
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented rearrangement of diketopyrrolopyrroles leads to structurally unique chromophores†
Olena
Vakuliuk
 a,
Shota
Ooi‡
a,
Irena
Deperasińska
b,
Olga
Staszewska-Krajewska
a,
Marzena
Banasiewicz
b,
Bolesław
Kozankiewicz
b,
Oksana
Danylyuk
c and
Daniel T.
Gryko
*a
a,
Shota
Ooi‡
a,
Irena
Deperasińska
b,
Olga
Staszewska-Krajewska
a,
Marzena
Banasiewicz
b,
Bolesław
Kozankiewicz
b,
Oksana
Danylyuk
c and
Daniel T.
Gryko
*a
aInstitute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dtgryko@icho.edu.pl
bInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland
cInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland
First published on 11th October 2017
Abstract
Diketopyrrolopyrroles possessing thienyl, furyl and benzofuryl substituents undergo unprecedented skeletal rearrangement in the presence of trimethylsilyl bromide resulting in the formation of thieno[2,3-f]isoindole-5,8-diones and furo[2,3-f]isoindole-5,8-diones. These relatively small dyes possess favorable photophysical properties with the emission maxima within the range of 573–624 nm, large fluorescence quantum yields, moderate sensitivity of emission to solvent polarity and a HOMO–LUMO gap of ca. 1.8 eV.
Organic electronics and fluorescence imaging require heterocycles with π-extended conjugation.1 Rational design, aided by combinatorial and diversity-orientated approaches, provides invaluable routes toward novel functional materials.2 Nonetheless, certain sets of chromophores evade even the best designed retrosynthetic analysis and the serendipity factor tends to play a role in the discovery of new dyes.3 Depending on the targeted application, fluorophores possessing different combinations of properties are desirable. Small red-emitters are pivotal for applications in live fluorescence imaging.4
As a part of a larger project aimed at the synthesis of novel fluorophores, we investigated oxidative aromatic coupling of diketopyrrolopyrroles (DPPs, Fig. S1, ESI†).5 We focused on iodine(III) reagents which efficiently promote such transformations, making them often the reagents of choice.6 During this study, we observed that subjecting 2,5-dimethyl-1,4-diketo-3,6-di(thien-3-yl)pyrrolo[3,4-c]pyrrole (1) to hydroxy(tosyloxy)iodobenzene (Koser's reagent, HTIB) and TMSBr led to the formation of a purple compound as the sole isolable product (Scheme 1 and Table 1, entry 1).7 Surprisingly, while the exact mol. mass of a new compound turned out to be equal to the mol. mass of the substrate, the 1H NMR data revealed the loss of symmetry in the product (Fig. S2, ESI†). Such findings are consistent only with the rearrangement of the DPP 1.
| Entry | Lewis acid | Additive, eq. | Solvent | Yield,b % |
|---|---|---|---|---|
| a Unless otherwise noted, reactions were performed with 1 (0.15 mmol), Lewis acid (0.15 mmol), solvent (1 ml), 50 °C, 17 h. b Isolated yields are reported. c Reaction was performed at 70 °C. d 2 eq. of the Lewis acid was utilized. | ||||
| 1 | TMSBr | HTIB (2) | HFIP | 36 |
| 2 | TMSBr | HTIB (0.05) | HFIP | 75 |
| 3 | TMSBr | — | HFIP | 73 |
| 4 | — | HTIB (2) | HFIP | 0 |
| 5 | TMSBr | — | TFEc | 75 |
| 6 | TMSBr | — | MeOH | 0 |
| 7 | TMSBr | — | DCM | 0 |
| 8 | BF3·Et2O | — | HFIP | 53 |
| 9 | BF3·Et2Od | — | TFEc | 72 |
NMR analysis provides structural information about the synthesized compound. The 13C NMR spectrum shows two signals in the carbonyl region (171.1 ppm and 165.6 ppm) along with two signals in the aliphatic region (32.8 ppm and 28.7 ppm). Moreover, the 1H NMR spectrum shows a singlet at 3.38 ppm and a doublet at 3.42 ppm, which we ascribe as methyl groups. The difference in the chemical shifts as well as the multiplicity of the signals reconfirms the formation of the asymmetric product. The character of the proton signal at 8.82 ppm (1H NMR, broad quartet) reveals the opening of one γ-lactam ring and the presence of an NH–CH3 moiety in the product. Moreover, COSY experiments support a 3 + 2 spin system with one uncorrelated downfield signal. The reaction on one of the carbons adjacent to the sulfur atom can rationalize this finding. Finally, thorough analysis of 1H13C HSQC and 1H13C HMBC spectra revealed that the structure of 2 possesses the thieno[2,3-f]isoindole-5,8-dione core, which has so far never been described in the literature (Scheme 1). The final proof was obtained from single crystal X-ray analysis (Fig. S2 and S3, ESI†).
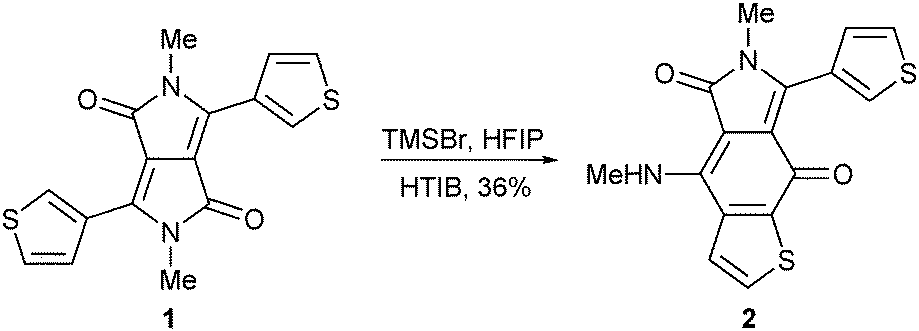 | ||
| Scheme 1 The rearrangement of DPP 1 into thieno[2,3-f]isoindole-5,8-dione 2. | ||
Encouraged by the unique structure of the prepared dye, we started a systematic study of the discovered transformation. The mixture of TMSBr and HTIB provides only moderate yields of the expected product, strongly emphasizing the need for optimization of the reaction conditions (Table 1, entry 1). In fact, the iodine(III) reagent does not support the formation of dye 2 product which suggests the non-oxidative character of this transformation. Rather, HTIB causes the decomposition of the desired product when added in excess (Table 1, entries 1–3). Conversely, the reaction does not proceed in the absence of TMSBr (Table 1, entry 4). We also elaborated the study of the discovered transformation to exhibit strong solvent dependence, which points to the importance of an acidic medium (Table 1, entries 3 and 5–7). In addition, improved solubility of the parent DPP aids a favorable outcome.
Aiming to increase the yield of the desired product, we tested a stronger Lewis acid, BF3·Et2O. Experimental investigations have proven the formation of dye 2 in up to 72% yield (Table 1, entries 8 and 9). It is noteworthy that the use of BF3·Et2O requires a higher loading and elevated temperature to achieve a reasonable yield of the rearranged product. Other Lewis acids such as POCl3, TfOH, TFA or BCl3 do not mediate this rearrangement (Table S1, ESI†).
By adopting conditions 2 and 5 (Table 1), we undertook the synthesis of analogues of heterocycle 2. Experimental findings showed a negligible effect of the alkyl substituents on the discovered transformation (Scheme 2). Notably, in this case the reaction medium plays an important role. We observed a significant improvement in the yield of dye 4 (60% versus 75%) when HFIP was used in place of TFE. Variations in the solubility of the substrate in the tested solvents can probably rationalize this finding. Surprisingly, the transformation of DPP 5, bearing additional tert-butyl groups, does not proceed as expected in either TFE or HFIP. Unexpectedly, the presence of HTIB in a catalytic amount leads to significant improvement in the reaction outcome (Scheme 2).
 | ||
| Scheme 2 Scope of the diketopyrrolopyrroles’ rearrangement. a Unless otherwise noted, reactions were performed with TMSBr (0.15 mmol), solvent (1 ml), for 17 h at 50 °C (for HFIP) or 70 °C (for TFE). Isolated yields are reported; b reaction was conducted for 2 days; c reaction was conducted for 3 days; d reaction was performed with 2 eq. of Lewis acid. | ||
2,5-Di(n-hexyl)-1,4-diketo-3,6-di(thien-2-yl)pyrrolo[3,4-c]pyrrole (7) affords the desired product 8 with a minuscule 6% yield even if the reaction is conducted for 3 days. The addition of Koser's reagent improves the reactivity of DPP 7, however, probably due to the oxidative character, this reagent induces the decomposition of the product or intermediates. Luckily, the replacement of TFE with HFIP and lowering the reaction temperature to 50 °C led to the desired product in 32% yield.
Diketopyrrolopyrroles bearing 2-furyl and 3-furyl groups (9 and 11) display a lower reactivity compared to 1 and 7, respectively. The desired product was obtained in low yield only for DPP 9. The attempts to improve the reaction outcome (HTIB, prolongation of the reaction time) did not bring about positive results. DPP 13, possessing a peripheral benzofuran moiety, displays poor reactivity as well. Fortunately, significant stability of the rearranged product allowed for the prolongation of the reaction time as well as utilization of the iodine(III) catalyst and, as a consequence, we could achieve a higher yield of 14. Under similar conditions, the rearrangement occurs for DPP 15 (Scheme 2), bearing 3,4-dimethoxyphenyl groups. N-Alkylated DPPs bearing less electron-rich aryl substituents do not undergo this rearrangement.
With the library of new dyes in hand, we conducted a series of photophysical studies, which reveal that the absorption bands corresponding to the lowest energy absorption are located in the 530–606 nm region (Table 2 and Fig. 1), displaying bathochromic shifts in comparison to the parent DPPs. Emission maxima are red-shifted to the orange-red part of the visible spectra. Compounds 2, 4 and 6 exhibit only small differences in absorption and emission maxima. Nevertheless, the type and position of the heteroatom influence λabs and λem significantly. Different charge distributions in the molecules can rationalize both the red-shift of the absorption and emission maxima when regioisomeric products 2 and 8 are superimposed and the blue-shift of λabs and λem for dyes possessing a furan unit (2versus10). Dye 14 possessing a π-expanded core is characterized by the most bathochromically shifted emission and absorption in the analyzed series.
| Dye | λ abs [nm] | λ em [nm] | ε max [M−1 cm−1] | Φ [%] | ΔS [cm−1] | τ [ns] | k r [107 s−1] |
|---|---|---|---|---|---|---|---|
| a Calculated with respect to Rh101 in MeOH. b Calculated with respect to Rh6G in EtOH. c Calculated with respect to SRh101 in EtOH. | |||||||
| 2 | 570 | 601 | 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
48a | 900 | 8.21 | 5.9 |
| 4 | 569 | 605 | 17200 |
49a | 1100 | 7.78 | 6.3 |
| 6 | 571 | 602 | 17300 |
47a | 900 | 6.45 | 7.3 |
| 8 | 582 | 619 | 16000 |
15a | 1000 | 2.25 | 6.7 |
| 10 | 549 | 573 | 17500 |
13b | 800 | 2.33 | 5.6 |
| 14 | 606 | 624 | 59400 |
20c | 500 | 2.24 | 8.9 |
| 16 | 541 | 582 | 18200 |
57b | 1300 | 7.25 | 7.9 |
 | ||
| Fig. 1 Absorption and fluorescence spectra for 8, 10, 14 and 16 in DCM. | ||
Fluorescence responses for the majority of new dyes are rather high (Table 2). Fluorescence quantum yields (Φ) and λem are modestly influenced by the solvent polarity (Table S2, ESI†). In the exemplary case of 2, Φ changes from 56% in toluene to 28% in methanol (Table 3). This outcome is due to their moderately polarized electronic structures.
| Solvent | λ abs [nm] | λ em [nm] | ΔS [cm−1] | Φ [%] |
|---|---|---|---|---|
| a Calculated with respect to Rh101 in MeOH. | ||||
| Hexane | 566 | 581 | 500 | — |
| Toluene | 573 | 599 | 800 | 56 |
| DCM | 570 | 601 | 900 | 48 |
| ACN | 566 | 603 | 1100 | 42 |
| MeOH | 565 | 617 | 1500 | 28 |
To rationalize these spectra, we performed additional quantum chemistry calculations for dye 2, which concurred with experimental data (Fig. 2). According to our findings, the second excited S2 state locates around 7000 cm−1 above the S1, which means that the lowest excited S1 state is well separated from the higher located, electronically excited states (Fig. 2 and Fig. S5 and Tables S3, S4, ESI†). Moreover, the electronic transition S0 → S1 is of π → π* character and corresponds to excitation from the HOMO to the LUMO level (Fig. 3).
 | ||
| Fig. 2 Experimental absorption and fluorescence spectra of 2 in DCM. Calculated transition energies from the ground S0 to the electronically excited states are given by vertical solid lines with the heights proportional to the oscillator strength of the transition (see Table S3, ESI† for details). Vibronic structure is smoothed by low frequency vibrations (for interpretation see Fig. S6, ESI†). | ||
 | ||
| Fig. 3 Energy diagram with transition energies for absorption from the ground S0 state to the SFC1 and fluorescence from the relaxed S1 state to SFC0 for 2. On the right – the orbitals HOMO and LUMO of 2. Electronic configuration HOMO → LUMO describes the transition S0 → S1. | ||
In order to get more information about the reasons for smoothing the fluorescence spectra (lack any well resolved vibrational structure even at 5 K) and the possible depopulation channels we simulated the vibrational structure of the fluorescence spectrum of 2 (Fig. S5, ESI†). Calculation of the Franck–Condon (FC) factors (Fig. S6, ESI†) shows that the vibrational structure of the absorption and fluorescence spectra are obscured by the three low frequency vibrations: 37, 59 and 100 cm−1. These vibrations are characterized by high shift parameters8 and contribute to the spectrum in their common combinations as well as in combination with other vibrations.
To elucidate the influence of the solvent polarity on the absorption and fluorescence spectra, we calculated dipole moments for 2 in the isolated form as well as in solution. The comparison of the HOMO and LUMO orbitals clearly depicts the charge shift in the excited S1 state (Fig. 3). This shift causes an increase of the dipole moment (for ca. 1.5–2.2 D) and the bathochromic shift of both absorption and emission maxima in the polar solvents (Table S5, ESI†).
The performed calculations also provide information on the origin of the remaining non-radiative depopulation channel. We found that the excited S1 state is located about 9500 cm−1 above the triplet T1 and 2500 cm−1 below the T2 states. Such an energetic situation precludes an efficient intersystem crossing pathway for the S1 → T1 depopulation. Thus, the main non-radiative depopulation channel of the S1 state should proceed via internal conversion. This result concurs with our unsuccessful effort to detect phosphorescence emission at 5 K.
Our calculations also indicate that 2 possesses a planar core with the peripheral thiophene ring rotated around a single bond by around 25° with respect to the molecular plane (Table S4, ESI†). Thus, new heterocycles are planar, structurally rigid fluorophores, the structure of which does not change substantially upon electronic excitation. As a consequence of the small conformational changes following the transition between S0 and S1 states in these compounds (additional computational data in Table S6, ESI†) the Stokes’ shifts are small.
To verify the potential use of the discovered dyes for optoelectronic applications we performed a series of cyclic voltammetry (CV) measurements, which depict the influence of structural changes on their susceptibility towards oxidation and reduction. In the majority of cases, the investigated molecules show reversible reduction and quasi-reversible oxidation waves (Table 4 and Fig. S5, ESI†). The exception to the rule is observed only for 10 (irreversible oxidation). It is noteworthy that no changes were perceived with increasing number of redox cycles and the direction of the CV-measurement.
| Dye | E paox, V | IP, eV | E 1/2red, V | E onsetred, V | EA, eV |
|---|---|---|---|---|---|
| a Measurement conditions: electrolyte (NBu4ClO4, c = 0.1 M); DCMdry, potential sweep rate: 100 mV s−1, working electrode: glassy carbon (GC); auxiliary electrode: Pt wire; reference electrode: Ag/AgCl; all measurements were conducted at room temperature. b E pcred in V for 10. | |||||
| 2 | 0.90 | −5.1 | −1.10 | −1.02 | −3.3 |
| 4 | 0.96 | −5.1 | −1.15 | −1.06 | −3.3 |
| 6 | 0.86 | −5.1 | −1.17 | −1.07 | −3.3 |
| 8 | 0.92 | −5.1 | −1.15 | −1.03 | −3.3 |
| 10 | 0.94 | −5.2 | −1.20b | −1.05 | −3.3 |
| 14 | 0.89 | −5.1 | −0.93 | −0.81 | −3.5 |
| 16 | 0.80 | −5.0 | −1.28 | −1.19 | −3.2 |
We calculated both ionization potential (IP) and electron affinity (EA) values from the corresponding onset potentials. The results clearly indicate that peripheral substituents, as well as the electronic nature of the aromatic rings participating in the rearrangement, have no significant influence on the ionic potential (−5.1 ± 0.1 eV) or on the electron affinity (−3.3 ± 0.2 eV).
Herein, we have introduced structurally unique dyes possessing an isoindoledione core, linearly fused with a thiophene, furan, or benzene ring. We have demonstrated that diketopyrrolopyrroles possessing moderately electron-rich aryl substituents undergo heretofore unknown rearrangements initiated by mild Lewis acids. Strong emission of red light combined with straightforward synthesis, relatively small molecular mass, and potential chemical reactivity may open doors for these dyes for applications in fluorescence microscopy (bioimaging).
The financial support of the Polish National Science Centre (MAESTRO-2012/06/A/ST5/00216), Foundation for Polish Science (TEAM/2016-3/22) and the Global Research Laboratory Program (2014K1A1A2064569) through the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (Korea) is cordially acknowledged. Theoretical calculations were performed at the Interdisciplinary Center of Mathematical and Computer Modeling (ICM) of the Warsaw University (Poland) under the computational grant No. G-32-10. S. O. acknowledges JSPS fellowships.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For the selected reviews, see: (a) H. Yao, L. Ye, H. Zhang, S. Li, S. Zhang and J. Hou, Chem. Rev., 2016, 116, 7397 CrossRef CAS PubMed; (b) M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833 CrossRef CAS PubMed; (c) Y. Tao, Ch. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC; (d) T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584 CrossRef CAS PubMed; (e) C. Preihs, J. F. Arambula, D. Magda, H. Jeong, D. Yoo, J. Cheon, Z. H. Siddik and J. L. Sessler, Inorg. Chem., 2013, 52, 12184 CrossRef CAS PubMed.
- (a) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC; (b) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef PubMed; (c) W. Chen, C.-L. Chen, Z. Zhang, Y.-A. Chen, W.-C. Chao, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2017, 139, 1636 CrossRef CAS PubMed; (d) S. L. Broman, A. U. Petersen, C. G. Tortzen, J. Vibenholt, A. Bond and M. B. Nielsen, Org. Lett., 2012, 14, 318 CrossRef CAS PubMed.
- Incidental discoveries of new fluorophores are as commonly known as they were 150 years ago: (a) A. von Bayer, Chem. Ber., 1871, 5, 255 Search PubMed; (b) M. Ishida, P. Kim, J. Choi, J. Yoon, D. Kim and J. L. Sessler, Chem. Commun., 2013, 49, 6950 RSC; (c) A. Osuka and H. Shimidzu, Angew. Chem., Int. Ed. Engl., 1997, 36, 135 CrossRef CAS; (d) A. Janiga, E. Głodkowska-Mrówka, T. Stokłosa and D. T. Gryko, Asian J. Org. Chem., 2013, 2, 411 CrossRef CAS.
- (a) H.-Y. Ahn, K. E. Fairfull-Smith, B. J. Morrow, V. Lussini, B. Kim, M. V. Bondar, S. E. Bottle and K. D. Belfield, J. Am. Chem. Soc., 2012, 134, 4721 CrossRef CAS PubMed; (b) A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D’Este, P.-T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed. Engl., 2016, 55, 3290 CrossRef CAS PubMed; (c) W. Xu, Z. Zeng, J.-H. Jiang, Y.-T. Chang and L. Yuan, Angew. Chem., Int. Ed., 2016, 55, 13568 Search PubMed; (d) C. Allain, F. Schmidt, R. Lartia, G. Bordeau, C. Fiorini-Debuisschert, F. Charra, P. Tauc and M. P. Teulade-Fichou, ChemBioChem, 2007, 8, 424 CrossRef CAS PubMed; (e) D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781 CrossRef CAS PubMed.
- (a) M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280 CrossRef CAS; (b) E. H. G. Zadeh, M. V. Bondar, I. A. Mikhailov and K. D. Belfield, J. Phys. Chem. C, 2015, 119, 8864 CrossRef.
- (a) Y. Kita and T. Dohi, Chem. Rec., 2015, 15, 886 CrossRef CAS PubMed; (b) V. V. Zhdankin, ARKIVOC, 2009, 1–62 CAS; (c) Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684 CrossRef CAS.
- Y. Kita, K. Morimoto, M. Ito, C. Ogawa, A. Goto and T. Dohi, J. Am. Chem. Soc., 2009, 131, 1668 CrossRef CAS PubMed.
- V. Barone, J. Bloino, M. Biczysko and F. Santoro, J. Chem. Theory Comput., 2009, 5, 540–554 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1574216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc07310k |
| ‡ Current address: Kyoto University, Department of Chemistry, Japan. |
| This journal is © The Royal Society of Chemistry 2017 |