
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative, ion-sensitive co-assembly of tripeptide hydrogels†
Yousef M.
Abul-Haija
 *ab,
Gary G.
Scott
a,
Jugal Kishore
Sahoo
a,
Tell
Tuttle
a and
Rein V.
Ulijn
*acde
*ab,
Gary G.
Scott
a,
Jugal Kishore
Sahoo
a,
Tell
Tuttle
a and
Rein V.
Ulijn
*acde
aWestCHEM/Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK. E-mail: Yousef.abul-haija@glasgow.ac.uk
bWestCHEM/School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
cAdvanced Science Research Center (ASRC) at the Graduate Center of the City University of New York (CUNY), New York, NY 10031, USA. E-mail: rein.ulijn@asrc.cuny.edu
dDepartment of Chemistry and Biochemistry, Hunter College of CUNY, 695 Park Ave., New York, NY 10065, USA
ePhD programs in Biochemistry and Chemistry, The Graduate Center of the City University of New York, NY 10016, USA
First published on 7th August 2017
Abstract
Peptide co-assembly is of interest for the development of functional supramolecular biomaterials. Herein, computational simulations were combined with experimental validation to aid the design and understanding of cooperative co-assembly of a structure-forming tripeptide (FFD) and a functional copper-binding tripeptide (GHK) leading to hydrogel formation in response to complexation with copper ions.
Supramolecular self-assembly1,2 provides simple functional mimics of biological materials.3–5 Co-assembly of two or more components provides options for control of morphological, physical and mechanical properties where the co-assembled material may have properties that are not found in the individual components. This cooperativity may be achieved by combining low molecular weight gelators with polymers6 or surfactants;7 or (aromatic) peptide amphiphiles with surfactants,8–10 polysaccharides,11,12 proteins13,14 or light responsive gelators.15 Co-assembly in these systems is typically governed by non-covalent interactions, in particular electrostatics.
While most existing supramolecular peptide biomaterials are based on long peptides,16 typically containing eight or more amino acids, it is increasingly recognized that di-17,18 or tripeptides contain sufficient molecular information to enable formation of self-assembling materials, including hydrogels,19–22 tunable emulsions,23 stable surface coatings24 and most recently, supramolecular substrates for formation of polymeric pigments with tunable properties.25 Thus, supramolecular structures based on short peptides hold promise as functional biomaterials.26–29
A number of short peptides are known to possess specific biological functions. These short peptides have been effectively introduced into supramolecular biomaterials through covalent linking to the structure-forming component of the system. Examples include materials bearing the fibronectin-derived RGDS,30,31 or RGD32,33 the laminin-derived IKVAV34 the tyrosine kinase nerve growth factor receptor-derived KFG.27 and the catalytically relevant35 histidine containing tripeptides (such as HFF).36 Another example is GHK, which is a copper-binding extracellular matrix derived peptide sequence that plays a role in wound healing.37,38
In this communication, we demonstrate how non-covalent incorporation of short functional peptides (exemplified by glycine–histidine–lysine, GHK) into supramolecular structures can be used to functionalise short peptide hydrogels. In particular, this work focuses on the design and co-assembly of a structure-forming tripeptide (diphenylalanine-aspartic acid, FFD)21,23 and GHK leading to hydrogel formation in response to complexation with copper ions.
GHK is itself not a gelator, and FFD and DFF were selected as candidate structural tripeptides due to their previously described self-assembly and charges that complement that of GHK. FFD and DFF were previously shown to form bilayer-like aggregates, but do not form gels in isolation at neutral pH (FFD has been shown to form a hydrogel at pH 5).23
30 mM of GHK and 30 mM of FFD were dissolved separately in PBS buffer (pH 7.4) at room temperature. Samples were sonicated and vortexed in order to improve solubility and homogeneity. Both samples formed clear solutions. To investigate whether electrostatic interactions of the positively charged lysine (K) and the negatively charged aspartic acid (D) could induce co-assembly, a mixture of GHK/FFD was prepared at the same conditions (i.e. 30 mM of each, 60 mM total) which formed a clear solution (no hydrogel was formed). Interestingly, spontaneous hydrogel formation was observed in the presence of 30 mM CuCl2, suggesting that GHK complexation with copper ions leads to the co-assembly and gelation observed.
Transmission electron microscopy (TEM) was used to observe the structures formed. As shown in Fig. 1, GHK on its own formed random aggregates, Fig. 1A. FFD (which was previously observed to form bi-layered nanotubes in acidic conditions)23 formed nanofibers at pH 7.4, Fig. 1B. Upon mixing of both FFD and GHK, the peptides co-assemble to form tape-like structures, Fig. 1C. However, this co-assembly did not lead to gelation as is typically observed for 2D self-assembled structures, such as tapes and sheets39 that are usually not able to entrap the solvent (in this case water) within their networks. When CuCl2 was mixed with the FFD/GHK mixture, networks of nanofibers were formed leading to hydrogel formation, Fig. 1D. The TEM images suggest a rapid nucleation and growth mechanism giving rise to spherulite-like structures.40 Additional TEM images with different magnifications are presented in Fig. S1 (ESI†).
 | ||
| Fig. 1 TEM images of all tripeptides showing random aggregates of GHK (A), self-assembly of FFD nanofibers (B), nanotape formation due to the co-assembly of FFD/GHK (C) and nanofiber formation due to the co-assembly of FFD/GHK after complexation with copper ions (D). Insets show the optical images of the corresponding peptides in inverted vials. | ||
Having established the co-assembly conditions for the successful formation of hydrogels, we used computational simulations to provide further understanding of the molecular co-assembly. Computational results were obtained using GROMACS 4.5.3 utilizing the MARTINI coarse grain (CG) force field. In each system, 300 peptide molecules are added (150 of each for mixed systems) to a 12.5 nm3 box solvated with MARTINI coarse grained water. This gives an approximate peptide concentration of 200 mM, which is a factor of 10 greater than normal experimental conditions, consistent with previous work in this field where the increased concentration is required to accelerate the simulated aggregation process.21,41,42 Each simulation is equilibrated for a total of 9.6 μs, with snapshots taken at initial, mid-point and final stages, Fig. 2. As can be seen, for GHK (Fig. 2A) the peptides show some clustering but overall maintain a random arrangement. For FFD, (Fig. 2B), the peptides arrange themselves with the phenylalanine rings interacting with aromatic side chains of other peptides. This induces the formation of a bilayer structure, with a hydrophobic core (FF) and negatively charged hydrophilic (D) surface. This formation is already observed at the start of the simulation, indicating a rapid hydrophobic collapse. When introducing GHK into a FFD system (Fig. 2C) a co-assembly is observed where GHK peptides are organized on the surface of the FFD structure. The computational results suggest that this occurs due to the electrostatic interaction between the lysine and the anionic surface of the FFD.
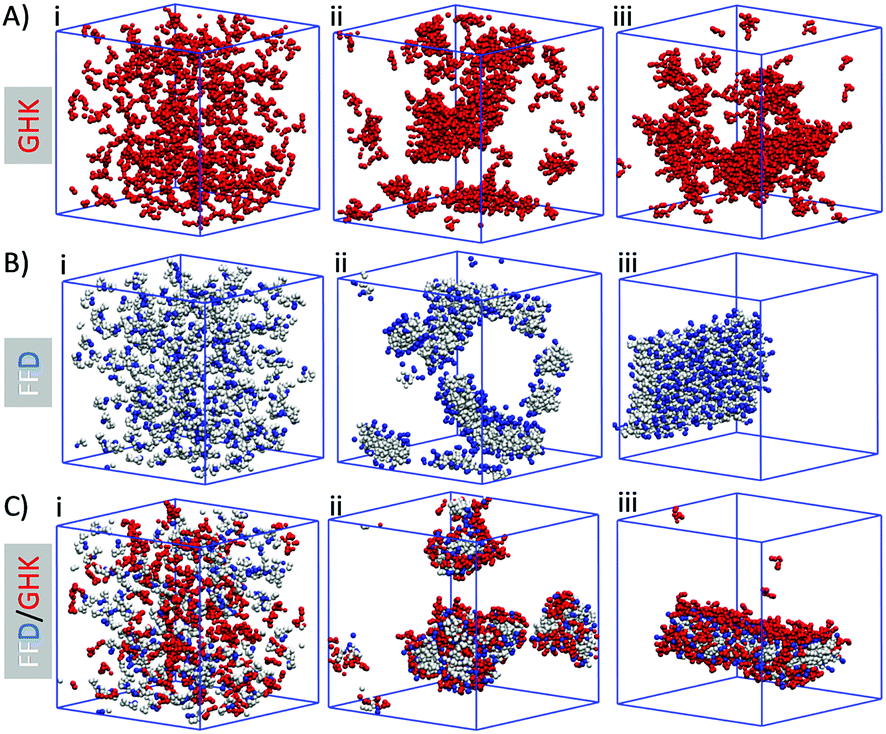 | ||
| Fig. 2 Computational time course for the self-assembly of the tripeptides in isolation (A and B) and co-assembled nanostructures (C). (i) Initial frame (0 μs), (ii) mid-point (∼3 μs), (iii) final frame (∼9.6 μs). | ||
To assess the effect of hydrogen bonding interactions experimentally, and to assess the formation of salt bridges between D and K residues, FTIR measurements were performed in D2O for individual components (GHK and FFD) and for mixtures of both before and after complexation with copper ions, Fig. 3A. For both GHK and FFD broad FTIR peaks at around 1560 cm−1 is assigned for the deprotonated COO−. The shift of this peak towards a lower wavenumber for FFD/GHK is indicative of the formation of more ordered structures with possible salt-bridge formation in co-assembled mixtures.21,23 It is worth noting that, although complexation with Cu2+ leads to fiber/hydrogel formation, the FTIR spectrum does not show a clear shift of peak at 1560 cm−1. This observation might be explained as that salt-bridge formation becomes less significant after complexation with copper ions, thus reducing the effective salt-bridge. The appearance of the peaks in the amide I region indicated the presence of hydrogen bonding between the amide carbonyls. However, the lower intensity of these peaks suggest that these are weak hydrogen bonds. As these peaks are present in only samples containing the copper, we can conclude that the presence of the copper helps the assembling propensity of the tripeptides resulting in hydrogelation, whilst forming highly ordered nanostructures. There is also a shift in the signal from FFD in the presence of both GHK and copper which indicates the formation of more stable structures.
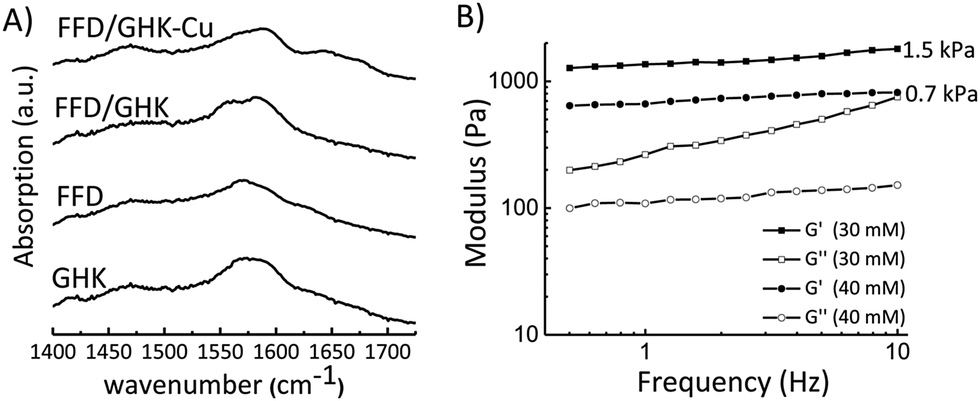 | ||
| Fig. 3 (A) FTIR spectra of tripeptides in isolation and after co-assembly, samples (30 mM of each component) were prepared in deuterated phosphate buffer pH 7.4 at room temperature. (B) Rheology data for the FFD/GHK–Cu hydrogel with associated storage and loss moduli. | ||
In order to assess the role of copper ions in specific binding and hydrogelation, we studied the gelation ability of GHK/FFD at different conditions. For instance, Cu2+ concentrations were varied (i.e. 20, 30, 40, 50 mM) and different metal ions (Zn2+, Co2+) were used as summarized in Table S1 (ESI†) (all added as chloride salts). We observed that gelation requires stoichiometric Cu2+ ions concentration (30 mM) and the gel was not formed when a lower Cu2+ ion concentration (20 mM) or much higher concentration (50 mM), most probably due to super saturation of the peptide-copper complexes in solution, thus preventing assembly. The lack of gelation for Zn2+ and Co2+ demonstrates that GHK binds specifically to Cu2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43,44 and this is required to enhance the peptide co-assembly leading to hydrogel formation. As previously reported, single crystal X-ray diffraction studies revealed that copper ions binding to GHK involves the N-terminal group of glycine, the nitrogen atom of the first amide bond, the unprotonated imidazole nitrogen from histidine, and two oxygen atoms in water (see also Scheme S1, ESI†).45
43,44 and this is required to enhance the peptide co-assembly leading to hydrogel formation. As previously reported, single crystal X-ray diffraction studies revealed that copper ions binding to GHK involves the N-terminal group of glycine, the nitrogen atom of the first amide bond, the unprotonated imidazole nitrogen from histidine, and two oxygen atoms in water (see also Scheme S1, ESI†).45
Having established the co-assembly conditions and ion-selectivity of gelation, we studied the mechanical properties of the formed hydrogels by rheology as shown in Fig. 3B. The rheology data shows that the hydrogel formed at 30 mM Cu2+ has an average loss modulus value of 1.5 kPa, while the hydrogel formed at 40 mM Cu2+ has a weaker gel with a storage modulus of about 0.7 kPa. The slope of the modulus might indicate thickening instability of the sample when 30 mM of CuCl2 was used as previously reported for similar systems.46
To assess the importance of peptide sequence, we also studied the co-assembly process in the presence of DFF instead of FFD, Fig. 4. Consistent with previously reported structure forming tripeptides (i.e., anionic amino acids are preferred at the C-terminus),21 no gel formation was observed for individual components as well as for mixtures before and after the addition of copper ions, Fig. 4A–C. TEM images (Fig. 4D–F and Fig. S2, ESI†) also suggest that both individual peptide solutions and mixtures are unable to self-assemble to form highly organized structures both in the presence and absence of copper ions. These results are in good agreement with our previous21 and current computational predictions (Fig. S3, ESI†).
 | ||
| Fig. 4 Optical images (upper panel) and TEM images (lower panel) of the control (DFF) aggregation (A and D), mixed with GHK (B and E) and co-assembled sample after complexation with copper ions (C and F). | ||
Molecular dynamics simulations of DFF and GHK mixtures show clear differences when compared with FFD/GHK, Fig. S3 (ESI†). The DFF structure compared with the FFD shows similar bilayer formation. The introduction of GHK appears to cause the collapse of the DFF structure resulting in random aggregates of DFF/GHK, as a result, no hydrogelation is observed in the presence of Cu2+. Similarly, MD simulations of FDF self-assembly have resulted in a structure formation similar to that of DFF and FFD (Fig. S4, ESI†). However, the presence of aspartic acid in the middle position in the co-assembled system disrupts the packing of the FF dyad within the tripeptide. The loss of the intramolecular π-interactions between the phenyl groups indicates that the FDF structure is not stable enough to form an elongated fibrillar structure when GHK is present.
It is worth noting that GHK has generated significant interest from the cosmetics industry, in particular as a complex with Cu2+ which facilitates copper intake by cells to enhance enzymatic processes, wound healing and promotes angiogenesis.47,48 It also improves skin firmness and texture, fine lines and hyperpigmentation.37,38 Thus, there is clear evidence of the biological activity of GHK and GHK–Cu complex and it is therefore considered as an additive for cosmetics and skin treatments. Formulating GHK into a biocompatible tripeptide hydrogel may be of direct interest for cosmetics industry.
In summary, the cooperative co-assembly of functional and structural tripeptides has been studied by combining both experimental validation and computational simulations. We have found that the co-assembled peptides can selectively complex with copper ions, leading to structural reconfiguration from clear solution of nanotapes to hydrogel formation of nanofibers. GHK is a known bioactive peptide and the hydrogels developed here may have applications in wound care and cell culture, which will require further investigation. The combined computation/experimental approach followed here provides a generally useful approach to identification of co-assembled structures of structural and functional components based on short peptides.
We thank the U.S. Air Force Office of Scientific Research for funding (grant FA9550-15-1-0192). We thank Dr Margaret Mullen for her help in performing the TEM imaging. Computational results were obtained using the EPSRC funded ARCHIE-WeSt High Performance Computer (http://www.archie-west.ac.uk) EPSRC grant no. EP/K000586/1.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- M. J. Webber, E. A. Appel, E. W. Meijer and R. Langer, Nat. Mater., 2016, 15, 13–26 CrossRef CAS PubMed.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165–13307 CrossRef CAS PubMed.
- S. Mondal, A. K. Barman and S. Verma, CHIMIA Int. J. Chem., 2012, 66, 930–935 CrossRef CAS PubMed.
- D. J. Cornwell and D. K. Smith, Mater. Horiz., 2015, 2, 279–293 RSC.
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- Y. M. Abul-Haija, S. Roy, P. W. J. M. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- E. V. Alakpa, V. Jayawarna, A. Lampel, K. V. Burgess, C. C. West, S. C. J. Bakker, S. Roy, N. Javid, S. Fleming, D. A. Lamprou, J. Yang, A. F. Miller, A. J. Urquhart, P. W. J. M. Frederix, N. T. Hunt, B. Péault, R. V. Ulijn and M. J. Dalby, Chem., 2016, 1, 298–319 CAS.
- R. M. Capito, H. S. Azevedo, Y. S. Velichko, A. Mata and S. I. Stupp, Science, 2008, 319, 1812–1816 CrossRef CAS PubMed.
- Y. M. Abul-Haija and R. V. Ulijn, Biomacromolecules, 2015, 16, 3473–3479 CrossRef CAS PubMed.
- K. E. Inostroza-Brito, E. Collin, O. Siton-Mendelson, K. H. Smith, A. Monge-Marcet, D. S. Ferreira, R. P. Rodríguez, M. Alonso, J. C. Rodríguez-Cabello, R. L. Reis, F. Sagués, L. Botto, R. Bitton, H. S. Azevedo and A. Mata, Nat. Chem., 2015, 7, 897–904 CrossRef CAS PubMed.
- N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef CAS PubMed.
- E. R. Draper, E. G. B. Eden, T. O. McDonald and D. J. Adams, Nat. Chem., 2015, 7, 848–852 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- H. Erdogan, E. Babur, M. Yilmaz, E. Candas, M. Gordesel, Y. Dede, E. E. Oren, G. B. Demirel, M. K. Ozturk, M. S. Yavuz and G. Demirel, Langmuir, 2015, 31, 7337–7345 CrossRef CAS PubMed.
- S. Marchesan, C. D. Easton, F. Kushkaki, L. Waddington and P. G. Hartley, Chem. Commun., 2012, 48, 2195–2197 RSC.
- S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752–6760 RSC.
- P. W. J. M. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30–37 CrossRef CAS PubMed.
- C. G. Pappas, P. W. J. M. Frederix, T. Mutasa, S. Fleming, Y. M. Abul-Haija, S. M. Kelly, A. Gachagan, D. Kalafatovic, J. Trevino, R. V. Ulijn and S. Bai, Chem. Commun., 2015, 51, 8465–8468 RSC.
- G. G. Scott, P. J. McKnight, T. Tuttle and R. V. Ulijn, Adv. Mater., 2016, 28, 1381–1386 CrossRef CAS PubMed.
- S. Maity, S. Nir, T. Zada and M. Reches, Chem. Commun., 2014, 50, 11154–11157 RSC.
- A. Lampel, S. A. McPhee, H.-A. Park, G. G. Scott, S. Humagain, D. R. Hekstra, B. Yoo, P. W. J. M. Frederix, T.-D. Li, R. R. Abzalimov, S. G. Greenbaum, T. Tuttle, C. Hu, C. J. Bettinger and R. V. Ulijn, Science, 2017, 356, 1064–1068 CrossRef CAS PubMed.
- S. Marchesan, Y. Qu, L. J. Waddington, C. D. Easton, V. Glattauer, T. J. Lithgow, K. M. McLean, J. S. Forsythe and P. G. Hartley, Biomaterials, 2013, 34, 3678–3687 CrossRef CAS PubMed.
- P. Moitra, K. Kumar, P. Kondaiah and S. Bhattacharya, Angew. Chem., Int. Ed., 2014, 53, 1113–1117 CrossRef CAS PubMed.
- A. V. Vargiu, D. Iglesias, K. E. Styan, L. J. Waddington, C. D. Easton and S. Marchesan, Chem. Commun., 2016, 52, 5912–5915 RSC.
- J. K. Sahoo, C. Nazareth, M. A. VandenBerg and M. J. Webber, Biomater. Sci., 2017, 5, 1526–1530 RSC.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS PubMed.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef CAS PubMed.
- V. Castelletto, R. J. Gouveia, C. J. Connon and I. W. Hamley, Eur. Polym. J., 2013, 49, 2961–2967 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- L. Duncan Krystyna and V. Ulijn Rein, Biocatalysis, 2015, 1, 67 Search PubMed.
- A. M. Garcia, M. Kurbasic, S. Kralj, M. Melchionna and S. Marchesan, Chem. Commun., 2017, 53, 8110–8113 RSC.
- F.-X. Maquart, L. Pickart, M. Laurent, P. Gillery, J.-C. Monboisse and J.-P. Borel, FEBS Lett., 1988, 238, 343–346 CrossRef CAS PubMed.
- S. Jose, M. L. Hughbanks, B. Y. K. Binder, G. C. Ingavle and J. K. Leach, Acta Biomater., 2014, 10, 1955–1964 CrossRef CAS PubMed.
- C. G. Pappas, Y. M. Abul-Haija, A. Flack, P. W. J. M. Frederix and R. V. Ulijn, Chem. Commun., 2014, 50, 10630–10633 RSC.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS PubMed.
- Y. Zheng, K. M. Gattas-Asfura, V. Konka and R. M. Leblanc, Chem. Commun., 2002, 2350–2351 RSC.
- Y. Zheng, Q. Huo, P. Kele, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, Org. Lett., 2001, 3, 3277–3280 CrossRef CAS PubMed.
- C. M. Perkins, N. J. Rose, B. Weinstein, R. E. Stenkamp, L. H. Jensen and L. Pickart, Inorg. Chim. Acta, 1984, 82, 93–99 CrossRef CAS.
- G. Cheng, V. Castelletto, C. Moulton, G. Newby and I. Hamley, Langmuir, 2010, 26, 4990–4998 CrossRef CAS PubMed.
- A. Siméon, Y. Wegrowski, Y. Bontemps and F.-X. Maquart, J. Invest. Dermatol., 2000, 115, 962–968 CrossRef PubMed.
- F. Buffoni, R. Pino and A. Dal Pozzo, Arch. Int. Pharmacodyn. Ther., 1995, 330, 345–360 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc04796g |
| This journal is © The Royal Society of Chemistry 2017 |