
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning photochemical properties of phosphorus(V) porphyrin photosensitizers†
Ivan N.
Meshkov
 ab,
Véronique
Bulach
a,
Yulia G.
Gorbunova
*bc,
Fedor E.
Gostev
d,
Victor A.
Nadtochenko
d,
Aslan Yu.
Tsivadze
bc and
Mir Wais
Hosseini
*a
ab,
Véronique
Bulach
a,
Yulia G.
Gorbunova
*bc,
Fedor E.
Gostev
d,
Victor A.
Nadtochenko
d,
Aslan Yu.
Tsivadze
bc and
Mir Wais
Hosseini
*a
aMolecular Tectonics Laboratory, UMR UDS-CNRS, 7140 & icFRC, Université de Strasbourg, F-67000, Strasbourg, France. E-mail: hosseini@unistra.fr
bA.N. Frumkin Institute of Physical Chemistry and Electrochemistry, Russian Academy of Sciences, Leninsky pr. 31-4, Moscow, 119071, Russia. E-mail: yulia@igic.ras.ru
cN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Leninsky pr. 31, Moscow, 119991, Russia
dSemenov Institute of Chemical Physics, Russian Academy of Sciences, Kosygina st. 4, Moscow, 119991, Russia
First published on 15th August 2017
Abstract
Photosensitizing and emission properties of P(V) porphyrins were studied. The nature of the axial ligands, occupying the apical position on the P centre adopting an octahedral coordination geometry, strongly influences singlet oxygen generation and charge transfer and allows switching between the two processes.
The very rich photochemical as well as photophysical properties of porphyrin derivatives have triggered extensive investigations including their role as photosensitizers in catalysis and medicines.1–5 In photosensitization, water-soluble derivatives, in particular cationic ones, are of interest, in particular in medicine. Indeed, owing to their charge nature, porphyrin based dyes by crossing the cell membrane bind to DNA molecules by intercalation.6–9 This process is of prime importance for antimicrobial photodynamic therapy owing to increasing resistance of bacteria to antibiotics. Usually, the generation of cationic species of porphyrin derivatives is achieved by the implementation of positive charges at their periphery.7,8,10 Alternatively, one may take advantage of the propensity of the porphyrin tetraaza core to bind metal centres to generate charged species by using metal cations with the oxidation state greater than 2. This approach has been explored using P(V)meso-tetraphenylporphyrins.9,11–15 For these species, the phosphorous atom, located at the centre of the porphyrin backbone, is hexacoordinated adopting a distorted octahedral coordination geometry surrounded by four N atoms and two axial ligands occupying the two apical positions. For some P(V)TPP derivatives, rather high quantum yields (up to 0.7) in water for singlet oxygen generation (SO) were reported.9,11,12 In a careful survey of the literature, it appeared that the nature of axial ligands plays a major role in their photophysical features. For example, for P(V) porphyrins bearing aryloxy moieties, the photoinduced electron transfer (PET) processes from the aromatic axial ligands to the porphyrin core may be modulated.13
However, to the best of our knowledge, relationships between singlet oxygen generation and the nature of axial ligands or peripheral substituents of the porphyrin core have not been explored to date.
Herein we report on the design and synthesis of two series of P(V) porphyrin (TPP and MPyP (meso-(5-pyridyl)-(10,15,20-triphenyl)porphyrin)) derivatives bearing four different types of axial ligands (Scheme 1) and their propensity to generate singlet oxygen in water and in chloroform and DMSO.
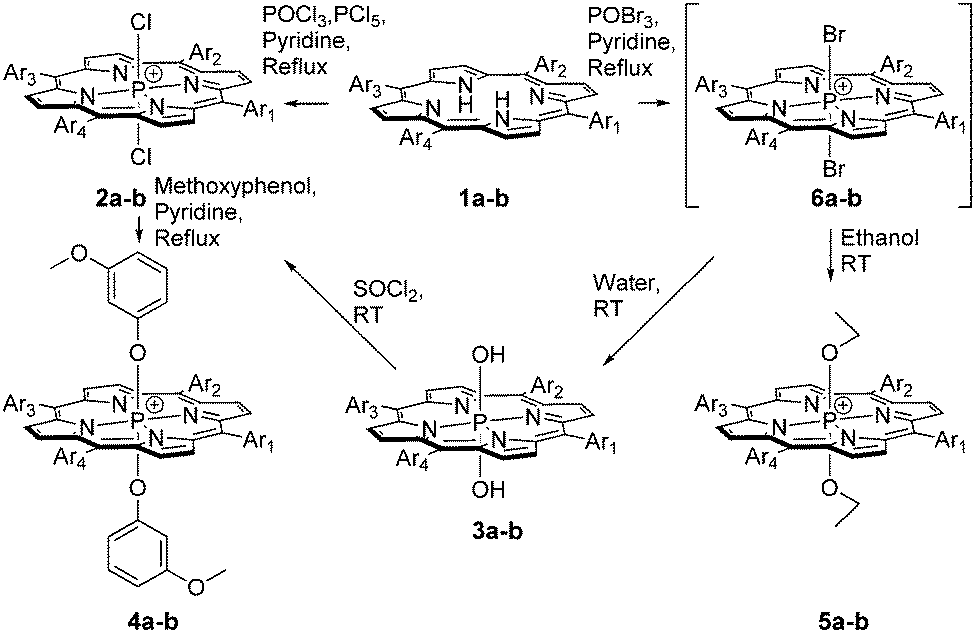 | ||
| Scheme 1 Syntheses of P(V) porphyrins in this communication; 1a–6a: Ar1, Ar2, Ar3, Ar4 = phenyl, 1b–6b: Ar1 = pyridyl, Ar2, Ar3, Ar4 = phenyl. | ||
The parent porphyrins 1a and 1b, as well as P(V) porphyrins 2a, 2b, 3a, 3b, 4a, and 4b15 and ethoxy derivatives 5a and 5b, were synthesized (for synthetic procedures and full characterization, see the ESI,† Fig. S2–S23).
For all described compounds, steady-state luminescence properties were measured in the same solvents (Table 1). Emission spectra and Stokes shifts are presented in Fig. 1 and in the ESI† (Table S1 and Fig. S24–S29). SO generation quantum yields were determined via chemical methods using SO sensitive traps (Fig. 2 and Fig. S30, S31) such as 1,3-diphenylisobenzofuran (DPBF) for non-aqueous media (chloroform and DMSO)1 and Singlet Oxygen Sensor Green (SOSG)16 for water (for experimental procedures and calculations, see the ESI†).
| Complex | Chloroform | DMSO | Water | |||
|---|---|---|---|---|---|---|
| Φ Δ | Φ f | Φ Δ | Φ f | Φ Δ | Φ f | |
| 2a | 0.93 | 0.014 | 0.36 | 0.016 | Hydrolysis | |
| 3a | 0.73 | 0.027 | 0.24 | 0.126 | 0.11 | 0.119 |
| 4a | 0.18 | 0.009 | 0.00 | 0.005 | 0.00 | 0.008 |
| 5a | 0.77 | 0.036 | 0.34 | 0.125 | 0.12 | 0.073 |
| 2b | 1.00 | 0.012 | 0.41 | 0.029 | Hydrolysis | |
| 3b | 0.99 | 0.029 | 0.31 | 0.119 | 0.29 | 0.113 |
| 4b | 0.16 | 0.005 | 0.00 | 0.006 | 0.00 | 0.007 |
| 5b | 0.99 | 0.060 | 0.37 | 0.104 | 0.46 | 0.083 |
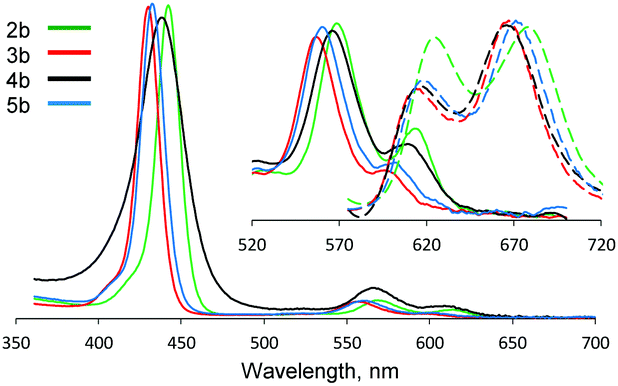 | ||
| Fig. 1 Normalized absorption (solid line) and fluorescence (dashed line, λex = 550 nm) spectra of 2b–5b in DMSO at room temperature. | ||
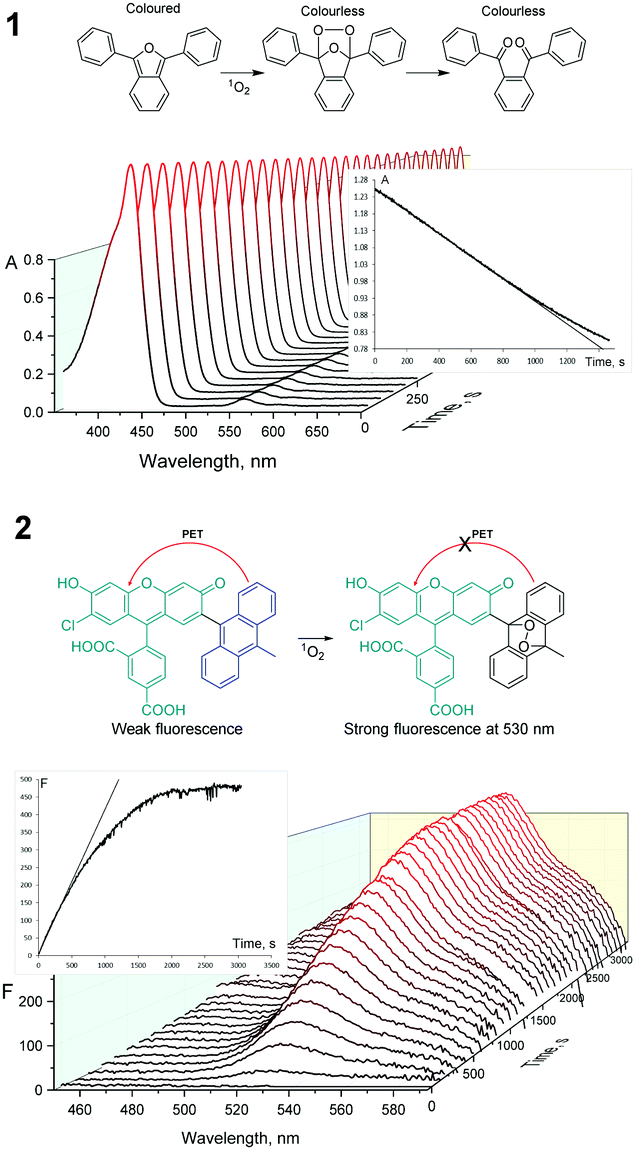 | ||
| Fig. 2 Oxidation of DPBF by 5b in chloroform (1) and SOSG in water (2) in the presence of singlet oxygen, monitored by UV-Vis spectroscopy and fluorimetry. Plots of DPBF and 5b overlapped absorption peaks (428 nm) and SOSG emission peaks (530 nm, λex = 405 nm) vs. time are presented in the insets. | ||
For all compounds investigated, quantum yields of SO generation are in the 0–100% range; however, up to 12% variation for the fluorescence quantum yield is observed (Table 1). Complexes 2a and 2b, bearing axial chloro ligands, undergo partial hydrolysis in water and thus were not investigated.
Analysis of the data gathered in Table 1 reveals three main dependences of both quantum yield of SO generation and steady-state fluorescence resulting from the nature of solvent, meso-substituents and axial ligands.
The photosensitizing efficiency and solvent nature are correlated through the SO lifetime: ∼2.5 × 10−4 s in chloroform,17 ∼5.5 × 10−6 s in DMSO18 and ∼4 × 10−6 s in water,19 indicating that, for example, for the same compound 5a, the quantum yield is decreased by ca. 6 times when replacing CHCl3 by H2O. At the same time, the fluorescence quantum yields for 5a and for 3a are increased by a factor of two and four, respectively.
The nature of substituents located at the meso positions of the porphyrin also plays a pertinent role in both the quantum yields of SO generation and steady-state fluorescence. Indeed, TPP derivatives (series a) are less efficient than MPyP complexes (series b). Although a difference of ca. 1.3 times in the quantum yield of SO generation between 5a and 5b is observed in chloroform, in water a factor of ca. 4 is obtained. Such an increase in water could result from a higher solubility of P(V)MPyP owing to the presence of the pyridyl unit. Although one would also expect a difference in the fluorescence quantum yield, almost the same values are observed in water and in organic media.
Depending on the nature of axial ligands, substantial changes in the quantum yields of SO generation and fluorescence are observed. Indeed, the presence of aromatic methylresorcinol at the apical positions (complexes 4a and 4b) dramatically decreases both values. These derivatives no longer generate singlet oxygen in water and in DMSO. As previously described, this should result from another competitive relaxation pathway such as charge-transfer (CT) processes.13 In order to verify this hypothesis, time-resolved transient absorption spectroscopy was used. Transient absorption spectra (TA) revealed the presence of a prominent bleaching band (BL) corresponding to the Soret absorption band close to ca. 440 nm. A wide excited absorption band (ESA) at 470–490 nm, partially overlapping with the weak BL Q-bands, is observed.
TA changes show kinetic components that correspond to three time scales (Fig. S32 and S33, ESI†): (1) 100–200 fs for intramolecular vibrational energy redistribution; (2) ∼1 ps for vibrational redistribution caused by elastic collision with solvent molecules; and (3) time scale from a dozen to hundreds ps and ns corresponding to the intramolecular transitions between different electronic states. TA experiments revealed the significant differences between the relaxation kinetics for 4a and 4b complexes as well as for compounds 2a, 2b, 3a, 3b, 5a and 5b. For example, Fig. 3 (see also Fig. S32–S35, ESI†) shows the transient absorption spectra of solutions 4b and 5b in acetonitrile at different time delays. The transient kinetics (see insets) display a clear double exponential decay for 4b at the time scale up to 500 ps, whereas for 5b the decay does not exceed 5% of the initial excitation level during 500 ps. The decay kinetics for 5b as well as for 2a, 2b, 3a, 3b and 5a may be approximately fitted with a monoexponential component:
Y0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−tτ−1) exp(−tτ−1) |
The time constant of the S1 decay is close to 1 ns for 2a, 2b, 3a, 3b, 5a and 5b (ESI†). This value is comparable to the time constants for different free-base or light-metal porphyrin complexes.21 The relaxation rate constant τ−1 is the sum of the rate constants of radiative transfer τfl−1, internal conversion in the S0 state – k1, and interconversion in the T1 state – k2 (Scheme 2). The quantum yield of interconversion into the T1 state can be estimated from the BI decay in the limit of t → ∞ as Y0/(Y0 + A). The double exponential decay observed for 4a and 4b suggests the relaxation of S1 into the S0 state through an intermediate electronic state, presumably the charge transfer state (CT). Kinetic equations related to Scheme 2 with the CT state have a solution of the BI Soret bleaching decay (see ESI†):

| Y0 + A1exp(−tτ1−1) + A2(−tτ2−1) |
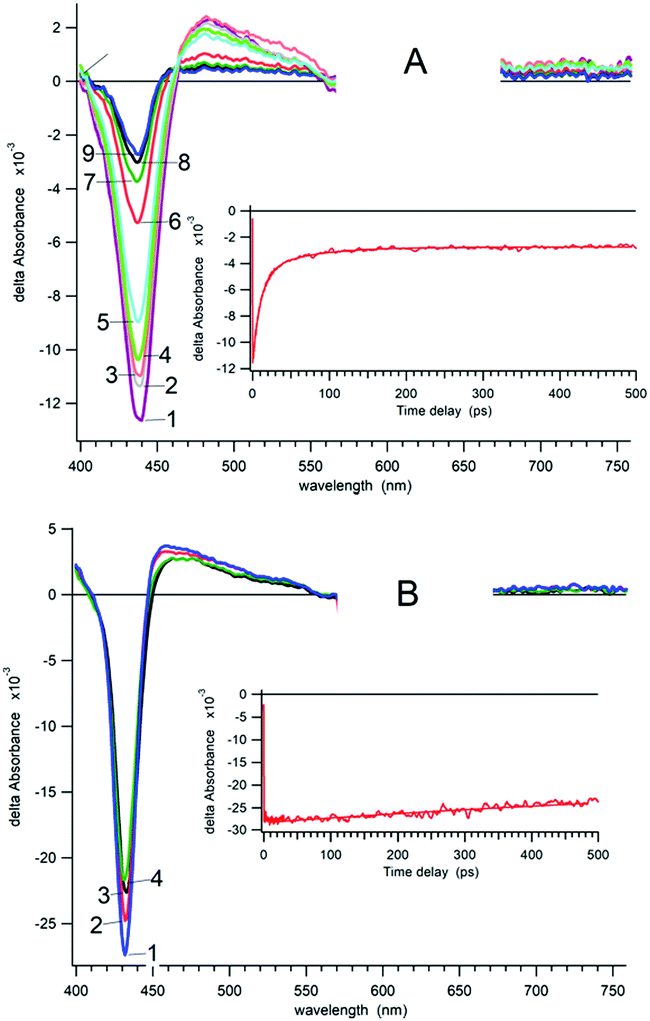 | ||
| Fig. 3 Transient absorption spectra of 4b (A) and 5b (B) in acetonitrile (λexc = 620 nm). Time delay (4b): (1) 100 fs; (2) 500 fs; (3) 900 fs; (4) 2.1 ps; (5) 5 ps; (6) 20 ps; (7) 50 ps; (8) 100 ps; (9) 450 ps; time delay (5b): (1) 100 fs; (2) 5 ps; (3) 50 ps; (4) 450 ps. Transient kinetics decay are presented in the insets. | ||
 | ||
| Scheme 2 Energy intramolecular transitions in 2, 3, 5 (1) and 4 (2). | ||
It is worth noting that taking into account the shift of the minima of the BL band at a very early stage requires the use of BI as a measure of a ground state depletion. Probably, conformational changes of a tetrapyrrolic ring may influence the shape of the absorption bleaching band. It has been shown that for P(V) meso-substituted derivatives, the porphyrin core is strongly distorted and adopts a “ruffled” deformation.15,22
The relaxation kinetics and processes for compounds 4a and 4b are similar (see ESI†). Kinetics curves for compounds 2 and 3 are similar to the one for 5 (see ESI†). Quantum yields of the T1 state for all compounds were calculated using the transient absorption data (Table S2, ESI†), which are in the good accordance with the data published by Harriman et al.23
The results obtained are in agreement with the observed differences in quantum yields of SO generation and fluorescence for P(V) porphyrins bearing aromatic and non-aromatic axial ligands.
In order to further confirm the above given explanations, quantum-chemical calculations have been also performed. Optimizations and calculations were performed via DFT (density functional theory) calculations using Spartan'14 software (Wavefunction Inc., CA, USA) with the B3LYP/6-31G* level of theory. Fig. 4 shows the energy minimized structure of the complex 4b with calculated four frontier molecular orbitals. Both HOMO+1 and HOMO orbitals are located on the electron donor axial aromatic ligands, while after excitation both LUMO and LUMO−1 move to the positively charged porphyrin core. Thus, a CT-state is strongly anticipated for such a molecule. Optimized structures with calculated frontier MO for other studied complexes are presented in the ESI† (Fig. S36–S43).
 | ||
| Fig. 4 Molecular orbitals, calculated for 4b. | ||
In conclusion, the experimental and theoretical investigations clearly demonstrated the possibility of tuning photochemical properties of P(V) porphyrins. The photochemical properties of P(V) porphyrin derivatives strongly depend on the nature of axial ligands. Indeed, the introduction of aromatic ligands at the two apical positions on the P atom activates CT processes and turns off SO generation and quenches fluorescence. In the absence of this type of ligands, fluorescent P(V) porphyrins are effective photosensitizers in water as well as in organic media.
This work was supported by the Russian Science Foundation (#14-13-01373), the University of Strasbourg, the CNRS, the International centre for Frontier Research in Chemistry (icFRC), the Labex CSC (ANR-10-LABX-0026 CSC) within the Investissement d'Avenir program ANR-10-IDEX-0002-02 and the Institut Universitaire de France. I. N. M. is grateful to the French Government for Joint Supervised PhD grants.
Conflicts of interest
There are no conflicts to declare.References
- T. Nyokong and V. Ahsen, Photosensitizers in Medicine, Environment and Security, Springer, 2012 Search PubMed.
- L. Jiang, C. R. R. Gan, J. Gao and X. J. Loh, Small, 2016, 12, 3609–3644 CrossRef CAS PubMed.
- J. M. Dabrowski, B. Pucelik, A. Regiel-Futyra, M. Brindell, O. Mazuryk, A. Kyzioł, G. G. Stochel, W. Macyk, L. G. Arnaut, J. M. Dąbrowski, B. Pucelik, A. Regiel-Futyra, M. Brindell, O. Mazuryk, A. Kyzioł, G. G. Stochel, W. Macyk and L. G. Arnaut, Coord. Chem. Rev., 2016, 325, 67–101 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- Y. M. Shaker, A. M. K. Sweed, C. Moylan, L. Rogers, A. A. Ryan, R. Petitdemange and M. O. Senge, Handbook of Photodynamic Therapy, 2016, pp. 95–149 Search PubMed.
- A. Minnock, D. I. Vernon, J. Schofield, J. Griffiths, H. Parish and S. B. Brown, J. Photochem. Photobiol., B, 1996, 32, 159–164 CrossRef CAS.
- P. Kubát, K. Lang, V. Král and P. Anzenbacher Jr., J. Phys. Chem. B, 2002, 106, 6784–6792 CrossRef.
- V. S. Chirvony, V. A. Galievsky, N. N. Kruk, B. M. Dzhagarov and P. Y. Turpin, J. Photochem. Photobiol., B, 1997, 40, 154–162 CrossRef CAS.
- K. Hirakawa, S. Kawanishi, T. Hirano and H. Segawa, J. Photochem. Photobiol., B, 2007, 87, 209–217 CrossRef CAS PubMed.
- N. G. Angeli, M. G. Lagorio, E. A. San Román and L. E. Dicelio, Photochem. Photobiol., 2000, 72, 49–56 CrossRef CAS PubMed.
- J. Matsumoto, T. Shinbara, S. I. Tanimura, T. Matsumoto, T. Shiragami, H. Yokoi, Y. Nosaka, S. Okazaki, K. Hirakawa and M. Yasuda, J. Photochem. Photobiol., A, 2011, 218, 178–184 CrossRef CAS.
- K. Hirakawa, N. Fukunaga, Y. Nishimura, T. Arai and S. Okazaki, Bioorg. Med. Chem. Lett., 2013, 23, 2704–2707 CrossRef CAS PubMed.
- (a) D. R. Reddy and B. G. Maiya, Chem. Commun., 2001, 117–118 RSC; (b) P. P. Kumar, G. Premaladha and B. G. Maiya, Chem. Commun., 2005, 3823–3825 RSC; (c) P. K. Poddutoori, P. Poddutoori, B. G. Maiya, T. K. Prasad, Y. E. Kandrashkin, S. Vasil'ev, D. Bruce and A. Van Der Est, Inorg. Chem., 2008, 47, 7512–7522 CrossRef CAS PubMed; (d) K. Hirakawa and H. Segawa, J. Photochem. Photobiol., A, 1999, 123, 67–76 CrossRef CAS; (e) M. Fujitsuka, D. W. Cho, S. Tojo, A. Inoue, T. Shiragami, M. Yasuda and T. Majima, J. Phys. Chem. A, 2007, 111, 10574–10579 CrossRef CAS PubMed; (f) K. Hirakawa and H. Segawa, Photochem. Photobiol. Sci., 2010, 9, 704 RSC; (g) P. K. Poddutoori, G. N. Lim, M. Pilkington, F. D'Souza and A. van der Est, Inorg. Chem., 2016, 55, 11383–11395 CrossRef CAS PubMed.
- A. A. Ryan, M. M. Ebrahim, R. Petitdemange, G. M. Vaz, E. Paszko, N. N. Sergeeva and M. O. Senge, Photodiagn. Photodyn. Ther., 2014, 11, 510–515 CrossRef CAS PubMed.
- I. N. Meshkov, V. Bulach, Y. G. Gorbunova, N. Kyritsakas, M. S. Grigoriev, A. Y. Tsivadze and M. W. Hosseini, Inorg. Chem., 2016, 55, 10774–10782 CrossRef CAS PubMed.
- H. Lin, Y. Shen, D. Chen, L. Lin, B. C. Wilson, B. Li and S. Xie, J. Fluoresc., 2013, 23, 41–47 CrossRef CAS PubMed.
- J. R. Hurst, J. D. McDonald and G. B. Schuster, J. Am. Chem. Soc., 1982, 104, 2065–2067 CrossRef CAS.
- C. Yu, T. Canteenwala, M. E. El-Khouly, Y. Araki, K. Pritzker, O. Ito, B. C. Wilson and L. Y. Chiang, J. Mater. Chem., 2005, 15, 1857 RSC.
- P. Ogilby and C. Foote, J. Am. Chem. Soc., 1981, 103, 3423–3430 CrossRef.
- S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, J. Chem. Phys., 2001, 115, 3256–3273 CrossRef CAS.
- (a) H.-Zh. Yu, J. S. Baskin and A. H. Zewail, J. Phys. Chem. A, 2002, 106, 9845–9854 CrossRef CAS; (b) Y. Venkatesh, M. Venkatesan, B. Ramakrishna and P. R. Bangal, J. Phys. Chem. B, 2016, 120, 9410–9421 CrossRef CAS PubMed.
- (a) S. Mangani, E. F. Meyer, D. L. Cullen, M. Tsutsui and C. J. Carrano, Inorg. Chem., 1983, 22, 400–404 CrossRef CAS; (b) Y.-H. Lin, M.-T. Sheu, C.-C. Lin, J.-H. Chen and S.-S. Wang, Polyhedron, 1994, 13, 3091–3097 CrossRef CAS; (c) Y.-H. Lin, C.-C. Lin, J.-H. Chen, W.-F. Zeng and S.-S. Wang, Polyhedron, 1994, 13, 2887–2891 CrossRef CAS; (d) Y.-H. Lin, S.-S. Tang, C.-C. Lin, J.-H. Chen, W.-F. Zeng, S.-S. Wang and H.-J. Lin, Aust. J. Chem., 1995, 48, 1367–1372 CrossRef CAS; (e) M. Sheu, I.-C. Ling, P.-C. Cheng, C.-C. Lin, J.-H. Chen, S.-S. Wang and W.-F. Zeng, J. Chem. Crystallogr., 1995, 25, 231–235 CrossRef CAS; (f) G. Jin Liang, S. Feng, L. Yong and N. Azuma, Polyhedron, 1995, 14, 1471–1476 CrossRef; (g) P.-C. Cheng, I.-C. Liu, T.-N. Hong, J.-H. Chen, S.-S. Wang, S.-L. Wang and J.-C. Lin, Polyhedron, 1996, 15, 2733–2740 CrossRef CAS.
- A. Harriman, J. Photochem., 1983, 23, 37–43 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc06052a |
| This journal is © The Royal Society of Chemistry 2017 |