
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single-molecule FRET sensor for monitoring DNA synthesis in real time†
Carel
Fijen
a,
Alejandro
Montón Silva‡
ab,
Alejandro
Hochkoeppler
b and
Johannes
Hohlbein
 *ac
*ac
aLaboratory of Biophysics, Wageningen University and Research, Stippeneng 4, Wageningen, 6708 WE, The Netherlands. E-mail: Johannes.Hohlbein@wur.nl; Fax: +31 317 482 725; Tel: +31 317 482 635
bDepartment of Pharmacy and Biotechnology, University of Bologna, Viale Risorgimento 4, Bologna, 40136, Italy
cMicrospectroscopy Centre, Wageningen University and Research, Stippeneng 4, Wageningen, 6708 WE, The Netherlands
First published on 18th January 2017
Abstract
We developed a versatile DNA assay and framework for monitoring polymerization of DNA in real time and at the single-molecule level. The assay consists of an acceptor labelled DNA primer annealed to a DNA template that is labelled on its single stranded, downstream overhang with a donor fluorophore. Upon extension of the primer using a DNA polymerase, the overhang of the template alters its conformation from a random coil to the canonical structure of double stranded DNA. This conformational change increases the distance between the donor and the acceptor fluorophore and can be detected as a decrease in the Förster resonance energy transfer (FRET) efficiency between both fluorophores. Remarkably, the DNA assay does not require any modification of the DNA polymerase and albeit the simple and robust spectroscopic readout facilitates measurements even with conventional fluorimeters or stopped-flow equipment, single-molecule FRET provides additional access to parameters such as the processivity of DNA synthesis and, for one of the three DNA polymerases tested, the detection of binding and dissociation of the DNA polymerase to DNA. We furthermore demonstrate that primer extensions by a single base can be resolved.
Introduction
DNA polymerases play a pivotal role in DNA replication and DNA repair.1–3 The complexity of the cellular environment and resultant requirements on adaptability and specificity led to a profound differentiation of DNA polymerases. In humans, 18 different DNA polymerases are currently known that all catalyse the stepwise addition of deoxyribonucleotides to a growing DNA chain4 but differ greatly in their specific properties. In addition to their fundamental role in living organisms, DNA polymerases have been utilised in many biotechnological applications ranging from polymerase chain reaction (PCR) for amplification of DNA fragments5 to DNA sequencing.6,7 Different techniques have been developed to characterize DNA polymerases in terms of their fidelity, processivity and rate of DNA synthesis. At the ensemble level, electrophoretic mobility shift assays (EMSA)8 and stopped-flow techniques are widely used for quantifying the kinetics of DNA polymerization. Recent fluorescence-based ensemble assays to monitor polymerase activity include monitoring the incorporation of dye-labelled nucleotides into DNA9 and surface-induced fluorescence quenching of a labelled DNA template on a chip.10In the last two decades, techniques with single-molecule sensitivity and resolution allowed to overcome temporal and spatial averaging inherent to ensemble based characterisations.11,12 For DNA polymerases, different experimental designs have been applied ranging from optical or magnetic traps13 (reviewed by Heller et al.14) to conductivity measurements on protein nanopores15,16 and applications utilising Förster resonance energy transfer (FRET), a process in which the distance-dependent energy transfer from a donor fluorophore to an acceptor chromophore allows to resolve changes of distances in the nanometre range.17,18 Single-molecule FRET experiments on DNA polymerases have been designed to measure the rate of DNA polymerization by strand-displacing DNA polymerases,19 to identify sliding characteristics and binding orientations of HIV reverse transcriptase,20,21 to identify translocation of DNA polymerases with single base pair resolution22 and to determine conformational dynamics and the free-energy landscapes of pre-chemistry nucleotide selection in E.coli DNA polymerase I.23,24 Most of the experimental approaches, however, required the positioning of one or two fluorescent labels on the polymerase, which is a time consuming and complicated procedure.
Moreover, DNA polymerase-based DNA sequencing was demonstrated at the single-molecule level using a sequential approach involving the successive addition of dye-labelled nucleotides25 and non-sequentially by utilising so-called zero-mode waveguides as optical waveguides that provide a confined excitation and detection volume which is small enough to monitor polymerization reactions in presence of all four nucleotides and in real time.6 As each of the four dNTPs is conjugated to a different fluorescent dye, the temporal immobilisation of the nucleotide during synthesis is sufficient for base calling. Alternative approaches for DNA sequencing utilise a polymerase–template complex attached to the tip of an atomic force microscope to sample different nucleotide pools26 or measuring nucleotide-dependent binding kinetics of singly labelled DNA polymerases.27
Despite the progress, a simple and versatile assay for characterizing DNA polymerization at the single-molecule level, in real time and without requiring fluorescently labelled DNA polymerases is still missing. Here, we propose a DNA assay that allows probing both the speed of DNA polymerization and processivity, and for one species, even the binding and dissociation constants of the DNA polymerases to DNA. Whereas a comparable assay has been successfully used to screen for HIV-1 reverse transcriptase on the ensemble level,28 further optimization of the assay and its application at the single molecule level allowed us to study dynamic and static heterogeneity with up to single base pair resolution. We compared different polymerases, namely the α subunit of E. coli DNA polymerase III (here after POLIIIα), E. coli DNA polymerase I Klenow fragment (KF) and human DNA polymerase beta (POLB). Albeit we performed the measurements at the single-molecule level, the design is suitable for any instrument featuring a fluorescent read out mode.
Material and methods
Design principles of the DNA sensor
Our DNA assay consists of a DNA primer labelled with an acceptor fluorophore annealed to a DNA template labelled with a donor fluorophore (Fig. 1a), inspired by DNA constructs featuring recessed DNA that have been used before to assess SSB protein binding,29 flexibility of single-stranded DNA30 or dynamics of the HIV transcriptase.20 As the single stranded overhang of the DNA template is randomly coiled, we expect a high FRET efficiency E* defined aswhere
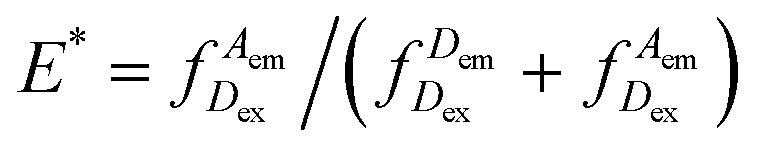
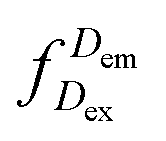 represents the donor emission intensity after donor excitation and
represents the donor emission intensity after donor excitation and 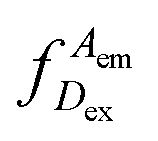 the acceptor emission intensity after donor excitation. Upon addition of DNA polymerase, interactions between the DNA polymerase and the single stranded template might change the distance between donor and acceptor, thereby modulating the FRET efficiency (Fig. 1b). The addition of dNTPs starts the DNA polymerization reaction that decreases the FRET efficiency as the distance between the fluorophores increases (Fig. 1c). After complete polymerization, the extended DNA molecule is rigid, yielding the lowest possible FRET signature depending on the chosen labelling positions (Fig. 1d and e). All experiments were performed at room temperature (21 °C).
the acceptor emission intensity after donor excitation. Upon addition of DNA polymerase, interactions between the DNA polymerase and the single stranded template might change the distance between donor and acceptor, thereby modulating the FRET efficiency (Fig. 1b). The addition of dNTPs starts the DNA polymerization reaction that decreases the FRET efficiency as the distance between the fluorophores increases (Fig. 1c). After complete polymerization, the extended DNA molecule is rigid, yielding the lowest possible FRET signature depending on the chosen labelling positions (Fig. 1d and e). All experiments were performed at room temperature (21 °C).
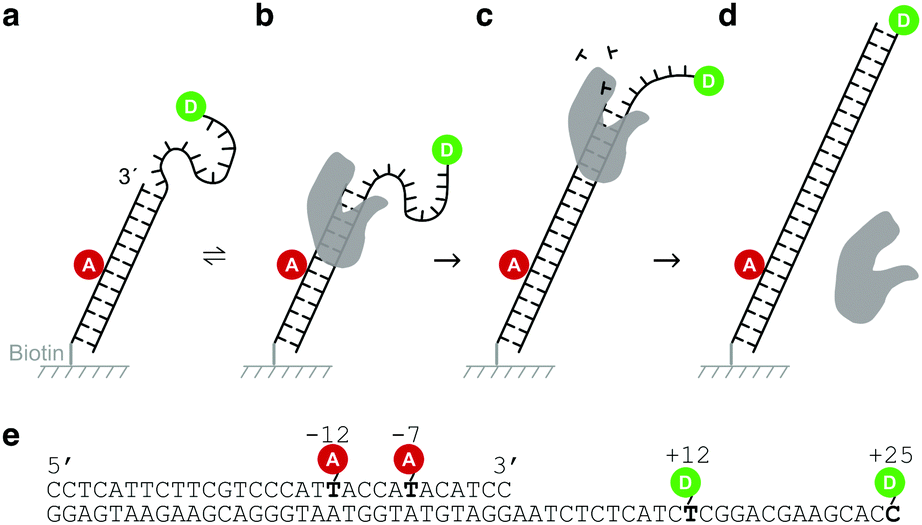 | ||
| Fig. 1 Design of the DNA sensor. (a) The DNA sensor consists of acceptor labelled DNA primer and a donor labelled DNA template immobilized on a cover slip using a biotin/neutravidin/biotin linkage. (b) Addition of polymerases establishes a dynamic equilibrium in which polymerases bind and dissociate from the DNA sensor. Interactions between the single-stranded overhang and the polymerase may cause a change in the distance between the donor and the acceptor. (c) Upon addition of nucleotides, the polymerase starts the polymerization reaction extending the DNA primer. During the process, the overhang is taken up in a rigid DNA helix, increasing the distance between donor and acceptor. (d) The final DNA shows a lower transfer efficiency between the donor and the acceptor than the native DNA sensor. (e) Sequence of the DNA sensor with indicated labelling positions. We used the combinations of +12/−12 and +25/−7 for our experiments. | ||
DNA polymerases
We used three different DNA polymerases: Klenow fragment from E. coli DNA Polymerase I (KF), human Polymerase Beta (POLB) and the α subunit from E. coli DNA Polymerase III (POLIIIα). Both KF and POLB are involved in DNA repair and have been studied extensively.1,31–34 DNA polymerase III, on the other hand, is a large enzyme complex responsible for DNA replication.35–37 Its α subunit (POLIIIα) possesses polymerase activity, but no exonuclease or proof-reading activity.DNA sequences and modifications
Oligonucleotides were ordered from IBA, Germany. 5-Amino-C6 modifications, internal (on dT) or terminal (on dC), were used to label positions with NHS-reactive dyes. A 30-mer primer sequence (biotin-5′-CCTCATTCTTCGTCCCATTACCATACATCC-3′) was labelled with ATTO647N at either position −12 or −7, counted from the 3′ end. This primer was annealed to a 55-mer template (3′-GGAGTAAGAAGCAGGGTAATGGTATGTAGGAATCTCTCATCTCGGACGAAGCACC-5′), labelled with Cy3B at either position +12 or +25 of the overhang. We used two labelling configurations: +25/−7 (donor/acceptor) and +12/−12 (donor/acceptor) (Fig. 1e).Total-internal reflection fluorescence (TIRF) microscopy
For imaging, we used the alternating-laser excitation scheme (ALEX)38–40 in combination with TIRF microscopy on a home-built microscope described in more detail elsewhere.41 Briefly, our microscope is equipped with a laser engine (Omicron, Germany), a 100× oil immersion objective with N.A. = 1.49 (Nikon, Japan), and an Ixon Ultra 897 emCCD camera for image acquisition (Andor, UK). The sensors were immobilized on PEGylated coverslips through a neutravidin–biotin interaction. All samples were measured in either gasket wells or flow channels formed by sticky-Slides VI0.4 (Ibidi, Germany). Images of immobilized DNA sensors were taken at a rate of 20 frames per second, using excitation powers of 0.75 mW (642 nm) and 1.5 mW (561 nm). Imaging was performed in a buffer containing 50 mM Tris HCl (pH7.5), 100 μg ml−1 BSA, 10 mM MgCl2, 5% glycerol, 1 mM DTT, 1 mM Trolox, 1% glucose oxidase/catalase and 1% glucose. Trolox is a triplet state quencher, while glucose oxidase and catalase serve as an oxygen scavenger system; both prevent premature bleaching of the fluorophores.42,43 After addition of the imaging buffer, DNA sensors were imaged for 50 seconds per movie (DNA binding studies) or for 500 seconds per movie (real-time polymerization studies). To increase the total number of observed molecules in binding studies, three consecutive movies were recorded for each sample condition.Analysis of completed DNA synthesis: end-point assays
Polymerases (40 nM), dNTPs (100 μM each) and sensors (10 nM) were mixed together in an aliquot using a buffer resembling our imaging buffer, but without the oxygen scavenger system. A mixture without any dNTPs was used as a negative control. To ensure the completion of all polymerization reactions, the mixtures were left to react at RT for 90 minutes, after which we stopped the reaction by adding EDTA to a final concentration of 0.1 M. For data acquisition, the DNA products were immobilized and imaged as described above.In a similar experiment, which was only performed for KF, polymerization reactions were stopped at specific positions along the template by adding only a subset of the dNTPs needed for full polymerization.
Monitoring DNA binding and DNA polymerization
For DNA binding studies, we mixed the respective polymerases into the imaging buffer (final concentrations: KF 500 pM, POLIIIα 2 nM and POLB 10 nM). The concentration of KF was chosen such that about 50% of all molecules are bound to DNA as verified using titrations of the DNA sensors with polymerase (Fig. S1, ESI†). The concentrations of POLIIIα and POLB for the DNA binding studies were chosen such that most time traces of the DNA polymerization experiments showed DNA synthesis implying that binding at these concentrations occurs.For real-time polymerization experiments, polymerases and dNTPs were directly provided in the imaging buffer. Since polymerization events can happen shortly after adding DNA polymerases and dNTPs, we started data acquisition of the immobilised molecules in standard imaging buffer before quickly adding polymerases and dNTPs during acquisition in a similar volume of imaging buffer. Final concentrations in the sample were identical to the ones chosen for the binding experiments. dNTPs were added to a concentration of 100 μM each, except for a set of experiments in which dNTPs were added to successively create artificial pausing sites.
At the ensemble level, the polymerization of the +25/−7 sensor by KF was recorded in a 96 wells plate, using a SpectraMax M2 plate reader. For experimental details, please refer to the ESI.†
Time trace analysis of single-molecule FRET data
Analysis of the acquired movies was performed in MATLAB (MathWorks, UK) using custom-built software packages44 yielding time traces of both donor and acceptor intensity, as well as FRET efficiency E* and stoichiometry S (ESI†) for each DNA. For calculation of exact distances within the polymerized DNA construct, E* was corrected for background, cross-talk between the channels, detection efficiencies of the dyes and quantum yield (ESI†). Theoretical distances were calculated based on a static model of the polymerized DNA construct45 (3D-DART). We used the FPS software developed by the Seidel lab46 to model the dye clouds representing possible positions of the fluorophores attached to modified bases around the DNA duplex and calculated the predicted inter-dye distance 〈RDA〉E.Hidden Markov modelling (HMM)
Single-molecule time traces of binding and polymerization experiments were analysed with the ebFRET software package.47 Using a Bayesian approach, ebFRET obtains the best fit via an iterative process after introducing a prior distribution of parameters such as mean E* values, dwell times and a general noise level. We used ebFRET to determine the number of steps involved in the polymerization of individual sensors as follows. HMM states lasting shorter than 1 s were removed from further analysis. To calculate the duration Δti = t2 − t1 of individual polymerization events i, we assigned the first intersection of the high FRET HMM state with the decreasing FRET efficiency as t1. The intersection of the decreasing FRET efficiency with the next lower FRET HMM state was taken as t2. We repeated that procedure for cases in which more than one polymerization step was visible and summed up the polymerization times for each individual time trace. For our +12/−12 polymerization sensor in combination with measurements using KF, we used a slightly modified approach to account for the presence of a short increase in E* just before each polymerization event. In these cases, t1 was defined as the last position of the high FRET peak before the FRET decrease due to DNA synthesis. For further information on HMM the reader is referred to the ESI.†Results
DNA sensors indicate completion of DNA synthesis
To test whether our DNA sensors can be polymerized by KF, POLB and POLIIIα, we mixed the sensors with the respective polymerases and dNTPs in an aliquot and imaged the DNA products after an incubation of 90 minutes at room temperature using our TIRF microscope (“end-point assays”). The native +25/−7 sensor showed a single peak at E* = 0.65, while the +12/−12 sensor shows a peak at E* = 0.52, both representing the respective E* value before DNA polymerization (Fig. 2a and b, row 1). Sensors that were incubated in presence of polymerases and dNTPs showed a peak at lower FRET efficiency than the native sensors (E* = 0.17 for +25/−7, and E* = 0.35 for +12/−12) (Fig. 2a and b, rows 2–4). The +25/−7 sensor showed the largest relative change in FRET efficiency of ΔE* = 0.48 (+12/−12: ΔE* = 0.17).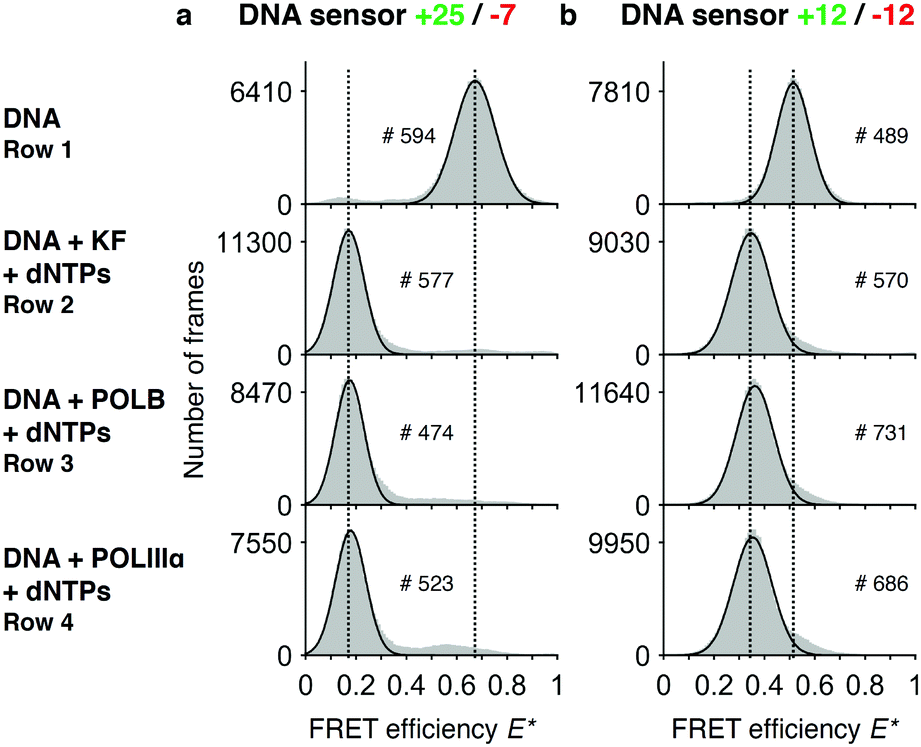 | ||
| Fig. 2 Assay to test completion of DNA synthesis. Histograms of FRET efficiencies E* for the +25/−7 (a) and the +12/−12 (b) DNA sensor. Each histogram (100 bins shown in grey) was derived from individual time traces obtained from fitting an indicated number of molecules (#). The histograms were fitted to a single-Gaussian model (black line) to indicate the mean peak position of the dominant FRET species. Vertical dashed lines were added for visual guidance. First row: FRET efficiencies of the native DNA sensor. Second to fourth row: FRET efficiencies after incubating the DNA sensor for 90 minutes in presence of dNTPs and DNA polymerases (second row: DNA polymerase I, Klenow fragment; third row: DNA polymerase beta; fourth row: DNA polymerase III alpha subunit). The histograms indicate successful polymerization of DNA as the FRET efficiencies are shifted towards lower values. Peak positions of the fits and the corresponding errors can be found in ESI† Table S1. | ||
As expected, we did not see any significant differences in the E* histograms of the various DNA polymerases after DNA synthesis and we note that >90% of all DNA molecules show a shift towards the lower FRET conformation. We calculated the distances between the fluorophores using accurate FRET (〈RDA〉E,+12/−12 = 7.1 nm, 〈RDA〉E,+25/−7 = 10.7 nm) and compared these values with the distances computationally derived from the structural model of a double stranded DNA helix (〈RDA〉E,mod,+12/−12 = 7.3 nm, 〈RDA〉E,mod,+25/−7 = 10.7 nm). Moreover, we found similar distances when measuring a separately ordered dsDNA construct with the same sequence as our fully polymerized DNA primer (ESI,† Table S4). These values suggest a successful and complete polymerization of the DNA sensors.
KF induces conformational changes upon binding to the DNA template strand
We asked whether the sensors show a change of FRET efficiency upon binding of DNA polymerases in the absence of nucleotides. Of the three DNA polymerases tested at the concentrations mentioned above, only binding of KF showed a new species in the FRET histogram with an increased E* (+25/−7: EDNA* = 0.65 and EDNA+KF* = 0.85; +12/−12: EDNA* = 0.52 and EDNA+KF* = 0.65) (Fig. 3a and b).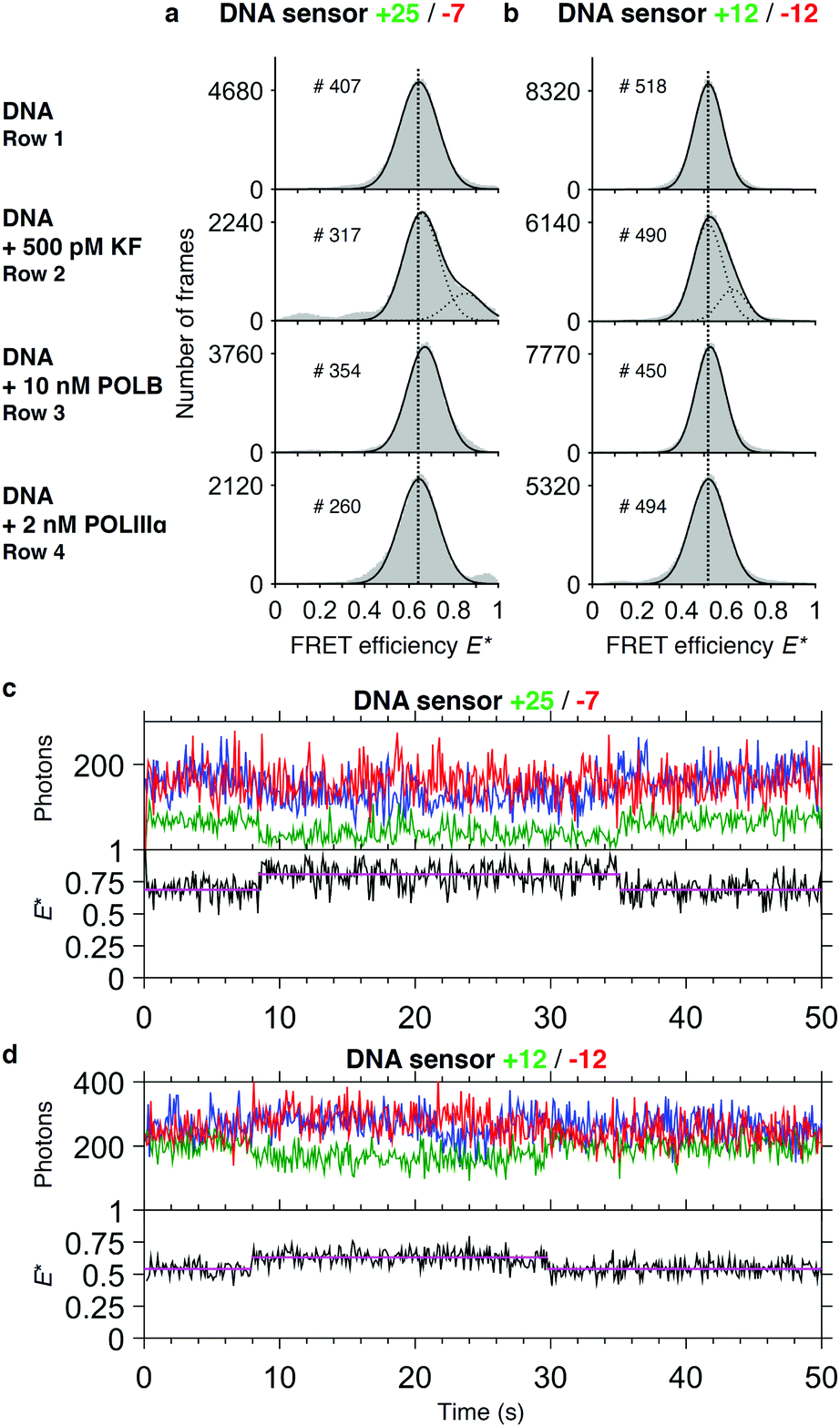 | ||
| Fig. 3 Binding studies. Histograms of FRET efficiencies E* for the +25/−7 (a) and the +12/−12 (b) DNA sensor. Each histogram (100 bins shown in grey) was derived from individual time traces obtained from fitting an indicated number of molecules (#). The histograms were fitted to a single-Gaussian model (black line) to indicate the mean peak position of the dominant FRET species. Vertical dashed lines were added for visual guidance. First row: FRET efficiencies of the native DNA sensor. Second to fourth row: FRET efficiencies after addition of DNA polymerases to the sensor (second row: DNA polymerase I, Klenow fragment; third row: DNA polymerase beta; fourth row DNA polymerase III alpha subunit). Addition of KF gives rise to a population of sensors that exhibit a higher E* than the native sensor. Peak positions of the fits and the corresponding errors can be found in ESI† Table S1. (c and d) Representative time traces of the DNA sensors +25/−7 (c) and +12/−12 (d) in presence of 500 pM KF. Upper panel: Acceptor fluorescence upon direct acceptor excitation (AA; blue trace). Donor (DD, green trace) and acceptor (DA, red trace) fluorescence upon donor excitation. Lower panel: FRET efficiency E* (black trace) with fitted HMM model (magenta trace). | ||
The increase in E* of POLB of the major species using the +25/−7 sensor is too small to be clearly attributed to a conformational change upon binding. For both POLB and POLIIIα we further tested higher concentrations of the polymerase without seeing any response on our DNA sensor (data not shown).
For KF, the population of sensors in the higher FRET state is concentration dependent: during titrations, the plateau value for the percentage of sensors bound to KF was found to be ∼50% for the +25/−7 sensor and ∼60% for the +12/−12 sensor (Fig. S1, ESI†), well below the expected maximum of 100% for the high-FRET state. We therefore hypothesise that different binding modes must exist which cannot be detected by a change in FRET efficiency. The titrations revealed apparent dissociation constants of 0.41 nM (sensor +25/−7) and 0.12 nM (sensor +12/−12) indicating strong binding of KF to the DNA (Fig. S1, ESI†).
For KF, analysis of the corresponding single-molecule time traces revealed dynamic switching between high and low FRET efficiencies (Fig. 3c and d), implying that binding and dissociation occurs at a timescale of seconds. As can be seen in the trace of the +25/−7 sensor, which was designed to maximize ΔE upon polymerization, the acceptor fluorescence after direct acceptor excitation appears to be slightly quenched upon binding of KF. During the binding event, the FRET signal (red line) remained constant, but the donor fluorescence signal decreased leading to an effective increase in FRET efficiency. As a consequence of this convolution of acceptor quenching and a change in the distances between the fluorophores, the correct interpretation of the FRET efficiency is complicated.48 We attribute the acceptor quenching to the proximity of the −7 labelling position to the polymerase binding site, which causes an interaction between the dye and the protein indicated by a slightly higher dissociation constant of KF for this sensor (Kd = 0.41 nM versus Kd = 0.12 nM for the +12/−12 sensor, Fig. S1, ESI†). Our attempts to perform global dwell time analysis on the binding events remained inconclusive, however, likely due to a combination of relatively long dwell times (>1 s) with small changes in ΔE* (<0.2) and base line FRET values changing slightly between individual traces.
Monitoring DNA synthesis in real time
As we have verified using the end-point experiments, the transfer efficiency E* decreases to stable values after successful DNA synthesis. For monitoring the synthesis in real time, we first focused on the +25/−7 sensor as it showed the largest change in E* upon polymerization. Polymerization of the sensor by KF showed a stepwise decrease of FRET efficiency (Fig. 4a): at around 140 s, we saw a short increase in FRET efficiency from the base level of E* ∼ 0.75 to E* ∼ 0.80, which was too small to be picked up by HMM, before dropping to E* ∼ 0.60, a level which was held for around 1.5 s. After that period, the FRET efficiency dropped to E* ∼ 0.20 representing fully polymerized DNA. The example time trace for POLIIIα shows fast synthesis requiring two synthesis steps (Fig. 4b). Polymerization by POLB, on the contrary, is found to be slow: time traces show a gradual decrease over a long time span (>100 s, Fig. 4c) in line with the slow DNA synthesis shown for POLB on recessed DNA substrates.49 In some cases, we observed an increase in the fluorescence of the acceptor after direct excitation upon polymerization. We attribute this to the close proximity of the dye to the incorporation site, where its local environment may change significantly during primer elongation. Our data showed that polymerization is in some cases not completed during the time of data acquisition. For KF and the +25/−7 sensor, 65% of all analysed time traces, representing 28 out of 43 DNA molecules in total, showed complete polymerization during data acquisition; 5 molecules completed DNA synthesis in one step and 23 molecules required two or three steps indicated by a considerable ‘lag time’ between regions with decreasing FRET efficiency (Fig. 4d). This lag time may be caused by either pausing or dissociation with subsequent re-association of the polymerase. Of the remaining 35% of time traces (15 molecules), 10 traces showed incomplete synthesis indicated by a final E* > 0.3 and 5 traces could not be analysed due to fluorophore blinking events or other complications (more example time traces are presented in Fig. S2, ESI†). Plotting the durations of all 28 completed polymerization events excluding the lag time revealed an average duration of 1.6 ± 0.5 s (mean ± standard deviation, Fig. 4e). The faster polymerization by POLIIIα is reflected in a lower average duration of 1.0 ± 0.4 s for the same sensor (N = 54, Fig. 4f). A similar analysis for POLB was not possible due to the low processivity, preventing us to fit the data with HMM.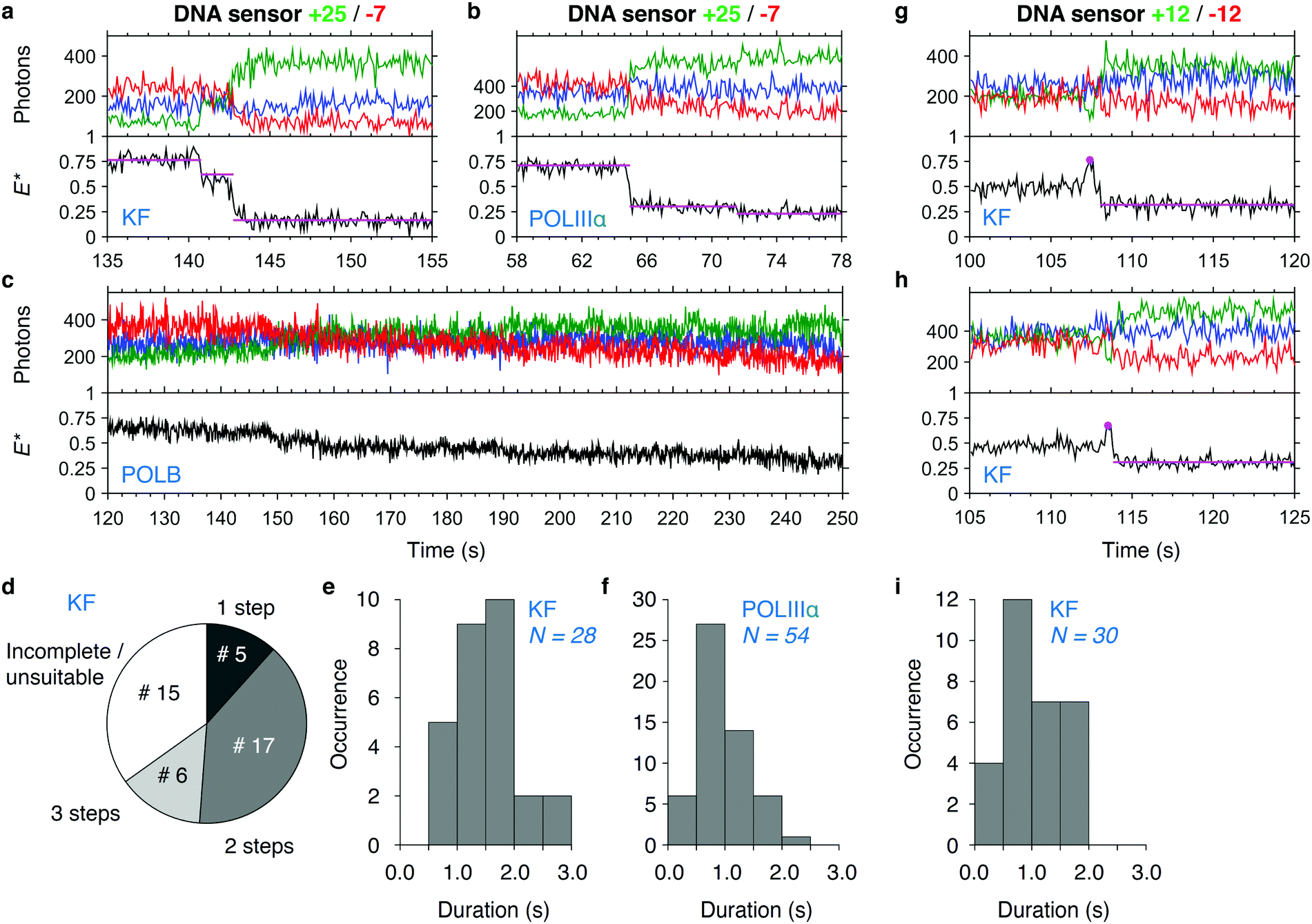 | ||
| Fig. 4 DNA synthesis monitored in real time. (a–c) Single-molecule time traces showing DNA polymerization in real time of sensor +25/−7 by KF (a), POLIIIα (b) and POLB (c). Upper panel: Acceptor fluorescence upon direct acceptor excitation (AA; blue trace). Donor (DD, green trace) and acceptor (DA, red trace) fluorescence upon donor excitation. Lower panel: FRET efficiency E* (black trace) with fitted HMM model (magenta trace). (d) Pie chart of the number of detectable steps in KF polymerization traces (sensor +25/−7). A total of 43 polymerization events were analysed. (e and f) Histograms of polymerization durations in KF (e) and POLIIIα (f) polymerization traces (sensor +25/−7). (g and h) Single-molecule time traces showing polymerization of sensor +12/−12 by KF. Peak positions and the HMM fit to the polymerized state (used for calculation of durations) are indicated. (i) Histogram of polymerization duration in KF polymerization traces (sensor +12/−12). More polymerization traces of KF, POLIIIα and POLB can be found in Fig. S2–S5 (ESI†). | ||
Using the +12/−12 sensor and KF, we observed that polymerization is often preceded by a clear increase in E* (Fig. 4g and h). We attribute this increase to reorganization of the template just before polymerization, similar to the increase in E* we observed in the binding experiments (Fig. 3). It is unclear, however, to what extent the polymerization reaction has already started before we begin to observe the signature decrease in E*. We determined the duration of the polymerization to be 1.1 ± 0.5 s (N = 30, Fig. 4h).
Additionally, we tested the performance of the +25/−7 sensor at the ensemble level for KF using a microwell plate reader (Fig. S6, ESI†). We further examined the influence of the fluorescent labels on polymerization reaction rates using gel-shift assays by comparing labelled and unlabelled DNA constructs (ESI,† Fig. S7–S9). From the data, we conclude that the rates of DNA synthesis are comparable, with a ∼2 fold decrease seen for the labelled constructs measured at 6 °C. Furthermore, we note that the general evaluation of processive polymerization remains challenging.50,51
Polymerization can be paused at specific points by tuning dNTP concentrations
As shown above, the limited processivity of KF often prevents the completion of DNA synthesis in a single step. We asked whether we could pause DNA synthesis at specific positions and whether we could resolve the addition of single bases to the primer template. We therefore designed the sequence of our template such that omitting certain dNTPs in the reaction mixture will prevent the polymerase from continuing DNA synthesis at well-defined positions. While dTTP is necessary to synthesise the first two bases, addition of dATP will lead to an extension of the primer by a single base. Additional dCTP allows for continuation of DNA synthesis halfway through the template. To determine the FRET efficiencies of such artificially created pausing sites, we first ran a series of polymerization reactions in a cuvette in which we left out different subsets of dNTPs. As expected, the E* histograms show partial polymerizations indicated by a decrease of the mean FRET value upon addition of a subset of dNTPs (Fig. 5a and b). For convenience, we added the template sequence and indicated the sites where synthesis can be halted (Fig. 5c). Remarkably, our data show for the +25/−7 sensor that extensions by two bases (addition of dTTP) and even by a single base (addition of dTTP and dATP) lead to a significant shift in the peak position (Fig. 5a, for a detailed analysis of the peak position and associated fitting errors please see the ESI†). An interesting deviation from the general trend is seen for the +12/−12 sensor, where the synthesis after addition of 3 out of 4 dNTPs leads to a peak position lower than after full synthesis (Fig. 5b). We attribute this to an altered mean position of the donor fluorophore when polymerization continues to form a double helix around the dye.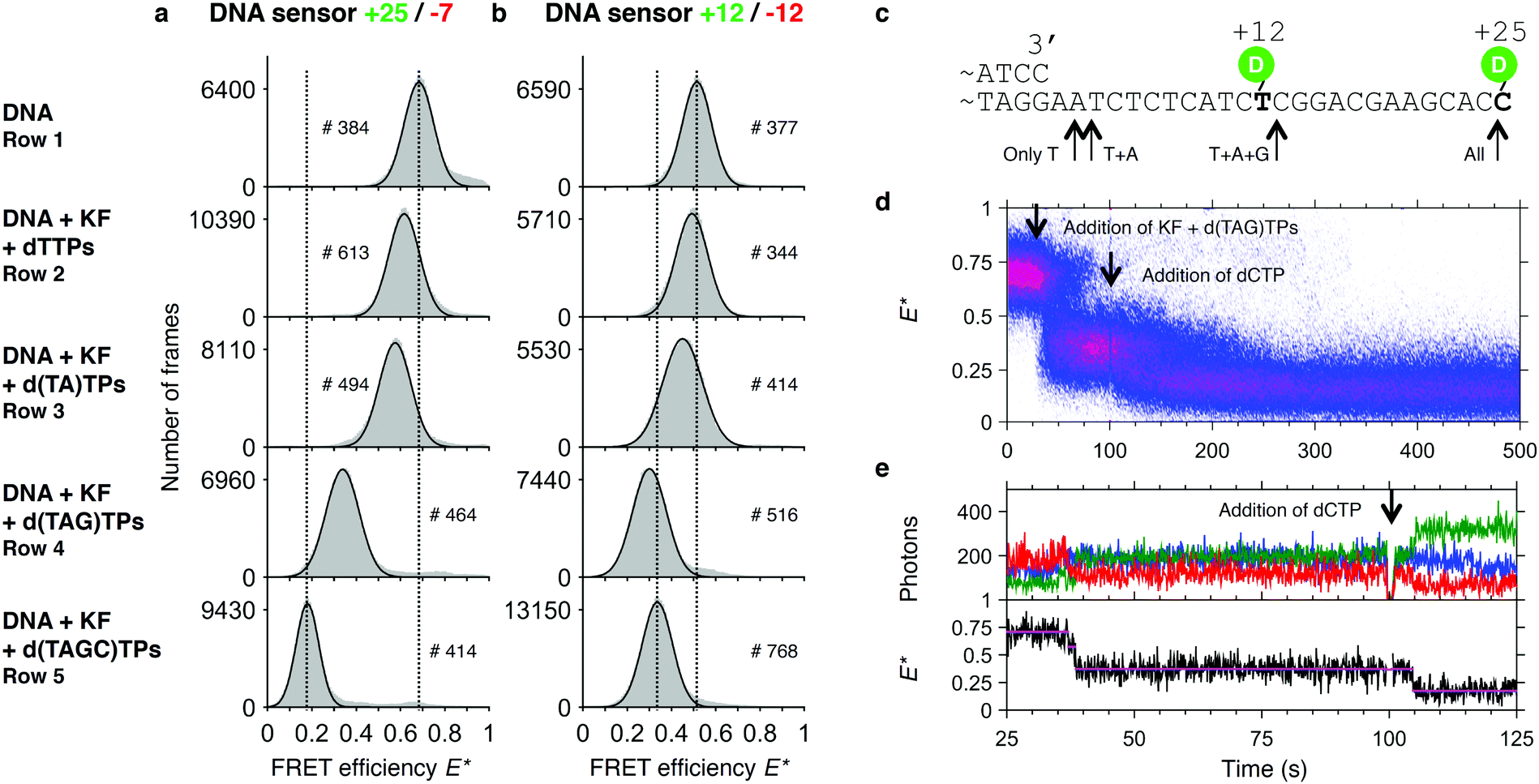 | ||
| Fig. 5 Assay to test pausing of DNA synthesis. Histograms of FRET efficiencies E* for the +25/−7 (a) and the +12/−12 (b) DNA sensor. Each histogram (100 bins shown in grey) was derived from individual time traces obtained from fitting an indicated number of molecules (#). The histograms were fitted to a single-Gaussian model (black line) to indicate the mean peak position of the dominant FRET species. Vertical dashed lines were added for visual guidance. First row: FRET efficiencies of the native DNA sensor. Second to fourth row: FRET efficiencies after incubating the DNA sensor for 90 minutes in presence of Klenow Fragment and the indicated subset of dNTPs. The histograms indicate that polymerization is only completed if all 4 dNTPs are provided, and that polymerization is otherwise halted at different positions along the template. Peak positions of the fits and the corresponding errors can be found in ESI† Table S1. (c) Part of the DNA sensor sequence with the stop positions on the template indicated by arrows. (d) Plot of 74 accumulated polymerization traces of the +25/−7 sensor, obtained from a single experiment. Polymerizations are only finished (E* < 0.25) after dCTP is added to the reaction mixture. (e) Example trace of a polymerization reaction from (d). | ||
In a real time polymerization experiment in which dCTP is initially omitted from the reaction mixture, we can show that polymerizations only finish after addition of dCTP at t = 100 s (Fig. 5d and e).
Discussion
We developed a fluorescently labelled DNA sensor reporting on DNA synthesis by a change in the FRET efficiency between two fluorophores placed on the DNA template and the DNA primer, respectively. Using single-molecule TIRF microscopy, we characterized three different polymerases that vary in speed and cellular function. We showed for all three polymerases (KF, POLIIIα and POLB) that the FRET efficiency of the sensor decreases upon polymerization of DNA. Examination of the inter-dye distances in the final products indicated that the sensors are fully polymerized. Real-time single-molecule experiments showed expected differences in polymerization speed and processivity. While polymerization by POLIIIα was fast, KF and POLB were noticeably slower. This finding can be explained by the role of these two polymerases in DNA repair, which does not require fast DNA synthesis as it is the case for DNA replication. Rates obtained from single-molecule studies for primer extension by KF vary between 1 and 30 nt s−1, but are known to depend strongly on template sequence, the base that is being incorporated and whether the synthesis is in the initial or steady state.13,19,22,52 Our assay finds broad distributions of polymerization durations, which is indicative of the large variation in speed even between polymerase molecules of the same species. We found the mean duration for polymerization of a 25 nt long template to be 1.6 ± 0.5 s for KF, which is in the same order of magnitude as the other single-molecule studies mentioned above. We found that most polymerization events required more than one step for completion caused by either pausing or dissociation of the DNA polymerase. Ensemble studies on a homopolymeric template showed that full-length POL I incorporates 40–50 nucleotides per binding time.53Our data also showed a conformational change in the single-stranded template overhang upon binding of KF, as indicated by a population of molecules at higher E*. Apparently, KF can cause a structural rearrangement that results in a decreased donor–acceptor distance. In fact, co-crystal structures of a structural homologue of KF suggest that the single stranded overhang of the templating DNA might be aligned with the help of residues of the fingers-subdomain.54 A potential explanation for this template folding could lie in the accessibility of the substrate allowing the incoming dNTP to be more easily incorporated.
Our assay provides an easy and robust way of testing the effect of polymerase inhibitors or compare polymerase mutants with each other. Recently, Sharma et al.28 proved with a comparable sensor design that dequenching of donor fluorescence is a reliable method to test the effect of reverse transcriptase inhibitors. We note that the sequence of the template strand may be modified to include sequence specific pausing sites such as Pyr-G-C motifs,55 triplet repeats,56,57 or secondary structures introduced for example as hairpins6 or G-quadruplexes.58,59 The extent to which these modifications itself may influence the change in FRET efficiency needs to be carefully assessed, but especially smFRET based on the alternating-laser excitation scheme provides an excellent opportunity to distinguish between FRET or intensity changes caused by either photophysical effects or conformational changes. We further note that even though placing a fluorophore close to the binding site can weaken the binding of the DNA polymerase, this spatial proximity could be used to develop assays based on protein induced fluorescence enhancement, which can indicate the presence of bound proteins without necessarily changing the FRET efficiency between the donor and the acceptor fluorophore.48,60,61 Additionally, using a dark quencher62 as an acceptor would allow experiments with single (continuous) colour excitation in which the extent of donor dequenching becomes the measure for the progression of the polymerization reaction.
In our current implementation, the achievable temporal resolution is limited by the rate of data acquisition to 100 ms as two consecutive camera frames of 50 ms duration are computationally merged to provide the three photon streams as discussed in the ESI.† Recently published work suggests, however, that smFRET measurements with 1–3 ms time resolution and simultaneous detection of hundreds to thousands of individual DNA molecules are feasible.41,63 In terms of spatial resolution, both the large FRET change upon full polymerization achievable with the +25/−7 sensor and the single-base pair sensitivity in particular regions of the templating DNA for both sensors are highlighting the robustness and the flexibility of the DNA assay. We note, however, that maximising the FRET change upon polymerisation requires one of the dyes to be placed closer to the 3′ end of the DNA primer. In fact, using the +25/−7 sensor revealed a weakening of the DNA polymerase binding to DNA compared to the +12/−12 sensor thereby potentially affecting the processivity of the polymerase for the first few added bases. We suggest to use a more conservative sensor design for applications aimed at synthesising only a few bases.
We expect that additional improvements on the design of the sensor can further increase the range of potential applications. Labelling the templating DNA, for example, with additional fluorophores on the single stranded overhang, thereby creating a multi-colour FRET arrangement, could increase the range of processivities beyond the 25 bases which we can currently monitor.
An additional advantage of the assay is that it does not require the labelling of DNA polymerases with fluorophores and that experiments on the ensemble level can be carried out with standard spectrophotometric lab equipment. With microwell plate imaging instruments reaching levels of sensitivity sufficient to detect fluorescence from samples present in femtomolar quantities, the time dependent imaging of increasing donor (or decreasing acceptor) intensity upon polymerization of DNA allows to evaluate large DNA polymerase libraries utilising an instantaneous readout. As such, this type of assay promises to be a simple and straightforward tool to characterize speed and processivity of DNA polymerases both at the ensemble and single-molecule level.
Funding
J. H. acknowledges support from a Marie Curie Career Integration Grant [630992]. Funding for open access charge: Marie Curie Career Integration Grant [630992].Acknowledgements
DNA Polymerase I KF was kindly supplied by Catherine Joyce, Tim Craggs and Achillefs Kapanidis. Human Polymerase Beta was kindly supplied by Jamie Towle-Weicksel and Joann Sweasy. We thank the Laboratory of Toxicology (Hans van den Berg) at Wageningen University and Research for providing access to the plate reader. J. H. acknowledges support from a Marie Curie Career Integration Grant (#630992).References
- T. A. Baker and A. Kornberg, in DNA replication., ed. A. Kornberg and T. A. Baker, W. H. Freeman, New York, 2nd edn, 1992 Search PubMed.
- U. Hübscher, S. Spadari, G. Villani and G. Maga, in DNA Polymerases, World Scientific, 2010, pp. 1–58 Search PubMed.
- U. Hübscher, S. Spadari, G. Villani and G. Maga, in DNA Polymerases, World Scientific, 2010, pp. 59–83 Search PubMed.
- S. García-Gómez, A. Reyes, M. I. Martínez-Jiménez, E. S. Chocrón, S. Mourón, G. Terrados, C. Powell, E. Salido, J. Méndez, I. J. Holt and L. Blanco, Mol. Cell, 2013, 52, 541–553 CrossRef PubMed.
- R. K. Saiki, D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis and H. A. Erlich, Science, 1988, 239, 487–491 CAS.
- J. Eid, A. Fehr, J. Gray, K. Luong, J. Lyle, G. Otto, P. Peluso, D. Rank, P. Baybayan, B. Bettman, A. Bibillo, K. Bjornson, B. Chaudhuri, F. Christians, R. Cicero, S. Clark, R. Dalal, A. Dewinter, J. Dixon, M. Foquet, A. Gaertner, P. Hardenbol, C. Heiner, K. Hester, D. Holden, G. Kearns, X. Kong, R. Kuse, Y. Lacroix, S. Lin, P. Lundquist, C. Ma, P. Marks, M. Maxham, D. Murphy, I. Park, T. Pham, M. Phillips, J. Roy, R. Sebra, G. Shen, J. Sorenson, A. Tomaney, K. Travers, M. Trulson, J. Vieceli, J. Wegener, D. Wu, A. Yang, D. Zaccarin, P. Zhao, F. Zhong, J. Korlach and S. Turner, Science, 2009, 323, 133–138 CrossRef CAS PubMed.
- J. Shendure and H. Ji, Nat. Biotechnol., 2008, 26, 1135–1145 CrossRef CAS PubMed.
- M. Fried and D. M. Crothers, Nucleic Acids Res., 1981, 9, 6505–6525 CrossRef CAS PubMed.
- M. T. Walsh, E. E. Roller, K.-S. Ko and X. Huang, Biochemistry, 2015, 54, 4019–4021 CrossRef CAS PubMed.
- A. Langer, M. Schräml, R. Strasser, H. Daub, T. Myers, D. Heindl and U. Rant, Sci. Rep., 2015, 5, 12066 CrossRef CAS PubMed.
- S. Farooq, C. Fijen and J. Hohlbein, Protoplasma, 2014, 251, 317–332 CrossRef CAS PubMed.
- A. Kapanidis and T. Strick, Trends Biochem. Sci., 2009, 34, 234–243 CrossRef CAS PubMed.
- B. Maier, D. Bensimon and V. Croquette, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12002–12007 CrossRef CAS PubMed.
- I. Heller, T. P. Hoekstra, G. A. King, E. J. G. Peterman and G. J. L. Wuite, Chem. Rev., 2014, 114, 3087–3119 CrossRef CAS PubMed.
- F. Olasagasti, K. R. Lieberman, S. Benner, G. M. Cherf, J. M. Dahl, D. W. Deamer and M. Akeson, Nat. Nanotechnol., 2010, 5, 798–806 CrossRef CAS PubMed.
- S. L. Cockroft, J. Chu, M. Amorin and M. R. Ghadiri, J. Am. Chem. Soc., 2008, 130, 818–820 CrossRef CAS PubMed.
- T. Förster, Ann. Phys., 1948, 2, 55–75 CrossRef.
- L. Stryer and R. P. Haugland, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 719–726 CrossRef CAS.
- J. J. Schwartz and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20294–20299 CrossRef CAS PubMed.
- S. Liu, E. A. Abbondanzieri, J. W. Rausch, S. F. J. L. Grice and X. Zhuang, Science, 2008, 322, 1092–1097 CrossRef CAS PubMed.
- E. A. Abbondanzieri, G. Bokinsky, J. W. Rausch, J. X. Zhang, S. F. J. L. Grice and X. Zhuang, Nature, 2008, 453, 184–189 CrossRef CAS PubMed.
- T. D. Christian, L. J. Romano and D. Rueda, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21109–21114 CrossRef CAS PubMed.
- G. W. Evans, J. Hohlbein, T. Craggs, L. Aigrain and A. N. Kapanidis, Nucleic Acids Res., 2015, 43, 5998–6008 CrossRef CAS PubMed.
- J. Hohlbein, L. Aigrain, T. D. Craggs, O. Bermek, O. Potapova, P. Shoolizadeh, N. D. F. Grindley, C. M. Joyce and A. N. Kapanidis, Nat. Commun., 2013, 4, 2131 Search PubMed.
- T. D. Harris, P. R. Buzby, H. Babcock, E. Beer, J. Bowers, I. Braslavsky, M. Causey, J. Colonell, J. DiMeo, J. W. Efcavitch, E. Giladi, J. Gill, J. Healy, M. Jarosz, D. Lapen, K. Moulton, S. R. Quake, K. Steinmann, E. Thayer, A. Tyurina, R. Ward, H. Weiss and Z. Xie, Science, 2008, 320, 106–109 CrossRef CAS PubMed.
- Y. Kim, E.-S. Kim, Y. Lee, J.-H. Kim, B. C. Shim, S. M. Cho, J. S. Lee and J. W. Park, J. Am. Chem. Soc., 2014, 136, 13754–13760 CrossRef CAS PubMed.
- M. J. R. Previte, C. Zhou, M. Kellinger, R. Pantoja, C.-Y. Chen, J. Shi, B. Wang, A. Kia, S. Etchin, J. Vieceli, A. Nikoomanzar, E. Bomati, C. Gloeckner, M. Ronaghi and M. M. He, Nat. Commun., 2015, 6, 5936 CrossRef CAS PubMed.
- K. K. Sharma, F. Przybilla, T. Restle, J. Godet and Y. Mély, Nucleic Acids Res., 2016, 44, e74–e74 CrossRef PubMed.
- R. Roy, A. G. Kozlov, T. M. Lohman and T. Ha, Nature, 2009, 461, 1092–1097 CrossRef CAS PubMed.
- M. C. Murphy, I. Rasnik, W. Cheng, T. M. Lohman and T. Ha, Biophys. J., 2004, 86, 2530–2537 CrossRef CAS PubMed.
- D. E. Barnes and T. Lindahl, Annu. Rev. Genet., 2004, 38, 445–476 CrossRef CAS PubMed.
- W. A. Beard and S. H. Wilson, Biochemistry, 2014, 53, 2768–2780 CrossRef CAS PubMed.
- J. B. Towle-Weicksel, S. Dalal, C. D. Sohl, S. Doublié, K. S. Anderson and J. B. Sweasy, J. Biol. Chem., 2014, 289, 16541–16550 CrossRef CAS PubMed.
- H. Klenow and I. Henningsen, Proc. Natl. Acad. Sci. U. S. A., 1970, 65, 168–175 CrossRef CAS.
- C. McHenry and A. Kornberg, J. Biol. Chem., 1977, 252, 6478–6484 CAS.
- P. J. Fay, K. O. Johanson, C. S. McHenry and R. A. Bambara, J. Biol. Chem., 1981, 256, 976–983 CAS.
- C. S. McHenry and W. Crow, J. Biol. Chem., 1979, 254, 1748–1753 CAS.
- J. Hohlbein, T. D. Craggs and T. Cordes, Chem. Soc. Rev., 2014, 43, 1156–1171 RSC.
- A. N. Kapanidis, N. K. Lee, T. A. Laurence, S. Doose, E. Margeat and S. Weiss, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8936–8941 CrossRef CAS PubMed.
- N. K. Lee, A. N. Kapanidis, Y. Wang, X. Michalet, J. Mukhopadhyay, R. H. Ebright and S. Weiss, Biophys. J., 2005, 88, 2939–2953 CrossRef CAS PubMed.
- S. Farooq and J. Hohlbein, Phys. Chem. Chem. Phys., 2015, 17, 27862–27872 RSC.
- T. Cordes, J. Vogelsang and P. Tinnefeld, J. Am. Chem. Soc., 2009, 131, 5018–5019 CrossRef CAS PubMed.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891–893 CrossRef CAS PubMed.
- S. J. Holden, S. Uphoff, J. Hohlbein, D. Yadin, L. Le Reste, O. J. Britton and A. N. Kapanidis, Biophys. J., 2010, 99, 3102–3111 CrossRef CAS PubMed.
- M. van Dijk and A. M. J. J. Bonvin, Nucleic Acids Res., 2009, 37, W235–W239 CrossRef CAS PubMed.
- S. Kalinin, T. Peulen, S. Sindbert, P. J. Rothwell, S. Berger, T. Restle, R. S. Goody, H. Gohlke and C. A. M. Seidel, Nat. Methods, 2012, 9, 1218–1225 CrossRef CAS PubMed.
- J.-W. van de Meent, J. E. Bronson, C. H. Wiggins and R. L. Gonzalez, Biophys. J., 2014, 106, 1327–1337 CrossRef CAS PubMed.
- E. Ploetz, E. Lerner, F. Husada, M. Roelfs, S. Chung, J. Hohlbein, S. Weiss and T. Cordes, Sci. Rep., 2016, 6, 33257 CrossRef PubMed.
- A. M. Chagovetz, J. B. Sweasy and B. D. Preston, J. Biol. Chem., 1997, 272, 27501–27504 CrossRef CAS PubMed.
- K. Datta, A. J. Wowor, A. J. Richard and V. J. LiCata, Biophys. J., 2006, 90, 1739–1751 CrossRef CAS PubMed.
- J. Rentergent, M. D. Driscoll and S. Hay, Biochemistry, 2016, 55, 5622–5634 CrossRef CAS PubMed.
- T. J. Olsen, Y. Choi, P. C. Sims, O. T. Gul, B. L. Corso, C. Dong, W. A. Brown, P. G. Collins and G. A. Weiss, J. Am. Chem. Soc., 2013, 135, 7855–7860 CrossRef CAS PubMed.
- F. R. Bryant, K. A. Johnson and S. J. Benkovic, Biochemistry, 1983, 22, 3537–3546 CrossRef CAS PubMed.
- S. J. Johnson, J. S. Taylor and L. S. Beese, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3895–3900 CrossRef CAS PubMed.
- D. S. Mytelka and M. J. Chamberlin, Nucleic Acids Res., 1996, 24, 2774–2781 CrossRef CAS PubMed.
- S. Kang, K. Ohshima, M. Shimizu, S. Amirhaeri and R. D. Wells, J. Biol. Chem., 1995, 270, 27014–27021 CrossRef CAS PubMed.
- G. M. Samadashwily, G. Raca and S. M. Mirkin, Nat. Genet., 1997, 17, 298–304 CrossRef CAS PubMed.
- S. L. Noer, S. Preus, D. Gudnason, M. Aznauryan, J.-L. Mergny and V. Birkedal, Nucleic Acids Res., 2016, 44, 464–471 CrossRef CAS PubMed.
- D. Schiavone, S. K. Jozwiakowski, M. Romanello, G. Guilbaud, T. A. Guilliam, L. J. Bailey, J. E. Sale and A. J. Doherty, Mol. Cell, 2016, 61, 161–169 CrossRef CAS PubMed.
- H. Hwang, H. Kim and S. Myong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 7414–7418 CrossRef CAS PubMed.
- E. Lerner, E. Ploetz, J. Hohlbein, T. Cordes and S. Weiss, J. Phys. Chem. B, 2016, 120, 6401–6410 CrossRef CAS PubMed.
- L. Le Reste, J. Hohlbein, K. Gryte and A. N. Kapanidis, Biophys. J., 2012, 102, 2658–2668 CrossRef CAS PubMed.
- M. F. Juette, D. S. Terry, M. R. Wasserman, R. B. Altman, Z. Zhou, H. Zhao and S. C. Blanchard, Nat. Methods, 2016, 13, 341–344 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05919h |
| ‡ Present address: Swammerdam Institute for Life Sciences, University of Amsterdam, Amsterdam, 1000 AB, The Netherlands. |
| This journal is © the Owner Societies 2017 |