
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiiodomethane as a halogen bond donor toward metal-bound halides†
Alexander S.
Novikov
 ,
Daniil M.
Ivanov
,
Margarita S.
Avdontceva
and
Vadim Yu.
Kukushkin
*
,
Daniil M.
Ivanov
,
Margarita S.
Avdontceva
and
Vadim Yu.
Kukushkin
*
Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg, 199034 Russian Federation. E-mail: v.kukushkin@spbu.ru
First published on 3rd April 2017
Abstract
A 1,3,5,7,9-pentaazanona-1,3,6,8-tetraenate (PANT) chloride platinum(II) complex (1) was obtained via the metal-mediated double coupling of 2,3-diphenylmaleimidine with both nitrile ligands in trans-[PtCl2(NCtBu)2]. Compound 1 was then co-crystallized with diiodomethane forming solvate 1·½CH2I2. The XRD experiment reveals that this solvate displays the halogen bonds H2C(I)–I⋯Cl–Pt and hydrogen bonds I2C(H)–H⋯Cl–Pt, which join two complex and one CH2I2 molecules in a heterotrimeric supramolecular cluster. Inspection of the CCDC database reveals only one example of the halogen bond H2C(I)–I⋯I–Pt between the CH2I2 molecule and metal-coordinated halide in the structure of VEMWOA. In VEMWOA, CH2I2 serves solely as a halogen bond donor with no hydrogen bond contribution. Results of the Hirshfeld surface analysis and DFT calculations (M06/DZP-DKH level of theory) followed by topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method) for both 1·½CH2I2 and VEMWOA confirmed the formation of these weak interactions. The evaluated energies of halogen bonds involving CH2I2 are in the 2.2–2.8 kcal mol−1 range.
Introduction
Halogen bonding1 (XB) is one of the most actively studied types of weak interactions2–4 insofar as along with hydrogen bonding, metallophilic interactions, and π-stacking, XB is efficiently used as a tool in crystal engineering and in supramolecular organization.2,3,5 Recent applications of XB include stabilization of explosives6 and synthesis of systems with tunable photophysical properties.7 XBs also play important roles in organocatalysis8 and in the metabolism of biologically active halogen-containing compounds.9In many reports devoted to XB, iodine-based bifunctional XB donors such as 1,2- and 1,4-diiodoperfluorobenzenes were employed because of their availability, large σ-holes, and high solubility in most common organic solvents.2,10 However, more and more attention is now paid to halomethanes, which can also behave as polyfunctional XB donors.11–20 Concurrently, halide complexes of d-metals can be employed as XB acceptors and their combination with halomethanes leads to supramolecular clusters, chains, and 3D networks.17,18,20–22
The halomethanes CBr4,11–13,18–20 CHI3,14,15,17,23 CHBr3,11,12,20 and CFBr3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12,19 are the most commonly used among other halomethanes in the formation of XBs with uncomplexed halides and polyhalometalate anions. Our recent works indicated that even weak XB donors such as CHCl3, CH2Cl2, and CH2Br2 (Fig. 1) are still able to form supramolecular clusters with chloride anions and chloride ligands in metal complexes held by XBs and HBs16,21,22 or by simultaneous XB and HB.16,21,22
12,19 are the most commonly used among other halomethanes in the formation of XBs with uncomplexed halides and polyhalometalate anions. Our recent works indicated that even weak XB donors such as CHCl3, CH2Cl2, and CH2Br2 (Fig. 1) are still able to form supramolecular clusters with chloride anions and chloride ligands in metal complexes held by XBs and HBs16,21,22 or by simultaneous XB and HB.16,21,22
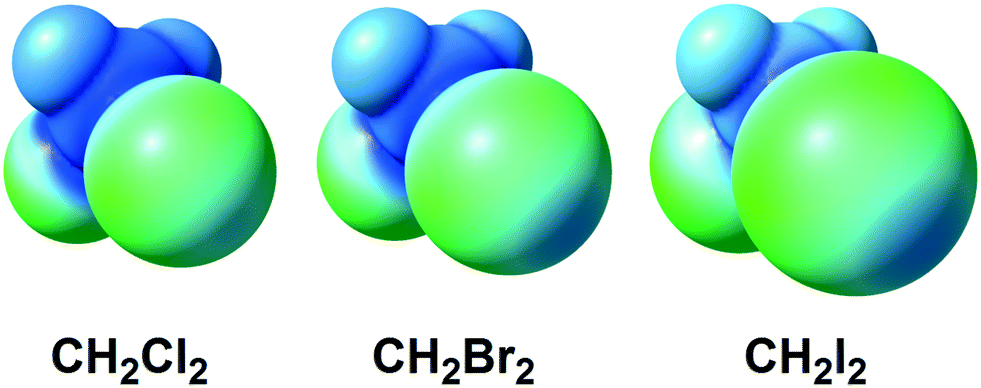 | ||
| Fig. 1 ESP distribution in the CH2X2 (X = Cl, Br, I) dihalomethane molecules (M06-2X/CEP-121G level of theory).24 Green color – negative ESP, blue color – positive ESP. | ||
In contrast to a rather broad application of halomethanes in crystal engineering, diiodomethane (CH2I2) – despite its relatively large σ-holes (Fig. 1; positive potential is up to 23.1 kcal mol−1 on the 0.001 a.u. molecular surface) – was poorly studied25–28 as an XB donor. Nassimbeni et al. reported a series of halomethane solvates of tetrakis(4-bromophenyl)- and tetrakis(4-iodophenyl)ethylenes, where diiodomethane forms H2C(I)–I⋯Br–C and H2C(I)–I⋯I–C contacts assigned25 to XB after comparison of their geometrical parameters with the IUPAC geometrical criteria1 for XB. The other three reports25–28 were focused on crystalline CH2I2 exhibiting numerous C–I⋯I–C XBs, where one diiodomethane molecule is an XB donor and another molecule behaves as an XB acceptor, again in full agreement with the IUPAC criteria.1 In addition, Geiser et al.29 observed weak H2C(I)–I⋯C60 interactions by XRD in the crystals of C60·C6H6·CH2I2, but these contacts were not treated as XBs.
Upon our studies of XB between platinum chloride complexes and halomethanes,17,22 we found that 1,3,5,7,9-pentaazanona-1,3,6,8-tetraenate (PANT) chloride platinum(II) complex301 (Fig. 2) co-crystallizes with diiodomethane forming solvate 1·½CH2I2. The XRD experiment reveals that this solvate displays H2C(I)–I⋯Cl–Pt XBs and I2C(H)–H⋯Cl–Pt HBs, which join two complexes and one CH2I2 molecule in one heterotrimeric supramolecular cluster. The nature of these weak interactions was also studied theoretically and their energies were evaluated in the range from 2.2 to 2.5 kcal mol−1 showing the ability of CH2I2 to behave as an XB donor. We also verified by theoretical calculations overlooked XBs associated with CH2I2 in the structure of VEMWOA (2.4–2.8 kcal mol−1). All these results are consistently disclosed in the sections that follow.
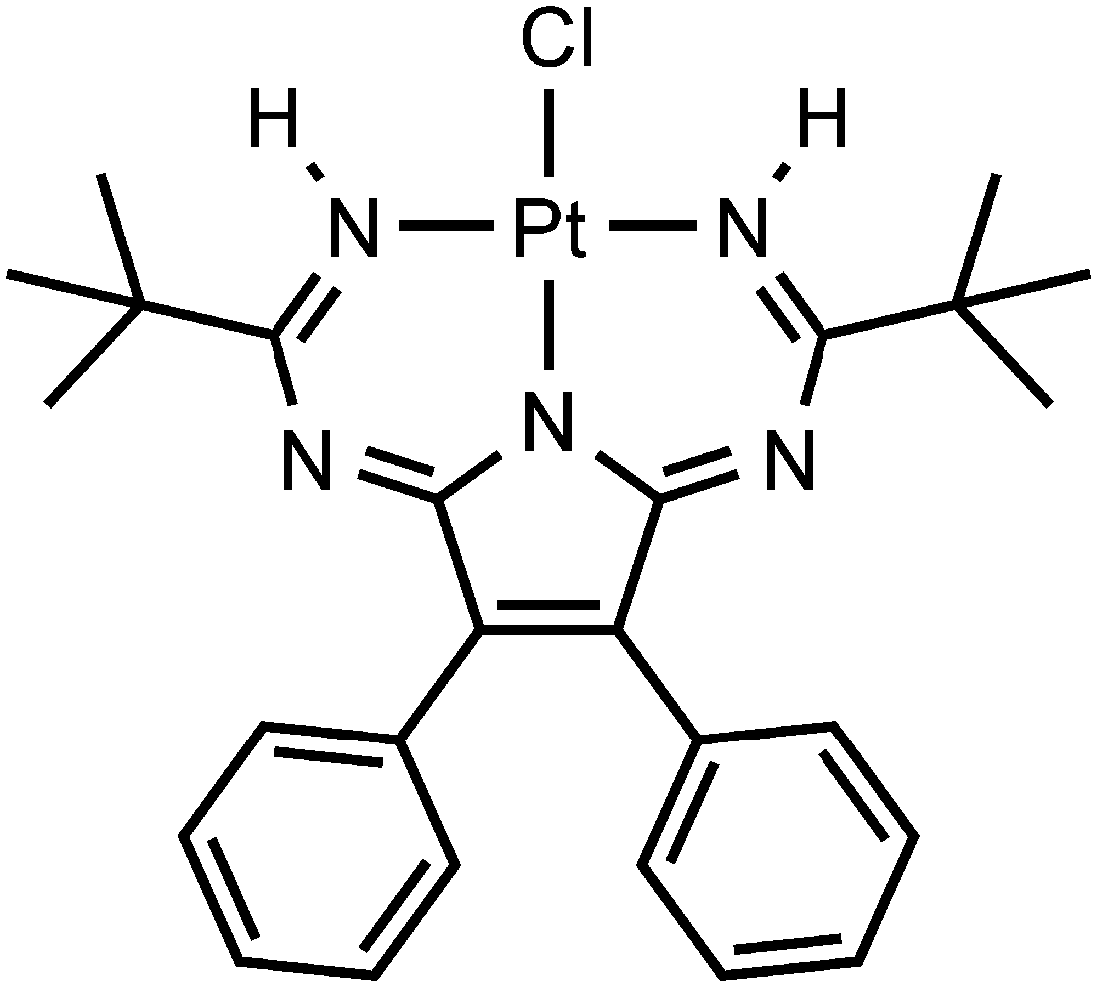 | ||
| Fig. 2 Schematic representation of 1. | ||
Experimental
X-ray structure determination
A suitable crystal of 1·½CH2I2 was studied using an Agilent Technologies Supernova Atlas diffractometer (monochromated MoKα radiation, λ = 0.71073 Å). The temperature was kept at 100 K. The structure has been solved by direct methods and refined by means of the SHELXL-9731 program incorporated in the OLEX2 program package32 using Least Squares minimization. The carbon-bound H atoms were placed in calculated positions and were included in the refinement in the ‘riding’ model approximation with Uiso(H) set to 1.5Ueq(C) and C–H = 0.96 Å for the CH3 groups, Uiso(H) set to 1.2Ueq(C) and C–H = 0.93 Å for the CH groups, and Uiso(H) set to 1.2Ueq(N) and N–H = 0.86 Å for the NH groups. Empirical absorption correction was applied in the CrysAlisPro33 program complex using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm.The crystallographic details are summarized in ESI† Table S2.
Computational details
Single point calculations based on the experimental X-ray geometry (quasi-solid-state approach) have been carried out at the DFT level of theory using the M06 functional34 (this functional was specifically developed to describe weak dispersion forces and non-covalent interactions) with the help of the Gaussian-0935 program package. The Douglas–Kroll–Hess 2nd-order scalar relativistic calculations requesting a relativistic core Hamiltonian were carried out using DZP-DKH basis sets36–40 for all atoms. The topological analysis of the electron density distribution with the help of the atoms-in-molecules (QTAIM) method developed by Bader41 has been performed by using the Multiwfn program (version 3.3.8).42We used the Chemcraft program (version 1.8)43 for visualization of electrostatic surface potential distribution. The Hirshfeld molecular surfaces were generated by the CrystalExplorer 3.1 program44,45 based on the results of the X-ray study. The normalized contact distances, dnorm,46 based on Bondi's van der Waals radii,47 were mapped into the Hirshfeld surfaces. In the color scale, negative values of dnorm are visualized by the red color indicating contacts shorter than the sum of van der Waals radii. The white color denotes intermolecular distances close to van der Waals contacts with dnorm equal to zero. In turn, contacts longer than the sum of van der Waals radii with positive dnorm values are colored with blue. The Cartesian atomic coordinates of model clusters are presented in Tables S3 and S4 (ESI†).
Results and discussion
General approach for verification of XB between organic and metal halides
Two types of short halogen⋯halogen contacts are commonly discussed in the literature (Fig. 3).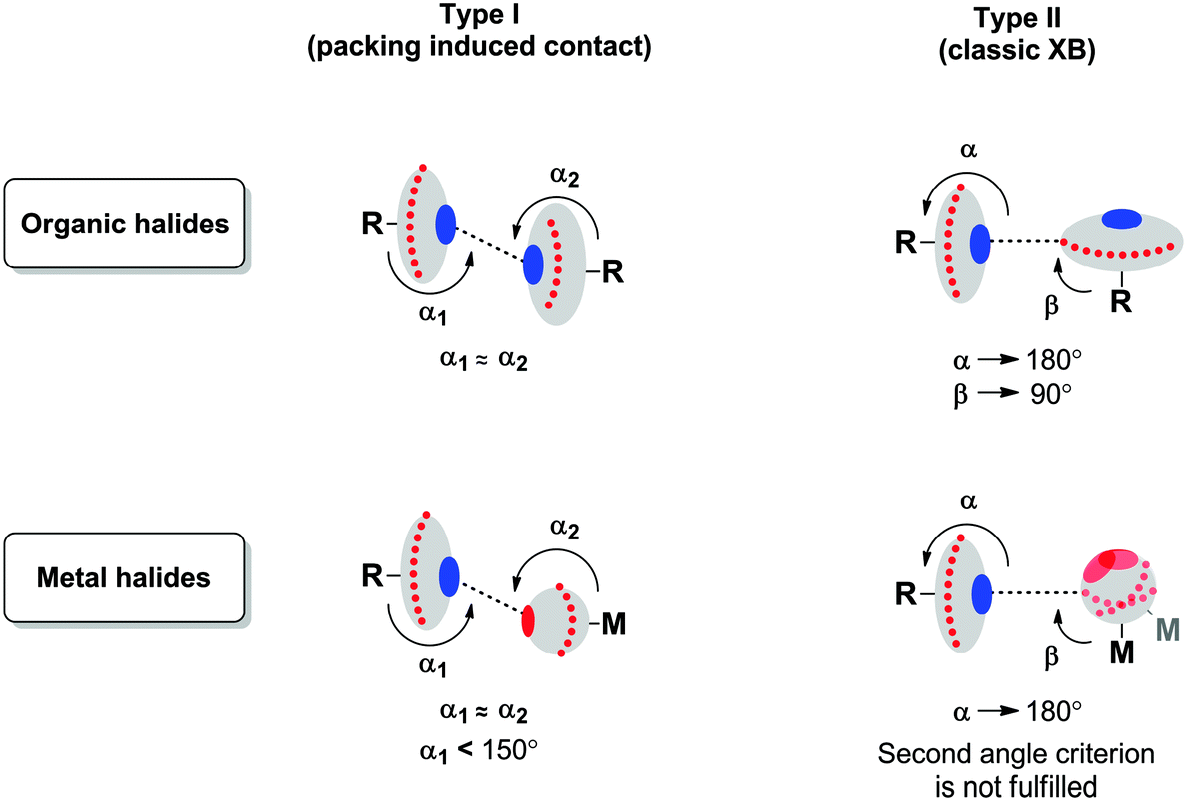 | ||
| Fig. 3 Illustration of the difference between carbon- (left) and metal-bound (right) halides. | ||
Type I is a packing-induced contact (which is actually not an XB, see ref. 48), whereas type II is due to a classic XB because a halogen atom with a 90° angle provides its lone pair for interaction and the other one provides its σ-hole (or, in other words, an area of electropositive potential). According to the IUPAC definition,1 XB is the real R–X⋯Y (X = halogen) contact, when, firstly, the interatomic distance between X and an appropriate Lewis base (Y = F, O, N, Cl, etc.) is less than the sum of their vdW radii and, secondly, the R–X⋯Y angle is close to 180°; exceptions from the linearity have been reviewed by Rissanen.49
It is not surprising that metal halide species – when they are non-covalently linked to organic R–X halogen bond donors – exhibit certain deviations from linearity of the R–X⋯X–[M] fragment. Fig. 4 schematically illustrates the difference in ESPs between organic- and metal-bound halides.
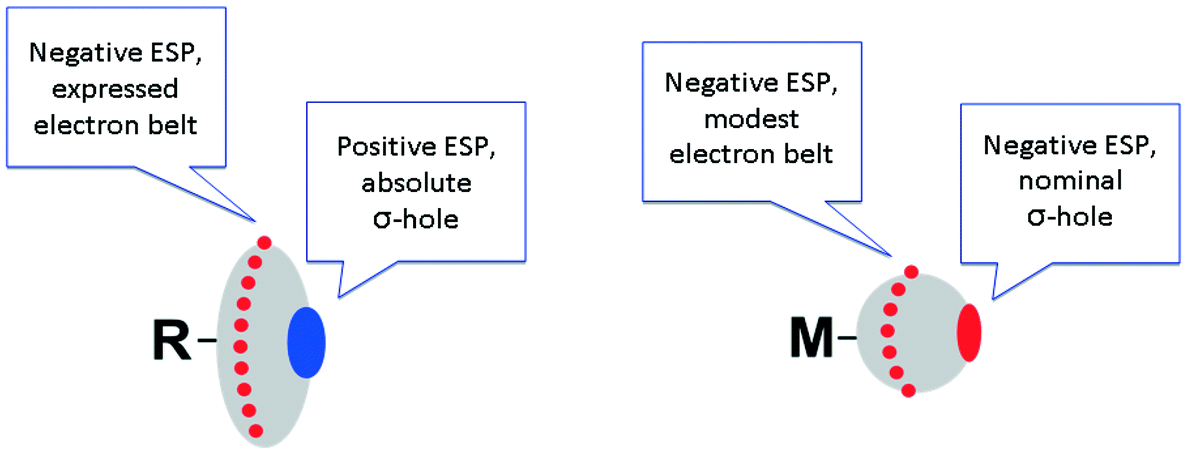 | ||
| Fig. 4 Schematic representation of different ESP distributions on the halogen atoms in organic (left) and metal halides (right). | ||
In contrast to conventional organic XB donors, R–X, the ESP of the complexed halides, as, e.g., calculated by Brammer and co-workers,50 is negative at both electron belt areas and also at the so-called σ-hole. This σ-hole is only nominal and its formulation has only relative, but not absolute sense. As an additional illustration (Fig. 5), we calculated the ESPs VS(r) on the molecular vdW surfaces of CH3I and cis-[PtI2(NH3)2] taken as model systems (optimization in the gaseous phase at the M06/6-311++G** level of theory, quasi-relativistic Stuttgart pseudopotentials and appropriate contracted basis sets MWB60 and MWB46 were used on the platinum and iodine atoms, respectively).
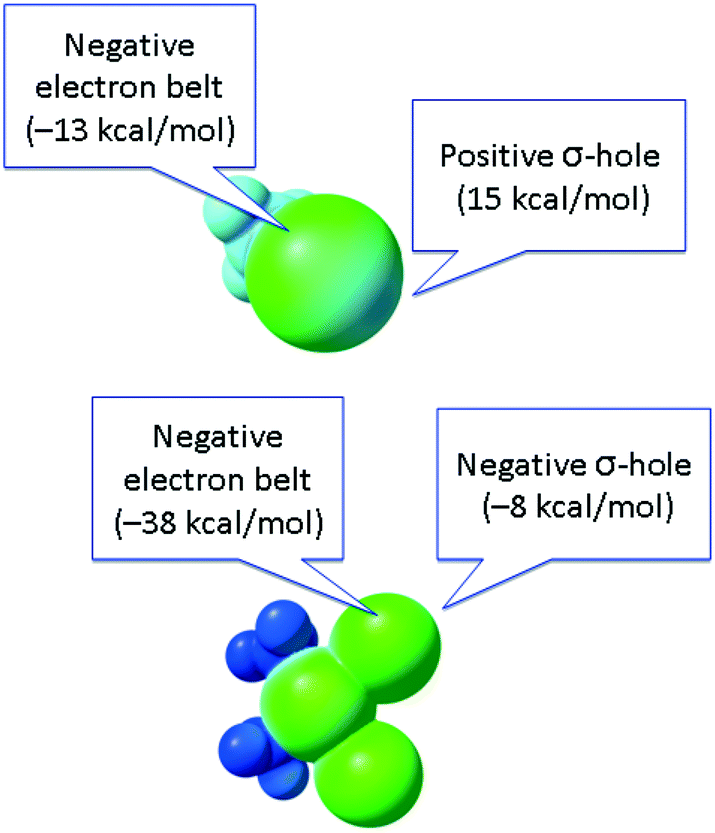 | ||
| Fig. 5 ESP distribution in the CH3I (top) and cis-[PtI2(NH3)2] (bottom) species. Green color – negative ESP, blue color – positive ESP. | ||
It is clear that the organic halide has an expressed negative electron belt and deep positive σ-hole. In contrast, the ESP of the platinum-bound iodides is entirely negative, whereas the σ-hole is “positive” only relatively to the electron belt area rather than in its absolute value. Fig. 3 (bottom) illustrates the R–X⋯X–[M] bonding with particular emphasis on the possibility of deviation from linearity due to the flotation of X–[M] shown in Fig. 3 by the semitransparent species. Hence, the linearity of the R–X⋯X–[M] angle is not a univocal criterion for verification of the attractive XB interaction.
All these considerations point out to the need of certain theoretical calculations for reliable identification of non-covalent R–X⋯X–[M] interactions. In this work, we follow this approach for verification of XBs, viz. we applied the vdW IUPAC criterion for identification of possible halogen⋯halogen interactions, whereupon we conducted theoretical calculations irrespectively of the applicability of the angle IUPAC criterion.
XRD structure of the diiodomethane solvate featuring mixed XB/HB bonding
Complex 1 was prepared as previously reported30via the metal-mediated coupling of two nitrile ligands in trans-[PtCl2(NCtBu)2] and 2,3-diphenylmaleimidine. Crystals of 1·½CH2I2 were obtained by slow evaporation of a solution of 1 in CH2I2 in air at RT. The crystallographic data and processing parameters for 1·½CH2I2 are listed in Table S2 (ESI†); the plots for the structure and selected bond lengths can be found in Fig. 6 and Table 1, respectively.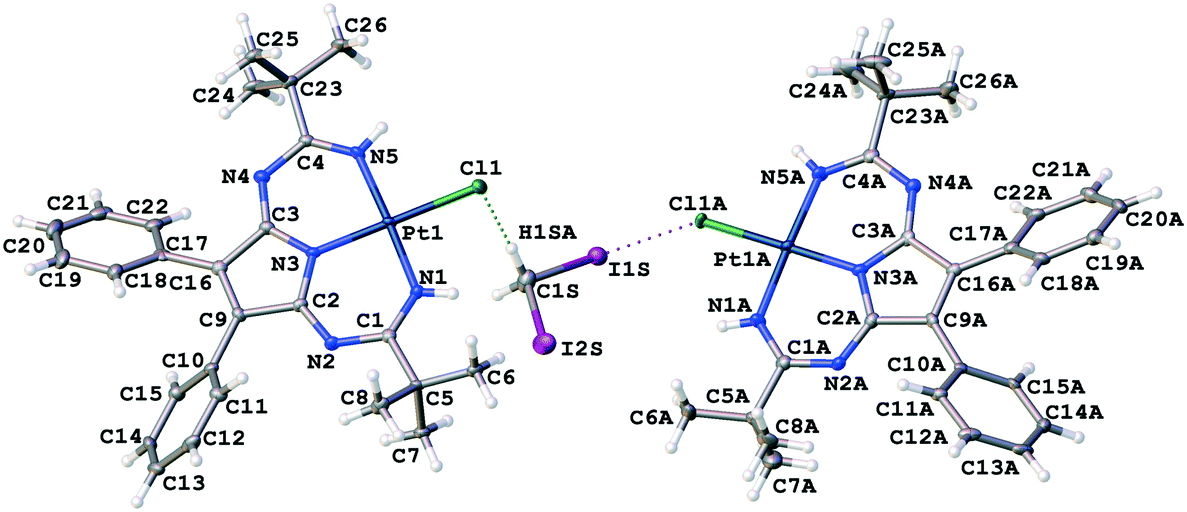 | ||
| Fig. 6 View of heterotrimeric cluster (1)2·CH2I2 in 1·½CH2I2 with the atomic numbering scheme. The H2C(I)–I⋯Cl–Pt XB and I2C(H)–H⋯I–Pt HB are shown by dotted lines. Hereinafter, thermal ellipsoids are drawn with 50% probability. | ||
| Molecule with Pt1 (left) | Molecule with Pt1A (right) | ||
|---|---|---|---|
| Bond | Length, Å | Bond | Length, Å |
| Pt1–Cl1 | 2.3232(11) | Pt1A–Cl1A | 2.3374(11) |
| Pt1–N1 | 1.967(4) | Pt1A–N1A | 1.966(3) |
| Pt1–N3 | 1.948(3) | Pt1A–N3A | 1.948(4) |
| Pt1–N5 | 1.969(4) | Pt1A–N5A | 1.968(4) |
| N1–C1 | 1.303(5) | N1A–C1A | 1.304(6) |
| C1–N2 | 1.371(6) | C1A–N2A | 1.357(6) |
| N2–C2 | 1.300(6) | N2A–C2A | 1.309(5) |
| C2–N3 | 1.360(5) | C2A–N3A | 1.366(6) |
| N3–C3 | 1.361(6) | N3A–C3A | 1.365(5) |
| C3–N4 | 1.312(6) | C3A–N4A | 1.303(6) |
| N4–C4 | 1.384(5) | N4A–C4A | 1.366(6) |
| C4–N5 | 1.283(6) | C4A–N5A | 1.306(6) |
Previously, we reported two solvates 1·1¼CH2Cl2 and 1·1⅖CH2Br2, which demonstrated close cell parameters and similar packing features.22 We found the same heterotetrameric clusters (1)2·(CH2X2)2 (X = Cl, Br) held by two simultaneous H2C(X)–X⋯Cl–Pt XBs and X2C(H)–H⋯I–Pt HBs. These two clusters demonstrate the isostructural CH2Cl2/CH2Br2 exchange. Therefore, the next logical step was an attempt to use diiodomethane as a solvent and either an XB or a mixed XB/HB donor. The CH2I2 solvate, 1·½CH2I2, however, exhibits another packing type different from those of 1·1¼CH2Cl2 and 1·1⅖CH2Br2, but some similarities with the dichloromethane and dibromomethane solvates can be detected even in this case (for details, see Fig. S1 in the ESI†).
In 1·½CH2I2, isolated heterotrimeric clusters (1)2·(CH2I2) (Fig. 6) are formed from two complex molecules and one solvent molecule linked together by the H2C(I)–I⋯Cl–Pt XB and I2C(H)–H⋯Cl–Pt HB. The corresponding parameters of both types of weak interactions are in accordance with the IUPAC definitions for XB1 and HB.51 The I1S⋯Cl1A distance is less than Rowland's52RvdW(I) + RvdW(Cl) sum [3.4071(12) vs. 3.79 Å]. The ∠(C1S–I1S⋯Cl1A) angle [173.11(13)°] is close to 180°, whereas the ∠(I1S⋯Cl1A–Pt1A) angle [108.83(4)°] is around 90°. All these mean that this interaction can be assigned to type II halogen⋯halogen contact, which is specific for XB.51 Theoretical calculations given in the next section fully supported our XRD-based conclusions.
The H1SA⋯Cl1 distance is less than the corresponding vdW sum (2.622 vs. 2.86 Å), and the ∠(C1S–H1SA⋯Cl1) angle (157.5°) is larger than 120°. The C1S⋯Cl1 separation is close to the corresponding vdW sum [3.538(5) vs. 3.53 Å] indicating that this HB should be treated as rather weak.
Two crystallographically independent molecules of 1 are almost the same (Table 1), and all the Pt–N bonds [1.948(4)–1.969(4) Å] as well as the C–N [1.357(6)–1.384(5) Å] and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N [1.283(6)–1.312(6) Å] bonds in the PANT ligand are equal within 3σ. However, the Pt1A–Cl1A bond [2.3374(11) Å] in the halogen-bonded molecule is slightly longer than the Pt1–Cl1 distance [2.3232(11) Å] in the hydrogen-bonded molecule.
N [1.283(6)–1.312(6) Å] bonds in the PANT ligand are equal within 3σ. However, the Pt1A–Cl1A bond [2.3374(11) Å] in the halogen-bonded molecule is slightly longer than the Pt1–Cl1 distance [2.3232(11) Å] in the hydrogen-bonded molecule.
This difference can be explained by more effective electron density redistribution caused by XB than that caused by HB (Fig. 7). The same elongation on the Pt–Cl bonds was found in the previously reported trans-[PtCl2(NCNR2)2] complexes linked with iodoform as an XB donor via C–I⋯Cl–Pt XBs.17
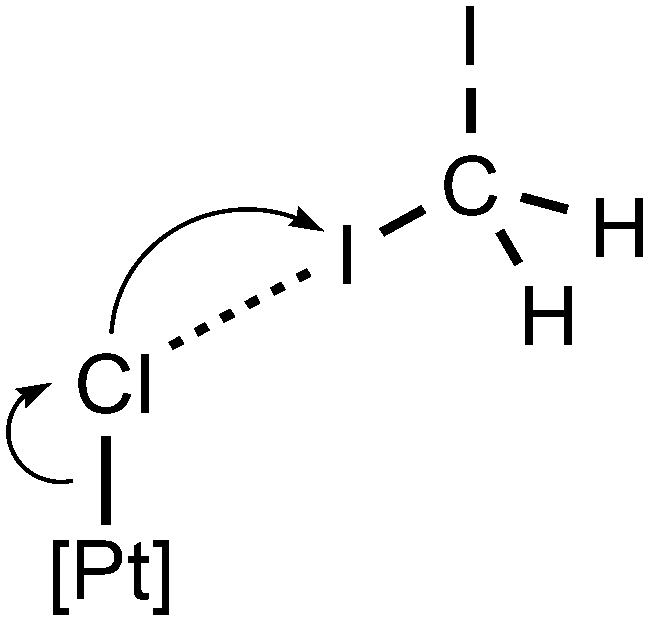 | ||
| Fig. 7 The XB effect on the Pt–Cl bond length. | ||
Halogen bond in the structure of VEMWOA
Although until now no XBs between diiodomethane and metal halide complexes have been reported, our inspection of the CCDC database verified the H2C(I)–I⋯I–Pt XBs in the structure of VEMWOA,53 which represents the platinum(II) associate [PtI2{S,S-![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) (Ph)2CH(Me)CH2CH(Me)Ph2}]·⅓MeCN·⅓CH2I2 (2·⅓MeCN·⅓CH2I2) (Fig. 8).
(Ph)2CH(Me)CH2CH(Me)Ph2}]·⅓MeCN·⅓CH2I2 (2·⅓MeCN·⅓CH2I2) (Fig. 8).
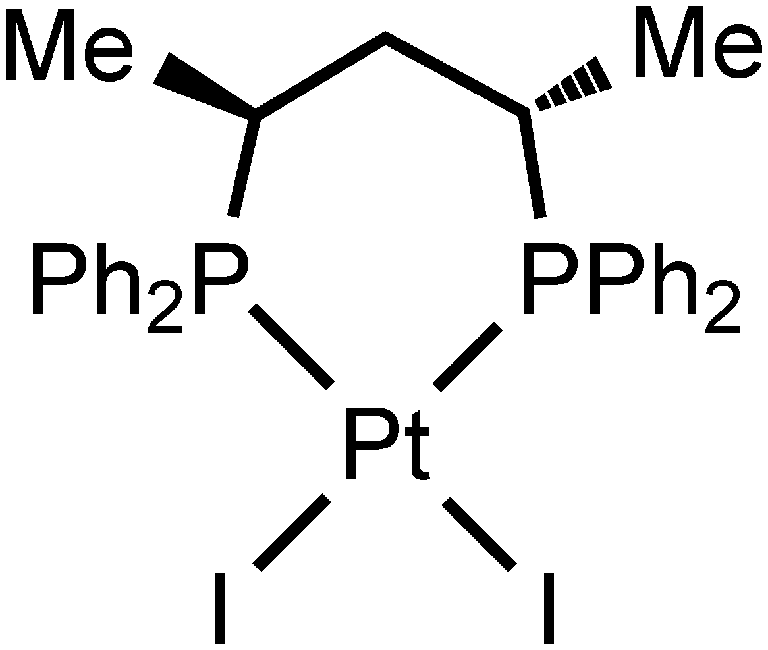 | ||
| Fig. 8 Graphical representation of the complex in 2·⅓MeCN·⅓CH2I2. | ||
In the corresponding work,53 it was pointed out that no anomalous van der Waals contacts were observed in 2·⅓MeCN·⅓CH2I2. By contrast, our inspection of this structure allows the identification of at least two H2C(I)–I⋯I–Pt short contacts (Fig. 9; d(I8⋯I6) = 3.5865(11) Å, d(I7⋯I2) = 3.6501(12) Å), which are substantially less than the vdW sum52 (2RvdW(I) = 4.06 Å).
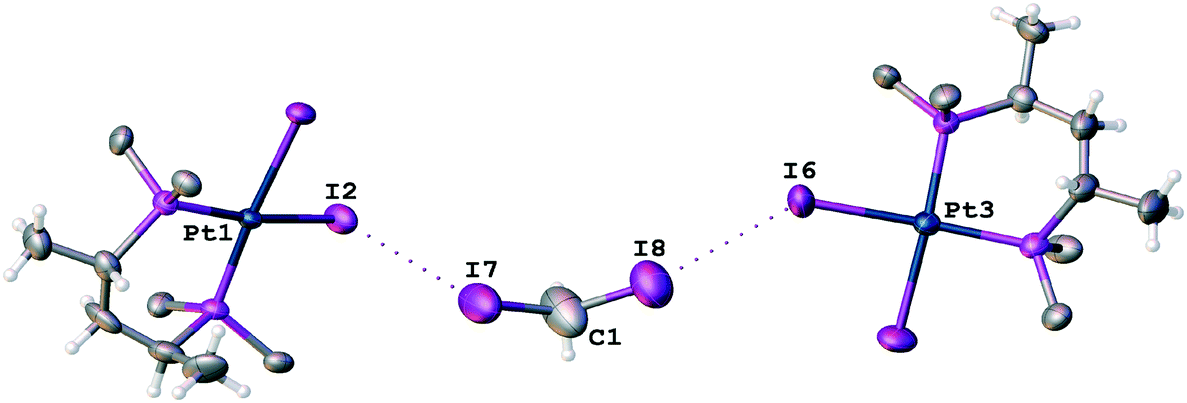 | ||
| Fig. 9 View of the H2C(I)–I⋯I–Pt short contacts in VEMWOA. Phenyl rings are omitted for clarity. | ||
The I8⋯I6 contact should be treated as a type II inter-halogen interaction.48 This can be attributed to the typical XB because the angle values around these atoms [159.0(4)° around I8 and 136.24(3)° around I6] fulfill the IUPAC criteria1 and the difference between these two angles is more than 20°. However, the type of the second I7⋯I2 contact is not clear. The very similar angle values around the iodine atoms [153.2(4)° around I7 and 144.56(3)° around I2] led to the idea that this contact could be assigned to type I halogen⋯halogen contact caused by the crystal packing. We decided to clarify the nature of the I7⋯I2 contact by ESP distribution at 2, calculated and analyzed at the M06/DZP-DKH level of theory in the Multiwfn program.42 In 2, the surface maxima with the negative absolute value of VS(r)max (−18 kcal mol−1) corresponding to “nominal σ-holes” (see above) were found on the iodine ligands (Fig. 10). The value of the I⋯I–Pt angle is acceptable for treatment of the iodide ligand as the nucleophilic partner in the non-covalent interaction. Taking into account the σ-hole on the diiodomethane I atom (Fig. 10, 23.1 kcal mol−1), the C–I⋯I–Pt contact also belongs to XB in agreement with the IUPAC definition.1
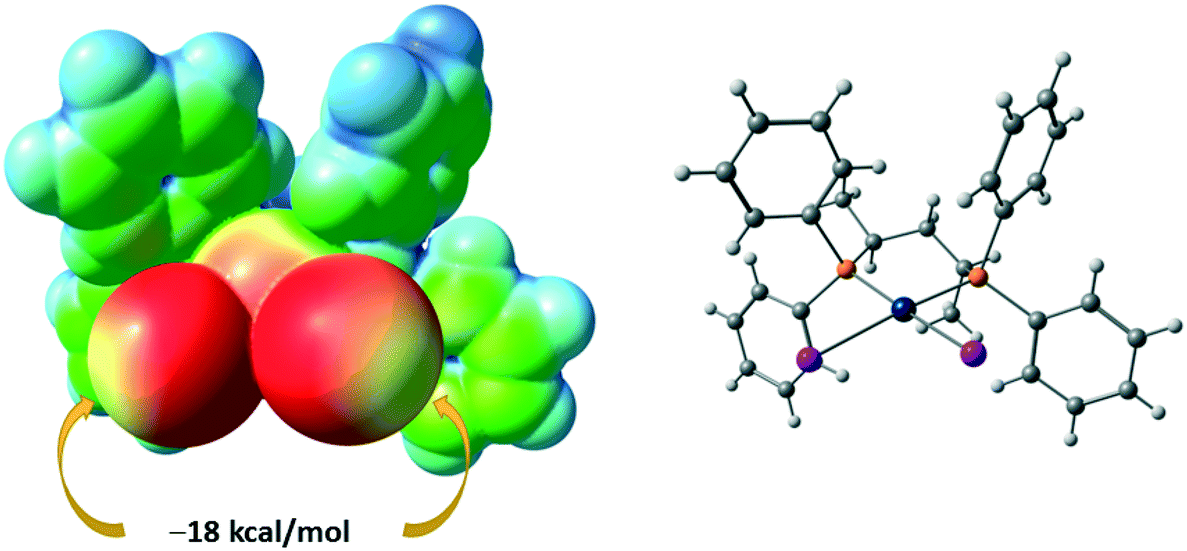 | ||
| Fig. 10 ESP distribution in 2 from the structure VEMWOA. Red color – negative ESP, blue color – positive ESP. | ||
Theoretical consideration of XB in the diiodomethane solvates
The H2C(I)–I⋯Cl–Pt XB and I2C(H)–H⋯Cl–Pt HB were found in the crystal structure of 1·½CH2I2 through the single-crystal XRD experiment. As always in such cases, in addition to the structural analysis, a detailed computational study directed toward understanding the nature of these non-covalent interactions is desirable. This study is particularly important when metal-bound halides are involved in XB (see above).In order to confirm or deny the hypothesis on the existence of these weak contacts in the solid state and to quantify their energies from a theoretical viewpoint, we carried out DFT calculations and performed topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method)54 for the (1)2·(CH2I2) heterotrimeric cluster as a model system (Table 2). This approach has already been successfully used by our group in studies of different non-covalent interactions (e.g., hydrogen, halogen and chalcogen bonding, metallophilic interactions, stacking) and properties of coordination bonds in various transition metal complexes.17,22,55–61 The contour line diagrams of the Laplacian distribution ∇2ρ(r), bond paths, and selected zero-flux surfaces for intermolecular H⋯Cl HBs and I⋯Cl XBs are shown in Fig. 11. To visualize the studied non-covalent interactions, reduced density gradient (RDG) analysis62 was carried out, and RDG isosurfaces were plotted (Fig. 11).
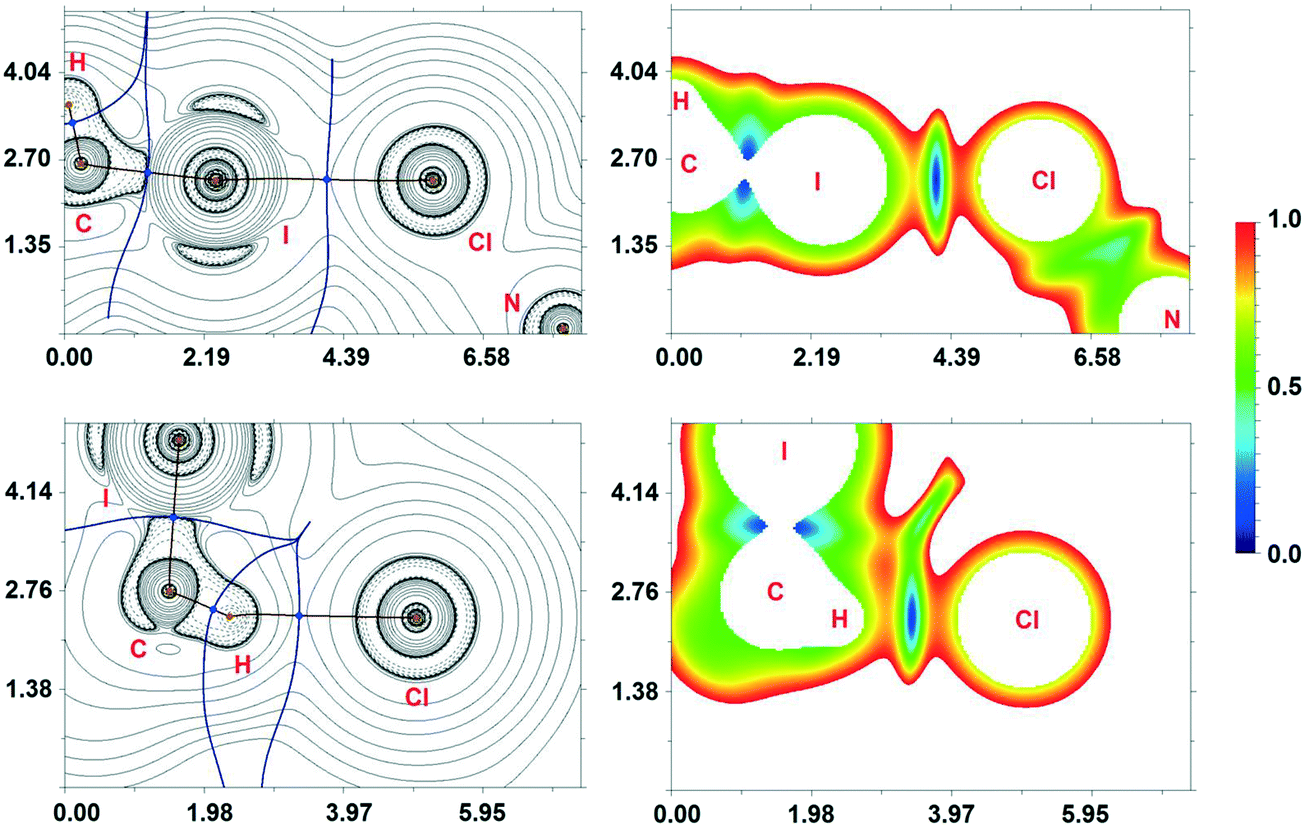 | ||
| Fig. 11 Contour line diagrams of the Laplacian distribution ∇2ρ(r), bond paths and selected zero-flux surfaces (left) and RDG isosurfaces referring to the non-covalent interactions (right) for the intermolecular H⋯Cl HBs and I⋯Cl XBs in (1)2·(CH2I2). Bond critical points (3, −1) are shown in blue, while nuclear critical points (3, −3) are shown in pale brown. Length units – Å, RDG isosurface values are given in Hartree. | ||
The same procedure was performed for cluster (2)2·(CH2I2) from VEMWOA to study the intermolecular XBs in this system (Table S1, Fig. S2†).
The QTAIM analysis demonstrates the presence of appropriate bond critical points (BCPs; 3, −1) for the intermolecular H⋯Cl HBs and I⋯Cl XBs in (1)2·(CH2I2) and the I⋯I XBs in (2)2·(CH2I2). The low magnitude of the electron density (0.011–0.013 Hartrees), positive values of the Laplacian (0.037–0.041 Hartrees), and close-to-zero positive energy density (0.001 Hartrees) in these BCPs are typical for both HB and XB.17,22,65,66 We have defined energies for these contacts according to the procedures proposed by Espinosa et al.63 and Vener et al.64 (Tables 2 and S1†) and one can state that the strength of these non-covalent interactions (2.2–2.8 kcal mol−1) is comparable with those of weak hydrogen bonds following the classification of Jeffrey (“strong”: 40–15 kcal mol−1, “moderate”: 15–4 kcal mol−1, “weak”: <4 kcal mol−1).67 The balance between the Lagrangian kinetic energy G(r) and potential energy density V(r) at the BCPs (3, −1) reveals the nature of these interactions. If the ratio −G(r)/V(r) > 1 is satisfied, then the nature of the appropriate interaction is purely non-covalent, whereas in the case of −G(r)/V(r) < 1, some covalent component takes place.68 Based on this criterion, we came to the conclusion that the covalent contribution is absent for intermolecular H⋯Cl HBs and I⋯Cl XBs in (1)2·(CH2I2) and I⋯I XBs in (2)2·(CH2I2). We additionally evaluated the magnitude of intermolecular interaction energies for HBs and XBs in (1)2·(CH2I2) and (2)2·(CH2I2) by the supermolecule method (Eint = EAB − EA − EB, i.e. the total electronic energy of the complex subtracting the energies of the monomers) using the crystallographic coordinates. The obtained values of intermolecular interaction energies (Eint(H⋯Cl) = 6.7 kcal mol−1, Eint(I⋯Cl) = 5.9 kcal mol−1, Eint(I⋯I) = 2.1–3.6 kcal mol−1) are higher than (for (1)2·(CH2I2)) or comparable to (for (2)2·(CH2I2)) those from QTAIM analysis. Thus, the QTAIM approach gives underestimated values of interaction energies for electrostatically favored HB and XB interactions.
Hirshfeld surface analysis of the crystal structures of 1·½CH2I2 and VEMWOA
The molecular Hirshfeld surface represents an area where molecules come into contact and its analysis gives the possibility of an additional insight into the nature of intermolecular interactions in the crystal state. For visualization, we have used a mapping of the normalized contact distance (dnorm); its negative value enables identification of molecular regions of substantial importance for detection of short contacts.Fig. 12A depicts the Hirshfeld surface of the CH2I2 molecule in the solvate (1)2·(CH2I2). In the Hirshfeld surface of CH2I2, the regions of intermolecular H⋯Cl and I⋯Cl contacts, visualized by large red circle areas, are seen near the hydrogen and iodine atoms, respectively. The following intermolecular contacts give the largest contributions to the Hirshfeld surface of CH2I2 in the solvate (1)2·(CH2I2): I–H 66.9%, H–H 22.5%, Cl–H 3.4%, I–Cl 3.3%, H–C 2.2%; contributions of other intermolecular contacts are less than 1%.
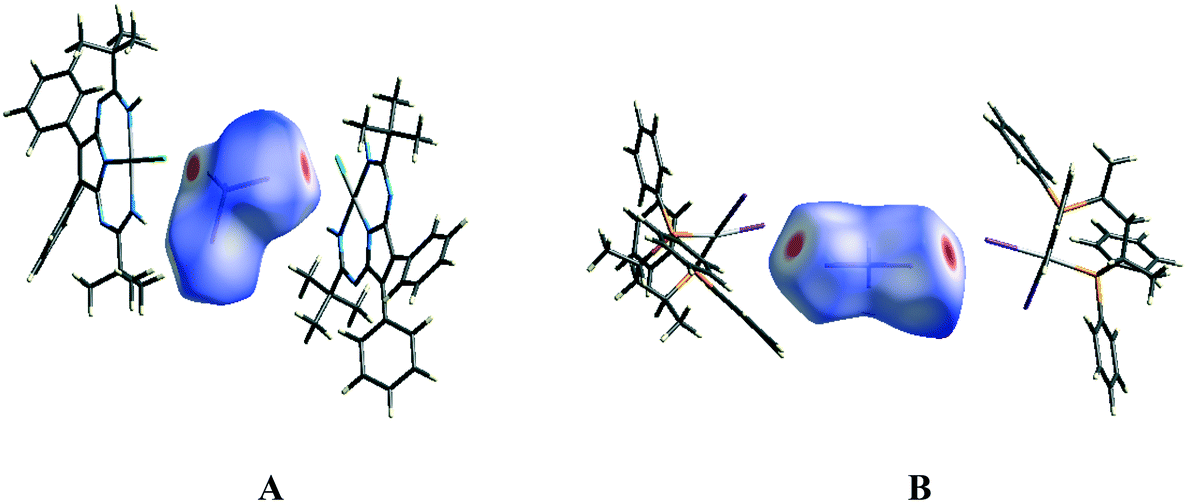 | ||
| Fig. 12 The Hirshfeld surfaces of the CH2I2 molecule in the solvates (1)2·(CH2I2) (A) and VEMWOA (B) with the colored scale, which corresponds to values ranging from −0.2 Å (red) to 1.5 Å (blue). | ||
Fig. 12B depicts the Hirshfeld surface of the CH2I2 molecule in VEMWOA. In the Hirshfeld surface of CH2I2, the regions of intermolecular I⋯I contacts, visualized by large red circle areas, are seen near the iodine atoms. The following intermolecular contacts give the largest contributions to the Hirshfeld surface of CH2I2: H–I 54.1%, H–H 20.8%, I–I 9.1%, C–H 7.9%, C–I 6.4%; contributions of other intermolecular contacts are less than 1%.
The Hirshfeld surface fingerprint plots (Fig. S2 and S3†) were generated using di (distance from the surface to the nearest atom in the molecule itself) and de (distance from the surface to the nearest atom in another molecule) as a pair of coordinates for each individual surface spot resulting in two-dimensional histograms. A color gradient in the plots ranging from blue to red represents the proportional contribution of contact pairs in the global surface.
Thus, the Hirshfeld surface analyses of 1·½CH2I2 and VEMWOA confirm that in the crystalline state CH2I2 behaves as an XB and HB donor.
Concluding remarks
In this work, we used multidisciplinary combined and complementary experimental and theoretical approaches that allow bypassing certain limitations of the XRD method in the studies of halogen bond systems. In particular, we succeeded in overcoming difficulties associated with the deviation from linearity of R–X⋯X–[M] linkage insofar as this deviation complicates the experimental identification of XBs in organic halide⋯metal-bound halide systems.We found that a weak XB donor such as diiodomethane can still form a halogen bond with metal-bound halides and CH2I2 can serve as either an XB or a mixed XB/HB donor. All these non-covalent interactions in 1·½CH2I2 and VEMWOA were confirmed by Hirshfeld surface analysis, and their nature was investigated by DFT calculations and topological analysis of the electron density distribution within the formalism of Bader's theory (QTAIM method). The evaluated energies of the HBs (2.2 kcal mol−1) and XBs (2.4–2.8 kcal mol−1) again indicate their non-covalent character.
The results reported in this work can be used as a starting point for further studies of XBs in dihalomethane solutions, where they may be sufficiently strong to contribute, along with HBs, to the overall solvation. It is expected that all these findings can find an application in crystal engineering because they expand the arsenal of supramolecular organizations which may be useful for solvent storage69 or separation70 and also in physical chemistry as they contribute to the understanding of solvation with halomethanes. We hope that our results open up an avenue to the generation of other diiodomethane associates and studies in this direction are underway in our group.
Acknowledgements
The experimental part of this work was conducted in the framework of the Russian Science Foundation project (14-13-00060), whereas the theoretical part was performed under the Russian Foundation for Basic Research grant (16-33-00212). XRD studies were performed at the Center for X-ray Diffraction Studies of Saint Petersburg State University. The authors declare no competing financial interests.References
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- H. Wang, W. Z. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072–5104 CrossRef CAS PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- V. V. Sivchik, A. I. Solomatina, Y.-T. Chen, A. J. Karttunen, S. P. Tunik, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2015, 54, 14057–14060 CrossRef CAS PubMed.
- D. Bulfield and S. M. Huber, Chem. – Eur. J., 2016, 22, 14434–14450 CrossRef CAS PubMed.
- P. S. Ho, in Halogen Bonding I: Impact on Materials Chemistry and Life Sciences, Springer-Verlag Berlin, Berlin, 2014, pp. 241–276 Search PubMed.
- G. Cavallo, S. Biella, J. A. Lu, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, J. Fluorine Chem., 2010, 131, 1165–1172 CrossRef CAS.
- S. V. Rosokha, I. S. Neretin, T. Y. Rosokha, J. Hecht and J. K. Kochi, Heteroat. Chem., 2006, 17, 449–459 CrossRef CAS.
- S. V. Rosokha, C. L. Stern and J. T. Ritzert, Chem. – Eur. J., 2013, 19, 8774–8788 CrossRef CAS PubMed.
- S. V. Lindeman, J. Hecht and J. K. Kochi, J. Am. Chem. Soc., 2003, 125, 11597–11606 CrossRef CAS PubMed.
- H. Bock and S. Holl, Z. Naturforsch., B: J. Chem. Sci., 2001, 56, 152–163 CAS.
- H. G. Lohr, A. Engel, H. P. Josel, F. Vogtle, W. Schuh and H. Puff, J. Organomet. Chem., 1984, 49, 1621–1627 Search PubMed.
- P. V. Gushchin, G. L. Starova, M. Haukka, M. L. Kuznetsov, I. L. Eremenko and V. Y. Kukushkin, Cryst. Growth Des., 2010, 10, 4839–4846 CAS.
- D. M. Ivanov, A. S. Novikov, I. V. Ananyev, Y. V. Kirina and V. Yu. Kukushkin, Chem. Commun., 2016, 52, 5565–5568 RSC.
- S. V. Rosokha, J. Lu, T. Y. Rosokha and J. K. Kochi, Chem. Commun., 2007, 3383–3385 RSC.
- S. V. Rosokha, C. L. Stern, A. Swartz and R. Stewart, Phys. Chem. Chem. Phys., 2014, 16, 12968–12979 RSC.
- S. V. Rosokha and M. K. Vinakos, Cryst. Growth Des., 2012, 12, 4149–4156 CAS.
- A. M. Afanasenko, M. S. Avdontceva, A. S. Novikov and T. G. Chulkova, Z. Kristallogr. - Cryst. Mater., 2016, 231, 435–440 CAS.
- D. M. Ivanov, A. S. Novikov, G. L. Starova, M. Haukka and V. Y. Kukushkin, CrystEngComm, 2016, 18, 5278–5286 RSC.
- W.-W. du Mont, V. Stenzel, J. Jeske, P. G. Jones, A. Sebald, S. Pohl, W. Saak and M. Baetcher, Inorg. Chem., 1994, 33, 1502–1505 CrossRef CAS.
- D. M. Ivanov, M. A. Kinzhalov, A. S. Novikov, I. V. Ananyev, A. A. Romanova, V. P. Boyarskiy, M. Haukka and V. Y. Kukushkin, Cryst. Growth Des., 2017, 17, 1353–1362 CAS.
- F. M. A. Noa, S. A. Bourne and L. R. Nassimbeni, Cryst. Growth Des., 2015, 15, 3271–3279 Search PubMed.
- M. Podsiadlo and A. Katrusiak, CrystEngComm, 2008, 10, 1436–1442 RSC.
- M. Podsiadlo, K. Dziubek, M. Szafranski and A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 1090–1098 CAS.
- M. Podsiadlo and A. Katrusiak, J. Phys. Chem. B, 2008, 112, 5355–5362 CrossRef CAS PubMed.
- U. Geiser, S. K. Kumar, B. M. Savall, S. S. Harried, K. D. Carlson, P. R. Mobley, H. H. Wang, J. M. Williams, R. E. Botto, W. Liang and M. H. Whangbo, Chem. Mater., 1992, 4, 1077–1082 CrossRef CAS.
- D. M. Ivanov, P. V. Gushchin, A. S. Novikov, M. S. Avdontceva, A. A. Zolotarev, G. L. Starova, Y.-T. Chen, S.-H. Liu, P.-T. Chou and V. Yu. Kukushkin, Eur. J. Inorg. Chem., 2016, 2016, 1480–1487 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- CrysAlisPro, Agilent Technologies Ltd., Version 1.171.136.120 (release 127–106-2012) Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, in Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- C. L. Barros, P. J. P. de Oliveira, F. E. Jorge, A. C. Neto and M. Campos, Mol. Phys., 2010, 108, 1965–1972 CrossRef CAS.
- F. E. Jorge, A. C. Neto, G. G. Camiletti and S. F. Machado, J. Chem. Phys., 2009, 130, 6 CrossRef PubMed.
- A. C. Neto and F. E. Jorge, Chem. Phys. Lett., 2013, 582, 158–162 CrossRef.
- C. L. Barros, P. J. P. de Oliveira, F. E. Jorge, A. C. Neto and M. Campos, Mol. Phys., 2010, 108, 1965–1972 CrossRef CAS.
- R. C. de Berredo and F. E. Jorge, J. Mol. Struct.: THEOCHEM, 2010, 961, 107–112 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- http://www.chemcraftprog.com.
- S. K. Wolff, D. J. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka and M. A. Spackman, CrystalExplorer (Version 3.1), University of Western Australia, 2012 Search PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- P. Metrangolo and G. Resnati, IUCrJ, 2014, 1, 5–7 CrossRef CAS PubMed.
- K. Rissanen, CrystEngComm, 2008, 10, 1107–1113 RSC.
- L. Brammer, E. A. Bruton and P. Sherwood, Cryst. Growth Des., 2001, 1, 277–290 CAS.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- L. Jánosi, L. Kollár, P. Macchi and A. Sironi, J. Organomet. Chem., 2006, 691, 2846–2852 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- A. S. Novikov, M. L. Kuznetsov and A. J. L. Pombeiro, Chem. – Eur. J., 2013, 19, 2874–2888 CrossRef CAS PubMed.
- V. N. Mikhaylov, V. N. Sorokoumov, K. A. Korvinson, A. S. Novikov and I. A. Balova, Organometallics, 2016, 35, 1684–1697 CrossRef CAS.
- T. V. Serebryanskaya, A. S. Novikov, P. V. Gushchin, M. Haukka, R. E. Asfin, P. M. Tolstoy and V. Yu. Kukushkin, Phys. Chem. Chem. Phys., 2016, 18, 14104–14112 RSC.
- E. Andrusenko, E. Kabin, A. S. Novikov, N. A. Bokach, G. L. Starova and V. Yu Kukushkin, New J. Chem., 2017, 41, 316–325 RSC.
- X. Ding, M. J. Tuikka, P. Hirva, V. Yu. Kukushkin, A. S. Novikov and M. Haukka, CrystEngComm, 2016, 18, 1987–1995 RSC.
- E. S. Yandanova, D. M. Ivanov, M. L. Kuznetsov, A. G. Starikov, G. L. Starova and V. Yu Kukushkin, Cryst. Growth Des., 2016, 16, 2979–2987 CAS.
- A. S. Mikherdov, M. A. Kinzhalov, A. S. Novikov, V. P. Boyarskiy, I. A. Boyarskaya, D. V. Dar'in, G. L. Starova and V. Yu Kukushkin, J. Am. Chem. Soc., 2016, 138, 14129–14137 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- M. V. Vener, A. N. Egorova, A. V. Churakov and V. G. Tsirelson, J. Comput. Chem., 2012, 33, 2303–2309 CrossRef CAS PubMed.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 102, 9925–9932 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Am. Chem. Soc., 2000, 122, 11154–11161 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- C. B. Aakeröy, T. K. Wijethunga, J. Benton and J. Desper, Chem. Commun., 2015, 51, 2425–2428 RSC.
- X. Q. Yan, Q. J. Shen, X. R. Zhao, H. Y. Gao, X. Pang and W. J. Jin, Anal. Chim. Acta, 2012, 753, 48–56 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data and structure refinement data. Cartesian atomic coordinates of model clusters. CCDC 1532142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ce00346c |
| This journal is © The Royal Society of Chemistry 2017 |