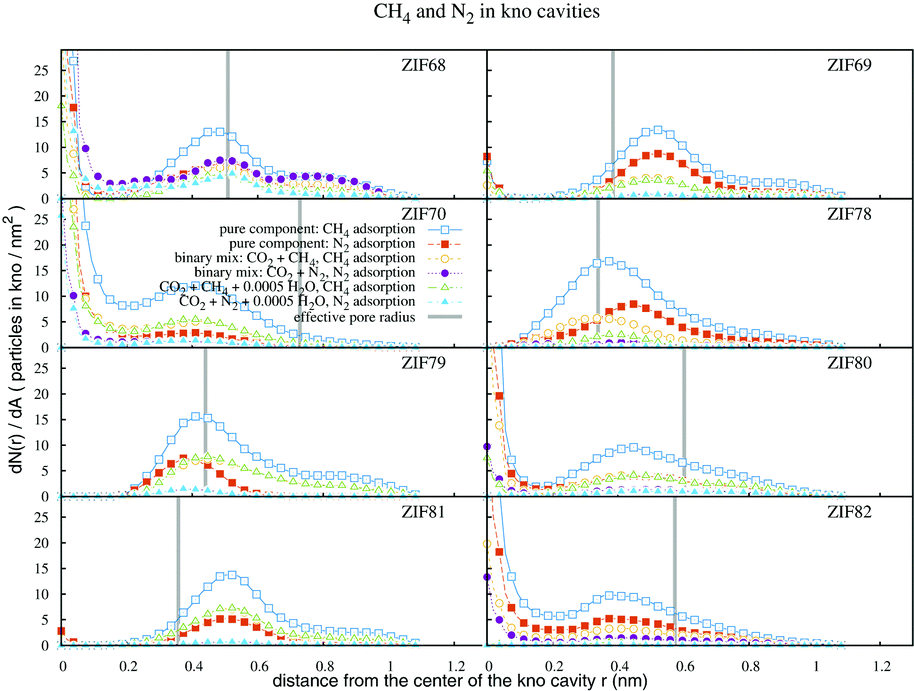 | ||
| Fig. 7 Profiles of CH4 and N2 densities within the kno channels, for single and multicomponent simulations. The bulk pressure is 20 bar. The effective pore radii are given in Table 1. | ||
The density profile curves of CH4 and N2 within the gme channels are shown in Fig. 8. Particles have been adsorbed in the wedge found at ≃0.6 nm for ZIF78 and ZIF79 although the pore filling process has not yet been completed in the rest of the particular cavity. This implies that the confinement formed by the tilted ligands along the gme surface, provides additional adsorption sites with stronger interaction energy than the common sites within this cavity.
 | ||
| Fig. 8 Profiles of CH4 and N2 densities within the gme and hpr channels, for single and multicomponent simulations. The bulk pressure is 20 bar. The effective pore radii are given in Table 1. | ||
Fig. 9 shows a configuration snapshot of CO2 adsorbed on ZIF78, where we gradually set the focus at the arc of a gme pore surface. The tilted orientation of the nbIm ligands and the wedge can be seen when we look at the gme surface from its axial direction.
 | ||
| Fig. 9 Configuration of CO2 particles within the (a) crystal (b) a gme pore and (c and d) an arc of a gme pore surface. The blue borderlines in (a) and (b) denote the focus area that we set at the next figure. | ||
 | ||
| Fig. 10 CO2 adsorption in the unit cell and the kno and gme channels of ZIF69, for single and multicomponent systems. | ||
| ZIF68 | ZIF69 | ZIF70 | ZIF78 | ZIF79 | ZIF80 | ZIF81 | ZIF82 | |
|---|---|---|---|---|---|---|---|---|
| Entire crystal | 8.38 | 10.22 | 13.40 | 4.89 | 12.15 | 9.73 | 13.12 | 5.86 |
| kno channel | 9.33 | 13.61 | 13.86 | 4.89 | 15.54 | 14.09 | 15.06 | 6.34 |
| gme channel | 7.34 | 9.25 | 6.43 | 4.41 | 5.38 | 6.34 | 3.92 | 2.96 |
The simulated isotherm densities of single component CO2 adsorption in ZIF69 were strongly correlated with a set of Raman spectroscopy intensity data presented in the work of Kontos et al.13 The experiment was performed in a cell chamber containing ZIF69 with different CO2 loadings. Kontos et al. quantified the adsorbed amount of CO2 by detecting the CO2–cbIm interactions from such spectra. In Fig. 10 we include the scaled Raman spectroscopy intensity values as a function of CO2 pressure. The experimental data are scaled suitably in order to be compared directly with the pure component CO2 simulated isotherms with respect to the entire crystal, and the kno and gme cavities. The correlation coefficients of the simulated and experimental isotherms are also listed in Fig. 10. The Raman spectroscopy isotherm is most strongly correlated with the isotherm of kno cavities. The cbIm ligands protruding from the surface inside the particular cavities are 360° degrees accessible to the CO2 adsorbate. In this respect the detected CO2–cbIm interactions have a greater impact on the adsorption energy within the kno than the other cavities.
4 Conclusions
We simulated multicomponent adsorption in eight gme zeolite imidazole frameworks at 298 K, using the GCMC method. We considered the adsorption of carbondioxide, methane and nitrogen, of their pair mixtures and of their ternary mixtures which also contained minute amounts of water. We presented adsorption density profiles regarded explicitly within the distinct pore channels of gme crystals, namely the kno and gme cavities. The profiles were with respect to high pressure systems where the adsorbate components are competitive. Using the profiles we detected an additional adsorption volume, which features strong interaction energies. This volume is found behind the surface of gme cavities only for the ZIFs with the longest ligands. Long ligands are connected to the framework in a slightly tilted orientation and are in a wedged shaped space in the axial direction of the gme cavity. The multicomponent isotherms were de-convoluted and compared directly in the kno and gme cavities of the samples. In all cases water prevailed in the adsorbed phase prohibiting different adsorbate components to enter the pore. The simulated CO2 adsorption isotherm of ZIF69 was compared with a set of Raman spectroscopy intensity values obtained for different CO2 loadings on the same sample. The Raman spectroscopy data and the kno CO2 isotherm were more correlated compared to the rest of the de-convoluted simulation isotherms. This was expected because Raman isotherm is based on the detection of CO2–ligand couplings and the ligands specifically within the kno channels are more exposed to the fluid.Acknowledgements
Calculations have been performed in the “High Performance Computing” facilities of the Environmental Research Laboratory in NCSR “Demokritos”. The authors thank Dr G. Romanos, Dr M. Kainourgiakis, Dr E. Tylianakis and Dr N. Papadimitriou for discussion.References
- B. Chen, Z. Yang, Y. Zhu and Y. Xia, J. Mater. Chem. A, 2014, 2, 16811–16831 CAS.
- B. R. Pimentel, A. Parulkar, E.-k. Zhou, N. A. Brunelli and R. P. Lively, ChemSusChem, 2014, 7, 3202–3240 CrossRef CAS PubMed.
- J. Yao and H. Wang, Chem. Soc. Rev., 2014, 43, 4470–4493 RSC.
- Y. Wu, H. Chen, D. Liu, Y. Qian and H. Xi, Chem. Eng. Sci., 2015, 124, 144–153 CrossRef CAS.
- F. Martínez, R. Sanz, G. Orcajo, D. Briones and V. Yángüez, Chem. Eng. Sci., 2016, 142, 55–61 CrossRef.
- K. M. Gupta, K. Zhang and J. Jiang, Langmuir, 2015, 31, 13230–13237 CrossRef CAS PubMed.
- C. Gücüyener, J. Van Den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- H. Amrouche, S. Aguado, J. Pérez-Pellitero, C. Chizallet, F. Siperstein, D. Farrusseng, N. Bats and C. Nieto-Draghi, J. Phys. Chem. C, 2011, 115, 16425–16432 CAS.
- J. V. Smith, Chem. Rev., 1988, 88, 149–182 CrossRef CAS.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O. Keeffe and O. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- R. B. Rankin, J. Liu, A. D. Kulkarni and J. K. Johnson, J. Phys. Chem. C, 2009, 113, 16906–16914 CAS.
- A. Kontos, V. Likodimos, C. Veziri, E. Kouvelos, N. Moustakas, G. Karanikolos, G. Romanos and P. Falaras, ChemSusChem, 2014, 7, 1696 CrossRef CAS PubMed.
- F. Cao, Y. Sun, L. Wang and H. Sun, RSC Adv., 2014, 4, 27571–27581 RSC.
- K. Zhang, K. Gupta, Y. Chen and J. Jiang, AIChE J., 2015, 61, 2763–2775 CrossRef CAS.
- J. McDaniel and J. Schmidt, J. Phys. Chem. C, 2012, 116, 14031–14039 CAS.
- B. Liu and B. Smit, J. Phys. Chem. C, 2010, 114, 8515 CAS.
- J. Xu, W. Xing, H. Wang, W. Xu, Q. Ding, L. Zhao, W. Guo and Z. Yan, J. Mater. Sci., 2016, 51, 2307–2319 CrossRef CAS.
- R. Krishna and J. M. van Baten, Phys. Chem. Chem. Phys., 2013, 15, 7994–8016 RSC.
- J. Pérez-Pellitero, H. Amrouche, F. Siperstein, G. Pirngruber, C. Nieto-Draghi, G. Chaplais, A. Simon-Masseron, D. Bazer-Bachi, D. Peralta and N. Bats, Chem. – Eur. J., 2010, 16, 1560–1571 CrossRef PubMed.
- K. Vogiatzis, A. Mavrandonakis, W. Klopper and G. Froudakis, ChemPhysChem, 2009, 10, 374–383 CrossRef CAS PubMed.
- L. Ding and A. Yazaydin, Phys. Chem. Chem. Phys., 2013, 15, 11856–11861 RSC.
- S. Biswas, D. E. P. Vanpoucke, T. Verstraelen, M. Vandichel, S. Couck, K. Leus, Y.-Y. Liu, M. Waroquier, V. V. Speybroeck, J. F. M. Denayer and P. V. D. Voort, J. Phys. Chem. C, 2013, 117, 22784–22796 CAS.
- M. G. Frysali, E. Klontzas, E. Tylianakis and G. E. Froudakis, Microporous Mesoporous Mater., 2016, 227, 144–151 CrossRef CAS.
- R. Babarao, S. Dai and D.-E. Jiang, J. Phys. Chem. C, 2011, 115, 8126–8135 CAS.
- Y. Liu, A. Kasik, N. Linneen, J. Liu and Y. Lin, Chem. Eng. Sci., 2014, 118, 32–40 CrossRef CAS.
- A. Sirjoosingh, S. Alavi and T. K. Woo, J. Phys. Chem. C, 2010, 114, 2171–2178 CAS.
- B. Li, S. Wei and L. Chen, Mol. Simul., 2011, 37, 1131–1142 CrossRef CAS.
- Q. Li, M. Ruan, B. Lin, M. Zhao, Y. Zheng and K. Wang, J. Porous Mater., 2016, 23, 107–122 CrossRef.
- M. Salah, K. Marakchi, S. Dalbouha, M. Senent, O. Kabbaj and N. Komiha, Comput. Theor. Chem., 2015, 1073, 1–8 CrossRef CAS.
- K. Ray, D. Olmsted, Y. Houndonougbo, B. Laird and M. Asta, J. Phys. Chem. C, 2013, 117, 14642–14651 CAS.
- D. Keffer, H. Davis and A. McCormick, Adsorption, 1996, 2, 9–21 CrossRef.
- E. D. Biase and L. Sarkisov, Carbon, 2015, 94, 27–40 CrossRef.
- T. Horikawa, T. Muguruma, D. Do, K.-I. Sotowa and J. R. Alcántara-Avila, Carbon, 2015, 95, 137–143 CrossRef CAS.
- T. Dixon, K. Yamaji, D. Marx, L. Joss, M. Hefti, R. Pini and M. Mazzotti, Energy Procedia, 2013, 37, 107–114 CrossRef.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS.
- J. J. Potoff and J. I. Siepmann, AIChE J., 2001, 47, 1676–1682 CrossRef CAS.
- N. Hansen, F. A. Agbor and F. J. Keil, Fluid Phase Equilib., 2007, 259, 180–188 CrossRef CAS.
- D. Paschek, J. Chem. Phys., 2004, 120, 6674–6690 CrossRef CAS PubMed.
- E. First and C. Floudas, Microporous Mesoporous Mater., 2013, 165, 32–39 CrossRef CAS.
- A. Gupta, S. Chempath, M. J. Sanborn, L. A. Clark and R. Q. Snurr, Mol. Simul., 2003, 29, 29–46 CrossRef CAS.
- T. Vuong and P. A. Monson, Langmuir, 1996, 12, 5425–5432 CrossRef CAS.
- A. Gotzias, G. Charalambopoulou and T. Steriotis, Mol. Simul., 2016, 42, 186–195 CrossRef CAS.
- E. J. Beckman, Chem. Commun., 2004, 1885–1888 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp06036f |
| This journal is © the Owner Societies 2017 |