
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bypassing the proline/thiazoline requirement of the macrocyclase PatG†
E.
Oueis
 a,
H.
Stevenson
a,
M.
Jaspars
b,
N. J.
Westwood
a and
J. H.
Naismith
*acde
a,
H.
Stevenson
a,
M.
Jaspars
b,
N. J.
Westwood
a and
J. H.
Naismith
*acde
aBiomedical Science Research Complex & School of Chemistry, University of St Andrews, BSRC, North Haugh, St Andrews, KY16 9ST, UK. E-mail: naismith@strubi.ox.ac.uk
bMarine Biodiscovery Centre, Department of Chemistry, University of Aberdeen, Old Aberdeen, AB24 3UE, UK
cState Key Laboratory of Biotherapy, Sichuan University, China
dDivision of Structural Biology, Oxford University, OX3 7BN, UK
eResearch Complex at Harwell, Didicot, Oxon, OX11 0FA, UK
First published on 1st November 2017
Abstract
Biocatalysis is a fast developing field in which an enzyme's natural capabilities are harnessed or engineered for synthetic chemistry. The enzyme PatG is an extremely promiscuous macrocyclase enzyme tolerating both non-natural amino acids and non-amino acids within the substrate. It does, however, require a proline or thiazoline at the C-terminal position of the core peptide which means the final product must contain this group. Here, we show guided by structural insight we have identified two synthetic routes, triazole and a double cysteine, that circumvent this requirement. With the triazole, we show PatGmac can macrocyclise substrates that do not contain any amino acids in the final product.
Enzymes provide useful tools as catalysts to achieve complex transformations in organic synthesis that either because of stereochemical variability or high activation energy are difficult to accomplish chemically. Concerns about environmental costs of organic solvents and waste streams have further driven the use of enzymes. Advances in recombinant DNA technology and directed evolution strategies have improved the availability, stability, and reactivity of enzymes.1 Innovations in protein immobilisation,2 microfluidic reactors,3 and protein design4 have further extended their utility. A wide range of transformations including hydrolytic reactions, reductive and oxidative reactions, transfer reactions, and carbon–carbon bond formation are catalysed by enzymes.5
Macrocyclisation is an important modification in the synthesis of many biologically active compounds, including not only natural products but also drug leads.6 It is argued that macrocyclic compounds (whether peptidic or not) are better drug leads for challenging molecular targets such as protein–protein interactions.7 Hence, the strong interest in these molecules as therapeutics.8 This has driven the development of a number of new technologies for the generation of diverse libraries of cyclic peptides in vivo (SICLOPPS: split-intein circular ligation of peptides and proteins)9 or the construction of non-standard peptide libraries in vitro (RaPID: random non-standard peptide integrated discovery)10 for example. Macrocyclic peptides are generally acknowledged as structurally diverse, rigid and stable (chemically and enzymatically), highly desirable properties for therapeutic applications, despite their size.11 Significant progress has been reported on predicting membrane permeability of large cyclic compounds that lie outside Lipinski's rule of 5, allowing for a more rational approach to macrocycle drug design.12 Several enzyme macrocyclases are known, and some have already been exploited for biocatalysis. Butelase 1 macrocyclises peptides and proteins (26–200 residues) at an extremely fast rate;13 PCY1 is a naturally occurring promiscuous macrocyclase for smaller peptides (5–9);14 PoPB macrocyclises the α-amanitin precursor peptide15 and PatGmac,16 is a highly promiscuous macrocyclase from the cyanobactins, a family of heterocycle-containing peptides.17 A principal limitation of these enzymes is that they operate on peptide substrates; yet a general enzyme catalysed synthesis of macrocycles is highly desirable.
PatGmac requires a C-terminal recognition sequence AYD (which is cleaved off during macrocyclisation) and either thiazoline or L-proline immediately preceding the recognition tag. The ring is thought to be essential as it adopts either a cis-(proline) or cis-like (thiazoline) conformation allowing the substrate (core) peptide to curve away from the enzyme.16a,b,d,e,17b Consequently, there are only a few interactions with the core peptide and very few restrictions on substrate. Those restrictions include no D-amino acids at either the N-terminus or either of the last two C-terminal positions of the core peptide.16a,e PatGmac has a broad substrate scope including non-natural amino acids,16a peptides with up to three 1,2,3-triazole rings,16d non-amino acidic scaffolds including sugars, benzene rings, alkyl and PEG chains16e (Fig. 1). Hybrid peptide non-peptide molecules with only three amino acids including the terminal L-Pro/thiazoline have been made.16e Structural biology has rationalised the requirement for this conformation-inducing terminal residue. The requirement does limit the scope of the enzyme and always results in a proline/thiazoline in the final product.
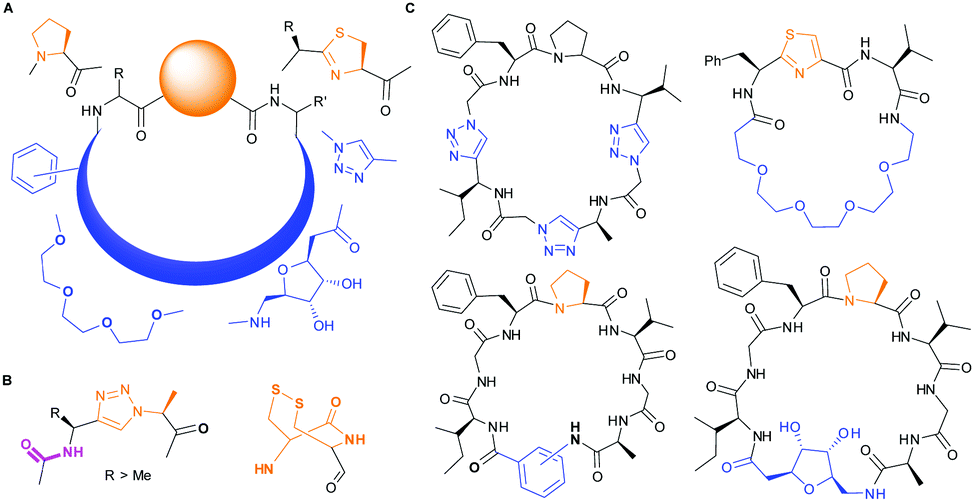 | ||
| Fig. 1 PatG substrates: (A) accepted substitutions within final product are shown in blue, but so far require proline or thiazoline (orange). (B) Triazole and double cysteine bypass this requirement. (C) Macrocycles synthesized with PatGmac so far.16d,e | ||
Here we explored options to remove the requirement for a thiazoline/L-Pro residue at the C-terminus of the core peptide. We have used 1,4-anti-1,2,3-triazole-alanine18 and vicinal cysteine disulphide bonds19 as replacements for L-Pro. The former has allowed the synthesis of fully non-peptidic macrocycles using enzymes, whilst the latter allows for the generation of non-heterocycle containing macrocycles that can vary conformation in response to redox conditions.
From structural analysis, we hypothesised that one or both hydrogen bonds between the C-terminus of the core peptide in the substrate and the enzyme was critical for recognition (Fig. 2).17b We designed, synthesised and tested range of substrates with an insertion of a 1,4-disubstituted anti-triazole-alanine at the C-terminus of the core peptide (position 8) (Table 1). The 1,4-anti-triazole is easily obtained using Cu(I) catalysis and can be achieved on solid phase during peptide synthesis.16d,20 We first used propargylamine for the triazole (Tz1–4) formation, as this glycine mimic was commercially available. The alanine azido-acid counterpart was synthesised in one step by a diazo transfer reaction from commercial alanine21 Peptides 1–3 were synthesised with Gly–Tz1–4–Ala dipeptide mimic at the C-terminus (positions 7 and 8) but all failed to yield the desired product. Only unreacted starting peptide remained in solution, indicating the peptide was not a PatGmac substrate. To explore if the 1,4-anti-triazole precluded a cis-like conformation, the 1,5-disubstituted syn-triazole (Tz1–5) was employed with Gly–Tz1–5–Ala dipeptide mimic at positions 7 and 8 of the precursor peptide. Neither Ru(Cp*Cl(PPh3)2) nor RuCp*(cod)Cl catalysts22 gave a useful amount of fully protected dipeptide Gly–Tz1–5–Ala. The thermal reaction between Fmoc-protected propargylamine and the alanine azido benzylic ester afforded a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio of syn- to anti-triazole. The carboxylic ester of the triazole-containing dipeptides was then hydrolysed and the regioisomeric mixture was used as a building block in the peptide synthesis to generate peptides 2/4 and 3/5. The regioisomeric triazole-containing peptides of both sequences were separated by HPLC but 1,5-syn-triazole peptides 4 and 5 gave the same negative result as their corresponding Tz1–4 peptides (cf. peptides 2 and 4, 3 and 5 in Table 1). The structure of PatGmac H618A in complex with a proline-containing substrate (PDB 4AKT)17b reveals a binding pocket for the side chain of position 7 (Fig. 2). We hypothesised that a side chain at position 7 might be necessary to rigidify the peptide and thus favour the hydrogen bond. In order to test this hypothesis, peptides 6 and 7 with a proline preceded by a glycine and alanine (phenylalanine 8 at this position has been previously shown to be a substrate)16d were tested. Peptide 6 (Gly) was unchanged by the enzyme but peptide 7 (Ala) was processed to afford cyclic peptide, at essentially the same rate and efficiency as PatGmac peptide substrates. We concluded a side chain at position 7 is required to properly orient substrate to make the second hydrogen bond. The requirement for both hydrogen bonds also rationalises why peptides containing β-alanine (9) or (R)-3-amino-2-methylpropanoic acid ((R)-β2-homoalanine) (10), which move the amide out of position due to an extra carbon, were not processed by the enzyme.
:2 ratio of syn- to anti-triazole. The carboxylic ester of the triazole-containing dipeptides was then hydrolysed and the regioisomeric mixture was used as a building block in the peptide synthesis to generate peptides 2/4 and 3/5. The regioisomeric triazole-containing peptides of both sequences were separated by HPLC but 1,5-syn-triazole peptides 4 and 5 gave the same negative result as their corresponding Tz1–4 peptides (cf. peptides 2 and 4, 3 and 5 in Table 1). The structure of PatGmac H618A in complex with a proline-containing substrate (PDB 4AKT)17b reveals a binding pocket for the side chain of position 7 (Fig. 2). We hypothesised that a side chain at position 7 might be necessary to rigidify the peptide and thus favour the hydrogen bond. In order to test this hypothesis, peptides 6 and 7 with a proline preceded by a glycine and alanine (phenylalanine 8 at this position has been previously shown to be a substrate)16d were tested. Peptide 6 (Gly) was unchanged by the enzyme but peptide 7 (Ala) was processed to afford cyclic peptide, at essentially the same rate and efficiency as PatGmac peptide substrates. We concluded a side chain at position 7 is required to properly orient substrate to make the second hydrogen bond. The requirement for both hydrogen bonds also rationalises why peptides containing β-alanine (9) or (R)-3-amino-2-methylpropanoic acid ((R)-β2-homoalanine) (10), which move the amide out of position due to an extra carbon, were not processed by the enzyme.
| Peptide | Substrate sequencea | Productb | Cyclicc | Yieldd (%) |
|---|---|---|---|---|
| a Underlined is the minimal recognition sequence AYD where the peptide is cleaved. Tz1–4 = 1,4-anti-triazole, Tz1–5= 1,5-syn-triazole, βA = β-alanine, β2-homoA = (R)-3-amino-2-methylpropanoic acid, D-amino acids in lower case, [CC] = disulphide bond. b NR = No reaction, C = Cyclic, L = Linear; major product shown in table, minor product shown in brackets. c Isolated macrocycles. d Their yields. | ||||
| 1 |

|
NR | ||
| 2 |

|
NR | ||
| 3 |

|
NR | ||
| 4 |

|
NR | ||
| 5 |

|
NR | ||
| 6 |
|
NR | ||
| 7 |

|
C | ||
| 8 |
|
C | 8c 16d | 32 |
| 9 |
|
NR | ||
| 10 |
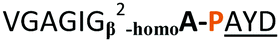
|
NR | ||
| 11a |

|
C | 11c/d | 40 |
| 11b | NR | |||
| 12 |
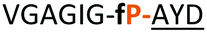
|
NR | ||
| 13a |

|
NR | ||
| 13b | NR | |||
| 14 |

|
C | 14c | 40 |
| 15 |

|
C | ||
| 16 |

|
C | 16c/d | 36 |
| 17 |
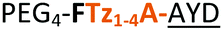
|
C | 17c | 34 |
| 18 |

|
C | ||
| 19 |
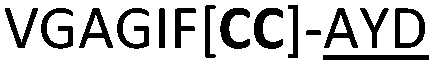
|
C (L) | 19c | 37 |
| 20 |
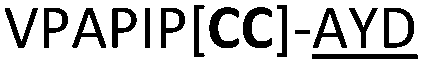
|
L | ||
| 21 |
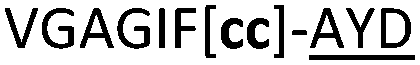
|
NR | ||
| 22 |
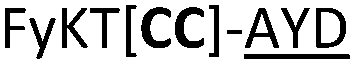
|
L | ||
| 23 |
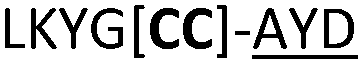
|
NR | ||
| 24 |
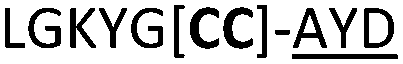
|
C | ||
| 25 |
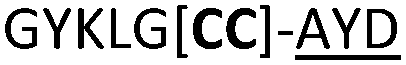
|
C (L) | ||
| 26 |
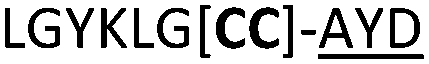
|
C (L) | ||
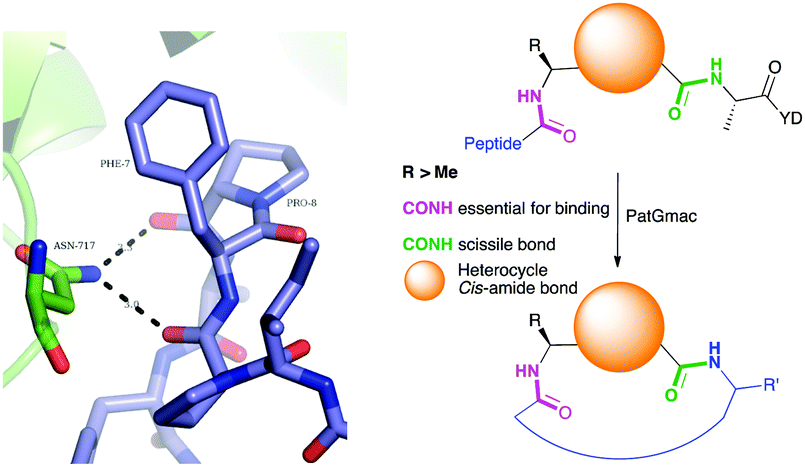 | ||
| Fig. 2 Left: Hydrogen bonding between the side chain of Asn717 and the carbonyls of substrate. We conclude these hydrogen bonds are critical in substrate recognition and explain the requirement for an L-configured non-glycine residue at position residues 7 of substrate (Phe is shown from PDB 4AKT). Right: PatGmac macrocyclisation substrate requirements: a heterocycle before AYD adopting a cis- or cis-like conformation, a side chain (R ≥ Me) preceding the heterocycle. We conclude two substrate carbonyls (coloured green & pink) hydrogen bonded with Asn717 are critical. | ||
Having established the importance of position 7, we modified our design strategy. We synthesized peptide 11 with Phe–Tz1–4–Ala dipeptide mimic at the C-terminus. The phenylalanine amino alkyne derivative was synthesised in five steps starting from the commercial Boc protected amino acid, as previously described.16d Epimerisation of phenylalanine amino alkyne occurred for the phenylalanine derivative during synthesis affording a mixture of inseparable enantiomers. The final diastereomeric peptides however were easily separated by HPLC affording peptides 11a and 11b. The major diastereomer 11a was macrocyclised by PatGmac, whereas the minor diastereomer 11b did not. Since D-amino acids at positions 7 (the control peptide 12 with a D-Phe at position 7 failed to give the corresponding macrocycle) and D amino acids at position 8 have been reported not to be tolerated,16a11b was assigned as having one D-amino acid at the triazole, either position 7 or 8 and does not process. The macrocyclisation of peptide 11a demonstrates for the first time that the L-Pro requirement can be dispensed with and consistent with knowledge of the system is assumed to be L-Phe–Tz1–4–L-Ala. Deletion of the methyl group to afford Phe–Tz1–4–Gly dipeptide mimic (peptides 13a, b) afforded no product in the presence of PatGmac, as expected. The dipeptide mimic Ala–Tz1–4–Ala 14 was a substrate as was peptide 15, which has two triazole units. This led us to conclude that replacing L-Pro with the triazole group did not diminish our ability to introduce other modifications elsewhere in the core peptide. In order to explore these limits, peptide 16 having one amino acid at the N-terminus linked by a PEG chain was tested and macrocyclised. Peptides 17 and 18, which have no α amino acids in the core, but have a free amine on a PEG group, gave macrocyclic compounds.
We then explored the use of vicinal L-cysteine disulphide bond as an alternative to proline, a defining motif of three important cyclic peptide families that are biologically active: malformins, somatostatins, and conotoxins.23 The disulfide bond containing substrate peptides (19–26) were synthesised by standard solid phase peptide synthesis and isolated as free cysteine-containing peptides and then oxidised overnight in 10% DMSO in TFA. Peptides 19 and 20 were designed as a positive and negative controls as 19 mimics 8, a good substrate, whilst 20 mimics VPAPIPFP a poor substrate that mainly gives linear peptide.17b Analysis after two days of incubation showed that peptide 19 gave mainly macrocyclic peptide whereas peptide 20 gave only cleaved linear peptide. Peptide 21 with the D-cysteine motif (akin to malformins)23 did not macrocyclise. We took these results as confirming that vicinal L-cysteine disulphide bond could replace L-Pro and we explored derivatives of the remaining natural product families with this motif. Cyclic hexapeptidic analogues of somatostatin-containing YwKT or FwKT and a vicinal cysteine disulphide bond were reported to be more potent than the natural product.23 Peptide 22 was designed to produce such an analogue, but mainly yielded cleaved peptide, most likely because of the PatGmac preference for longer peptides.16e As a final test we explored conotoxin cyclic analogues2423–26. The hexapeptide 23 did not react; however the other hepta- and octa-peptide substrates all gave the desired cyclic peptides. PatGmac does afford facile access to analogues of conotoxin. We confirmed that reduction of the disulfide bond of macrocyle 19c by TCEP in methanol/water was possible after macrocyclisation to give peptide 19d (ESI†). This result opens the possibility of redox control of the macrocycle structure.
Products were identified by MALDI-MS and further verified by MS–MS fragmentation data (ESI†). Where the reactions were carried out at scale, macrocycles (Fig. 3) were isolated, purified by HPLC, and characterised using HRMS and NMR (ESI†). We observed moderate yields (32–40%) with an incubation of two weeks at 37 °C and pH = 8.1. Cyclic peptides derived from 11a and 16 purified as two HPLC separable peaks with identical chemical composition determined by NMR and HRMS. EXSY NMR analysis indicates that they are not readily exchangeable conformers, suggesting these are either diasteriomers or structurally rigid conformers unable to interconvert (atropisomers). Spontaneous epimerization of macrocycles is known,25 but seems less likely for alpha position of a triazole. PatGmac does not process peptide substrates with D-amino acids at either position 7 or 8, it maybe the triazole leads to PatGmac tolerating substrate D-configuration. Cyclic peptide 17c purified as a single peak, but proton NMR identified a minor (<5%) conformer. Macrocycle 14c has in addition to the predominant conformer other minor conformers. Macrocycle 19c gave rise to a complex spectrum, showing one major compound with at least two other minor conformers that were not fully identified.
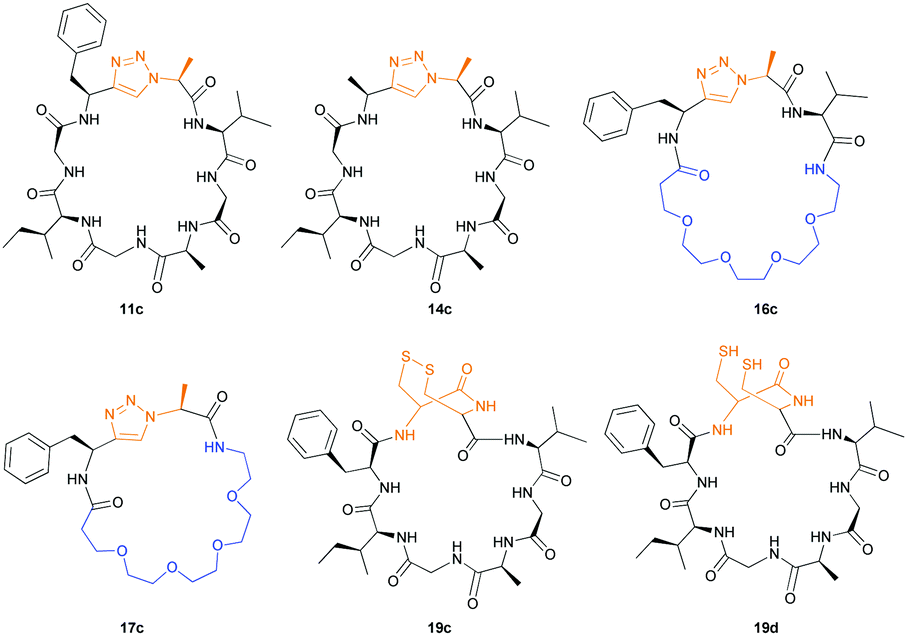 | ||
| Fig. 3 Non-proline containing macrocycles macrocyclised by PatGmac. | ||
Macrocyclisation is an important and essential transformation in nature in general, and for the synthesis of biologically active molecules. Many enzymes in nature are involved in ring closure reactions, but are usually restricted to their biosynthetic pathway. Indeed, biocatalysis in general can be very efficient in conducting certain reactions, however this usually comes at a very high price in terms of substrate specificity. Hence, the ability to macrocyclise a range of structurally different compounds is a huge advantage allowing for a wider diversity. Herein we show that PatGmac is able to macrocyclise cyclic peptides containing non-natural and natural proline mimics at the C-terminus of the core peptide, a position that was thought to be restricted to natural heterocycles. We were able to synthesize macrocyclic compounds with no amino acids or no heterocycles. This very broad substrate range of PatGmac expands the scope of its applications, despite its slower rate and moderate yields. Nonetheless, further improvement of the catalytic efficiency of the enzyme achieved by directed evolution26 or protein engineering would be valuable. To the best of our knowledge, PatGmac is the first described peptidic ligase capable of macrocyclising non-peptidic precursors, making it the first enzymatic tool employed for the generation of diverse macrocyclic libraries.
This work was supported by the European Research Council (339367), UK Biotechnology and Biological Sciences Research Council (K015508/1), the Wellcome Trust (TripleTOF 5600 mass spectrometer (094476), the MALDI TOF–TOF Analyser (079272AIA), 700 NMR) and the EPSRC UK National Mass Spectrometry Facility at Swansea University. J. H. N. is a Royal Society Wolfson Merit Award Holder and 1000 talent scholar at Sichuan University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480 CrossRef CAS PubMed.
- S. Cynthia and D. M. Shelley, Recent Pat. Eng., 2008, 2, 195 CrossRef.
- E. Laurenti and A. dos Santos Vianna Jr., Biocatalysis, 2016, 1, 148 Search PubMed.
- Y. Li and P. C. Cirino, Biotechnol. Bioeng., 2014, 111, 1273 CrossRef CAS PubMed; U. T. Bornscheuer, Biotechnol. J., 2007, 2, 155 CrossRef PubMed.
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585 RSC.
- A. T. Bockus, C. M. McEwen and R. S. Lokey, Curr. Top. Med. Chem., 2013, 13, 821–836 CrossRef CAS PubMed; L. A. Wessjohann, E. Ruijter, D. Garcia-Rivera and W. Brandt, Molecular Diversity, 2005, 9, 171 CrossRef PubMed; X. Yu and D. Sun, Molecules, 2013, 18, 6230 CrossRef PubMed; A. K. Yudin, Chem. Sci., 2015, 6, 30 RSC.
- T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787–794 CrossRef CAS PubMed; J. R. Corte, T. Fang, H. Osuna, D. J. P. Pinto, K. A. Rossi, J. E. Myers, S. Sheriff, Z. Lou, J. J. Zheng, T. W. Harper, J. M. Bozarth, Y. Wu, J. M. Luettgen, D. A. Seiffert, C. P. Decicco, R. R. Wexler and M. L. Quan, Journal of Medicinal Chemistry, 2017, 60, 1060–1075 CrossRef PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- A. Tavassoli, Curr. Opin. Chem. Biol., 2017, 38, 30–35 CrossRef CAS PubMed.
- T. Passioura and H. Suga, Chem. Commun., 2017, 53, 1931–1940 RSC.
- D. J. Newman and G. M. Cragg, Macrocycles in Drug Discovery, The Royal Society of Chemistry, 2015 Search PubMed.
- F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278–295 CrossRef CAS PubMed; B. Over, P. Matsson, C. Tyrchan, P. Artursson, B. C. Doak, M. A. Foley, C. Hilgendorf, S. E. Johnston, M. D. Lee Iv, R. J. Lewis, P. McCarren, G. Muncipinto, U. Norinder, M. W. D. Perry, J. R. Duvall and J. Kihlberg, Nat. Chem. Biol., 2016, 12, 1065–1074 CrossRef PubMed; C. R. Pye, W. M. Hewitt, J. Schwochert, T. D. Haddad, C. E. Townsend, L. Etienne, Y. Lao, C. Limberakis, A. Furukawa, A. M. Mathiowetz, D. A. Price, S. Liras and R. S. Lokey, J. Med. Chem., 2017, 60, 1665–1672 CrossRef PubMed.
- G. K. T. Nguyen, A. Kam, S. Loo, A. E. Jansson, L. X. Pan and J. P. Tam, J. Am. Chem. Soc., 2015, 137, 15398 CrossRef CAS PubMed.
- J. R. Chekan, P. Estrada, P. S. Covello and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6551 CrossRef CAS PubMed; C. J. Barber, P. T. Pujara, D. W. Reed, S. Chiwocha, H. Zhang and P. S. Covello, J. Biol. Chem., 2013, 288, 12500 CrossRef PubMed.
- C. M. Czekster and J. H. Naismith, Biochemistry, 2017, 56, 2086 CrossRef CAS PubMed; H. Luo, S.-Y. Hong, R. M. Sgambelluri, E. Angelos, X. Li and J. D. Walton, Chem. Biol., 2014, 21, 1610 CrossRef PubMed.
- (a) J. A. McIntosh, C. R. Robertson, V. Agarwal, S. K. Nair, G. W. Bulaj and E. W. Schmidt, J. Am. Chem. Soc., 2010, 132, 15499–15501 CrossRef CAS PubMed; (b) E. Oueis, C. Adamson, G. Mann, H. Ludewig, P. Redpath, M. Migaud, N. J. Westwood and J. H. Naismith, ChemBioChem, 2015, 16, 2646 CrossRef CAS PubMed; (c) D. Sardar, Z. Lin and Eric W. Schmidt, Chem. Biol., 2015, 22, 907–916 CrossRef CAS PubMed; (d) E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., Int. Ed., 2016, 55, 5842–5845 CrossRef CAS PubMed; (e) E. Oueis, B. Nardone, M. Jaspars, N. J. Westwood and J. H. Naismith, ChemistryOpen, 2017, 6, 11 CrossRef CAS PubMed.
- (a) E. W. Schmidt, J. T. Nelson, D. A. Rasko, S. Sudek, J. A. Eisen, M. G. Haygood and J. Ravel, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7315–7320 CrossRef CAS PubMed; (b) J. Koehnke, A. Bent, W. E. Houssen, D. Zollman, F. Morawitz, S. Shirran, J. Vendome, A. F. Nneoyiegbe, L. Trembleau, C. H. Botting, M. C. M. Smith, M. Jaspars and J. H. Naismith, Nat. Struct. Mol. Biol., 2012, 19, 767–772 CrossRef CAS PubMed; (c) J. Koehnke, A. F. Bent, W. E. Houssen, G. Mann, M. Jaspars and J. H. Naismith, Curr. Opin. Struct. Biol., 2014, 29, 112–121 CrossRef CAS PubMed.
- A. Tam, U. Arnold, M. B. Soellner and R. T. Raines, J. Am. Chem. Soc., 2007, 129, 12670–12671 CrossRef CAS PubMed.
- F. A. Etzkorn, Advances in Amino Acid Mimetics and Peptidomimetics, JAI Press, 1st edn, 1999 Search PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800 CrossRef CAS PubMed.
- B. C. Boren, S. Narayan, L. K. Rasmussen, L. Zhang, H. Zhao, Z. Lin, G. Jia and V. V. Fokin, J. Am. Chem. Soc., 2008, 130, 14900 CrossRef CAS; L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, J. Am. Chem. Soc., 2005, 127, 15998 CrossRef PubMed.
- B. K. W. Chung and A. K. Yudin, Org. Biomol. Chem., 2015, 13, 8768 CAS.
- A. Brust, C.-I. A. Wang, N. L. Daly, J. Kennerly, M. Sadeghi, M. J. Christie, R. J. Lewis, M. Mobli and P. F. Alewood, Angew. Chem., Int. Ed., 2013, 52, 12020 CrossRef CAS PubMed.
- P. Wipf, P. C. Fritch, S. J. Geib and A. M. Sefler, J. Am. Chem. Soc., 1998, 120, 4105–4112 CrossRef CAS.
- M. S. Packer and D. R. Liu, Nat. Rev. Genet., 2015, 16, 379–394 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc06550g |
| This journal is © The Royal Society of Chemistry 2017 |