
Structural evolution of a metal–organic framework and derived hybrids composed of metallic cobalt and copper encapsulated in nitrogen-doped porous carbon cubes with high catalytic performance†
Hui
Li
a,
Fan
Yue
a,
Chao
Yang
a,
Peng
Xue
a,
Nannan
Li
a,
Yi
Zhang
b and
Jide
Wang
*a
aKey Laboratory of Oil and Gas Fine Chemicals, Ministry of Education & Xinjiang Uygur Autonomous Region, College of Chemistry and Chemical Engineering of Xinjiang University, Urumqi, 830046, China. E-mail: awangjd@sina.cn
bKey Laboratory of Resources Chemistry of Nonferrous Metals (Ministry of Education), College of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, China
First published on 21st November 2016
Abstract
We present here the structural evolution of a Prussian blue analogue CuII3[CoIII(CN)6]2 (Cu–Co PBA) controlled by sodium citrate as a function of time at room temperature. We identified the different stages of Cu–Co PBA formation and elucidated its transformation kinetics. Moreover, 3D hybrid composites of nitrogen-doped porous carbon cubes, with cobalt and copper nanoparticles embedded within the well-graphitized shells, were rationally fabricated using Cu–Co PBA as the single precursor. The resulting Cu/Co@NPCC catalysts showed superior catalytic activity in the reduction of 4-nitrophenol to 4-aminophenol. Metallic cobalt and copper cores were leached out by FeCl3 and HCl treatment to determine the catalytically active sites. Results showed that the nitrogen-doped porous carbon still exhibited excellent catalytic performance after the metallic cores were removed. Based on the experimental results, we speculate that the unique structure of metal residues encapsulated inside the graphitic carbon layers may have higher catalytic activity than the metal particles themselves.
1. Introduction
Metal–organic frameworks (MOFs) have attracted great interest due to the variety of their applications, which include gas adsorption and storage,1–5 chemical separation,6,7 drug delivery,8,9 and chemical catalysis.10–12 In particular, some MOFs have been utilized to generate porous carbons,13–15 metal oxides,16–19 metal sulfides,20–24 and uniformly dispersed metal nanoparticles25–28 in a carbon matrix by dealing with the as-prepared MOF crystals. They have also been applied in electrocatalysis,24,29 batteries,15,30,31 supercapacitors,32–34 and heterogeneous catalytic reactions.35–37Recently, non-noble metallic nanoparticle catalysts (such as Cu, Co, and Ni) have attracted widespread attention because they are much more economical than palladium and gold nanoparticles.27,38,39 Some recent reports have demonstrated that encapsulating non-noble metallic nanoparticles in graphene layers or carbon nanotubes is a promising method of fabricating functional materials.40–42 Bao's group proposed that the interaction between the metallic core and nitrogen-doped ultrathin graphitic shells could change the local work function of the shell, thereby providing it with surprisingly high chemical activities.43 Meanwhile, Li and co-workers demonstrated that metallic cores encapsulated in multilayered graphene shells might not be directly involved in catalytic reactions, but M–N–C moieties.44 Although many convincing works have been accomplished, identifying the nature of the catalytically active sites is still an existing fundamental challenge.
In MOFs, the highly ordered metal ions are regularly isolated by organic ligands, which would emerge as metallic cores coated with graphitic shells just during thermolysis. After Xu's group45 firstly employed MOF-5 as a template and precursor with furfuryl alcohol (FA) as an additional carbon source to prepare porous carbons, it has been shown that the direct carbonization of a MOF precursor without an additional carbon source is another way to prepare highly porous carbon and carbon–metal hybrids.46,47 This strategy is fascinating because carbons or metal nanoparticles with extremely porous structures can be obtained directly from MOFs without extra templates.
Aside from being devoted to the synthesis of new MOF structures and the exploration of new properties, researchers have started to give increasing attention to the control of crystal size and morphology.22,48,49 In particular, the size control of MOF crystals into the nanoscale or mesoscale regime is certainly necessary. In order to prepare the MOFs with large specific surface area, controlling the smallest possible single individual is obligatory, but, unfortunately, very small particles exhibit extremely high agglomeration tendencies. Monodisperse crystals with distinct polyhedral morphologies have been prepared from various families of MOF materials. However, compared to the huge number of reported MOFs, they are still very limited for controlling the growth of crystals nanometers to micrometers in size.
In this work, we developed a facile method to control the growth and crystallization process of a Prussian blue analogue CuII3[CoIII(CN)6]2 (Cu–Co PBA) by tuning the addition of sodium citrate. To the best of our knowledge, this is one of the rarest works reporting the control of growth and structural evolution of MOFs,50 although there have been many papers reporting that sodium citrate controls the growth of metals and metal oxides.51–54 More importantly, such a method would provide a much more feasible solution in bulkier MOF substrates when they are unable to reach a uniform size in the nanoscale or mesoscale regime. In addition, Co and Cu metal nanoparticles embedded in 3D nitrogen-doped porous carbon cube hybrids (Cu/Co@NPCC) were obtained through direct annealing of pre-synthesized Cu–Co PBA nanocubes. In the resulting structure, Co and Cu nanoparticles are embedded in the graphene shells. The as-prepared Cu/Co@NPCC complexes exhibit excellent catalytic activity toward the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in the presence of NaBH4. After removing the metallic cores with FeCl3 and HCl treatment (Cu/Co–NPCC), no obvious deterioration, or even promotion present in certain circumstances, was observed for the catalytic activity. We therefore propose that the main catalytic activity of Cu/Co@NPCC comes from the unique structure of metal residues encased inside the graphitic layers.
2. Experimental
2.1 Synthesis of Cu–Co PBA cubes
Copper nitrate, potassium hexacyanocobaltate(III), and sodium citrate were purchased from Sinopharm Chemical Reagent Co. (Shanghai, China), while 4-NP was purchased from Acros. In a typical procedure for the synthesis of Cu–Co PBA nanocubes with sizes of about 500 nm, 0.6 mmol of copper nitrate and 0.9 mmol of sodium citrate were dissolved in 15 mL of deionized water (DI). Meanwhile, 0.4 mmol of potassium hexacyanocobaltate(III) was dissolved in another 15 mL of DI water. Afterwards, the two solutions were mixed fleetly, and the obtained mixed solution was aged for 24 h at room temperature. After collection by centrifugation and washing with water and ethanol, the precipitates were dried in a vacuum at 60 °C.2.2 Synthesis of Cu/Co@NPCC and Cu/Co–NPCC
Cu/Co@NPCC-600 was synthesized by direct carbonization of Cu–Co PBA at 600 °C in an Ar atmosphere. The temperature of the oven was increased gradually at a heating rate of 2 °C min−1. After the target temperature was reached, the powders were annealed for 3 h and then cooled to room temperature. Similarly, compounds Cu/Co@NPCC-700 and Cu/Co@NPCC-800 were obtained by annealing Cu–Co PBA at 700 °C and 800 °C, respectively.Cu/Co–NPCC-600 and Cu/Co–NPCC-700 were prepared by leaching out the metallic copper and cobalt cores of Cu/Co@PCC-600 and Cu/Co@PCC-700 using FeCl3 and HCl, respectively.
2.3 Instruments and characterization
XRD patterns were collected on a Bruker D8 Advance powder X-ray diffractometer using Cu Kα radiation (Bruker, Germany). Thermogravimetric analysis (TGA) was performed on a Shimadzu DTG-50 thermal analyzer from room temperature to 800 °C at a heating rate of 10 °C min−1. Scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS) were conducted on a Hitachi S-8010, while high-resolution transmission electron microscopy (HR-TEM) investigations were performed using a JEOL-2010 (JEOL, Japan). X-ray photoelectron spectroscopy (XPS) was conducted using an ESCALAB 250Xi spectrometer (Thermo Fisher Scientific, USA). The surface areas of the samples were calculated using the Brunauer–Emmett–Teller (BET) method, whereas the pore volume and average pore diameter were determined by applying the Barrett–Joyner–Halenda (BJH) method to the desorption branches of nitrogen isotherms. The changes in absorbance were recorded using a UV-vis spectrophotometer (Shimadzu, UV-2550), while the products were characterized on a Xevo G2-XS QTof liquid chromatograph mass spectrometer (LC-MS, Waters, USA).2.4 Catalytic activity of Cu/Co@ NPCC and Cu/Co–NPCC for 4-NP reduction in the presence of NaBH4
The catalytic redox reaction process was set up in a standard quartz cuvette with 1.0 cm path length and 4 mL volume. Initially, a sufficient excess of NaBH4 (0.24 mmol) was added to the above quartz cuvette containing 4-NP (3 mL, 0.1 mM) with stirring at 25 °C before adding Cu/Co@NPCC or Cu/Co–NPCC (1 mg mL−1, Cu/Co@NPCC or Cu/Co–NPCC was dispersed in DI). The change of the mixture solution was monitored using UV-vis spectroscopy, while the decrease in absorbance at 400 nm was recorded by time-scanning of the UV-vis spectrometer. The product was analyzed using LC-MS.3. Results and discussion
The time-lapse observations on the formation and growth of Cu–Co PBA frameworks were illustrated by SEM images, as shown in Fig. 1 and S1.† The SEM images revealed that the sample synthesized at 10 min exhibited polyhedral structures with six surfaces obviously layered in the structure (Fig. 1a). The layers presented an orderly arrangement indicating the initial stages of crystallization. At 60 min, the layered structure in each direction exhibited significant growth along the two-dimensional direction (Fig. 1b). Furthermore, the crystals were found to be cube-shaped at this time point. As shown in Fig. 1c and d, the number and thickness of these layers gradually shrank along all surfaces at the 6 h to 12 h time point, while more homogeneous top surfaces were observed. After 24 h of incubation, the crystals grew into a well-shaped cube, and the crystals retained the traces of layering (Fig. 1e). | ||
| Fig. 1 SEM images of Cu–Co PBA as a function of synthesis time: (a) 10 min; (b) 1 h; (c) 6 h; (d) 12 h; (e) 24 h. | ||
Control experiments were performed to understand the formation mechanism of Cu–Co PBA cubes. Without adding or reducing the dosage of sodium citrate, the reaction was completed very rapidly, with only irregular nanoparticles obtained (Fig. S2†). Obviously, sodium citrate is the crucial factor driving the 2D layers to grow along the six surfaces of the crystals and generate Cu–Co PBA cubes. The UV-vis spectra indicate that, after the addition of sodium citrate, the intensity of the maximum absorption peak of the Cu(NO3)2 solution significantly increased and the position was shifted (Fig. S3†). This absorbance variation was caused by the coordination between citrate ions and Cu2+ ions.55 In addition, this chelation effectively inhibited the rapid coordination reaction between Cu2+ and [Co(CN)6]3−, resulting in the deceleration of the crystallization process of the Cu–Co PBA cubes.
The growth mechanism is shown in Fig. 2. In the initial stages of the reaction, the Cu–Co PBA grew mainly along the longitudinal direction layer by layer. Following a lengthy process, lateral growth dominated the growth process until crystal growth stopped. In general, rapid crystallization develops small, irregularly shaped nanoparticles, while slow crystallization develops nanoparticles with a well-defined morphology. In our case, Cu–Co PBA showed a cube-shaped morphology. The structure of Cu–Co PBA as a function of time was evaluated by XRD, while all diffraction peaks corresponded to pure Cu–Co PBA and matched well with the simulation results (Fig. 3).
 | ||
| Fig. 2 Schematic illustration of the synthesis procedure of Cu–Co PBA cubes. | ||
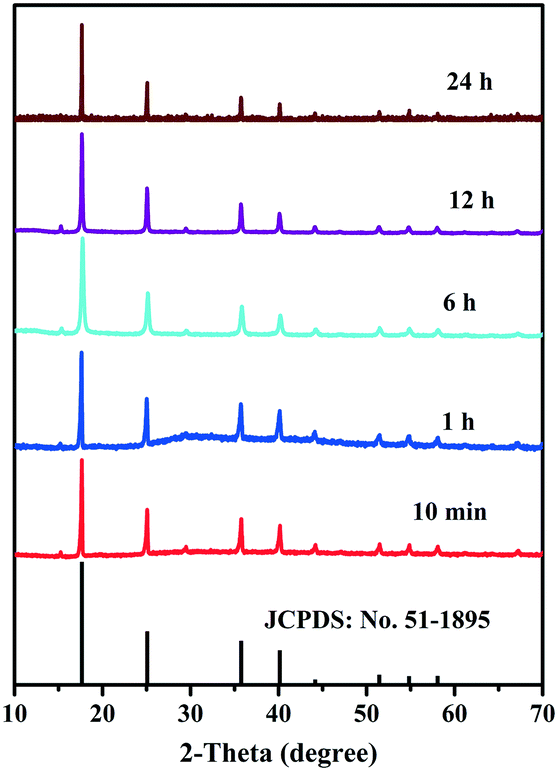 | ||
| Fig. 3 XRD patterns illustrating the structural evolution of Cu–Co PBA as a function of synthesis time. | ||
Subsequent carbonizations were applied to produce carbon-coated bimetal nanoparticles at 600 °C, 700 °C and 800 °C. Our experimental results confirmed that after carbonization, the obtained carbons inherited the original morphology of the Cu–Co PBA crystals, with the surfaces of the cubes slightly distorted (Fig. 4). Furthermore, during the carbonization process, copper and cobalt ions were reduced to form metallic copper and cobalt nanoparticles (Fig. 5), which subsequently acted as catalysts to promote the in situ growth of carbon nanotubes on the surface of the carbon cube.56 The TEM images reveal strong contrasting differences between the darker marginal region and the brighter central region, further suggesting the porous structure of Cu/Co@NPCC (Fig. 4b, d and f). The pores in the Cu/Co@NPCC structures were caused by the carbonization of the cyano group and the original channel of Cu–Co PBA. When the temperature was increased to 800 °C, the obtained Cu/Co@NPCC-800 product still retained its cubic shape, while the metal nanoparticles embedded in the carbon apparently agglomerated when compared to Cu/Co@NPCC-600 and Cu/Co@NPCC-700.
 | ||
| Fig. 4 SEM images of (a) Cu/Co@NPCC-600, (c) Cu/Co@NPCC-700, and (e) Cu/Co@NPCC-800; TEM images of (b) Cu/Co@NPCC-600, (d) Cu/Co@NPCC-700, and (f) Cu/Co@NPCC-800. | ||
 | ||
| Fig. 5 XRD patterns of metallic cobalt and copper encapsulated in nitrogen-doped porous carbon cubes obtained at different calcination temperatures. | ||
The specific surface area and pore size distribution of Cu/Co@NPCC-600, Cu/Co@NPCC-700, and Cu/Co@NPCC-800 were analyzed using N2 adsorption and desorption isotherms, respectively (Fig. 6). All three samples displayed typical type IV adsorption isotherms with an H3-type hysteresis loop. This indicated the presence of a mesoporous structure which was inherited from Cu–Co PBA and the decomposition of the samples during heat treatment. Cu/Co@NPCC-600 and Cu/Co@NPCC-700 had high BET-specific surface areas of approximately 45.7 m2 g−1 and 42.3 m2 g−1, respectively. The specific surface area decreased to 22.6 m2 g−1, when the thermal treatment temperature increased to 800 °C. The pore size distribution (calculated via the BJH method) shows that both Cu/Co@NPCC-600 and Cu/Co@NPCC-700 were porous in a narrow distribution range that centered at approximately 3.7 nm and had a relatively wide distribution of less than 20 nm (Fig. 6b). This is beneficial for improving the throughput rate of the reactants.
 | ||
| Fig. 6 N2 adsorption–desorption isotherms (a) and the corresponding pore size distributions (b) of Cu/Co@NPCC-600, Cu/Co@NPCC-700, and Cu/Co@NPCC-800. | ||
To evaluate the catalytic activity of Cu/Co@NPCC-600, reduction of 4-NP to 4-AP by NaBH4 in solution was selected as the model reaction. The reaction process, as well as the reaction time, was monitored by UV-vis spectroscopy (Fig. 7a). The absorption peak of the 4-NP aqueous solution was located at 317 nm while this characteristic peak red-shifted to 400 nm after NaBH4 was added because of the formation of 4-nitrophenolate ions.57 Furthermore, the characteristic peak of the 4-NP ion at 400 nm decreased in in tensity rapidly and almost disappeared after 1 min along with the appearance of a new peak at 300 nm, corresponding to the formation of 4-AP. Without catalysts, the maximum absorption peak remained unchanged, indicating that NaBH4 alone cannot initiate the conversion reaction even with a large excess in amount (Fig. 7b). When Cu/Co@NPCC-600 was dispersed in the solution of 4-NP and NaBH4, a linear relationship was found between ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (Ct/C0) (Ct and C0 are 4-NP concentrations at time t and 0, respectively) and reaction time, which matches well with first-order reaction kinetics, where ln(Ct/C0) = −kt (k: apparent first-order rate constant [s−1]). The corresponding kinetic rate constant k was estimated to be 0.029, 0.050, and 0.086 s−1 when the added catalyst volume was increased from 30 μl (0.01 mg ml−1), to 60 μl (0.02 mg ml−1), and then to 90 μl (0.03 mg ml−1), respectively. The Cu/Co@NPCC-600 in our experiments exhibits superior catalytic activity, even better than those of some noble metal nanoparticles for the catalytic reduction of 4-NP.58–60 Cu/Co@NPCC-700 and Cu/Co@NPCC-800 had lower catalytic activity than Cu/Co@NPCC-600 (Fig. 7c), with their corresponding kinetic rate constant k estimated to be 0.012 s−1 and 0.005 s−1, respectively, when the catalyst dosage was 0.02 mg ml−1. Although Cu/Co@NPCC-600 showed superior catalytic activity in the reduction of 4-NP to 4-AP, the catalytically active sites are still a mystery, and further research is very necessary.
(Ct/C0) (Ct and C0 are 4-NP concentrations at time t and 0, respectively) and reaction time, which matches well with first-order reaction kinetics, where ln(Ct/C0) = −kt (k: apparent first-order rate constant [s−1]). The corresponding kinetic rate constant k was estimated to be 0.029, 0.050, and 0.086 s−1 when the added catalyst volume was increased from 30 μl (0.01 mg ml−1), to 60 μl (0.02 mg ml−1), and then to 90 μl (0.03 mg ml−1), respectively. The Cu/Co@NPCC-600 in our experiments exhibits superior catalytic activity, even better than those of some noble metal nanoparticles for the catalytic reduction of 4-NP.58–60 Cu/Co@NPCC-700 and Cu/Co@NPCC-800 had lower catalytic activity than Cu/Co@NPCC-600 (Fig. 7c), with their corresponding kinetic rate constant k estimated to be 0.012 s−1 and 0.005 s−1, respectively, when the catalyst dosage was 0.02 mg ml−1. Although Cu/Co@NPCC-600 showed superior catalytic activity in the reduction of 4-NP to 4-AP, the catalytically active sites are still a mystery, and further research is very necessary.
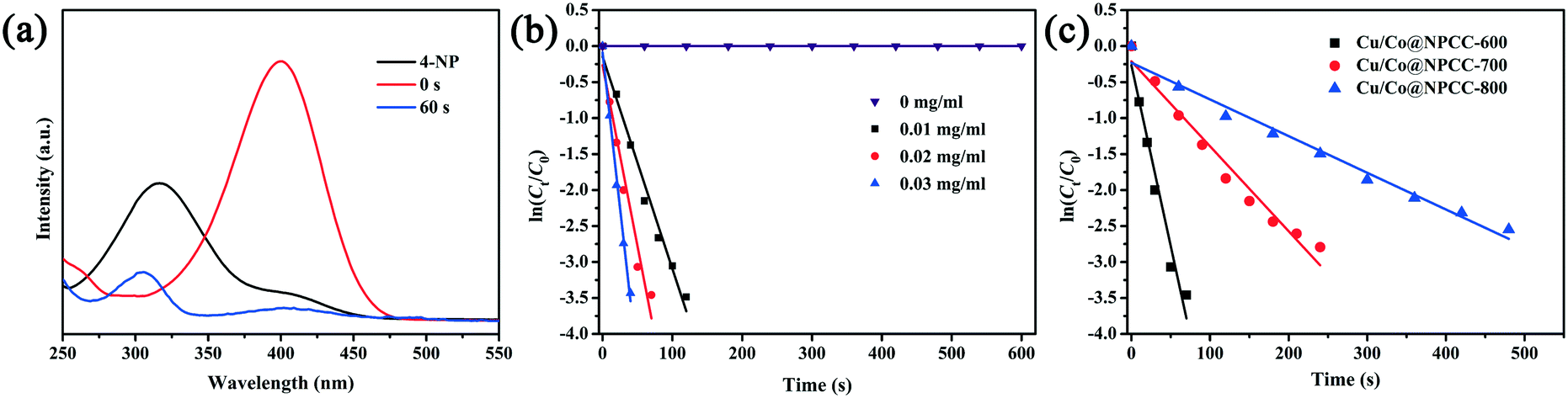 | ||
| Fig. 7 (a) Time-dependent UV-vis absorption spectra of 4-NP reduced by NaBH4 with 0.02 mg ml−1 Cu/Co@NPCC-600; (b) plot of ln(Ct/C0) against the reaction time for the reduction reaction of 4-NP at different Cu/Co@NPCC-600 dosages; (c) plot of ln(Ct/C0) against the reaction time for the reduction reaction with 0.02 mg ml−1 Cu/Co@NPCC-600, Cu/Co@NPCC-700, and Cu/Co@NPCC-800 as the catalysts (T = 25 °C). | ||
To understand the superior catalytic activity of Cu/Co@NPCC-600 in the reduction of 4-NP to 4-AP, further research was performed to reveal the catalytically active sites of this complex. Moreover, the multi-layered graphitic carbon should be taken as part of the porous carbon cube. The high-resolution TEM images of Cu/Co@NPCC-600 show that most of the metallic cores were less than 20 nm in size (Fig. 8a–c and S6†). Furthermore, the shells completely coated the metallic cores with an interlayer distance of about 0.34 nm, indicating the existence of a graphene layer.61 On the outside, it is sheathed by dozens of layers (Fig. 8c and e).
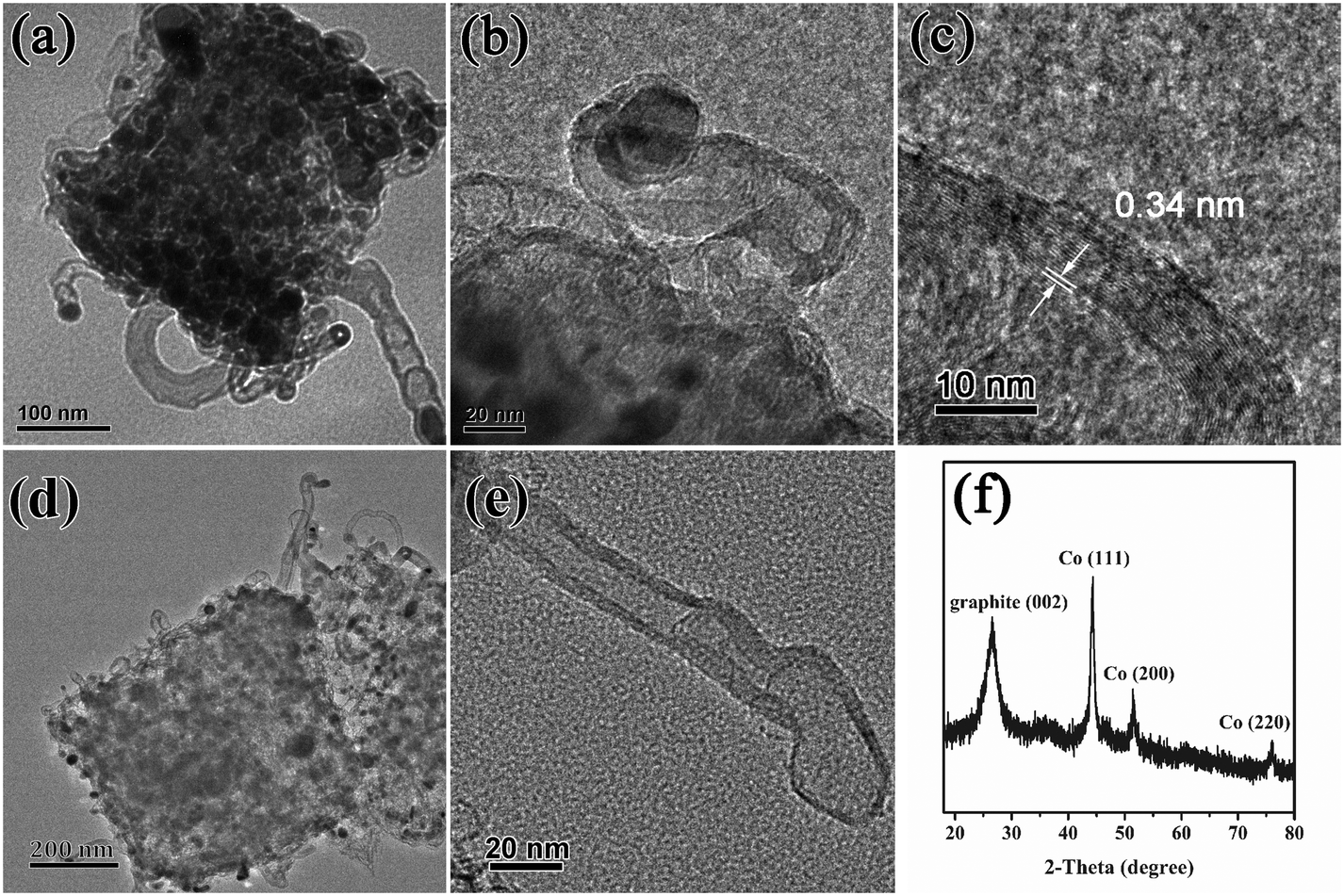 | ||
| Fig. 8 (a–c) HR-TEM images of Cu/Co@NPCC-600, (d and e) TEM images of Cu/Co–NPCC-600, and (f) XRD pattern of Cu/Co–NPCC-600. | ||
The copper and cobalt cores can be leached out (denoted as Cu/Co–NPCC-600) by FeCl3 and HCl treatment (2Fe3+ + M → M2+ + 2Fe2+, M = Cu and Co). The TEM images revealed that Cu/Co–NPCC-600 retains its previous morphology, leaving a large number of hollow graphene shells (Fig. 8d and e). Moreover, even after several cycles of FeCl3 oxidation and concentrated HCl treatment, respectively, metal residues were still cannot been completely removed (Table S1†). The XRD pattern revealed that the Cu peaks vanished and the Co peaks diminished, while the (002) diffraction peak of graphene gained relative intensity. The Cu peaks disappeared because most of the metal Cu was leached out. The results of the characterization were in good agreement with each other. After leaching out the copper and cobalt cores, a notable enhancement of the specific surface areas from 45.7 m2 g−1 to around 201 m2 g−1 was observed (Fig. S7†). The Cu/Co–NPCC-600 complexes with pore sizes of less than 20 nm were mainly distributed in the area, which is consistent with the metal core of Cu/Co@NPCC-600.
X-ray photoemission spectroscopy (XPS) was carried out to further probe the chemical environment and bonding configuration of Cu/Co@NPCC-600 and Cu/Co–NPCC-600 (Fig. 9). The XPS data further confirmed the formation of C, N, Co, and Cu composites. In addition, the C 1s spectra were fitted to five individual peaks centered at 284.6, 285.0, 286.6, 288.4, and 289.1 eV, corresponding to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N, C–O, CO, and OC–O bonds, respectively.62 The existence of the C–N bond further confirmed the nitrogen doping in the carbon cubes. The N 1s spectrum can be deconvoluted into four peaks: pyridinic N (398.6 eV), pyrrolic N (399.5 and 400.7 eV), and quaternary N (402.4 eV).15 After Fe3+ and acid treatment, the nitrogen concentration increased to 5.63% (Cu/Co@NPCC-600, 3.83%). The two strong peaks at 778.5 and 793.2 eV were assigned to Co 2p3/2 and Co 2p1/2 of metallic Co.38 Major contributions from Cu 2p3/2 at 932.7 eV and Cu 2p1/2 at 952.6 eV confirmed the presence of zero valent copper.63 Since XPS is a surface-sensitive technique, the appearance of Co(II) and Cu(II) species in the Co 2p and Cu 2p spectra is understandable.
C, C–N, C–O, CO, and OC–O bonds, respectively.62 The existence of the C–N bond further confirmed the nitrogen doping in the carbon cubes. The N 1s spectrum can be deconvoluted into four peaks: pyridinic N (398.6 eV), pyrrolic N (399.5 and 400.7 eV), and quaternary N (402.4 eV).15 After Fe3+ and acid treatment, the nitrogen concentration increased to 5.63% (Cu/Co@NPCC-600, 3.83%). The two strong peaks at 778.5 and 793.2 eV were assigned to Co 2p3/2 and Co 2p1/2 of metallic Co.38 Major contributions from Cu 2p3/2 at 932.7 eV and Cu 2p1/2 at 952.6 eV confirmed the presence of zero valent copper.63 Since XPS is a surface-sensitive technique, the appearance of Co(II) and Cu(II) species in the Co 2p and Cu 2p spectra is understandable.
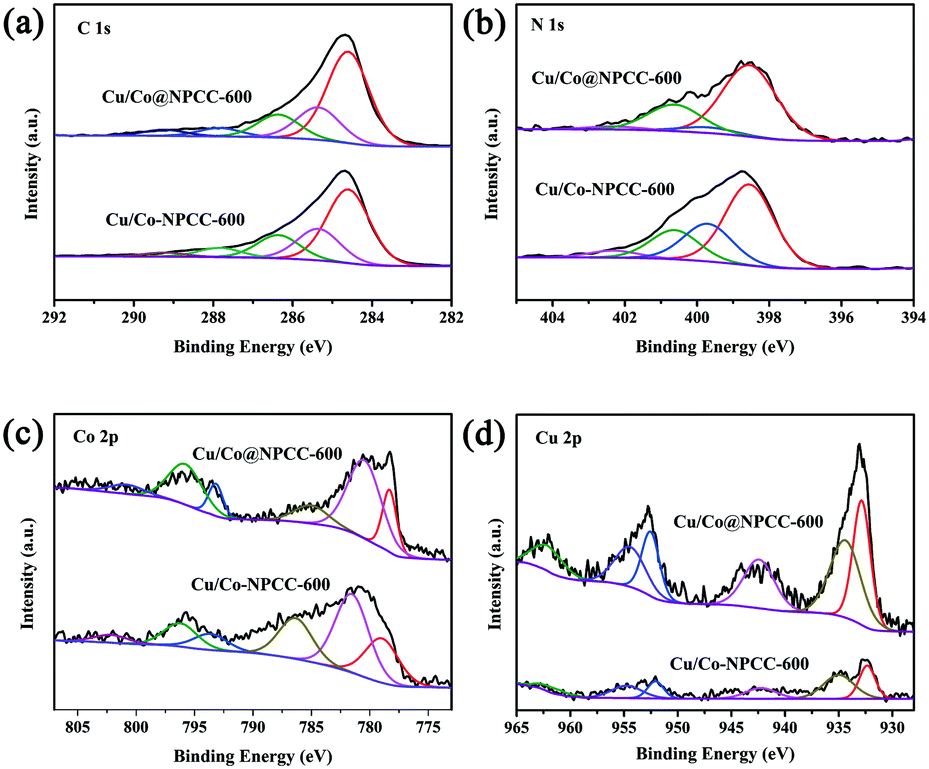 | ||
| Fig. 9 XPS characterization of Cu/Co@NPCC-600 and Cu/Co–NPCC-600. (a) C 1s XPS spectra, (b) N 1s XPS spectra, (c) Co 2p XPS spectra, and (d) Cu 2p XPS spectra. | ||
Similar methods were used to evaluate the catalytic activity of Cu/Co–NPCC-600 and Cu/Co–NPCC-700 (Fig. 10). The continued superior catalytic activity of Cu/Co–NPCC-600 and Cu/Co–NPCC-700 was a big surprise. The corresponding kinetic rate constant k was estimated to be 0.036 s−1 and 0.024 s−1. To prove that the process is a catalytic reaction rather than simple adsorption for 4-NP, UV-vis was conducted to monitor the peak change of the 4-NP solution in the absence of NaBH4 (Fig. S4†). LC-MS was used to characterize the product, which was further proven to be the target product (Fig. S5†). Although the reaction rate driven by Cu/Co–NPCC-600 was slightly lower than that of Cu/Co@NPCC-600, it was still much higher than those of both Cu/Co@NPCC-700 (k = 0.012 s−1) and Cu/Co@NPCC-800 (k = 0.005 s−1), which were embedded with large metallic cores. It is easily deduced that the catalytic rate of the reaction is very much dependent on the amount of catalyst, as derived from Fig. 7b. If the metallic Cu and Co cores were the main active sites of Cu/Co@NPCC, the reaction rate would dramatically decrease or even cease after leaching out the metallic cores.
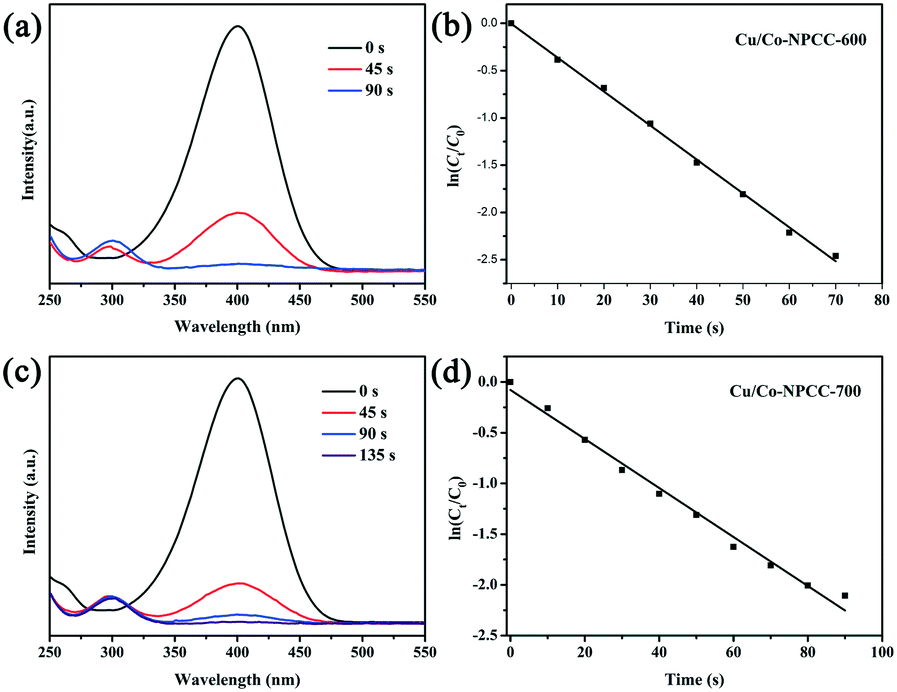 | ||
| Fig. 10 UV-vis spectra showing the gradual reduction of 4-NP over (a) Cu/Co–NPCC-600 and (c) Cu/Co–NPCC-700; plot of ln(Ct/C0) against the reaction time with (b) Cu/Co–NPCC-600 and (d) Cu/Co–NPCC-700 as the catalyst (catalyst dosage = 0.02 mg ml−1; T = 25 °C). | ||
According to our proposal, the main active sites of the Cu/Co@NPCC complexes may not be the metal cores embedded in the graphitic carbon shells. Some Cu and Co atoms may diffuse into the carbon shells and bind with nitrogen atoms, resulting in Co(Cu)–N–C. This may be one of the reasons why Cu/Co–NPCC-600 and Cu/Co–NPCC-700 still have excellent catalytic activity. The theory on the existence of M–N–C moieties as the catalytically active sites has also been widely accepted, especially when metal and nitrogen precursors are pyrolyzed together with carbon under high temperature.44,64 However, in our case, the N atom content in Cu/Co–NPCC-700 was just very limited; the reason why the catalytic activity of Cu/Co–NPCC-700 did not deteriorate, but rather increased, when compared to Cu/Co@NPCC-700, is difficult to explain using this theory. Based on the results, we conclude that Co(Cu)–N–C was not the only catalytically active site in Cu/Co–NPCC-600 and Cu/Co–NPCC-700. XRD, XPS, and EDS results confirm the existence of metallic residues in both Cu/Co–NPCC-600 and Cu/Co–NPCC-700. This is because of the metal residues encased inside the graphitic carbon layers, which could not be removed by Fe3+ and acid. These metal residues are different from the metal cores which are coated by graphitic carbon layers in the original porous carbon cubes. In light of the aforementioned facts, the metal residues encased inside the carbon layers may have higher catalytic activity. Their catalytic activity may be derived from the electron transfer from the metal layer to the graphite layer leading to a decrease in the local work function on the carbon surface (confinement effect).42,43 Meanwhile, despite having almost the same type of encased carbon, and almost the same surface area and pore size distribution, Cu/Co–NPCC-600 (Cu/Co@NPCC-600) exhibits better catalytic activity compared to Cu/Co–NPCC-700 (Cu/Co@NPCC-700) (Fig. 6 and S7†). The reason may be because Cu/Co–NPCC-600 and Cu/Co@NPCC-600 have relatively abundant nitrogen atoms in the porous carbon cubes. Hence, improving the catalytic activity in the presence of N–C might be beneficial. Nitrogen atoms are inherently better than carbon atoms in interacting with reactants due to the presence of lone-pair electrons. Moreover, the relatively higher electronegativity of nitrogen atoms gives a higher positive charge density on their adjacent carbon atoms.65,66
4. Conclusions
We have monitored the structural evolution of Cu–Co PBA as a function of time at room temperature while identifying the different stages of Cu–Co PBA formation. We employ the Cu–Co PBA MOF in deriving Cu and Co bimetal nanoparticles embedded in 3D nitrogen-doped porous carbon cubes through direct annealing. The highly efficient catalyst represents a promising step toward the practical applications of MOF-derived non-noble metal catalysts in catalytic hydrogenation reaction systems. More importantly, our results reveal that Cu and Co nanoparticles were not the main active sites. The unique structure of metal residues confined inside the graphitic carbon layers might change the local work function of the shell, thereby providing it with surprisingly high chemical activities. Different from previous excellent works about electrocatalytic activities for ORR and HER, which were based on thin enough graphene shells or abundant M–N–C, we propose that both the thick graphitic layer and M–N–C are not significantly affected by the catalytic activity in our system. Meanwhile, further attention to understanding the chemical nature of their active sites is called for. We believe that these findings will provide inspiration to those who are interested in developing a new kind of structurally enhanced catalyst.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21162027 and 21261022), the Graduate Student Research Innovation Project of Xinjiang (No. XJGRI2013016), and the Outstanding Doctoral Innovation Project of Xinjiang University (XJUBSCX-2012020) is gratefully acknowledged.Notes and references
- D. Gygi, E. D. Bloch, J. A. Mason, M. R. Hudson, M. I. Gonzalez, R. L. Siegelman, T. A. Darwish, W. L. Queen, C. M. Brown and J. R. Long, Chem. Mater., 2016, 28, 1128–1138 CrossRef CAS.
- L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- X. Han, X. Han, R. Li, X. J. Wang and Y. Zhao, CrystEngComm, 2016, 18, 1277–1281 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- Y. S. Bae, C. Y. Lee, K. C. Kim, O. K. Farha, P. Nickias, J. T. Hupp, S. T. Nguyen and R. Q. Snurr, Angew. Chem., Int. Ed., 2012, 51, 1857–1860 CrossRef CAS PubMed.
- H. Wu, Q. Gong, D. H. Olson and J. Li, Chem. Rev., 2012, 112, 836–868 CrossRef CAS PubMed.
- T. Kundu, S. Mitra, P. Patra, A. Goswami, D. Díaz Díaz and R. Banerjee, Chem. – Eur. J., 2014, 20, 10514–10518 CrossRef CAS PubMed.
- M. Nazari, M. Rubio Martinez, G. Tobias, J. P. Barrio, R. Babarao, F. Nazari, K. Konstas, B. W. Muir, S. F. Collins and A. J. Hill, Adv. Funct. Mater., 2016, 26, 3244–3249 CrossRef CAS.
- H. L. Nguyen, F. Gándara, H. Furukawa, T. L. H. Doan, K. E. Cordova and O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 4330–4333 CrossRef CAS PubMed.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed.
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef CAS.
- R. R. Salunkhe, C. Young, J. Tang, T. Takei, Y. Ide, N. Kobayashi and Y. Yamauchi, Chem. Commun., 2016, 52, 4764–4767 RSC.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718–724 CrossRef CAS PubMed.
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 1–10 Search PubMed.
- L. Lux, K. Williams and S. Ma, CrystEngComm, 2015, 17, 10–22 RSC.
- H. Pang, J. Deng, J. Du, S. Li, J. Li, Y. Ma, J. Zhang and J. Chen, Dalton Trans., 2012, 41, 10175–10181 RSC.
- Y. Z. Zhang, Y. Wang, Y. L. Xie, T. Cheng, W. Y. Lai, H. Pang and W. Huang, Nanoscale, 2014, 6, 14354–14359 RSC.
- B. Ramaraju, C. H. Li, S. Prakash and C. C. Chen, Chem. Commun., 2016, 52, 946–949 RSC.
- Q. Wang, R. Zou, W. Xia, J. Ma, B. Qiu, A. Mahmood, R. Zhao, Y. Yang, D. Xia and Q. Xu, Small, 2015, 11, 2511–2517 CrossRef CAS PubMed.
- R. Wu, D. P. Wang, V. Kumar, K. Zhou, A. W. K. Law, P. S. Lee, J. Lou and Z. Chen, Chem. Commun., 2015, 51, 3109–3112 RSC.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 1–6 CrossRef.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- Z. F. Huang, J. Song, K. Li, M. Tahir, Y. T. Wang, L. Pan, L. Wang, X. Zhang and J. J. Zou, J. Am. Chem. Soc., 2016, 138, 1359–1365 CrossRef CAS PubMed.
- N. L. Torad, M. Hu, S. Ishihara, H. Sukegawa, A. A. Belik, M. Imura, K. Ariga, Y. Sakka and Y. Yamauchi, Small, 2014, 10, 2096–2107 CrossRef CAS PubMed.
- J. Wang, D. Gao, G. Wang, S. Miao, H. Wu, J. Li and X. Bao, J. Mater. Chem. A, 2014, 2, 20067–20074 CAS.
- W. Zhong, H. Liu, C. Bai, S. Liao and Y. Li, ACS Catal., 2015, 5, 1850–1856 CrossRef CAS.
- A. Kong, C. Mao, Q. Lin, X. Wei, X. Bu and P. Feng, Dalton Trans., 2015, 44, 6748–6754 RSC.
- Y. Z. Chen, C. Wang, Z. Y. Wu, Y. Xiong, Q. Xu, S. H. Yu and H. L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS PubMed.
- N. Yan, L. Hu, Y. Li, Y. Wang, H. Zhong, X. Y. Hu, X. K. Kong and Q. W. Chen, J. Phys. Chem. C, 2012, 116, 7227–7235 CAS.
- B. Liu, X. Zhang, H. Shioyama, T. Mukai, T. Sakai and Q. Xu, J. Power Sources, 2010, 195, 857–861 CrossRef CAS.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- L. Wang, Y. Han, X. Feng, J. Zhou, P. Qi and B. Wang, Coord. Chem. Rev., 2016, 307(Part 2), 361–381 CrossRef CAS.
- X. Rui, H. Tan and Q. Yan, Nanoscale, 2014, 6, 9889–9924 RSC.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- J. B. Zhu, K. Li, M. L. Xiao, C. P. Liu, Z. J. Wu, J. J. Ge and X. Wei, J. Mater. Chem. A, 2016, 4, 7422–7429 CAS.
- K. Meyer, M. Ranocchiari and J. A. van Bokhoven, Energy Environ. Sci., 2015, 8, 1923–1937 CAS.
- C. Bai, X. Yao and Y. Li, ACS Catal., 2015, 5, 884–891 CrossRef CAS.
- P. Deka, R. C. Deka and P. Bharali, New J. Chem., 2014, 38, 1789–1793 RSC.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- X. Zou, X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmeková and T. Asefa, Angew. Chem., 2014, 126, 4461–4465 CrossRef.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- D. Deng, L. Yu, X. Chen, G. Wang, L. Jin, X. Pan, J. Deng, G. Sun and X. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- M. Zeng, Y. Liu, F. Zhao, K. Nie, N. Han, X. Wang, W. Huang, X. Song, J. Zhong and Y. Li, Adv. Funct. Mater., 2016, 26, 4397–4404 CrossRef CAS.
- B. Liu, X. B. Zhang, H. Shioyama, T. Mukai, T. Sakai and Q. Xu, J. Power Sources, 2010, 195, 857–861 CrossRef CAS.
- J. K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 CAS.
- K. Shen, X. D. Chen, J. Y. Chen and Y. W. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- M. H. Pham, G. T. Vuong, A. T. Vu and T. O. Do, Langmuir, 2011, 27, 15261–15267 CrossRef CAS PubMed.
- Q. Xu, H. Li, F. Yue, L. Chi and J. D. Wang, New J. Chem., 2016, 40, 3032–3035 RSC.
- S. R. Venna, J. B. Jasinski and M. A. Carreon, J. Am. Chem. Soc., 2010, 132, 18030–18033 CrossRef CAS PubMed.
- Z. Yang, H. Qian, H. Chen and J. N. Anker, J. Colloid Interface Sci., 2010, 352, 285–291 CrossRef CAS PubMed.
- J. Han, Z. Zhou, Y. Yin, X. Luo, J. Li, H. Zhang and B. Yang, CrystEngComm, 2012, 14, 7036–7042 RSC.
- I. C. Chang, P. C. Chen, M. C. Tsai, T. T. Chen, M. H. Yang, H. T. Chiu and C. Y. Lee, CrystEngComm, 2013, 15, 2363–2366 RSC.
- D. Han, P. Xu, X. Jing, J. Wang, P. Yang, Q. Shen, J. Liu, D. Song, Z. Gao and M. Zhang, J. Power Sources, 2013, 235, 45–53 CrossRef CAS.
- M. Hu, S. Ishihara and Y. Yamauchi, Angew. Chem., Int. Ed., 2013, 52, 1235–1239 CrossRef CAS PubMed.
- R. Wu, D. P. Wang, X. Rui, B. Liu, K. Zhou, A. W. K. Law, Q. Yan, J. Wei and Z. Chen, Adv. Mater., 2015, 27, 3038–3044 CrossRef CAS PubMed.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- Y. Deng, Y. Cai, Z. Sun, J. Liu, C. Liu, J. Wei, W. Li, C. Liu, Y. Wang and D. Zhao, J. Am. Chem. Soc., 2010, 132, 8466–8473 CrossRef CAS PubMed.
- L. Hu, R. Zhang, L. Wei, F. Zhang and Q. Chen, Nanoscale, 2015, 7, 450–454 RSC.
- N. Yan, Z. Zhao, Y. Li, F. Wang, H. Zhong and Q. Chen, Inorg. Chem., 2014, 53, 9073–9079 CrossRef CAS PubMed.
- J. Liu, C. Wu, D. D. Xiao, P. Kopold, L. Gu, P. A. van Aken, J. Maier and Y. Yu, Small, 2016, 12, 2354–2364 CrossRef CAS PubMed.
- T. H. Cai, W. Xing, Z. Liu, J. B. Zeng, Q. Z. Xue, S. Z. Qiao and Z. F. Yan, Carbon, 2015, 86, 235–244 CrossRef CAS.
- S. Ghosh, R. Das, I. H. Chowdhury, P. Bhanja and M. K. Naskar, RSC Adv., 2015, 5, 101519–101524 RSC.
- J. Masa, W. Xia, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2015, 54, 10102–10120 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- X. X. Zou, X. X. Huang, A. Goswami, R. Silva, B. R. Sathe, E. Mikmekovã and T. Asefa, Angew. Chem., 2014, 126, 4461–4465 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce01995a |
| This journal is © The Royal Society of Chemistry 2017 |