
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric radical alkylation of N-sulfinimines under visible light photocatalytic conditions†‡
Alberto F.
Garrido-Castro
 a,
Houcine
Choubane
ab,
Mortada
Daaou
b,
M. Carmen
Maestro
*a and
José
Alemán
*ac
a,
Houcine
Choubane
ab,
Mortada
Daaou
b,
M. Carmen
Maestro
*a and
José
Alemán
*ac
aDepartamento de Química Orgánica (Módulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, 28049, Madrid, Spain. E-mail: jose.aleman@uam.es; Web: www.uam.es/jose.aleman
bL.S.P.B.E, Département de Chimie Organique Industrielle, Faculté de Chimie, Université des Sciences et de la Technologie d'Oran- Mohamed Boudiaf- B.P 1505 El Mnaouer, Oran, 31000, Algeria
cInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 21st June 2017
Abstract
In this communication, a new photocatalytic strategy for the addition of alkyl-radical derivatives to N-sulfinimines with complete diastereoselectivity and moderate to good yields is presented. This is the first asymmetric photocatalytic addition to N-sulfinimines under visible light irradiation with smooth conditions and functional group tolerance.
Enantiopure sulfinimines, or N-sulfinyl imines, have been used in the asymmetric synthesis of diverse chiral amino derivatives, e.g. α-branched amines, α- and β-amino acids, and α- and β-amino phosphonic acids.1 The N-sulfinyl group is one of the best chiral auxiliaries for imines because the C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond is activated for nucleophilic addition, diastereofacial selectivity is provided, and it is easily removable under mild acidic conditions. However, most reactions with N-sulfinimines employ organometallic compounds such as organolithium or Grignard reagents under very restrictive conditions (low temperature and minimal functional group tolerance).
N bond is activated for nucleophilic addition, diastereofacial selectivity is provided, and it is easily removable under mild acidic conditions. However, most reactions with N-sulfinimines employ organometallic compounds such as organolithium or Grignard reagents under very restrictive conditions (low temperature and minimal functional group tolerance).
In this regard, radical reactions offer a better functional group tolerance under more suitable reaction conditions. However, only a few examples involving radical-mediated intermolecular additions to N-sulfinimines have been reported,2–4 which are impractical because of the large amount of radical precursors and the use of toxic reagents (Bu3SnH). These issues are impeding a simple gateway to valuable chiral amines, and therefore a new straightforward methodology capable of overcoming these limitations is required.
On the other hand, visible light photoredox catalysis has received significant attention over the past decade due to its ability to achieve bond constructions that are not possible using established procedures.5 At the same time, photoredox catalysts can also be employed to generate radicals to be used in a diverse range of proven radical strategies. This approach is based on the adeptness transition metal complexes6 and organic dyes7 display by engaging in SET processes following photoexcitation with visible light. Numerous photocatalytic examples of racemic radical transformations performed with imines have been published.8 However, only one asymmetric approach has been disclosed by Ooi and co-workers (Scheme 1a).9 In Ooi's work, the addition of α-alkylamine derivatives to N-sulfonylimines to afford diamine derivatives was described. Nonetheless, no examples of visible light photocatalytic reactions with N-sulfinimines exist to the best of our knowledge.
 | ||
| Scheme 1 Previous reaction and present work of this manuscript. | ||
Therefore, our goal is to deliver a novel radical addition to N-sulfinimines, using photocatalysis and visible light irradiation as a viable radical-generating process, and the N-sulfinyl group as a chiral auxiliary to achieve complete stereoselectivity in the synthesis of amine derivatives (Scheme 1b).
In order to achieve this goal, we performed an initial study in which the reactivity of the enantiopure N-sulfinimine 1a (R1 = p-Tol) was tested with different radical donors (see ESI‡ for more details). Reactions performed with isobutyric acid (2a)10 and isopropyl oxalate (2b)11 were unsuccessful; probably caused by the effect the required additives (K2HPO4 and water) inflicted upon the N-sulfinimine, observing its hydrolysis in the crude mixture. However, under fairly anhydrous conditions, the reaction of N-(isobutyryloxy)phthalimide 2c12 with N-sulfinimine 1a catalyzed by [Ru(bpy)3]2+ (3a) gave rise to our desired product in moderate conversion after 14 hours (entry 1, Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Photocatalyst | Solvent | Conv.b (%) | drb |
| a Reaction conditions: 0.1 mmol scale using 1.5 equiv. of N-sulfinimine 1a, 1.0 equiv. of phthalimide 2c, 2.0 equiv. of DIPEA, 1.5 equiv. of Hantzsch ester (HE) and 1 mL of solvent. b Conversion and diastereomeric ratio were determined by 1H NMR analysis of the crude mixture. c Reliable data could not be obtained due to partial N-sulfinimine hydrolysis. | ||||
| 1 | [Ru(bpy)3]Cl2·6H2O (3a) | DCM | 39 | 73![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 :27 |
| 2 | [Ir(ppy)2(bpy)]PF6 (3b) | DCM | —c | — |
| 3 | Ir(ppy)3 (3c) | DCM | 67 | 72:28 |
| 4 | Ir(dFppy)3 (3d) | DCM | —c | — |
| 5 | [Ir{dF(CF3)ppy}2(dtbpy)]PF6 (3e) | DCM | —c | — |
| 6 | Eosin Y (3f) | DCM | 58 | 68:32 |
| 7 | Rose bengal (3g) | DCM | 46 | 75:25 |
| 8 | Rhodamine 6G (3h) | DCM | 29 | 67:33 |
| 9 | Ir(ppy)3 (3c) | Tol | 30 | 71:29 |
| 10 | Ir(ppy)3 (3c) | DMF | 100 | 69:31 |
| 11 | Ir(ppy)3 (3c) | MeCN | 100 | 66:34 |
| 12 | Ir(ppy)3 (3c) | DMSO | 100 | 68:32 |
Following this positive result, we tried to identify the optimal photocatalyst that would allow us to increase the final conversion. As revealed in Table 1, the use of some metal photocatalysts (entries 2, 4 and 5) occasionally led to N-sulfinimine hydrolysis, whereas reactions performed with organic dyes (entries 6–8) yielded 4a without hydrolysis of 1a. Nicely, Ir(ppy)33c displayed the highest conversion in DCM (entry 3), and it was consequently used as photocatalyst during the solvent screening (entries 9–12). Gratifyingly, reactions performed with Ir(ppy)3 in highly polar solvents (entries 10–12) led to complete conversion to sulfinamide 4a/4a′.
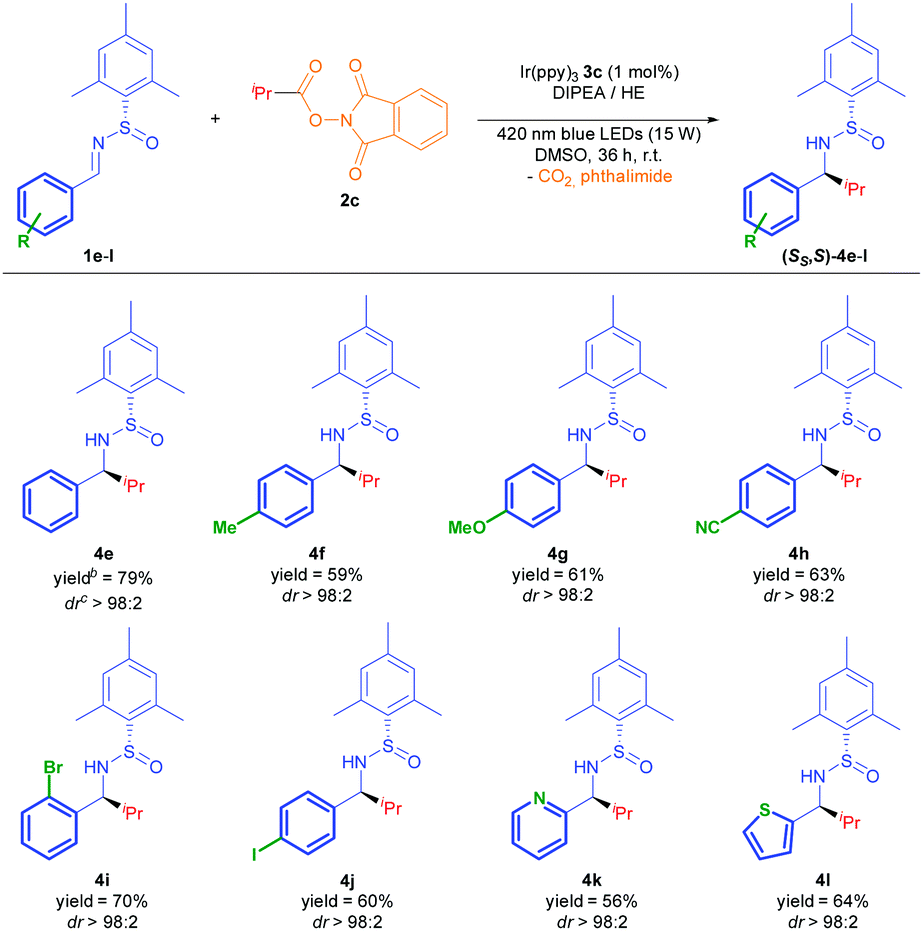
Despite reaching optimal conversion, dr values obtained during screening studies confirmed that the reaction displayed low diastereoselectivity under the optimized conditions. Variation of different reaction parameters such as temperature (0, −30 °C instead of r.t.) and concentration (0.03 M instead of 0.10 M) seemed to have no effect upon diastereoselectivity. We performed several tests with N-tert-butanesulfinimine 1b, aiming to improve this feature by taking advantage of the bulkier tBu group. However, none of these trials provided the desired product, obtaining a complex mixture under the present conditions (Table 2). These results prompted us to conduct an evaluation of three different N-sulfinimines bearing bulkier aryl groups (1c–e), anticipating that the stereoinduction would improve upon increase of the steric size of the N-sulfinyl moiety (Table 2). To our delight, all three of them delivered excellent diastereoselectivity, especially the 2-methoxy-1-naphthyl- and mesityl-substituted N-sulfinimines (1d and 1e, respectively). Since 1d was less reactive than 1e after 18 hours, we selected the latter as the optimal substrate to complete the scope with different imines (Table 3), and different radical donors (Table 4), using DMSO as solvent because higher yields were achieved after isolation by flash chromatography (see ESI‡).

| a Reaction conditions: 0.1 mmol scale using 1.0 equiv. of N-sulfinimine 1, 1.5 equiv. of phthalimide 2c, 3.0 equiv. of DIPEA, 0.5 equiv. of Hantzsch ester (HE) and 1 mL of DMSO. b Isolated yields after flash chromatography. c Determined by 1H NMR. |
|---|

|
The reaction was found to be compatible with a large variety of aromatic rings (Table 3).13 Therefore, electron donating and electron withdrawing groups underwent the alkylation with excellent diastereoselectivity (4f–4h), and good chemoselectivity in the case of 1h, where no addition to the nitrile group was detected. Pleasantly, this photocatalytic protocol tolerated the use of halogen substituents at para and ortho positions without reduction or loss of the bromide and iodide atoms (4i and 4j). Different electron rich and poor heterocycles were used, yielding 4k and 4l with an excellent diastereoselectivity.
The simple one-step preparation of the phthalimide reagent (see ESI‡) led us to conduct a study of the addition of different alkyl radicals derived from 2 to the imine 1e (Table 4). As a result, secondary alkyl groups were easily added with excellent yields (74–79%) and diastereoselectivity, as well as the tert-butyl group (4n). Primary alkyl radicals have been traditionally problematic in terms of their generation, and other methods have failed to successfully complete the addition to the N-sulfinimine.4a Remarkably, primary alkyl radical addition was achieved with an excellent dr, albeit in moderate yield due to its difficult isolation (4o).
The absolute configuration of compound 4e was determined by comparison of the spectroscopic data of a known compound in the literature (see ESI‡),14 assuming the same stereochemical behaviour for all compounds 4. We carried out the desulfinylation under acidic conditions of 4e, giving rise to amine 5 which has been correlated with a known (S)-amine in the literature (see ESI‡). The stereoselectivity of these reactions can be rationalized from the more stable s-cis arrangement conformation of the sulfinylimine (enforced by the intramolecular hydrogen bond interaction).4a The approach of the radical to the less hindered si-face of imines explains the S configuration assigned to the obtained compounds 4e–o (bottom-left, Scheme 2).
 | ||
| Scheme 2 Proposed mechanism for the decarboxylative alkylation. | ||
A plausible mechanism of the radical addition to N-sulfinimines is depicted in Scheme 2. The radical formation of the reaction pathway is based on mechanistic studies performed on our system along with fundamental concepts related to photoredox catalytic mechanisms.5,6 Photoredox catalysts can reach an excited state upon irradiation with visible light. Therefore, Ir(ppy)3 absorbs irradiation with blue light, populating the highly reducing excited *Ir(ppy)3 species (E1/2 IrIV/*IrIII = −1.73 V vs. SCE).6 The photoexcited catalyst engages in a SET process with phthalimide 2c (Ered1/2 = −1.14 V vs. SCE, see cyclic voltammetry in ESI‡), generating anion radical I through an oxidative quenching pathway (see Scheme 2). The photocatalytic cycle is completed with the reduction of the IrIV species (E1/2 IrIV/IrIII = +0.77 V vs. SCE)6 by an electron donor such as DIPEA (Eox1/2 = +0.94 V vs. SCE, see cyclic voltammetry in ESI‡), regenerating the photocatalyst. The resultant reductive elimination of the phthalimide group leads to decarboxylation and subsequent formation of radical II. Next, N-sulfinimine 1 undergoes radical addition to afford N-centered radical III. Finally, Hantzsch ester donates a hydrogen atom to III, giving rise to sulfinamide 4via hydrogen atom transfer (HAT). The reaction also proceeds adequately in absence of Hantzsch ester, although requiring longer reaction times, because the oxidized species of DIPEA IV can transfer a hydrogen atom. This step is part of the widely-known DIPEA decomposition pathway in photocatalytic procedures following electron donation.5,6
In order to validate this proposal, different mechanistic studies were performed. Stern–Volmer quenching evaluation allowed us to establish the first step of the mechanism (see ESI‡). The excited state of our photocatalyst 3c can decay via phosphorescence (λem = 518 nm).15 However, we observed that phthalimide 2c interfered with this process, quenching the excited state of the photocatalyst.
On the other hand, DIPEA and N-sulfinimine 1 did not display this quenching ability. Therefore, the mechanism should follow an oxidative quenching pathway, starting with reduction of phthalimide 2.
Finally, to ensure whether this reaction is governed by a radical chain propagation process or a visible light-photocatalytic one, we carried out light/dark experiments, (Fig. 1).16 In addition, the radical chain mechanism seems to be incoherent since the N-centered radical III and phthalimide 2c should both reduced.17 Predictably, we observed that product formation (4e) only occurs during visible light irradiation periods, indicating that radical formation is taking place. Moreover, the quantum yield (ϕ) of this reaction was measured (see ESI‡) to provide further information in this regard. Since ϕ (420 nm) = 0.077, the photocatalyst must absorb 13 photons to afford one equivalent of product, indicating that a chain propagation-type mechanism should not be taking place.
 | ||
| Fig. 1 Light/dark experiment for the decarboxylative alkylation of N-sulfinimines. Conversion determined by 1H NMR. Areas shaded in blue indicate irradiation and those shaded in black indicate darkness. | ||
In conclusion, the first highly diastereoselective photocatalytic addition to N-sulfinimines is presented. This is an efficient transformation with full conversion under mild conditions in absence of toxic reagents. The reaction presents a wide scope, affording different sulfinamides with high diastereomeric control. We propose a mechanism based on the reductive decarboxylation of the phthalimide to generate the alkyl radical which is added to the N-sulfinimine via oxidative quenching of the photocatalyst, as it is confirmed by Stern–Volmer studies. Furthermore, the quantum yield indicates that the reaction should not involve a radical chain mechanism.
Spanish Government (CTQ2015-64561-R) and the European Research Council (ERC-CG, contract number: 647550) are acknowledged.
Notes and references
- (a) P. Zhou, B.-C. Chen and F. A. Davis, Tetrahedron, 2004, 60, 8003 CrossRef CAS; (b) F. A. Davis, J. Org. Chem., 2006, 71, 8993 CrossRef CAS PubMed; (c) F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Chem. Soc. Rev., 2009, 38, 1162 RSC; (d) M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- Although Lin's reductive cross-coupling with aldehydes is not a radical alkyl addition, it can be considered a radical mediated process, see: Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef CAS PubMed.
- T. Akindele, Y. Yamamoto, M. Maekawa, H. Umeki, K.-I. Yamada and K. Tomioka, Org. Lett., 2006, 8, 5729 CrossRef CAS PubMed.
- (a) J. A. Fernández-Salas, M. C. Maestro, M. M. Rodríguez-Fernández, J. L. García-Ruano and I. Alonso, Org. Lett., 2013, 15, 1658 CrossRef PubMed . For works on intramolecular radical additions to N-sulfinimines, see: ; (b) E. M. Rochette, W. Lewis, A. G. Dossetter and R. A. Stockman, Chem. Commun., 2013, 49, 9395 RSC; (c) E. Lacôte and M. Malacria, C. R. Acad. Sci., Ser. IIc: Chim., 1998, 1, 191 CrossRef.
- (a) M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- (a) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97 RSC; (b) S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561 RSC; (c) D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688 RSC; (d) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed.
- For racemic photocatalytic addition to sulfonylimines, see: (a) J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef CAS PubMed . For racemic photocatalytic reaction with N-arylimines, see: ; (b) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef CAS PubMed; (c) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312 CrossRef CAS PubMed . For racemic photocatalytic reactions with ketimines, see: ; (d) J. L. Jeffrey, F. R. Petronijević and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404 CrossRef CAS PubMed; (e) For an intramolecular photocatalytic racemic example, see: S.-Y. Hsieh and J. W. Bode, Org. Lett., 2016, 18, 2098 CrossRef PubMed.
- For asymmetric additions, see: (a) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef CAS PubMed; (b) T. Kizu, D. Uraguchi and T. Ooi, J. Org. Chem., 2016, 81, 6953 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef CAS PubMed.
- C. C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef CAS PubMed.
- J. Yang, J. Zhang, L. Qi, C. Hu and Y. Chen, Chem. Commun., 2015, 51, 5275 RSC.
- Alkyl N-sulfinimines were found to be unreactive under the present conditions. Alkenyl N-sulfinimines yielded a complex mixture in which 1,2- and 1,4-additions were detected in the crude NMR.
- (a) C. Roe, T. M. Solá, L. Sasraku-Neequaye, H. Hobbs, I. Churcher, D. MacPherson and R. A. Stockman, Chem. Commun., 2011, 47, 7491 RSC; (b) C. Roe, H. Hobbs and R. A. Stockman, J. Org. Chem., 2011, 76, 9452 CrossRef CAS PubMed.
- J.-H. Seo, N.-S. Han, H.-S. Shim, J.-H. Kwon and J.-K. Song, Bull. Korean Chem. Soc., 2011, 32, 1415 CrossRef CAS.
- (a) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC ; For fundamental review, see: ; (b) D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803 RSC.
- Intermediate III (see Scheme 2) would be reduced to the corresponding anion followed by protonation to yield 4.
Footnotes |
| † Authors dedicate this work to Prof. Jose Luis García Ruano on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cc03724d |
| This journal is © The Royal Society of Chemistry 2017 |