
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methyl substituent effect on structure, luminescence and semiconducting properties of furan/phenylene co-oligomer single crystals†
Maxim S.
Kazantsev
 *ab,
Alina A.
Beloborodova
ab,
Ekaterina S.
Frantseva
ab,
Tatyana V.
Rybalova
ab,
Vladislav G.
Konstantinov
c,
Inna K.
Shundrina
ab,
Dmitry Yu.
Paraschuk
c and
Evgeny A.
Mostovich
ab
*ab,
Alina A.
Beloborodova
ab,
Ekaterina S.
Frantseva
ab,
Tatyana V.
Rybalova
ab,
Vladislav G.
Konstantinov
c,
Inna K.
Shundrina
ab,
Dmitry Yu.
Paraschuk
c and
Evgeny A.
Mostovich
ab
aN.N. Vorozhtsov Novosibirsk Institute of Organic Chemistry, Lavrentieva 9, Novosibirsk, 630090, Russian Federation. E-mail: maximkazantsev1988@gmail.com
bNovosibirsk State University, Pirogova 2, Novosibirsk, 630090, Russian Federation
cFaculty of Physics and International Laser Center, Lomonosov Moscow State University, Leninskie Gory 1/62, Moscow, 119991, Russian Federation
First published on 28th February 2017
Abstract
Single crystals of furan/phenylene co-oligomers are among the most promising highly-emissive materials for applications in various optoelectronic devices. In this work, we synthesized and studied furan/phenylene co-oligomers with the same conjugated core 1,4-bis(5-phenylfuran-2-yl)benzene and methyl substituents at p- and m-positions of the terminal phenyls. The effect of substituents on the crystal packing, charge transport and luminescence of the single crystals was studied. Compared to the unsubstituted compound, the methyl-substituted co-oligomers demonstrated improved thermostability and enhanced photoluminescence, which we assign to J-aggregation resulting from the strong inclination of the molecules against the main crystal facet. The charge mobility in single crystal organic field-effect transistors decreased upon the inclination of the molecules. We conclude that the molecular tilt angle, intermolecular distances and interactions in crystals of heteroaryl-containing linear conjugated oligomers can be controlled by the introduction of end methyl groups in the appropriate positions.
1. Introduction
Organic highly emissive semiconducting single crystals attract much attention because of their high performance in organic light-emitting devices, such as light-emitting transistors and lasers.1–4 Crystals for light-emitting transistors should have, on the one hand, high charge mobility, which is highest for the perpendicular orientation of the molecules against the crystal basal plane along which the electrical current flows and, on the other hand, efficient light outcoupling. The latter is usually impeded by the strong waveguide effect, which is first of all dictated by the orientation of the molecular transition dipole moments defining the direction of primary emission.1,5 Evidently, for linear conjugated molecules the light outcoupling efficiency will be maximal for the horizontal molecular orientation with respect to the basal plane. Therefore, one may expect that there is an inclination of the molecules corresponding to the optimum balance between the charge mobility and luminescence. Accordingly, control of molecular inclination against the main crystal facet is a promising tool for the development of high-performance single crystal light-emitting devices.Single crystals of small conjugated molecules, due to their highly ordered and low-defect structure, also provide a unique opportunity for the investigation of the fundamental structure–property relationship via systematic design and the study of libraries of related compounds with peculiar molecular packing. For example, by varying a number of aromatic rings in thiophene/phenylene co-oligomers, single crystals with high mobility6 and high luminescence efficiency were found.7,8 The introduction of terminal substituents in thiophene/phenylenes was demonstrated to be a powerful tool for solubility and morphology control.9,10 Distyrylbenzenes (DSB) were also shown to give highly emissive single crystals and besides terminal substitution could be modified using vinylene units resulting in a tremendous library of DSB derivatives having different intra- or intermolecular interactions and demonstrating specific optical and charge transport properties.1 Aggregation motifs in DSB single crystals were intensively explored by substitution of the backbone by methyl,11,12 tertbutyl,13 methoxy,14 cyano,14,15 fluorine,13,14,16 trifluoromethyl15,17 and other1 groups. Among the variety of substituents, the methyl group is the most compact and minimally disturbs the molecular electronic structure; therefore, end-group methylation might be a tool for fine tuning the crystal packing, and hence the intermolecular interactions. For example, 4,4′-methyl-substituted DSB single crystals demonstrated high photoluminescence quantum yields (PL QYs), amplified spontaneous emission and efficient charge transport.11 Varghese et al. reported unusual co-facial arrangement, high PL QYs and stimulated emission in 3,5,3′,5′-tetramethyl DSB single crystals.12 However, methyl-substituted heteroaryl-containing co-oligomers have not yet been reported.
Recently, we have demonstrated a furan/phenylene co-oligomer, 1,4-bis(5-phenylfuran-2-yl)benzene (BPFB, Fig. 1a), outperforming its thiophene/phenylene analogue in PL efficiency.18 Here, we report on the dramatic effect of methyl substituents on the crystal packing and optoelectronic properties of the furan/phenylene co-oligomer BPFB. We compare vapor-grown single crystals of co-oligomers substituted at p- and m-positions of the terminal phenyls and demonstrate that the tilt angle and intermolecular distances could be effectively tuned without distortion of the molecular electronic structure.
 | ||
| Fig. 1 a) Chemical formulae of the studied furan/phenylene co-oligomers; b) thermogravimetric (dashed lines) and differential scanning calorimetry (solid lines) analyses of BPFB (olive), 2Me-BPFB (blue) and 4Me-BPFB (red) in an inert (He) atmosphere. | ||
2. Experimental
2.1 Synthesis
All reagents and solvents were purchased from commercial sources (Sigma-Aldrich, Acros) and were used without additional purification. Reaction mixtures were monitored by TLC using Sorbfil HPTLC-AF-V-UV plates and hexane as the eluent. For column chromatography, Masherey-Nagel Kieselgel 60 and pure hexane as the eluent were used. The detailed synthetic procedures are presented in the ESI.†2.2 Characterization
Combustion analysis was performed using a CHN-analyzer (EURO EA). 1H NMR spectra were recorded on a Bruker AV 400 (400.13 MHz) spectrometer in CDCl3. Mass spectra were obtained with a Thermo Electron Corporation DFS mass spectrometer (70 eV) using direct injection; the temperature of the ionization chamber was 220–270 °C. IR spectra were recorded in transmission mode on a Bruker Tensor 27 FT-IR spectrometer in potassium bromide pellets. Thermogravimetric analysis and differential scanning calorimetry measurements were performed using a NETZSCH STA 409 instrument at a heating rate of 10 °C min−1 in an inert (He) atmosphere. Cyclic voltammetry measurements were performed in CH2Cl2 solution with a computer-controlled P-8nano potentiostat/galvanostat (Elins, Russia) in combination with a three-electrode cell (Gamry), and 0.1 M tetrabutylammonium hexafluorophosphate was used as a supporting electrolyte. Pt, a Pt wire and Ag/AgCl were used as the working, counter and reference electrodes, respectively. The reference electrode was calibrated by measuring the redox potential of ferrocene. UV/VIS spectra were recorded in diluted (10−5 M) THF solutions and solid samples in potassium bromide pellets on a Varian Cary 5000 UV-VIS-NIR spectrophotometer. The PL QY in THF solution was measured with a Varian Cary Eclipse fluorescence spectrophotometer according to a standard procedure, and a diluted solution of 1,4-bis-(5-phenyloxazole-2-yl)benzene in ethanol (PL QY = 0.885) was used as a reference standard.19 The PL QY of solid samples was measured using a calibrated 3.3 inch diameter integrating sphere (Newport 819C-SL-3.3) coupled to a spectrometer (InVia, Renishaw). Solid samples on glass substrates were excited at 405 nm using a semiconductor laser. The detailed experimental procedure was described in our previous work.182.3 Crystal growth and analysis
For the crystal growth, physical vapour transport in a stream of high-purity helium (99.995%) was used. Powders were sublimed in a glass tube (13 mm i.d.) placed into a copper tube with a heater to create a temperature gradient.20 The temperature was set near the T10% of weight loss (Table 1). The crystal growth time typically was overnight (∼14 h.) The crystals were examined using an optical microscope (MP-7, “Lomo”, Russia) using transmitted light through crossed polarizers and under blue laser irradiation (405 nm). The X-ray diffraction experiments were performed using a Bruker KAPPA APEX II diffractometer with graphite monochromated MoKα radiation. Integration and scaling of the intensity data were accomplished by means of SAINT.21 Absorption corrections were applied using SADABS.22 The structures were solved by direct methods with SHELXS-97.23 Refinement was carried out by the full-matrix least-squares technique with SHELXL.23 All non-hydrogen atoms were refined anisotropically. The hydrogen atom positions were calculated geometrically and refined isotropically according to the riding model. The crystal structures were analysed for intermolecular interactions using PLATON.24,25 The asymmetric unit in all the three cases contains half of a molecule because of a molecule lying on the centre of symmetry. Crystallographic data for the structures have been deposited at the Cambridge Crystallographic Data Centre as supplementary publication CCDC no. 1520469–1520471.2.4 Single crystal field-effect transistors
Organic field-effect transistors (OFETs) were fabricated on glass substrates using a top-contact/top-gate configuration reported elsewhere.26 The crystals were attached to the substrates using cyanoacrylate glue. Source and drain contacts were painted on the surface of the crystals using a colloidal graphite suspension (PELCO, TED PELLA, INC). The OFET channel width and length were measured using an optical microscope. As a top dielectric layer, 1.5 μm thick parylene N (C = 1.6 nF cm−2) was deposited from [2,2]paracyclophane (Sigma Aldrich), as described elsewhere.20 Finally, the gate electrode was painted using graphite paste. The OFET current–voltage characteristics were recorded using a source-meter instrument (Agilent B2902A) under dark conditions, ambient atmosphere and room temperature. The voltage sweep rate was 0.3 V s−1. For the determination of field-effect mobility, the saturation regime was used, and the source-drain current (Isd) was fitted according to the equation:27where C is the capacitance per unit area of the insulating layer; W and L are the channel width and length; μ is the carrier mobility; Vg and Vth are the gate and threshold voltages, respectively.

3. Results and discussion
3.1 Synthesis and characterization
Fig. 1a demonstrates the chemical formulae of the studied 1,4-bis(5-phenylfuran-2-yl)benzene (BPFB) and its derivatives with multiple methyl substituents in the 4,4′- (2Me-BPFB) and 3,5,3′,5′- (4Me-BPFB) positions of the terminal phenyls. The target compounds were synthesized according to a previously described procedure18 (see the ESI†). The methyl substituents only slightly affect the electronic structure (Table 1), as monitored from the cyclic voltammetry measurements (Fig. S8†) and optical spectroscopy data (Fig. S9a†). Therefore, mainly intermolecular interactions affect the properties and optoelectronic performance of substituted BPFB single crystals.Thermogravimetric analysis and differential scanning calorimetry (Fig. 1b) reveal a significant increase in the melting and sublimation temperatures for 2Me- and 4Me-BPFB compared to the parent compound (Table 1). The melting enthalpy also increases upon the addition of methyl groups, suggesting the amplification of intermolecular interactions. Indeed, 2Me-BPFB and 4Me-BPFB are unexpectedly very poorly soluble in toluene (lower than 0.01 g l−1) in contrast to the previously reported 1 g l−1 for unsubstituted BPFB.18 As a result, we used physical vapor transport for the crystal growth.
3.2 Single crystal analysis
Fig. 2 demonstrates the optical images of 2Me- (a and b) and 4Me-BPFB (c and d) single crystals which have an elongated plate shape in contrast to solution-18 and vapor-grown BPFB single crystals grown for comparison (Fig. 2e and f). In-plane rotation of the crystals in crossed polarizers results in completely dark images demonstrating linear birefringence (Fig. S10†). The lengths of 2Me- and 4Me-BPFB single crystals (up to 1 cm) are typically much longer than their width (∼200 μm) and thickness (∼50–100 μm). PL in crystals was observed mainly at the edges and defects (Fig. 2b and d) due to a strong waveguide effect previously reported for thiophene/ and furan/phenylene co-oligomer single crystals.8,18,28,29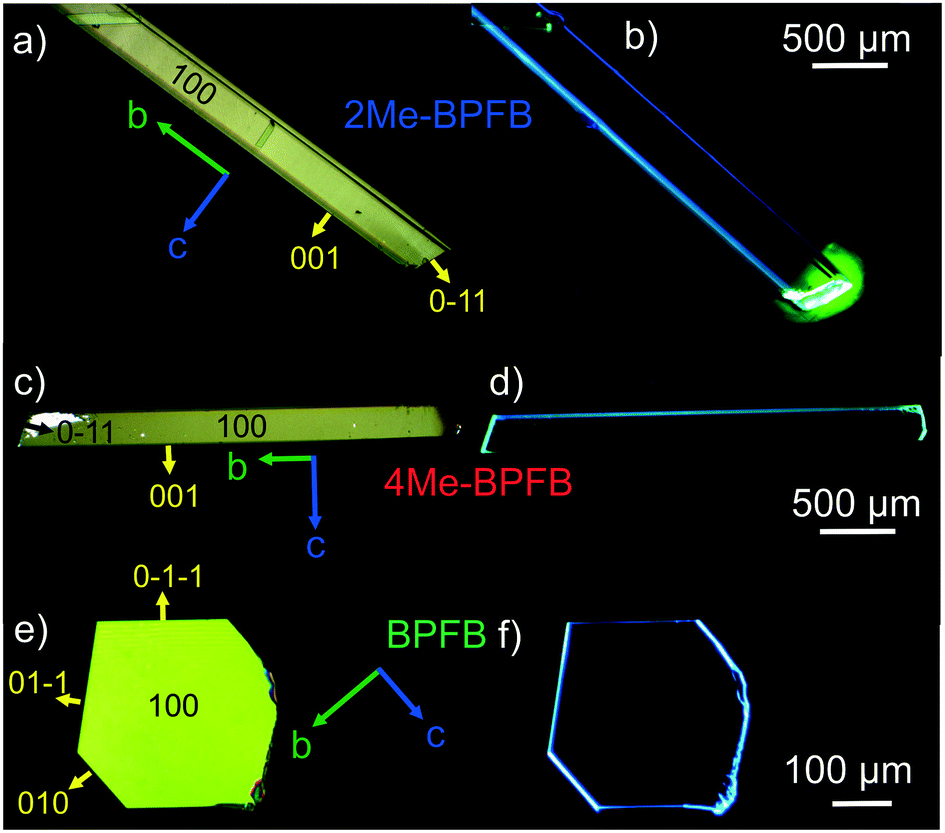 | ||
| Fig. 2 Microscopy images of single crystals: 2Me-BPFB (attached to a substrate by cyanoacrylate glue) a) in crossed polarizers and b) under blue irradiation; 4Me-BPFB c) in crossed polarizers and d) under blue irradiation; BPFB e) in crossed polarizers and f) under blue irradiation; the arrows indicate the orientation of crystallographic axes and Miller indices of the crystal facets. | ||
X-ray diffraction experiments on the single crystals revealed the same crystal group P21/c for all three compounds. The main crystal facet corresponds to the (100) plane for all the crystals, and the 2Me- and 4Me-BPFB ones are extended along the b-axis (Fig. 2a and c). The π-system of 2Me-BPFB is the most planar, as monitored from the values of torsion angles ϕ1 and ϕ2 and the maximal deviation dmax from the average plane of the π-system demonstrated by the C10 atom (Table S1†). The molecules have a typical herringbone packing motif controlled mainly by C–H⋯π interactions. While BPFB has an almost right tilt angle between the long molecular axes (connecting two carbons of terminal phenyls) and the main crystal facet, the methyl-substituted compounds demonstrate remarkable inclination of the molecules (Fig. 3). This inclination results in the decrease in crystal density and increase in intermolecular distances (Table S1†). Therefore, one may expect a decrease in charge mobility for methyl-substituted BPFBs (vide infra).
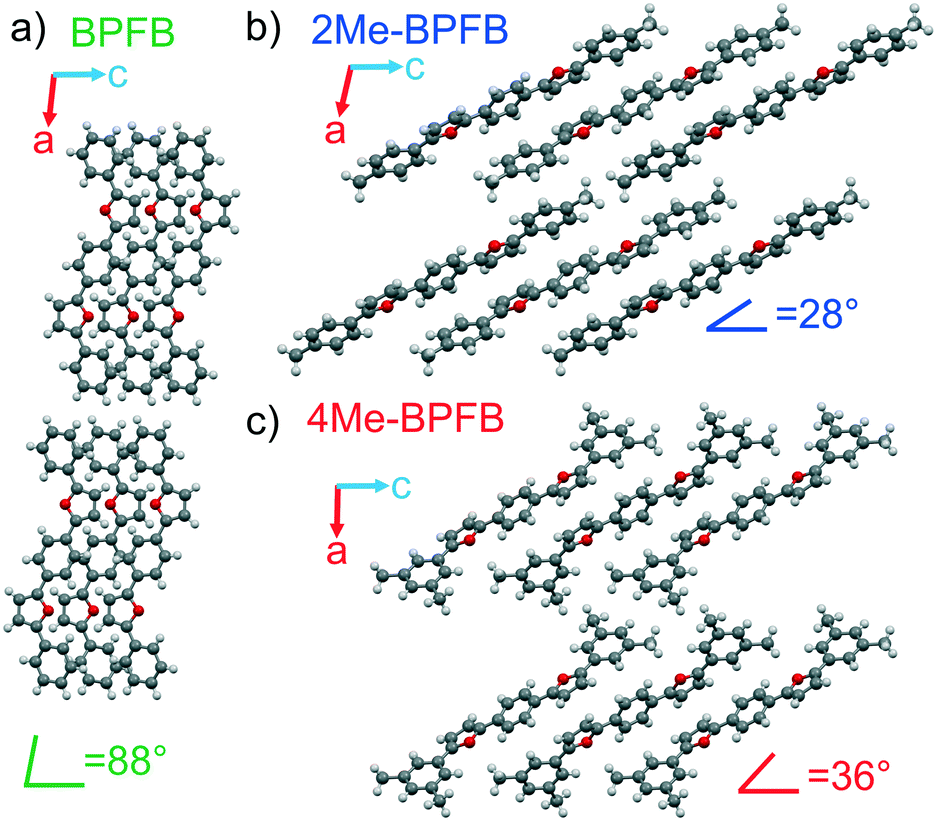 | ||
| Fig. 3 Crystal structures of the studied co-oligomers: a) BPFB, b) 2Me-BPFB and c) 4Me-BPFB; view along the b-axis. | ||
In contrast to BPFB crystals, where the C–H⋯π interactions are within the layer, in 2Me-BPFB crystals similar interactions between the neighboring layers are observed (Fig. 4). These interlayer interactions in 2Me-BPFB crystals stabilize the structure and increase the melting temperature (Table 1). In 4Me-BPFB, due to steric hindrance, similar interlayer interactions are restricted. Nevertheless, the additional interactions (Fig. 4d) between the methyl groups and the π-systems of neighboring molecules within the layer contribute to the increase in thermal stability (Table 1).
 | ||
| Fig. 4 a) Molecular structure, atom and cycle numbering of BPFB; intermolecular C–H⋯π interactions in the crystals of: b) BPFB, c) 2Me-BPFB, and d) 4Me-BPFB (the corresponding parameters are presented in Table S2†); the numbering for 2Me-BPFB and 4Me-BPFB is the same as for BPFB, except for methyl groups marked as C14 for 2Me-BPFB and C14 and C15 for 4Me-BPFB; ϕ1 and ϕ2 correspond to the twisting angles between phenyl and furan rings. | ||
Note that 2Me- and 4Me-BPFB have tilt angles as low as 28 and 36°, respectively, which are among the smallest for the thiophene/phenylene6,10,30–32 and furan/phenylene29 co-oligomers. Note that a similar inclination is not so pronounced in methyl-substituted DSBs.11,12 Such inclination of molecules is usually achieved by the introduction of bulky substituents31,33 or highly electron-withdrawing groups1,14,16,32,34 leading to strong intermolecular interactions. It is very unusual that in the case of furan/phenylene co-oligomers, terminal methyl substituents lead to high inclination which could be exploited for the design of materials with desired orientation of luminophores without distortion of their molecular electronic structure. Remarkably, by introducing the methyl groups in appropriate positions, the inclination angle could be varied. And this effect is not directly interrelated with the bulkiness of the substituent because the relatively small terminal methyl substituents in 2Me-BPFB induce extremely high inclination (tilt angle 28°) which is even higher than those in more sterically hindered 4Me-BPFB (tilt angle 36°). Note that assuming the direction of transition dipole moments is parallel to the long molecular axes, the tilt angles of 2Me- and 4Me-BPFB correspond to J-aggregation,35 which is beneficial for high luminescence.1
3.3 Luminescence and charge transport properties
The optical properties of BPFB, 2Me- and 4Me-BPFB were studied in diluted solutions and the solid state. The solution absorption and emission spectra of all the co-oligomers are almost identical (Fig. S9a†) except for a small bathochromic shift for the methyl-substituted compounds caused presumably by a weak electron-donor effect of methyl groups. All the compounds are highly emissive in diluted solutions with a PL QY of about 90% (Table 2). The crystals of methyl substituted BPFBs were also strongly emissive with high quantum yields of 76 and 80% for 2Me- and 4Me-BPFB, respectively (Table 2 and Fig. S9b†), in contrast to BPFB single crystals showing a PL QY of only 45% (Table 2). Therefore, the methyl substituents enhance the PL QY of BPFB-based single crystals. The high PL QY and red shift of the PL spectra of 2Me- and 4Me-BPFB compared to BPFB single crystals (Table 2) are consistent with the J-aggregation model. On the other hand, this red shift is partially caused by a strong reabsorption effect36 so that the longer the crystal, the more red-shifted the emission observed. For example, green emission was observed at the ends of the long crystals (Fig. 2b). To highlight the effect of aggregation on optical absorption, we recorded the optical density spectra of the ground crystals pressed in potassium bromide pellets (Fig. S9c†). All the spectra of the solid samples show red shifts of the 0–0 transition compared to the solution spectra. These shifts can be partially assigned to the higher polarizability of the molecular environment in crystals. Meanwhile, 2Me- and 4Me-BPFB demonstrate considerably higher red shifts in agreement with their J-aggregation, and the values of the red shifts are correlated with the tilt angles of the molecules.| Solution | Single crystal | ||||
|---|---|---|---|---|---|
| λ abs, nm | λ em, nm | PL QY, % | λ em, nm | PL QY, % | |
| BPFB | 376 | 410, 434 | 91 | 498 | 45 |
| 2Me-BPFB | 379 | 413, 438 | 86 | 502 | 76 |
| 4Me-BPFB | 379 | 413, 438 | 89 | 495, 521 | 80 |
To study the charge transport properties of the crystals, we fabricated top-contact/top-gate OFETs (due to the high thickness of the crystals, they could not be laminated on hard SiO2 substrates to study devices in common top-contact/bottom-gate architectures). An optical image of a typical single crystal with painted source-drain contacts is shown in Fig. S11.† We did not select any specific charge transport direction for the BPFB single crystals, whereas for the 2Me- and 4Me-BPFB crystals the charge transport was studied along the b-axis. Note that this charge transport direction corresponds to the shortest intermolecular distances (Table S1†). Fig. 5 demonstrates the output (a and c) and transfer (b and d) current–voltage characteristics corresponding to p-type conductivity in 4Me-BPFB and BPFB single crystals, respectively.
 | ||
| Fig. 5 a) Output and (b) transfer characteristics of a 4Me-BPFB single crystal OFET; c) output and d) transfer characteristics of a BPFB single crystal OFET; Vsd, Vg and Vth are the source-drain, gate and threshold voltages, respectively; μ is the field-effect mobility extracted in the saturation regime. | ||
The mobility of a 4Me-BPFB single crystal OFET is 0.039 cm2 V−1 s−1, which is almost 3 times lower than that of an unsubstituted BPFB single crystal (Fig. 5d). Therefore, despite the inclination of the molecules, 4Me-BPFB still demonstrates reasonable charge transport. We fabricated a series of 3 devices for BPFB and 4Me-BPFB (Table S3†) demonstrating reproducible OFET characteristics. However, for all 6 fabricated devices on 2Me-BPFB single crystals no field-effect was observed at all. This lack of OFET performance could be due to high inclination of the molecules (Fig. 3). This inclination in methyl-substituted BPFBs results in an increase in intermolecular distances as well as a decrease in the molecular stacking angle along the charge transport direction (Table S1†). Both lower the overlapping of electronic wavefunctions and hence should reduce the charge mobility.
Note that in contrast to previous studies of bulky substituent effect on charge transport,33,37 the charge mobility of substituted BPFB single crystals does not correlate with the steric effect of the substituents. Indeed, the relatively small methyl group at the p-position of the terminal phenyl in 2Me-BPFB results in complete loss of mobility, whereas the sterically hindered 4Me-BPFB still demonstrates the field-effect in devices.
4. Conclusions
In summary, we have demonstrated a series of highly-emissive single crystals based on methyl-substituted furan/phenylene co-oligomers. The introduction of terminal methyl groups lowers the solubility, enhances the thermal stability and results in high inclination of the molecules against the basal crystal plane. Accordingly, the crystal packing of methyl-substituted derivatives corresponds to J-aggregation, which is beneficial for high luminescence. The tilt angles as low as 28 and 36° for 2-Me and 4Me-BPFB, respectively, are among the lowest for thiophene/ and furan/phenylene co-oligomers and very unusual for linear conjugated molecules. We have shown that the charge mobility in the crystals decreases upon the inclination of the molecules. Therefore, the crystal packing of heteroaryl-containing linear conjugated oligomers can be effectively tuned by methyl substituents, and this is a useful approach for obtaining desired intermolecular distances and interactions without distortion of the molecular electronic structure.Acknowledgements
This work was supported by the Russian Foundation for Basic Research project 16-33-60011 mol_a_dk. The work on PL in crystals was supported by Russian Science Foundation (project 15-12-30031). This work was done using equipment purchased under the Lomonosov Moscow State University Program of Development. The authors acknowledge Novosibirsk State University program “5-100” and Multi-Access Chemical Service Center SB RAS for the spectral and analytical measurements.Notes and references
- J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818–5832 RSC
.
- H.-H. Fang, J. Yang, J. Feng, T. Yamao, S. Hotta and H.-B. Sun, Laser Photonics Rev., 2014, 8, 687–715 CrossRef
- S. Hotta, T. Yamao, S. Z. Bisri, T. Takenobu and Y. Iwasa, J. Mater. Chem. C, 2014, 2, 965–980 RSC
- J. Gierschner, S. Varghese and S. Y. Park, Adv. Opt. Mater., 2016, 2, 348–364 CrossRef
- S. Varghese and S. Das, J. Phys. Chem. Lett., 2011, 2, 863–873 CrossRef CAS PubMed
- S. Hotta and T. Yamao, J. Mater. Chem., 2011, 21, 1295–1304 RSC
- T. Komori, H. Nakanotani, T. Yasuda and C. Adachi, J. Mater. Chem. C, 2014, 2, 4918–4921 RSC
- L. G. Kudryashova, M. S. Kazantsev, V. A. Postnikov, V. V. Bruevich, Y. N. Luponosov, N. M. Surin, O. V. Borshchev, S. A. Ponomarenko, M. S. Pshenichnikov and D. Y. Paraschuk, ACS Appl. Mater. Interfaces, 2016, 8, 10088–10092 CAS
- R. Hirase, M. Ishihara, T. Katagiri, Y. Tanaka, H. Yanagi and S. Hotta, Org. Electron., 2014, 15, 1481–1492 CrossRef CAS
- V. A. Postnikov, Y. I. Odarchenko, A. V. Iovlev, V. V. Bruevich, A. Y. Pereverzev, L. G. Kudryashova, V. V. Sobornov, L. Vidal, D. Chernyshov, Y. N. Luponosov, O. V. Borshchev, N. M. Surin, S. A. Ponomarenko, D. A. Ivanov and D. Y. Paraschuk, Cryst. Growth Des., 2014, 14, 1726–1737 CAS
- R. Kabe, H. Nakanotani, T. Sakanoue, M. Yahiro and C. Adachi, Adv. Mater., 2009, 21, 4034–4038 CrossRef CAS
- S. Varghese, S. K. Park, S. Casado, R. C. Fischer, R. Resel, B. Milian-Medina, R. Wannemacher, S. Y. Park and J. Gierschner, J. Phys. Chem. Lett., 2013, 4, 1597–1602 CrossRef CAS PubMed
- J. Gierschner, M. Ehni, H. J. Egelhaaf, B. M. Medina, D. Beljonne, H. Benmansour and G. C. Bazan, J. Chem. Phys., 2005, 123, 144914 CrossRef PubMed
- G. P. Bartholomew, G. C. Bazan, X. Bu and R. J. Lachicotte, Chem. Mater., 2000, 12, 1422–1430 CrossRef CAS
- S. K. Park, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An and S. Y. Park, Chem. Mater., 2012, 24, 3263–3268 CrossRef CAS
- M. L. Renak, G. P. Bartholomew, S. Wang, P. J. Ricatto, R. J. Lachicotte and G. C. Bazan, J. Am. Chem. Soc., 1999, 121, 7787–7799 CrossRef CAS
- S. Varghese, S. K. Park, S. Casado, R. Resel, R. Wannemacher, L. Lüer, S. Y. Park and J. Gierschner, Adv. Funct. Mater., 2016, 26, 2349–2356 CrossRef CAS
- M. S. Kazantsev, E. S. Frantseva, L. G. Kudriashova, V. G. Konstantinov, A. A. Mannanov, T. V. Rybalova, E. V. Karpova, I. K. Shundrina, G. N. Kamaev, M. S. Pshenichnikov, E. A. Mostovich and D. Y. Paraschuk, RSC Adv., 2016, 6, 92325–92329 RSC
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS
- R. W. I. De Boer, M. E. Gershenson, A. F. Morpurgo and V. Podzorov, Phys. Status Solidi, 2004, 201, 1302–1331 CrossRef CAS
-
SAINT, Data Reduction and Frame Integration Program for the CCD Area-Detector System, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, 1997–2006 Search PubMed
-
G. M. Sheldrick, SADABS, Program for area detector adsorption correction, Institute for Inorganic Chemistry, University of Goettingen, Goettingen, Germany, 1996 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, version 10M, Utrecht University, Netherlands, 2003 Search PubMed
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- V. Podzorov, V. M. Pudalov and M. E. Gershenson, Appl. Phys. Lett., 2003, 82, 1739–1741 CrossRef CAS
- V. Podzorov, MRS Bull., 2013, 38, 15–24 CrossRef
- Y. Yomogida, T. Takenobu, H. Shimotani, K. Sawabe, S. Z. Bisri, T. Yamao, S. Hotta and Y. Iwasa, Appl. Phys. Lett., 2010, 97, 173301 CrossRef
- K. Oniwa, T. Kanagasekaran, T. Jin, M. Akhtaruzzaman, Y. Yamamoto, H. Tamura, I. Hamada, H. Shimotani, N. Asao, S. Ikeda and K. Tanigaki, J. Mater. Chem. C, 2013, 1, 4163–4170 RSC
- S. Hotta, Y. Shimizu, T. Yamao, M. Goto and R. Azumi, Chem. Lett., 2009, 38, 294–295 CrossRef CAS
- T. Nicolini, A. Famulari, T. Gatti, J. Martí-Rujas, F. Villafiorita-Monteleone, E. V. Canesi, F. Meinardi, C. Botta, E. Parisini, S. Valdo Meille and C. Bertarelli, J. Phys. Chem. Lett., 2014, 5, 2171–2176 CrossRef CAS PubMed
- H. Mizuno, T. Maeda, H. Yanagi, H. Katsuki, M. Aresti, F. Quochi, M. Saba, A. Mura, G. Bongiovanni, F. Sasaki and S. Hotta, Adv. Opt. Mater., 2014, 2, 529–534 CrossRef CAS
- G. Schweicher, V. Lemaur, C. Niebel, C. Ruzié, Y. Diao, O. Goto, W. Lee, Y. Kim, J.-B. Arlin, J. Karpinska, A. R. Kennedy, S. R. Parkin, Y. Olivier, S. C. B. Mannsfeld, J. Cornil, Y. H. Geerts and Z. Bao, Adv. Mater., 2015, 27, 3066–3072 CrossRef CAS PubMed
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed
- E. G. Mcrae and M. Kasha, J. Chem. Phys., 1958, 28, 721–722 CrossRef CAS
- R. Katoh, K. Suzuki, A. Furube, M. Kotani and K. Tokumaru, J. Phys. Chem. C, 2009, 113, 2961–2965 CAS
- C. Reese, M. E. Roberts, S. R. Parkin and Z. Bao, Adv. Mater., 2009, 21, 3678–3681 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectra of synthesized compounds, cyclic voltammetry and optical spectroscopy data, microscopy images, additional X-ray data and OFET data. CCDC 1520469–1520471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02565j |
| This journal is © The Royal Society of Chemistry 2017 |