
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigating interfacial electron transfer in dye-sensitized NiO using vibrational spectroscopy†
Fiona A.
Black
a,
Charlotte A.
Clark
b,
Gareth H.
Summers
ab,
Ian P.
Clark
c,
Michael
Towrie
cd,
Thomas
Penfold
a,
Michael W.
George
bde and
Elizabeth A.
Gibson
 *ab
*ab
aSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: elizabeth.gibson@newcastle.ac.uk
bSchool of Chemistry, The University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cCentral Laser Facility, Research Complex at Harwell, Science and Technology Facilities Council, Rutherford Appleton Laboratory, Harwell Oxford, Didcot, Oxfordshire OX11 0QX, UK
dDynamic Structural Science Consortium, Research Complex at Harwell, Didcot, Oxfordshire OX11 0FA, UK
eDepartment of Chemical and Environmental Engineering, University of Nottingham Ningbo China, 199 Taikang East Road, Ningbo 315100, China
First published on 22nd February 2017
Abstract
Understanding what influences the formation and lifetime of charge-separated states is key to developing photoelectrochemical devices. This paper describes the use of time-resolved infrared absorption spectroscopy (TRIR) to determine the structure and lifetime of the intermediates formed on photoexcitation of two organic donor–π–acceptor dyes adsorbed to the surface of NiO. The donor and π-linker of both dyes is triphenylamine and thiophene but the acceptors differ, maleonitrile (1) and bodipy (2). Despite their structural similarities, dye 1 outperforms 2 significantly in devices. Strong transient bands in the fingerprint region (1 and 2) and nitrile region (2300–2000 cm−1) for 1 enabled us to monitor the structure of the excited states in solution or adsorbed on NiO (in the absence and presence of electrolyte) and the corresponding kinetics, which are on a ps–ns timescale. The results are consistent with rapid (<1 ps) charge-transfer from NiO to the excited dye (1) to give exclusively the charge-separated state on the timescale of our measurements. Conversely, the TRIR experiments revealed that multiple species are present shortly after excitation of the bodipy chromophore in 2, which is electronically decoupled from the thiophene linker. In solution, excitation first populates the bodipy singlet excited state, followed by charge transfer from the triphenylamine to the bodipy. The presence and short lifetime (τ ≈ 30 ps) of the charge-transfer excited state when 2 is adsorbed on NiO (2|NiO) suggests that charge separation is slower and/or less efficient in 2|NiO than in 1|NiO. This is consistent with the difference in performance between the two dyes in dye-sensitized solar cells and photoelectrochemical water splitting devices. Compared to n-type materials such as TiO2, less is understood regarding electron transfer between dyes and p-type metal oxides such as NiO, but it is evident that fast charge-recombination presents a limit to the performance of photocathodes. This is also a major challenge to photocatalytic systems based on a “Z-scheme”, where the catalysis takes place on a µs–s timescale.
Introduction
Understanding the dynamics of donor–acceptor systems is vital for a number of applications, including harvesting solar energy. This manuscript focuses on photoelectrochemical cells in which charge separation is fundamentally important for the efficiency. Upon illumination of a dye-sensitized solar cell (DSC), charge is passed from the sensitizer to the semiconductor, generating a charge-separated state.1–3 For an efficient device, electron transfer between the excited dye and the redox couple (regeneration) must be faster than recombination between charges in the semiconductor surface and charges in the dye molecules.4–6 Therefore, electron transfer in TiO2-based solar cells and photocatalytic systems has been widely investigated and there has been a great deal of research into increasing the lifetime of the dye excited state and the dye|semiconductor charge-separated state lifetime.7–9 The donor–acceptor nature of some molecules is a beneficial property for photosensitizers in dye-sensitized solar cells. This is because it is anticipated that increasing the spatial separation between the charge in the semiconductor and the dye slows down recombination.10,11 However, Troisi et al. recently questioned whether donor–acceptor character significantly improved dye-sensitized solar cell performance as a statistical analysis of the literature did not reveal a correlation.12Compared to TiO2-based dye-sensitized photoelectrochemical devices (e.g. DSCs), there has been less research into NiO photocathodic systems, despite the promise of these systems for p/n tandem DSCs and dye-sensitized water splitting cells (Fig. 1).10,13 Since dye molecules are better oxidising and reducing agents in their excited state, they can transfer an electron into n-type TiO2 or receive an electron from p-type NiO. The dye−|NiO+ recombination reaction is thought to be one of the main barriers to increasing the efficiency of photoelectrochemical devices based on dye-sensitized NiO electrodes.14–16 Push–pull dyes (e.g.1 in Fig. 2) are designed to encourage charge separation and slow the recombination at the dye|NiO interface.17 To achieve this, 1 is comprised of a carboxylic acid anchoring group for adsorption to a semiconductor surface, an electron-donating triphenylamine, a conjugated thiophene linker group to increase the separation and charge-acceptor groups to shift electron density away from the semiconductor surface (see Fig. 2). p-DSCs incorporating this dye have reached 64% IPCE, making it one of the best performing NiO photosensitizers, and 1 has also been incorporated in proof-of-concept dye-sensitized water-splitting cells with promising results.18–20
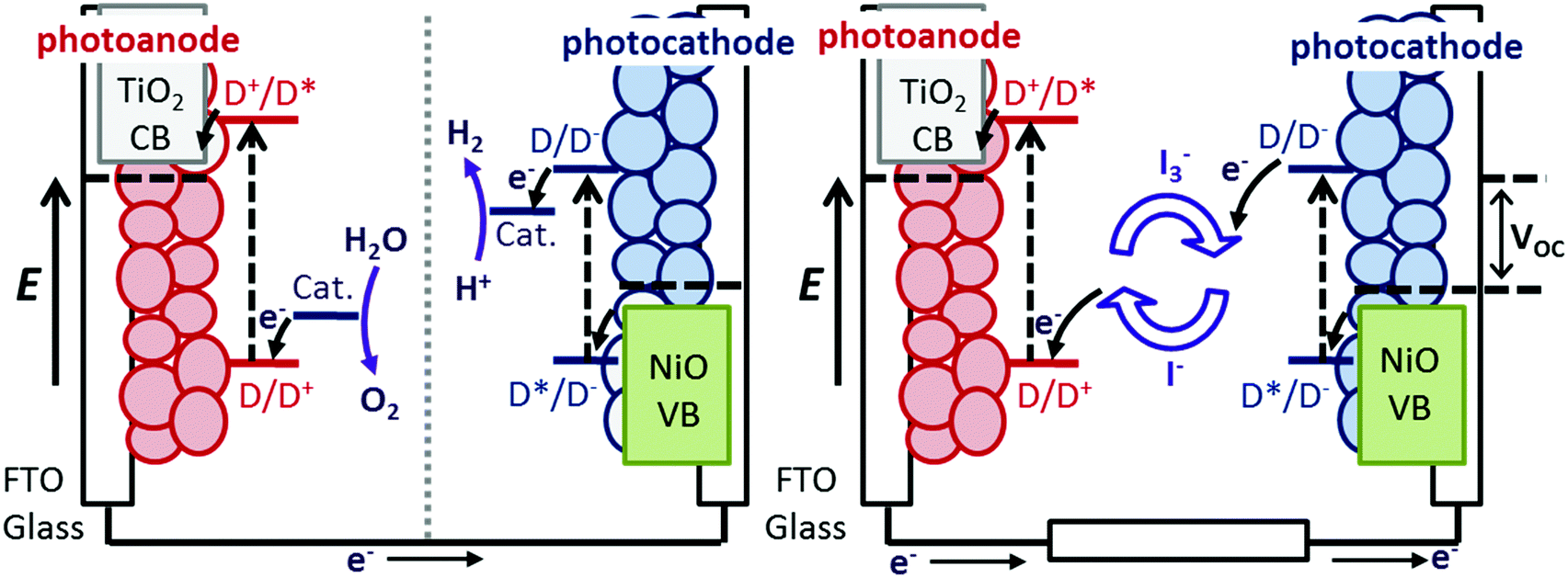 | ||
| Fig. 1 Schematic energy level diagram of a photoelectrosynthesis cell for splitting H2O (left) and a tandem DSC where a n-type photoanode and a p-type photocathode are incorporated into one device (right). | ||
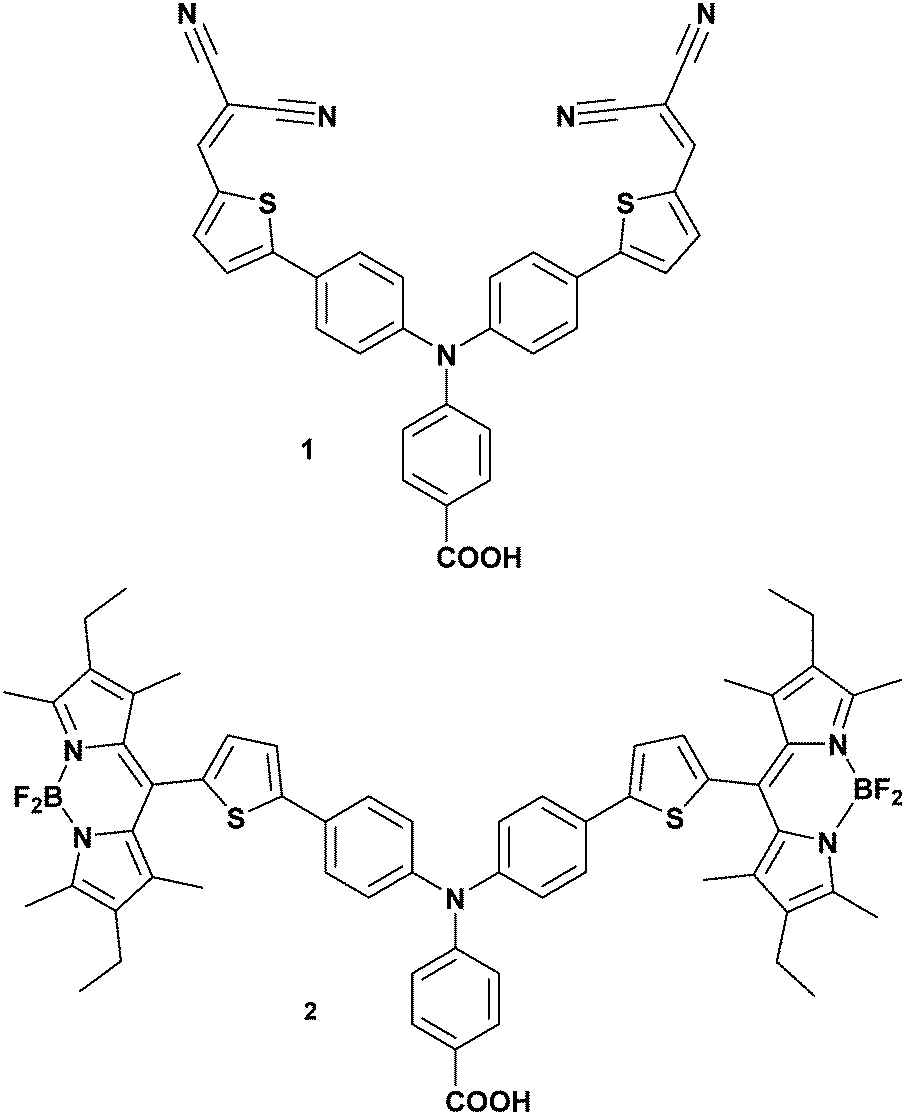 | ||
| Fig. 2 Chemical structure of the dyes investigated in this work. | ||
Dye 2 contains a boradiazacene chromophore rather than nitrile electron withdrawing groups and has twice the molar absorption coefficient of 1. The photoinduced charge-separated state formed with 2-sensitized NiO was previously shown to be three orders of magnitude longer compared to 1|NiO.21 This is possibly due to electronic decoupling of the chromophore from the triphenylamine/thiophene donor motif because of the steric demand of the methyl substituents on the dipyrrene (Fig. 2). Despite the better optoelectronic properties of 2, the IPCE of the corresponding p-DSC was only 28% (Fig. S1, ESI†).21 Indeed, since 1 was reported in 2008, the IPCE has been matched by only one dye, the perylenemonoimide–oligothiophene–triphenylamine triad reported by Nattestad et al.22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ‡ The reason why 1 is a better dye in p-DSCs compared to the numerous variants reported since 2008 is not clear. Furthermore, we have found that dye 2 generated a substantially lower photocurrent than 1 when incorporated in a photoelectrochemical water splitting device. Consequently, to shed new light onto the contrasting performance of these two dyes and the interfacial electron transfer, we have used ground state and time resolved vibrational spectroscopy with a focus upon determining if the success of 1 is linked to the electronic structure of the photoinduced charge-separated state formed at the NiO surface.
‡ The reason why 1 is a better dye in p-DSCs compared to the numerous variants reported since 2008 is not clear. Furthermore, we have found that dye 2 generated a substantially lower photocurrent than 1 when incorporated in a photoelectrochemical water splitting device. Consequently, to shed new light onto the contrasting performance of these two dyes and the interfacial electron transfer, we have used ground state and time resolved vibrational spectroscopy with a focus upon determining if the success of 1 is linked to the electronic structure of the photoinduced charge-separated state formed at the NiO surface.
Experimental method
1 was synthesised using the method reported by Qin et al.17 and 2 was prepared according to Lefebvre et al.21Preparation of dyed NiO films
The NiO films were prepared using a similar method to the preparation of photocathodes for dye-sensitized solar cells and photoelectrosynthesis cells in our group. A saturated solution of NiCl2 in acetylacetone was sprayed onto one side of a CaF2 window, which was pre-heated to 450 °C on a hotplate; this was then allowed to cool slowly to room temperature to give a compact film of NiO. The mesoporous layer was then deposited on top of the compact layer using an F108-templated precursor solution containing NiCl2 (1 g), Pluronic® co-polymer F108 (1 g), Milli-Q water (3 g) and ethanol (6 g) using a Scotch tape mask and the excess was removed by doctor blade. The film was annealed at 450 °C for 30 minutes and an additional layer of precursor solution was applied and annealed to increase the film thickness. The films were then submerged in a dye-bath containing a solution of dye (approx. 0.3 mM) in CH2Cl2 (2) or CH3CN (1) overnight. Prior to each measurement the surfaces were rinsed with solvent and UV-visible absorption spectra were taken.Sample preparation
Samples on NiO that we ran in the presence of electrolyte comprised of 3 mM I2 and 0.1 M LiI (a tenth of the LiI concentration and a third of the concentration of I2 used in a typical solar cell) dissolved in the specified solvents. This particular combination was chosen for comparison with previous work.21Time-resolved infrared spectroscopy
TRIR spectra were recorded using the ULTRA instrument located in the Central Laser Facility at the Rutherford Appleton Laboratory. The data were recorded using time-resolved multiple probe spectroscopy (TRMPS).23 Briefly, two Ti:sapphire amplifiers 10 kHz and 1 kHz were synchronized using a common 65 MHz oscillator. The 1 kHz output was used as a pump and the 10 kHz as probe, to permit a pump–probe–probe–probe… data recording scheme. The pump laser was tuned to 532 nm by optical parametric amplification (OPA). The mid-IR probe was generated using an OPA with difference frequency mixing. The pump–probe time delay was controlled up to 100 µs using a combination of electronic and optical delay. Full details of the TRIR setup has been provided in ref. 23. The spot sizes in the sample region were ca. 150 µm and 50 µm for the pump and probe respectively, with pump energies of 200 nJ. For all measurements the pump polarization was set to magic angle relative to the probe.All spectra were recorded in solution cells (Harrick Scientific Products Inc.) with CaF2 windows and PTFE spacers. For samples prepared in solution a 390 µm path-length was used. In all experiments the cell was rastered in the two dimensions orthogonal to the direction of beam propagation to minimise localised sample decomposition. Single point analysis was used to fit the evolution of the peak maxima over time. Peaks were fitted to multiexponential decay functions using the minimum number of parameters required. Lifetimes discussed in the text are amplitude weighted averages calculated using the formula:
| τavg = ((A1 × τ1) + (A2 × τ2)/(A1 + A2)). |
Results
In the following sections time-resolved infrared spectroscopy (TRIR) is used to provide both structural information regarding the excited state and the lifetimes of the transient species. The kinetics are determined for the dyes in solution, adsorbed on NiO films in the presence and absence of an I−/I3− electrolyte. We also tested our dye-sensitized NiO films in the presence of a hydrogen evolving catalyst, [Co(dmg-BF2)2(OH2)2].24 In our experiments, no difference in spectral shape or lifetime was observed for either dye-sensitized NiO film in the presence of the catalyst compared to the dye-sensitized films in the absence of the catalyst. Therefore, we have focused our discussion on dye-sensitized solar cells. The bands have been assigned by comparison with ground state ATR FT-IR spectra of the components (Fig. S2, ESI†) and DFT calculations. Lifetimes discussed in the text are amplitude weighted averages, as described above. The charge transfer processes discussed are shown in Fig. 3.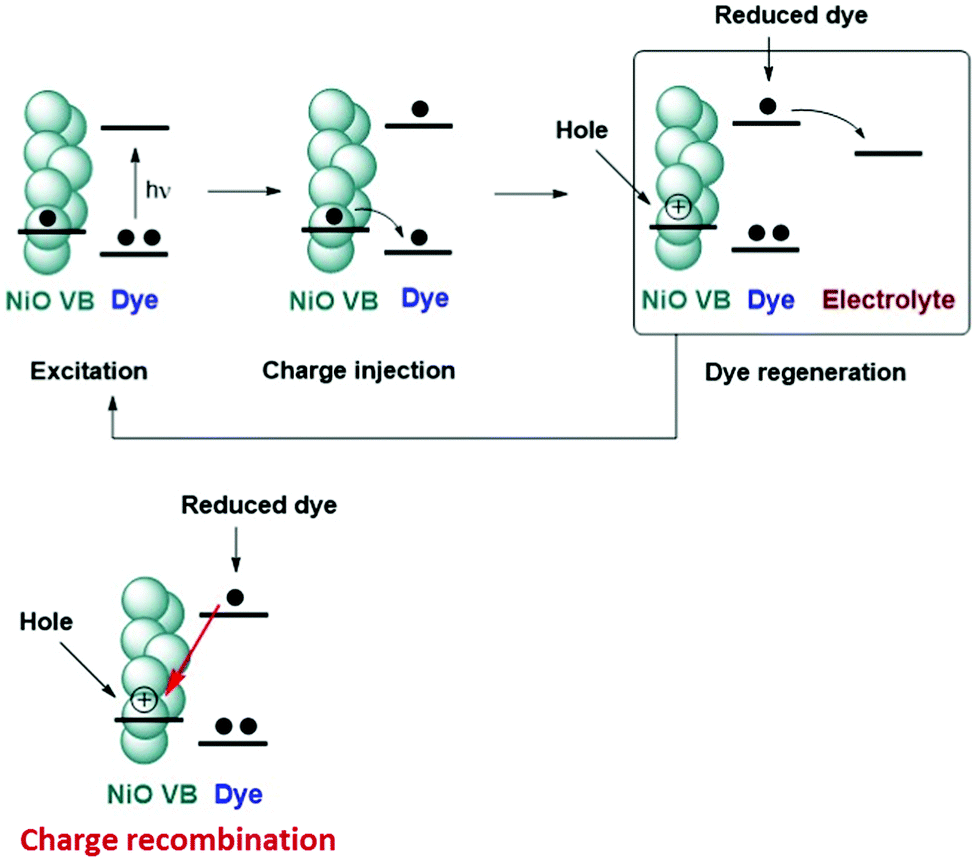 | ||
| Fig. 3 Key excitation and electron transfer processes in a p-type dye-sensitized solar cell. | ||
Solution experiments
Initially we investigated the photophysical behaviour of 1 in CH2Cl2, Fig. 4(a) shows the TRIR spectra for 1 in CH2Cl2 at a range of time delays. The transient features are formed within the time resolution of the instrument and decay to reform the parent bands within τ = ca. 1.1 ns. In the fingerprint region, a number of key features are present. Following photolysis the parent band at 1719 cm−1 assigned to the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) of the CO2H anchoring group bleaches and a band at 1742 cm−1 is formed in this region. This shift to higher energy is consistent with the expected charge transfer from the anchoring CO2H group to the CN acceptors. The direction and magnitude of the shift (+23 cm−1) are similar to those reported previously for structurally similar dyes and are in agreement with the density difference plots shown in Fig. 6.25 A shift in band position is also observed for the band at 1407 cm−1, which we have assigned as associated with the CC stretch of the thiophene. This shift to lower energy relative to the ground state is consistent with an increase in electron density on the π-linker upon photoexcitation. Positive features at 1583 to 1561 cm−1 and 1498 to 1447 cm−1 are assigned to phenyl stretches associated with the triphenylamine. The spectral shape is consistent with electrochemical oxidation of polytriphenylamine reported by Kvarnström et al.26 Further evidence for the charge-transfer excited state formed following photolysis can be seen in the ν(CN) region. Photolysis results in bleaching of the 2234 cm−1 band and appearance of new broad bands at 2178 and 2146 cm−1. The shift to lower energy in the position of the transient bands in relation to the parent provides further evidence for increased electron density on the maleonitrile acceptor groups following irradiation. The presence of multiple transient bands is indicative of a reduction in molecular symmetry in the excited state. The breadth of these bands is indicative of multiple conformations, however each was fit best to a monoexponential decay and the time constants for each agreed within the error margins.
O) of the CO2H anchoring group bleaches and a band at 1742 cm−1 is formed in this region. This shift to higher energy is consistent with the expected charge transfer from the anchoring CO2H group to the CN acceptors. The direction and magnitude of the shift (+23 cm−1) are similar to those reported previously for structurally similar dyes and are in agreement with the density difference plots shown in Fig. 6.25 A shift in band position is also observed for the band at 1407 cm−1, which we have assigned as associated with the CC stretch of the thiophene. This shift to lower energy relative to the ground state is consistent with an increase in electron density on the π-linker upon photoexcitation. Positive features at 1583 to 1561 cm−1 and 1498 to 1447 cm−1 are assigned to phenyl stretches associated with the triphenylamine. The spectral shape is consistent with electrochemical oxidation of polytriphenylamine reported by Kvarnström et al.26 Further evidence for the charge-transfer excited state formed following photolysis can be seen in the ν(CN) region. Photolysis results in bleaching of the 2234 cm−1 band and appearance of new broad bands at 2178 and 2146 cm−1. The shift to lower energy in the position of the transient bands in relation to the parent provides further evidence for increased electron density on the maleonitrile acceptor groups following irradiation. The presence of multiple transient bands is indicative of a reduction in molecular symmetry in the excited state. The breadth of these bands is indicative of multiple conformations, however each was fit best to a monoexponential decay and the time constants for each agreed within the error margins.
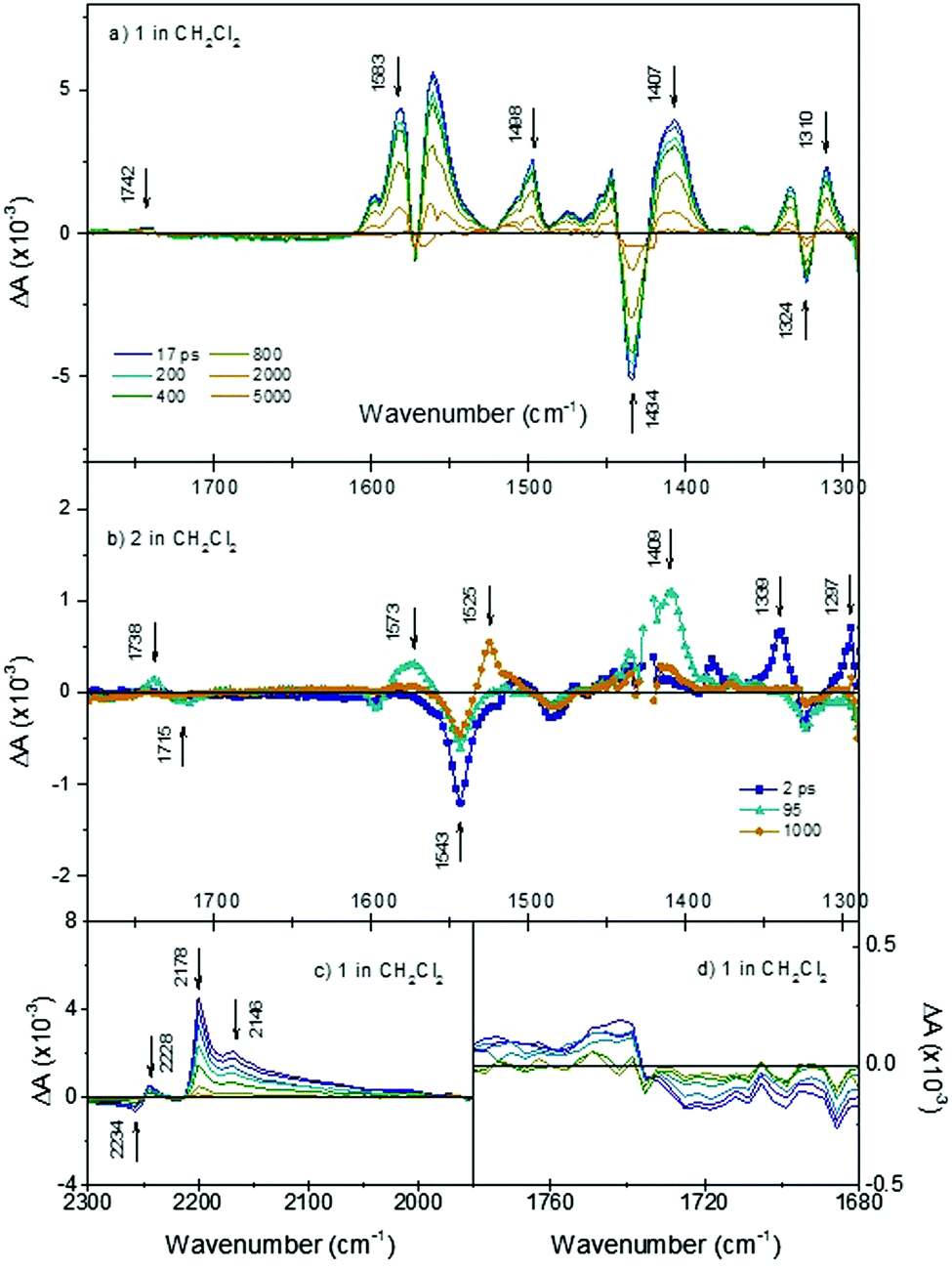 | ||
Fig. 4 TRIR spectra after excitation at 532 nm of (a) 1 in CH2Cl2 across the fingerprint regions at various time delays; (b) 2 in CH2Cl2 fingerprint region at 2 ps (blue line and squares); 95 ps (cyan line and triangles) and 100 ps (orange line and circles); (c) 1 in CH2Cl2 in the ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) region; (d) 1 in CH2Cl2 in the ν(CO) region. N) region; (d) 1 in CH2Cl2 in the ν(CO) region. | ||
Upon changing the solvent to MeCN, the observed spectral features were identical, but the lifetime is shortened to ∼35 ps, in agreement with the transient absorption spectra reported by Qin et al.18 In the previous publication, this was attributed to aggregation and intramolecular charge separation, generating P1−/P1+ in polar solution. The TA signals for 1 are extremely broad and shift over time which makes their analysis challenging. In the TRIR, there is no difference in the spectral shape for 1 in CD3CN compared to 1 in CH2Cl2, which we would have expected if 1−/1+ was generated. Therefore, the interpretation in ref. 18 is inconsistent with the present data. The absorption and emission spectra for 1 in MeCN are strongly red-shifted compared to THF or CH2Cl2 (E00 for 1 in CH2Cl2 is 2.24 eV, λabs = 485 nm & λem = 620 nm). Therefore, we propose that the origin of the decrease in lifetime is likely to be a lowering in energy of the lowest excited states, which have charge transfer character, due to the higher polarity of the solvent environment.
The dynamics of the TRIR spectra indicate more complex photophysical processes for 2 than observed for 1. Fig. 4(b) shows the TRIR spectra at selected time-delays for 2 in CH2Cl2 (detailed spectra are shown in Fig. S5, ESI†). Immediately following photolysis, within ca. 1 ps, a negative bleach at 1543 cm−1 and positive bands at 1339, 1297 and 1507 (weak) cm−1 are apparent and subsequently decay with a time constant of τ = 25 (±5) ps. We have assigned these as the vibrational bands of the singlet excited state of bodipy, based on the similarity with the FT-IR of the ground state of bodipy, and the lifetime is consistent with previous transient absorption spectroscopy.21 On the same timescale as this decay, there is a rise of positive features relating to the carbonyl band of the anchoring group at 1738 cm−1 (shifted +23 cm−1 relative to the ground state bleach at 1715 cm−1) and assigned phenyl band at 1573 cm−1 (shifted −25 cm−1 relative to the ground state bleach) and an intense band at 1409 cm−1. The weaker band which was initially at 1507 cm−1 shifts ca. +10 cm−1. These shifts are similar to those observed for 1 and are indicative of formation of a charge-transfer state (TPA+–bodipy−). The bands associated with this secondary transient species decay with τ = ca. 500 (±70) ps. The band at 1409 cm−1 is in a similar region to a band which we associated with the thiophene linker in 1 (above), based on a comparison with the ground state absorption spectrum (Fig. S2, ESI†). However, we also observe a band grow in at this frequency when a simple bodipy dye is reduced electrochemically (see Fig. S8, ESI†). Finally, a third transient grows in over ca. 400 ps with a sharp band at 1525 cm−1, and decays with τ2 = 760 (±130) ns. This is consistent with a long-lived triplet excited state of bodipy previously observed with ns transient optical absorption spectroscopy.21
The spectral features for 2 are slightly altered when changing the solvent from CH2Cl2 to CD3CN (see Fig. S6, ESI†). In CD3CN the transient band associated with the carbonyl group, shifts +11 cm−1 relative to 2 in CH2Cl2, and the sharp band associated with the thiophene is shifted +17 cm−1 in CD3CN. In addition, no formation of the band at 1525 cm−1 assigned as the locally excited triplet (3LE) state of the bodipy is observed. In general, the overall band shift is less than for the CH2Cl2 case. The change of solvent has a distinct effect on the dynamics. In CD3CN, three bands at 1507, 1340 and 1297 cm−1 were observed to form within the time resolution of the experiment. These bands, which we have assigned to the singlet excited state of bodipy, decay quickly (τ = 7 to 18 ps). Subsequently, bands at 1729, 1570, 1497 and 1416 cm−1 grow within ∼10 ps and decay with τ = ca. 60 ps. These bands are assigned to the charge-transfer state and decay around 10 times faster than in CH2Cl2 (τ = ca. 500 ps).
Experiments using dye-sensitized films
Fig. 5(a) shows the TRIR of 1 adsorbed on NiO (1|NiO) in a solvent free environment. In contrast to 1 in solution, the kinetics associated with each band is typically heterogeneous with 95% of the signal decaying with an average τ = ca. 75 ps. This lifetime which is shorter than the two solvent measurements is in agreement with previous transient optical absorption spectroscopy.18 The transient signal of 1|NiO exhibits a number of distinct differences from the solution measurements shown in Fig. 4(a). The positive features at 1583 to 1553 cm−1 corresponding to phenyl stretches are narrower than the bleaches at 1595 and 1572 cm−1, masking the transients. The band at 1407 cm−1 is absent, instead a strong positive band at 1509 cm−1 and a smaller band at 1382 cm−1 are present. The size of the signals for 1|NiO are considerably smaller than those recorded in solution. Comparing the two conditions, this is most apparent for the broad absorption bands at 2178 cm−1 and 2146 cm−1 in CH2Cl2 solution, which are significantly reduced in intensity. For 1|NiO the negative band at 2228 cm−1, corresponding to the bleach of the ground state, and the absorption at 2217 cm−1 are shifted to lower energy by 6 cm−1 relative to the ground and excited state spectra of 1 in CH2Cl2 solution (Fig. S4, ESI†). Additionally, a transient band at 2171 cm−1 is also present. These slight differences in the shape of the transients corresponding to the nitrile bands could be due to slight differences in the localisation of electron density in the dye|NiO charge-separated state compared to the intramolecular charge-transfer state (vide infra). Zanni et al. have resolved multiple absorption bands, which were closely spaced, due to multiple conformations of a Re dye on the surface of TiO2 by applying 2D IR spectroscopy.27 This technique is well suited to dye-sensitized semiconductors because signals due to injected charges, which absorb broadly across the IR region, are not present. It is possible that 2D IR could be used to resolve the shape and kinetics of the components in the nitrile region in our system.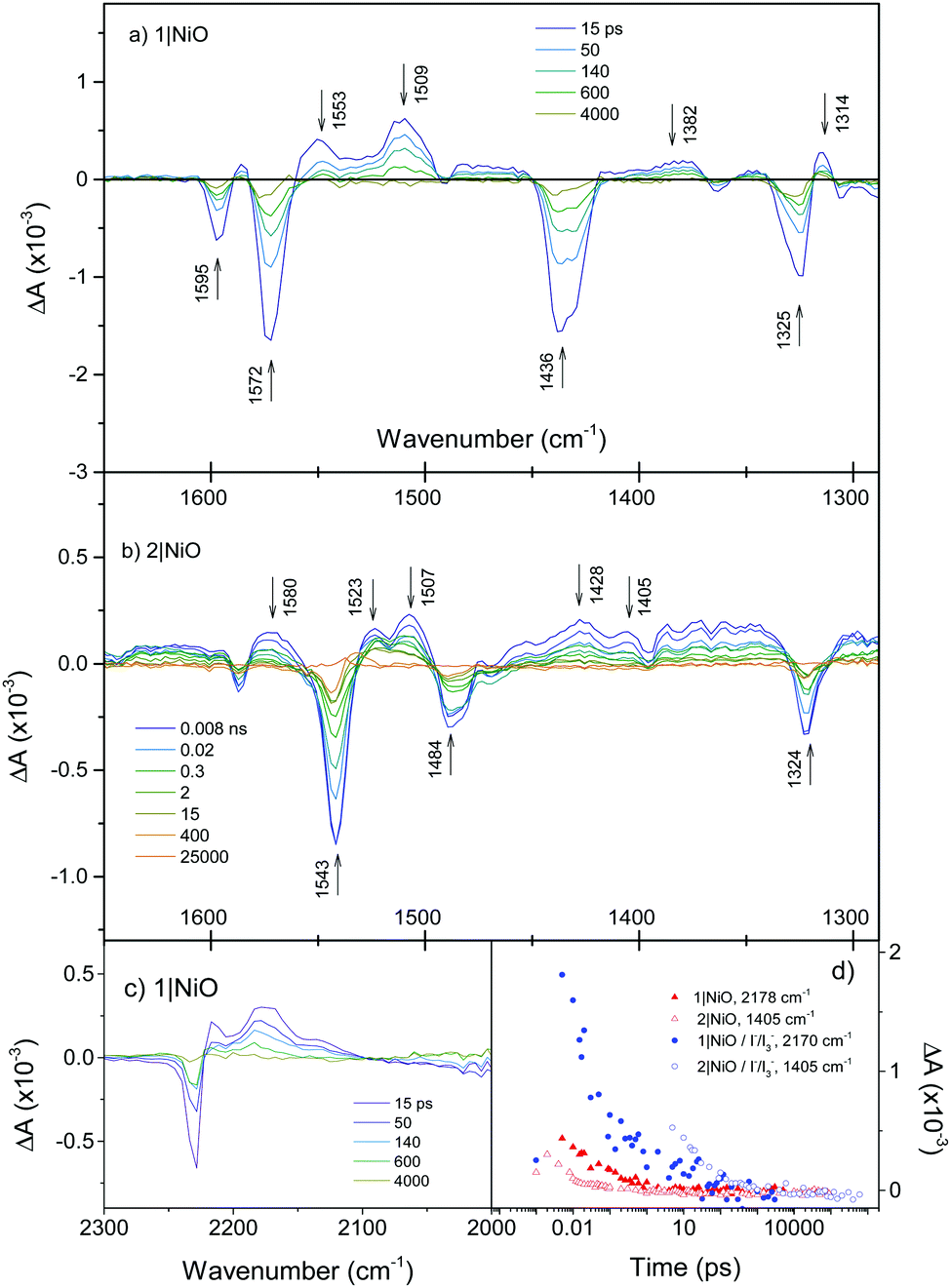 | ||
| Fig. 5 (a) TRIR spectra of 1|NiO; (b) 2|NiO; (c) 2|NiO in the ν(CN) region. (d) Decay of reduced species 1−|NiO+ and 2−|NiO+ (red), decay of reduced species 1−|NiO+ and 2−|NiO+ in the presence of I−/I3− (blue). We have chosen 1405 cm−1 as a small absorption band grows in here when a similar bodipy is reduced electrochemically (Fig. S8, ESI†). | ||
When 1|NiO was excited in the presence of I3−/I− electrolyte in CD3CN and also repeated in CH3CN, the spectral features were unchanged apart from subtle differences at ca. 1370 and 1470 cm−1 and an increase in intensity at 2173 and 2130 cm−1 (Fig. S4, ESI†). However, slower kinetics were exhibited, with 96% of the signal decaying with τ = ca. 180 ps (Fig. 5(d)). Switching the solvent to propylene carbonate did not appear to change the dynamics significantly. The band at 1555 cm−1 decays faster than the others consistently in 1|NiO and 1|NiO|I3−.
The TRIR spectra of 2|NiO at selected time delays are shown in Fig. 5(b). As for 2 in solution, a strong bleach at 1543 cm−1 (τ ≈ 2–3 ns) corresponds to the ground state of the bodipy. The sharp, long-lived absorption band at 1525 cm−1 observed in the CH2Cl2 solution of 2 is not present in the spectra for 2|NiO. Unlike the solution measurements (Fig. 4(b)), the spectral shape in Fig. 5(b) does not change substantially over time. For example, the bands at 1339 and 1297 cm−1 observed within the first few ps in the solution experiments were not present in the spectra for 2|NiO. A number of features in the 2|NiO spectra are similar to 1|NiO, for example the negative bands at 1595 and 1324 cm−1 and positive features at 1507 and 1580 cm−1. The general spectral shape is similar to that of 2 in solution after ca. 20 ps (i.e. the charge transfer excited state TPA+–bodipy−). However, as was observed with 1, the strong absorption band at 1409 cm−1 observed in the solution spectra for 2 is not apparent in the spectra for 2|NiO. A small feature at 1405 cm−1 is present. Unlike 1|NiO, the kinetics of each band are not consistent with each other, for example, τ ≈ 30 ps at 1580 cm−1, which is similar to the charge-transfer exited state of 2 in CH3CN, whereas τ ≈ 2 ns at 1507 cm−1, which is in better agreement with the bleach at 1543 cm−1. This suggests that there could be more than one species present in the spectrum of 2|NiO. However, because there is no strong absorption band corresponding to the reduced bodipy (Fig. S8, ESI†), it is not possible to estimate the yield of the charge-separated state, 2−|NiO+. This also causes a discrepancy between the TRIR results and the optical transient absorption spectroscopy, which we reported previously.21 In those experiments, an intense and long-lived band corresponding to the reduced bodipy was observed, which enabled us to monitor the formation and decay of the charge-separated state. In the TRIR, while there are regions which contain long-lived signals (e.g. between 1540–1500 cm−1, 12% of the absorption lives for τ ≥ 30 ns) these are broad and difficult to fit reliably. However, the bands which correspond to the charge-transfer excited state of 2 are strong in the TRIR. Therefore, a combination of TRIR and TA spectroscopy gives a clearer picture of the photophysics, enabling us to resolve the dynamics of the charge-separated state and the charge-transfer state.
The TRIR spectra of 2|NiO in the presence of I−/I3− in CD3CN have similar spectral features to 2|NiO (Fig. S7, ESI†) apart from an increase in intensity of the bands at 1405 and 1570 cm−1. The presence of the electrolyte generally extends the lifetime of the transient (Fig. 5(d) and Fig. S11, ESI†) with the exception of the phenyl CC band at 1570 cm−1, which decays with an average lifetime of τ = ca. 380 ps. Neighbouring bands at 1520 and 1505 cm−1 decay on a longer timescale (τ = 10–30 ns). These values are in broad agreement with those measured using transient absorption spectroscopy.
Discussion
In the previous sections, time-resolved IR spectra for two dye molecules have been shown. These studies were performed under three distinct experimental conditions, which systematically approach a realistic dye-sensitized solar cell setup. Importantly, the dyes chosen for the present study have similar redox potentials (i.e. similar driving force for charge separation, recombination and regeneration) and method of binding to NiO. E0–0 = 2.25 eV for 1 compared to E0–0 = 2.27 eV for 2 and ED/D– = −1.46 V vs. FeCp2+/FeCp2.21 However, despite these similarities, they give very different performances in a DSC or dye-sensitized photosynthesis cell. Indeed, with the absorption coefficient of 2 being almost twice that of 1 it is surprising that the quantum efficiency of a device using 2 is approximately half that of 1.Photoexcitation of 1 leads to the lowest (S1) excited state, which as shown by the density difference plot in Fig. 6 is largely a charge transfer excitation, with a small overlap between the electron and the hole in the region of the thiophene bridging groups. In Fig. 6 there is relatively little change in electron density on the carboxylic anchoring group between the ground state and the excited state. This is consistent with the small amplitude of the carbonyl band in Fig. 4(d). Importantly, the close proximity of the hole on the amine group and the electron on the 4-[5-(2,2-dicyanovinyl)thiophene-2-yl]phenyl group promotes charge-separation when the dye is adsorbed to NiO.17,18 For the reduced dye (1−), the electron density in the singly occupied molecular orbital (SOMO) is localised somewhat asymmetrically over one 4-[5-(2,2-dicyanovinyl)thiophene-2-yl]phenyl arm. This localisation is driven by the structural change of 1 both in the excited and reduced states (Fig. 6(c) and (d)).
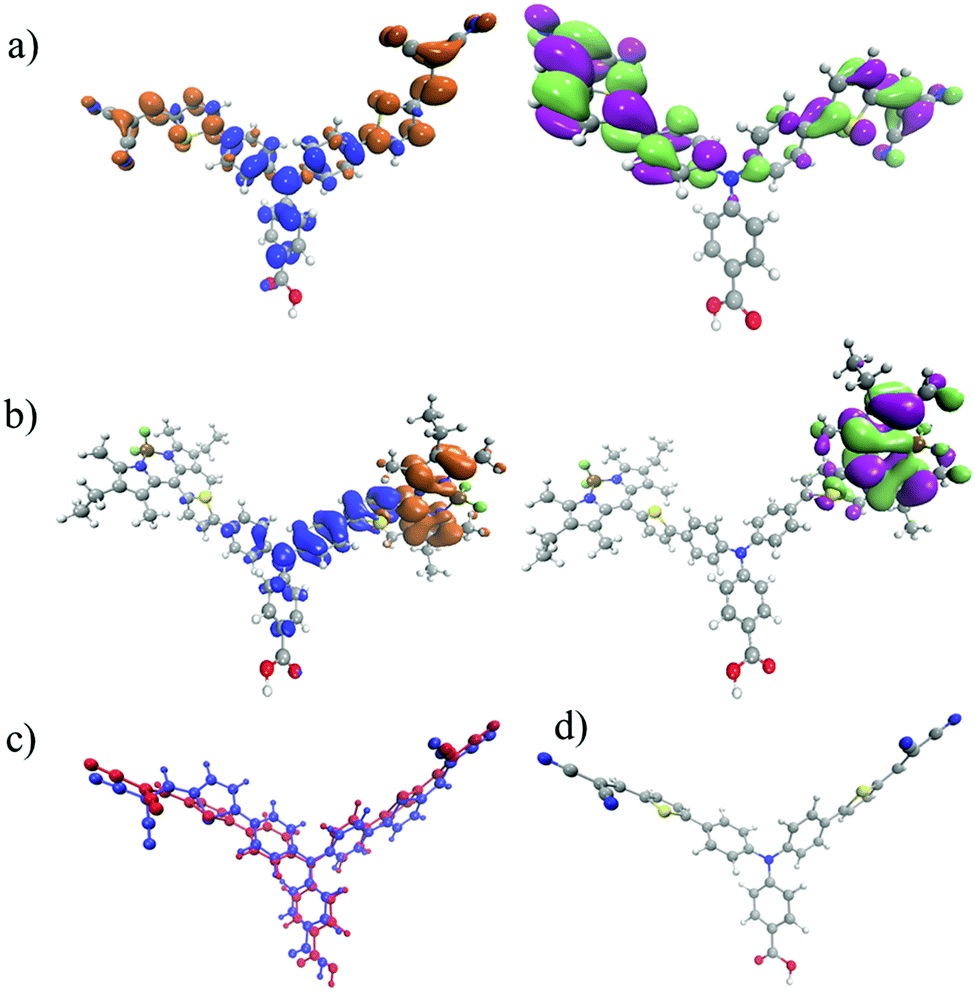 | ||
| Fig. 6 (a) Density difference plot (Δρ = ρS1 − ρS0) between the ground and first excited states of 1 (left) and singly occupied molecular orbital (SOMO) of 1−; (b) density difference plot between the ground and first excited states of 2 (left) and singly occupied molecular orbital (SOMO) of 2−; (c) an overlay of the ground state of 1 (blue) and excited state 1* (red) geometry; (d) a full colour image of 1*. On the left hand side of the molecule there is a large symmetry-breaking structural change. | ||
For 2 the lowest bands visible in the absorption spectrum (Fig. S10, ESI†) are associated with local excitation upon the bodipy unit. However, as shown in Fig. 4(b), when in solution, the TRIR spectra reveal three transient species. The first is the decay of the bodipy localised excited state into an intramolecular charge transfer from aminobenzoic acid to bodipy. This requires the transfer of a hole from the bodipy to the aminobenzoic acid, which occurs with τ = ca. 25 ps. This is rather long, but is unsurprising given the strongly uncoupled (i.e. spatially separated) electron and hole in the charge transfer state, as highlighted in the density difference plot of Fig. 6.
Previous transient absorption measurements in the UV-visible region,21 indicated that this charge-transfer state recombines to form the lowest triplet state, which is a local excitation on the bodipy unit (3LE). The long-lived bleach at 1543 cm−1 and absorption at 1523 cm−1 support that assignment. Importantly this is only observed in low polarity solvents, i.e. dichloromethane. When the same measurements are performed in acetonitrile the 3LE state is not observed, this is because the higher polarity will stabilise the charge transfer state meaning that it is likely to fall below the 3LE state, similar observations have recently been reported for D–A charge transfer complexes.28
As shown in the results above, there are key differences between the solution spectra and those recorded for (i) the dyes adsorbed on the NiO surface and (ii) of films in the presence of electrolyte or an electron acceptor. For example, the triplet state is not observed with 2|NiO. A number of features are present in the fingerprint region of the spectra, which are difficult to resolve fully and we propose that the spectra for the charge-separated state lies underneath the spectra for the excited charge-transfer state. The lifetime of the bands corresponding to the charge-transfer state are extremely short and electron transfer from NiO to the dye must occur within a few 1–2 ps in order to compete with decay to the ground state. The lifetime of the charge-transfer state in both dyes is strongly solvent dependent. While the formation of the charge-transfer state is consistently faster for 1 compared to 2, in CH2Cl2 the decay of the charge-transfer state is faster for 2 than 1 but in MeCN the charge-transfer state decays faster for 1 than 2. Both dyes have shorter lifetimes in MeCN compared to CH2Cl2. Excitation of 1|NiO leads to the rapid formation of 1−|NiO+ and we do not observe the excited state within the time-resolution of our experiments. In contrast, the presence of the charge transfer state of 2 when 2|NiO is excited suggests that electron transfer from the NiO to the dye is slower for 2 than for 1. This is surprising given the identical structure of the anchoring group in both dyes and the similar oxidation potential.
Fig. S10 (ESI†) shows the absorption spectrum of 2 in solution and the π–π* transition thiophene–triphenylamine unit is the origin of the intense band below 450 nm. The results from our prior emission and resonance Raman studies suggest that the 532 nm pump in our TRIR experiments should exclusively excite the bodipy chromophore.21,29,30 We have previously shown that dyes anchored through non-conjugated linkers can exhibit ultrafast photoinduced charge separation at the NiO surface (Process C in Fig. S12, ESI†).31 Paoprasert et al. observed rapid injection from bithiophene to TiO2 in addition to the Re chromophore in their “multi-injecting” system, where the thiophene bridge was decoupled from the Re bipyridine complex.32 However some of the stronger features in the spectra for 2|NiO (Fig. 5(b)) are consistent with those of 1|NiO (Fig. 5(a)) which suggests that they correspond to the triphenylamine moiety. One explanation could be that intramolecular charge transfer occurs before electron transfer from NiO to 2 (Process B in Fig. S12, ESI†). Since the lifetime of the charge-transfer excited state is so short, the charge-separation efficiency is low and this gives rise to the poor solar cell efficiency. An alternative explanation is that this signal corresponds to dyes which do not inject (i.e. they are not reduced at all and simply decay to the ground state without contributing to the photocurrent). Even the combination of optical TA and TRIR does not enable us to confirm whether processes B, C or D in Fig. S12 (ESI†) are preferred. What the TRIR has revealed, that the TA alone did not, is that charge separation is less efficient for 2|NiO than 1|NiO.
In the presence of electrolyte, a general increase in lifetime and amplitude of the signals corresponding to the NiO+|Dye− was observed for both dyes. This makes the evaluation of the regeneration efficiency difficult. There are two possible causes of the increased lifetime. Firstly, the electron acceptor could associate with the dye and stabilise the reduced species. Non-diffusion limited electron transfer on a <ns timescale would be feasible if the electron acceptor is already associated with the dye or the surface pre-excitation.33 Alternatively, I− species could react with NiIII sites known to be present on the electrode surface, effectively “passivating it” so that recombination at the NiO+|1− interface is slowed down. The higher quantum efficiency of the solar cell sensitized with 1 is consistent with more efficient regeneration with 1 than with 2. The longer lifetime of NiO|2|I3− relative to NiO|1|I3− is consistent with this interpretation.
These results confirm that strong electronic coupling is required between the chromophore and the NiO in the ground state for efficient charge transfer to take place. An alternative approach is that of Odobel et al. who appended an electron acceptor to opposite side of the chromophore to the anchoring group.34,35 Our system requires a slight overpotential for charge-separation, whereas theirs requires a slight overpotential for dye-regeneration. Despite their short-lived charge-separated states, the best results we have obtained with I3−/I− have been with charge-transfer dyes, but we note that these dyes have only worked with iodine-based electrolytes.36 The results presented here have prompted us to reconfigure our dyes to increase the electronic coupling in the ground state. TRIR spectroscopy has highlighted the small change in electron density in the anchoring group in both dyes upon excitation. Using this technique we should be able to correlate the shift in electron density away from the anchor with the performance of the dyes in photoelectrochemical devices. This should enable us to optimise our dyes to maximise light induced charge-separation at the dye|semiconductor interface in the future.
Conclusions
Although the charge transfer and recombination kinetics have previously been described for 1 and 2 based on optical methods, the use of TRIR spectroscopy provides a far more detailed picture of the complex series of events in the dyes in solution and adsorbed on NiO following initial excitation. The transient optical absorption spectra of charge-transfer dyes like 1 contain broad features that make it difficult to resolve the excited state from the reduced dye. The infrared-active nitrile groups on 1 make it ideal for probing in the infrared and we confirmed that charge-transfer from NiO to the excited dye occurs rapidly (<1 ps) and efficiently. In contrast, the bodipy groups were more difficult to probe in the TRIR, possibly because of the small structural reorganisation associated with excitation and reduction. The bodipy excited state and reduced state absorb strongly in the visible region. However, the triphenyl amine donor and thiophene linker absorb strongly in the IR. The TRIR experiments have revealed that multiple species are present shortly after excitation of the bodipy chromophore, including the charge-transfer excited state and the fully charge-separated state. This suggests that charge separation is less efficient in 2|NiO than in 1|NiO and this is consistent with the difference in performance between the two dyes in dye-sensitized solar cells and photoelectrochemical water splitting devices.We have also shown that the environment around the dyes affects the photophysics. Therefore, it is important to measure the photoelectrodes under conditions as close as possible to working devices. The I3−/I− electrolyte slowed down the decay of the signals corresponding to the reduced dye, which makes estimating the regeneration efficiency difficult. Further work will investigate the dependence of these dynamics on concentration, light intensity and Fermi-level in the NiO.
Acknowledgements
We thank STFC for access to the CLF ULTRA facility at the Rutherford Appleton Laboratories. EAG thanks the Royal Society for a Dorothy Hodgkin Research Fellowship and 2nd Year Research Grant, the Leverhulme Trust for a Project Grant, the University of Nottingham for an Anne McLaren Research Fellowship and the EPSRC and Newcastle University for a studentship for Fiona A. Black (EPJ5002881). Data supporting this publication is openly available under an ‘Open Data Commons Open Database License’. Additional metadata are available at: http://dx.doi.org/10.17634/154300-42. Please contact Newcastle Research Data Service at rdm@ncl.ac.uk for access instructions.References
- J. B. Asbury, R. J. Ellingson, H. N. Ghosh, S. Ferrere, A. J. Nozik and T. Lian, J. Phys. Chem. B, 1999, 103, 3110–3119 CrossRef CAS.
- J. B. Asbury, Y.-Q. Wang, E. Hao, H. N. Ghosh and T. Lian, Res. Chem. Intermed., 2001, 27, 393–406 CrossRef CAS.
- T. A. Heimer and E. J. Heilweil, J. Phys. Chem. B, 1997, 101, 10990 CrossRef CAS.
- A. Y. Anderson, P. R. F. Barnes, J. R. Durrant and B. C. O. Regan, J. Phys. Chem. C, 2010, 114, 1953–1958 CAS.
- T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925–16928 CrossRef CAS PubMed.
- K. C. D. Robson, K. Hu, G. J. Meyer and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1961–1971 CrossRef CAS PubMed.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- B. C. O’Regan, K. Walley, M. Juozapavicius, A. Anderson, F. Matar, T. Ghaddar, S. M. Zakeeruddin, C. Klein and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 3541–3548 CrossRef PubMed.
- J. N. Clifford, E. Martínez-Ferrero and E. Palomares, J. Mater. Chem., 2012, 22, 12415 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245–2247 RSC.
- C. M. Ip and A. Troisi, J. Phys. Chem. Lett., 2016, 7, 2989–2993 CrossRef PubMed.
- L. Li, L. Duan, Y. Xu, M. Gorlov, A. Hagfeldt and L. Sun, Chem. Commun., 2010, 46, 7307 RSC.
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS.
- A. L. Smeigh, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2012, 48, 678–680 RSC.
- S. Marjorie, C. Houarner-rassin, Y. Pellegrin, E. Blart, F. Odobel, J. Fortage and M. Séverac, J. Photochem. Photobiol., A, 2008, 197, 156–169 CrossRef.
- P. Qin, H. Zhu, T. Edvinsson, G. Boschloo, A. Hagfeldt and L. Sun, J. Am. Chem. Soc., 2008, 130, 8570–8571 CrossRef CAS PubMed.
- P. Qin, J. Wiberg, E. A. Gibson, M. Linder, L. Li, T. Brinck, A. Hagfeldt, B. Albinsson and L. Sun, J. Phys. Chem. C, 2010, 114, 4738–4748 CAS.
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988–990 RSC.
- L. Li, E. A. Gibson, P. Qin, G. Boschloo, M. Gorlov, A. Hagfeldt and L. Sun, Adv. Mater., 2010, 22, 1759–1762 CrossRef CAS PubMed.
- J.-F. Lefebvre, X.-Z. Sun, J. A. Calladine, M. W. George and E. A. Gibson, Chem. Commun., 2014, 50, 5258–5260 RSC.
- A. Nattestad, A. J. Mozer, M. K. R. Fischer, Y.-B. Cheng, A. Mishra, P. Bäuerle and U. Bach, Nat. Mater., 2010, 9, 31–35 CrossRef CAS PubMed.
- G. M. Greetham, P. Burgos, Q. Cao, I. P. Clark, P. S. Codd, R. C. Farrow, M. W. George, M. Kogimtzis, P. Matousek, A. W. Parker, M. R. Pollard, D. A. Robinson, Z.-J. Xin and M. Towrie, Appl. Spectrosc., 2010, 64, 1311–1319 CrossRef CAS PubMed.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D’Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- C. J. Wood, M. Cheng, C. A. Clark, R. Horvath, I. P. Clark, M. L. Hamilton, M. Towrie, M. W. George, L. Sun, X. Yang and E. A. Gibson, J. Phys. Chem. C, 2014, 118, 16536–16546 CAS.
- C. Kvarnström, A. Petr, P. Damlin, T. Lindfors, A. Ivaska and L. Dunsch, J. Solid State Electrochem., 2002, 6, 505–512 CrossRef.
- W. Xiong, J. E. Laaser, P. Paoprasert, R. A. Franking, R. J. Hamers, P. Gopalan and M. T. Zanni, J. Am. Chem. Soc., 2009, 131, 18040–18041 CrossRef CAS PubMed.
- P. L. Santos, J. S. Ward, P. Data, A. S. Batsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- G. H. Summers, J.-F. Lefebvre, F. A. Black, E. Stephen Davies, E. A. Gibson, T. Pullerits, C. J. Wood and K. Zidek, Phys. Chem. Chem. Phys., 2016, 18, 1059–1070 RSC.
- G. H. Summers, G. Lowe, J.-F. Lefebvre, S. Ngwerume, M. Bräutigam, B. Dietzek, J. E. Camp and E. A. Gibson, ChemPhysChem, 2016 DOI:10.1002/cphc.201600846.
- Y. Hao, C. J. Wood, C. A. Clark, J. A. Calladine, R. Horvath, M. W. D. Hanson-Heine, X.-Z. Sun, I. P. Clark, M. Towrie, M. W. George, X. Yang, L. Sun and E. A. Gibson, Dalton Trans., 2016, 45, 7708–7719 RSC.
- P. Paoprasert, J. E. Laaser, W. Xiong, R. A. Franking, R. J. Hamers, M. T. Zanni, J. R. Schmidt and P. Gopalan, J. Phys. Chem. C, 2010, 114, 9898–9907 CAS.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef CAS PubMed.
- L. Le Pleux, A. L. Smeigh, E. Gibson, Y. Pellegrin, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, Energy Environ. Sci., 2011, 4, 2075 CAS.
- J. Warnan, J. Gardner, L. Le Pleux, J. Petersson, Y. Pellegrin, E. Blart, L. Hammarström and F. Odobel, J. Phys. Chem. C, 2014, 118, 103–113 CAS.
- C. J. Wood, G. H. Summers and E. A. Gibson, Chem. Commun., 2015, 51, 3915–3918 RSC.
- Q.-Q. Zhang, K.-J. Jiang, J.-H. Huang, C.-W. Zhao, L.-P. Zhang, X.-P. Cui, M.-J. Su, L.-M. Yang, Y.-L. Song and X.-Q. Zhou, J. Mater. Chem. A, 2015, 3, 7695–7698 CAS.
- K. A. Click, D. R. Beauchamp, B. R. Garrett, Z. Huang, C. M. Hadad and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 26103–26111 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp05712h |
| ‡ Dyes which have achieved higher short-circuit photocurrent density, but lower IPCE, than 1 include an indolium dye (JSC = 8.2 mA cm−2),36 a quinoidal thiophene dye (8.2 mA cm−2)37 and a perylene dye (7.4 mA cm−2).38 |
| This journal is © the Owner Societies 2017 |