
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusual reactivity of rhodium carbenes with allenes: an efficient asymmetric synthesis of methylenetetrahydropyran scaffolds†
Òscar
Torres
 ,
Miquel
Solà
,
Anna
Roglans
and
Anna
Pla-Quintana
*
,
Miquel
Solà
,
Anna
Roglans
and
Anna
Pla-Quintana
*
Institut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Facultat de Ciències, C/Maria Aurèlia Capmany, 69, E-17003-Girona, Catalunya, Spain. E-mail: anna.plaq@udg.edu
First published on 15th August 2017
Abstract
A RhI/(S)-BINAP catalytic system is able to promote carbene alkyne metathesis and cascade this elemental step with an stereoselective reaction with allenes. An unusual carbene/allene reactivity is discovered that, through a formal addition of p-toluenesulfinic acid to a Rh-bound trimethylenemethane intermediate, affords 4-methylenetetrahydropyran compounds in good yields and excellent enantioselectivities.
Metal carbenes are versatile reaction intermediates that can react with a wide variety of functional groups to synthesize complex organic molecules with high levels of chemo-, diastereo-, and enantioselectivity.1 The reaction of metal carbenes with alkenes to afford cyclopropanes has been extensively and systematically studied.2 In contrast, their reaction with allenes has received much less attention. In these latter reactions, alkylidenecyclopropanes are normally formed through a formal [2+1] cycloaddition.3 However, formal [3+2] cycloadditions, which afford cyclopentenes containing an exocyclic double bond, are observed when vinyl carbenes are reacted.4
We recently reported that RhI/BINAP is an efficient catalytic system for base-free generation of rhodium carbenes from N-tosylhydrazones and their subsequent carbene/alkyne metathesis.1h,5 This reaction provides rhodium vinylcarbenes that can react further in an enantioselective manner. When an alkyne is present in the molecule, a double carbene alkyne metathesis6 takes place, providing enantiomerically pure sulfones.5a On the other hand, when there is an alkene in the molecule, an intramolecular cyclopropanation takes place, efficiently affording chiral vinylcyclopropanes.5b Continuing with our interest in the cascade cyclization reactions involving rhodium vinylcarbenes, we report that N-tosylhydrazone/alkyne/allene substrates generate a rhodium vinylcarbene that reacts with an allene. Unexpectedly the reaction does not follow any of the already reported reaction modes but rather generates a rhodium trimethylenemethane intermediate that formally adds arylsulfinic acid to allow for stereoselective construction of methylenetetrahydropyran scaffolds.
At the outset of this study, linear substrate 1 with NTs tethers (Scheme 1) was synthesized. Under the conditions previously optimized in our earlier studies,5 a 28% yield of an inseparable mixture of two trienic isomers 2 and 2′ in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio was isolated (see Scheme S1 in the ESI† for the mechanistic proposal).
:1 ratio was isolated (see Scheme S1 in the ESI† for the mechanistic proposal).
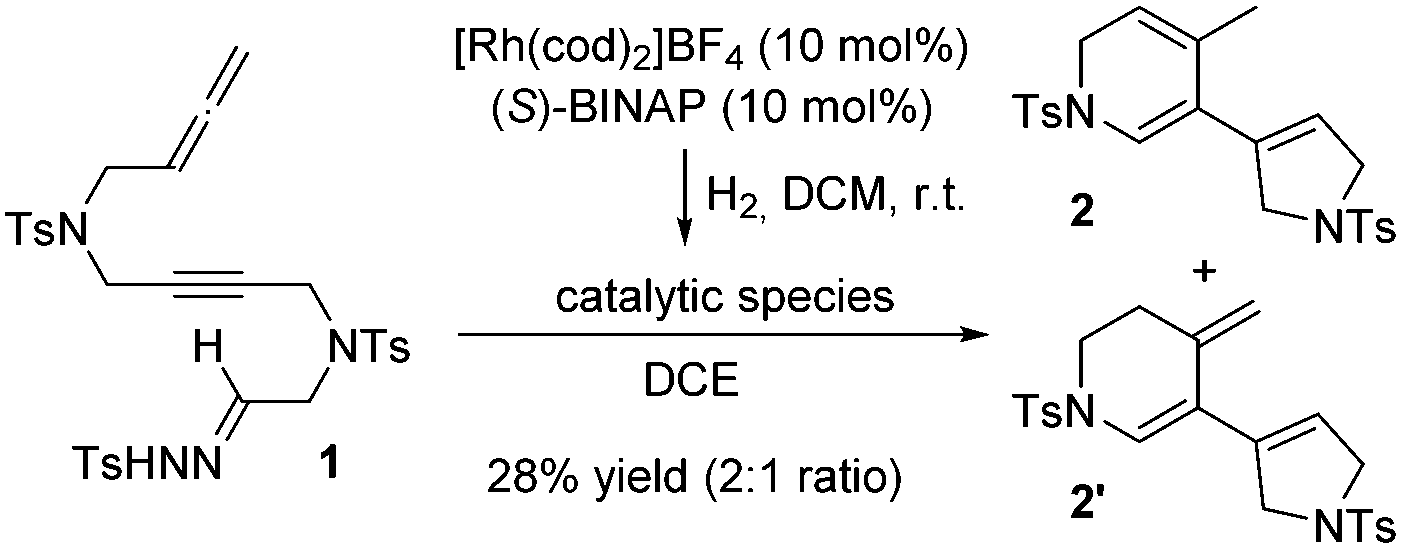 | ||
| Scheme 1 Cyclization of NTs-tethered substrate 1. | ||
As attempts to improve the reaction conditions, either by decreasing the temperature or by adding a base, were unsuccessful, we decided to place an oxygen tether between the alkyne and the allene. The resulting compound, 3a (R = p-C6H4CH3), was then synthesized and its reactivity was evaluated under the previously described reaction conditions. After one hour of reaction, two products were isolated that, upon complete characterization including X-ray diffraction,7 were identified as the 4-methylenetetrahydropyran derivative (R,R)-4a and the cyclopentenone derivative 5 (Scheme 2). Although 4a was obtained with excellent enantioselectivity, the reaction showed a low selectivity since the two products were isolated in equal quantities. Due to the relevance of chiral functionalized tetrahydropyran rings in natural products and their use as fragrances and pharmaceuticals,8 we sought to develop an efficient synthesis of the methylenetetrahydropyran core taking advantage of the unusual rhodium carbene/allene reactivity that we had discovered.
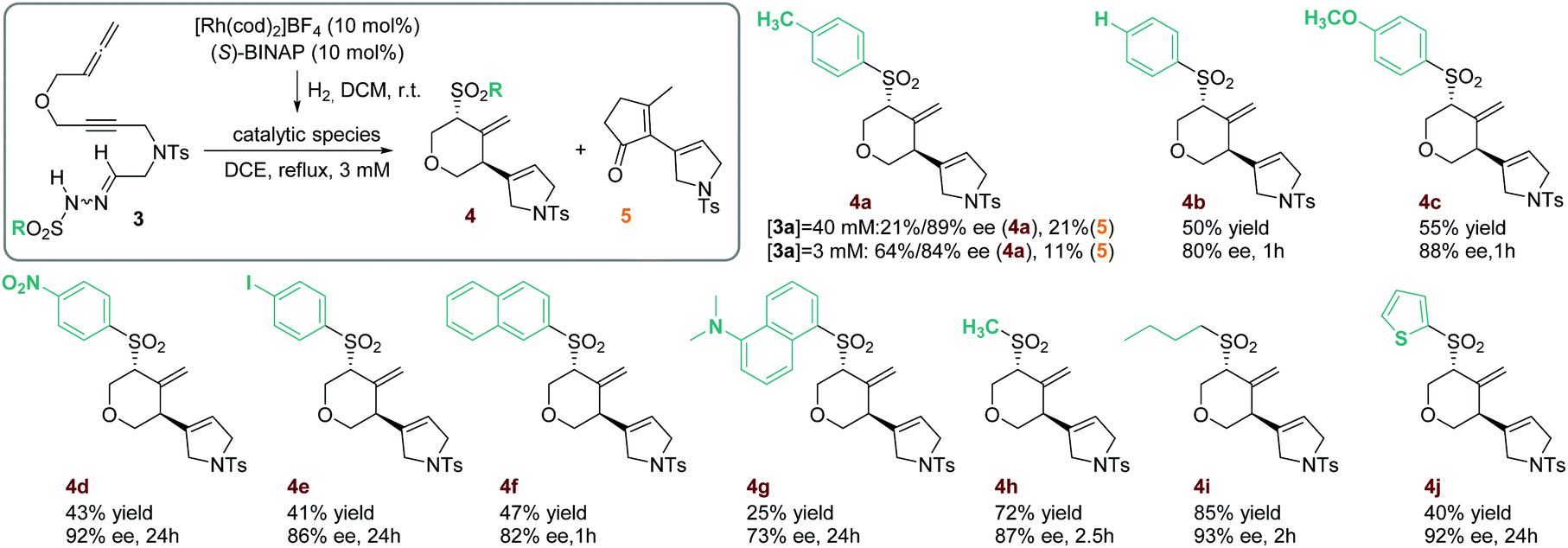 | ||
| Scheme 2 Rhodium-catalyzed asymmetric cyclization of 3. Reaction conditions: 0.21 mmol of substrate 3 in 70 mL of dichloroethane (DCE). Cited yields are of isolated products. The stereochemistry of products 4 is assigned with analogy to 4a. Traces of cyclopentenone derivative 5 were obtained in the formation of 4f and 4g. | ||
We therefore proceeded to optimize the reaction conditions, finally finding that the most critical parameter for selectivity was the concentration (see Table S2 in the ESI† for the optimization). When the reaction was carried out for one hour in lower concentrations (3 mM versus 40 mM in our first test), the 4-methylenetetrahydropyran derivative 4a was obtained in 64% yield whilst excellent enantioselectivity and a decrease in cyclopentenone derivative 5 formation were obtained (Scheme 2). We postulate that the cyclopentenone derivative 5 is formed from a product analogous to 2 (Scheme 1) from which an allyl Claisen rearrangement, followed by a RhI-catalyzed alkene hydroacylation, gives the cyclopentenone derivative 5 (see Scheme S1 in the ESI† for the detailed mechanistic proposal).
The scope of the process was then evaluated (Scheme 2). First, a series of substrates with several substituents in the phenyl ring of the sulfonyl hydrazone were reacted to give the corresponding 4-methylenetetrahydropyran derivatives (4b–4e). In all cases the reaction occurred with high enantioselectivity and the sulfonyl group migration was regioselective.9 However, when an electron-withdrawing substituent such as nitro (4d) or iodine (4e) was present, the yield dropped and there was a considerable increase in the reaction time. It is noteworthy that a p-C6H4I-sulfonyl group is efficiently involved in the reaction (4e), opening the possibility of further transformations of the cyclized compound.
More sterically hindered products, containing either a 2-naphthylsulfonyl (4f) or dansyl (4g) group, were also efficiently obtained although the yields were lower, especially in the case of dansyl. In these two examples, traces of derivative 5 were detected, indicating that the less favorable attack of a more sterically hindered sulfinate favors the formation of cyclopentanone derivative 5. Two aliphatic sulfonylhydrazones were also reacted under the optimized conditions. Cyclized products 4h and 4i were enantioselectively obtained in high yields. Finally, a 2-thiophenylsulfonylhydrazone-yne-allene substrate 3j was reacted affording the heteroaromatic derivative 4j after 24 hours of reaction.
The Ramberg–Bäcklund rearrangement, a base-mediated conversion of α-halosulfones, is an excellent option to synthesize regiodefined alkenes. Having synthesized aliphatic sulfones we decided to test their conversion to alkenes. Methylsulfone 4h was reacted under the Ramberg–Bäcklund rearrangement conditions, by forming in situ the α-halosulfones, to generate conjugated triene 6 in 69% yield (Scheme 3).
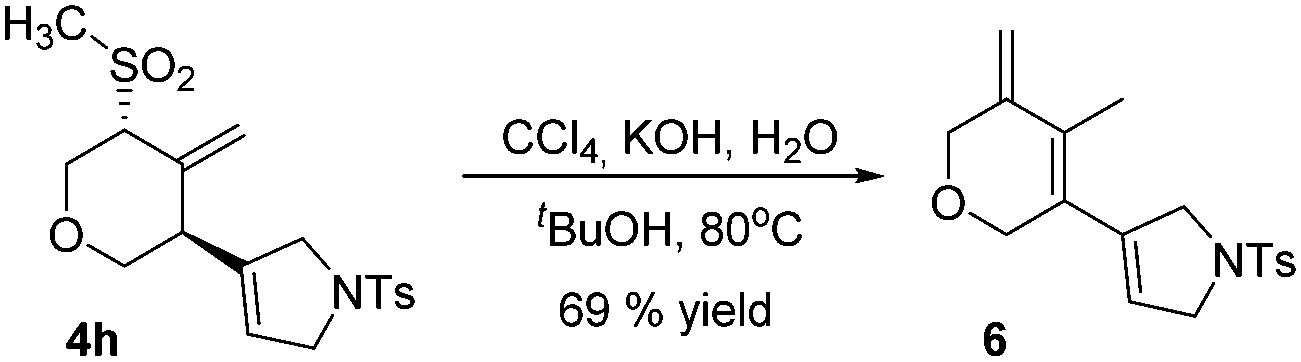 | ||
| Scheme 3 Ramberg–Bäcklund rearrangement of 4h. | ||
To get further insight into the reaction mechanism of the transformation of 3a to 4a, we performed B3LYP/aug-cc-pVDZ-PP//B3LYP/cc-pVDZ-PP calculations using reasonable models of the RhI/BINAP catalyst and reactant 3a to reduce the computational cost (see Scheme 4 and Computational methods in the ESI† for more detailed information). The reported energies are Gibbs energies that incorporate the effect of dichloroethane solution.
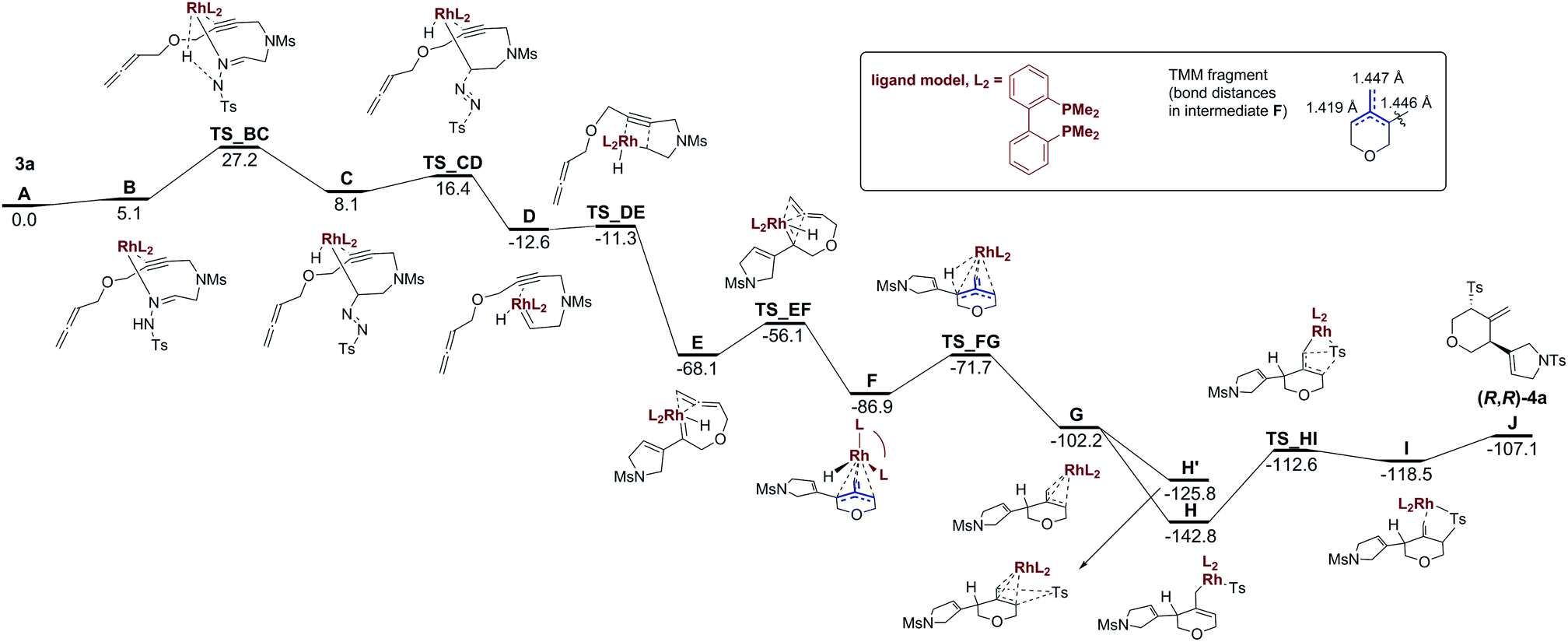 | ||
| Scheme 4 Gibbs energy profile (kcal mol−1) for the RhI catalyzed transformation of 3a into (R,R)-4a. Intermediates and TS from B to TS_CD and from H and H′ to I have +1 charge. Intermediates and TS from D to G have +2 charge. | ||
Scheme 4 shows the Gibbs energy profile for the transformation of the reactant 3a into the final product (R,R)-4a. In the first step, the RhI/BINAP catalyst coordinates to 3a through the N electron pair and η2 with the π-system of the alkyne group to form intermediate B in an endergonic process (5.1 kcal mol−1). B evolves to C by means of a H transfer combined with the breaking of the Rh–N initial coordination and the formation of a N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond and a Rh–C bond. The Gibbs energy barrier for the conversion of 3a to C is 27.2 kcal mol−1 (see Fig. S70 in the ESI† for the molecular structure of TS_BC). C yields D through TS_CD with a barrier of 8.3 kcal mol−1 in a complex process that involves the release of N2, the generation of the Ts− group, and the formation of a RhC bond. Then, an almost barrierless carbene/alkyne metathesis of D leads to intermediate E.
N double bond and a Rh–C bond. The Gibbs energy barrier for the conversion of 3a to C is 27.2 kcal mol−1 (see Fig. S70 in the ESI† for the molecular structure of TS_BC). C yields D through TS_CD with a barrier of 8.3 kcal mol−1 in a complex process that involves the release of N2, the generation of the Ts− group, and the formation of a RhC bond. Then, an almost barrierless carbene/alkyne metathesis of D leads to intermediate E.
The relative Gibbs energy of E with respect to our model of 3a and RhI/BINAP is −68.1 kcal mol−1 in dichloroethane. E is a rhodium(I) species in a square pyramidal coordination geometry in which the π-system of the external double bond of the allene group interacts with the d orbitals of the metal in a basal position and the hydride occupies the apical position. Ring closure through a [2+2] cycloaddition of the double bond of the external allene and the rhodium–carbene double bond develops a strained rhodacyclobutane that is not stable and rearranges to form intermediate F in an exergonic process (18.8 kcal mol−1) that takes place through a barrier of 8.0 kcal mol−1 (see Fig. S73 in the ESI† for the molecular structure of TS_EF). Intermediate F had an electronic structure resembling that of trimethylenemethane (fragment in blue in Scheme 4). The biradical π-system of the 4-methylenetrihydropyran ring is stabilized by a facial η3-coordination to the metal so that the intermediate F formed has a closed-shell electronic ground state. The binding is essentially symmetric with respect to the three C–C bonds (see the C–C bond distances in the inset of Scheme 4) with the rhodium located in a central position.10 Moreover, the similar Rh–C bond distances of 2.17, 2.21, and 2.29 Å confirm this almost symmetric arrangement.
Trimethylenemethane (TMM) diyl intermediates have been invoked in the tandem cycloaddition reaction of allenyl diazo compounds forming triquinane structures.11 In this metal-free process, the TMM diyl, constrained as a part of a ring, intramolecularly reacts with a tethered alkene in a formal [3+2] cycloaddition. The TMM reactivity can also be exploited in synthetic applications when complexed to a metal centre.12 Rhodium-bound TMM has also been invoked by Sarpong et al. in the synthesis of 3,4-fused pyrroles by the reaction of an imino rhodium carbene – generated by the decomposition of N-sulfonyl-1,2,3-triazoles – and a tethered allene.4a The rhodium-bound TMM again reacts in a formal [3+2] cycloaddition. An intermolecular variation has also been reported by Murakami et al. using nickel as the catalyst.4b In the reaction under study, the rhodium-bound trimethylenemethane F evolves through a reverse β-H-elimination viaTS_FG that transfers the H atom from Rh to the C atom attached to the 2,5-dihydropyrrole substituent to yield G. This process is exergonic by 15.3 kcal mol−1 and has to surpass a barrier of 15.2 kcal mol−1. In the next step, the Ts− group coordinates to Rh to yield intermediate H, which has a square pyramidal geometry with the diphosphine ligand and the Ts− group, coordinating in a bidentate fashion, occupying the basal positions. This process is exergonic by 40.6 kcal mol−1. An alternative pathway through H′ (Scheme 4) in which the Ts− group directly attacks the other side of the 4-methylenetrihydropyran ring of intermediate G without prior coordination to Rh is also possible, although this alternative is energetically less favourable. From H, an intramolecular nucleophilic attack of the Ts− group to C5 of the dihydropyran ring takes place to yield I, in which the final trans-disubstituted product (R,R)-4a coordinated to the metal has already been formed. This attack is endergonic by 24.3 kcal mol−1 and has to surmount a barrier of 30.2 kcal mol−1 (see Fig. S79 in the ESI† for the molecular structure of TS_HI). This, therefore, is the rate-determining step (rds) of the reaction mechanism. It would be expected that this step would have higher barriers for systems with electron-withdrawing substituents such as 4d or 4e or sterically hindered sulfinates such as 4f or 4g. The final release of 4a costs an additional energy of 11.4 kcal mol−1. However, it is likely that the release of 4a would be assisted by the addition of 3a to reduce or even remove this energetic cost. The transformation of reactants 3a to the product (S,S)-4a (the enantiomer of the experimentally obtained product (R,R)-4a) via an alternative diastereomeric route is also possible (see Section S.14 and Scheme S2 in the ESI†). However, the route that leads to (S,S)-4a has an rds with a barrier that is 2.7 kcal mol−1 higher in energy than that of the pathway shown in Scheme 4. This energy difference in the barrier of the rds justifies the experimental formation of only (R,R)-4a.13
Further evidence supporting the postulated mechanism was gained by carrying out a deuterium-labelling experiment. The treatment of substrate 3e with deuterated water afforded the deuterium-labelled compound 3e-D. Upon cyclization, NMR characterization confirmed that the deuterium was selectively incorporated into the 5-position of the tetrahydropyran ring (see ESI†).
In conclusion, we have developed an enantioselective synthesis of 4-methylenetetrahydropyran derivatives that rely on the reaction of rhodium vinyl carbenes and allenes as a key step. This cyclization reaction represents an efficient synthesis of trans-disubstituted methylenetetrahydropyrans that has revealed a highly unusual reactivity pattern in carbene/allene chemistry. DFT calculations show that a rhodium-bound trimethylenemethane intermediate is involved which has formally added arylsulfinic acid in an enantioselective manner. It is important to highlight that asymmetric variants of the transition-metal catalyzed [3+2] cycloaddition involving TMM remained elusive for a long period of time14 and that enantioselective additions of pronucleophiles are even rarer. The rate-determining step is the nucleophilic intramolecular attack of the Ts− group to the tetrahydropyran ring.
We are grateful for the financial support by the Spanish Ministry of Economy and Competitiveness (MINECO) (Project CTQ2014-54306-P) and the Generalitat de Catalunya (Project 2014-SGR-931, Xarxa de Referència en Química Teòrica i Computacional, ICREA Academia 2014 prize for M. S., and FI predoctoral grant to Ò. T.). The EU under the FEDER grant UNGI10-4E-801 has also funded this research. We thank one of the anonymous reviewers for his/her constructive and helpful suggestion on the two diastereomeric pathways.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews, see: (a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (b) A. Padwa, Chem. Soc. Rev., 2009, 38, 3072 RSC; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (d) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC; (e) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed; (f) D. Gillingham and N. Fei, Chem. Soc. Rev., 2013, 42, 4918 RSC; (g) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed; (h) Ò. Torres and A. Pla-Quintana, Tetrahedron Lett., 2016, 57, 3881 CrossRef.
- A. B. Charette, M.-N. Roy and V. N. G. Lindsay, in Cyclopropanation Reactions in Stereoselective Reactions of Carbon–Carbon Double Bonds, ed. J. G. de Vries, Science of Synthesis Series, Thieme, Stuttgart, 2011, vol. 1, ch. 1.14, pp. 731–817 Search PubMed.
- (a) G. Audran and H. Pellissier, Adv. Synth. Catal., 2010, 352, 575 CrossRef CAS; (b) C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker, Chem. Soc. Rev., 2014, 43, 3136 RSC; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed.
- (a) E. E. Schultz and R. Sarpong, J. Am. Chem. Soc., 2013, 135, 4696 CrossRef CAS PubMed; (b) T. Miura, K. Hiraga, T. Biyajima, Y. Nakamuro and M. Murakami, Org. Lett., 2013, 15, 3298 CrossRef CAS PubMed; (c) E. López, G. Lonzi, J. González and L. A. López, Chem. Commun., 2016, 52, 9398 RSC; (d) R. R. Singh, S. K. Pawar, M.-J. Huang and R.-S. Liu, Chem. Commun., 2016, 52, 11434 RSC.
- (a) Ò. Torres, T. Parella, M. Solà, A. Roglans and A. Pla-Quintana, Chem. – Eur. J., 2015, 21, 16240 CrossRef PubMed; (b) Ò. Torres, A. Roglans and A. Pla-Quintana, Adv. Synth. Catal., 2016, 358, 3512 CrossRef.
- (a) A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS PubMed; (b) A. Padwa, J. Organomet. Chem., 2000, 610, 88 CrossRef CAS.
- CCDC 1493176 (compound (R,R)-4a) and 1493175 (compound 5) contains the supplementary crystallographic data for this paper. The thermal ellipsoids in the ORTEP plot of the X-ray structure of compound (R,R)-4a and 5 are drawn at 50% probability.
- P. A. Clarke and S. Santos, Eur. J. Org. Chem., 2006, 2045 CrossRef CAS.
- The sulfonyl group is stereo- and regioselectively incorporated in the cyclized product independently of the electronics of the substituent. This contrasts with the decreased regioselectivity observed in our previous study (see ref. 5a) when electron-withdrawing substituents were present on the sulfonylhydrazone moiety.
- S. Mazumder, D. Shang, D. E. Negru, M.-H. Baik and P. A. Evans, J. Am. Chem. Soc., 2012, 134, 20569 CrossRef CAS PubMed.
- H.-Y. Lee, Acc. Chem. Res., 2015, 48, 2308 CrossRef CAS PubMed.
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1986, 25, 1 CrossRef; (b) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111 CrossRef CAS.
- An alternative mechanism involving similar steps to those found in the [2+2+2] cycloadditions was also studied (see Scheme S3 in the ESI†) but was ruled out since the Gibbs energy barrier for one of the steps was higher than 40 kcal mol−1.
- B. M. Trost, S. M. Silverman and J. P. Stambuli, J. Am. Chem. Soc., 2011, 133, 19483 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1493175 and 1493176. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc04803c |
| This journal is © The Royal Society of Chemistry 2017 |