
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conjunctive functionalization of vinyl boronate complexes with electrophiles: a diastereoselective three-component coupling†
Roly J.
Armstrong
 ,
Christopher
Sandford
,
Cristina
García-Ruiz
and
Varinder K.
Aggarwal
*
,
Christopher
Sandford
,
Cristina
García-Ruiz
and
Varinder K.
Aggarwal
*
School of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: v.aggarwal@bristol.ac.uk
First published on 20th April 2017
Abstract
A method for the conjunctive functionalization of vinyl boronate complexes with electrophiles is described. The overall process represents a three-component coupling between a vinyl boronic ester, carbon nucleophile and an electrophile, thus affording complex multifunctionalized products from simple starting materials. The diastereoselectivity (syn or anti) of this process is strongly dependent upon the nature of the electrophile.
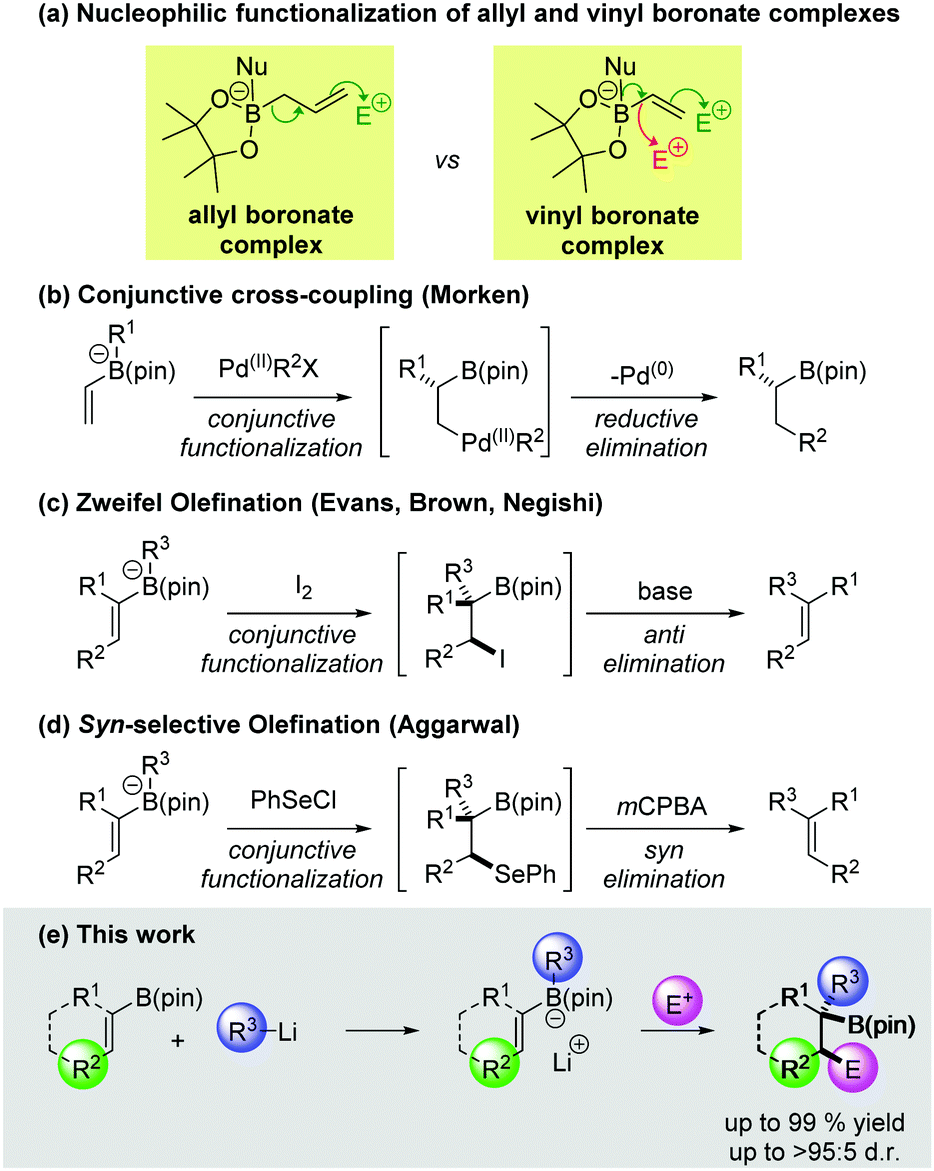
Boronic esters are versatile reagents which have been employed in a wide array of synthetic transformations.1 They are easy to handle, bench stable materials that can be readily activated by the addition of a suitable nucleophile to form a boronate complex.2 It is well known that the trivalent boron atom of allylic boronic esters can undergo activation with a Lewis base, leading to strongly nucleophilic character at the γ-position. Consequently, allylic boronic esters can be combined with a range of electrophiles, most notably in allylborations of aldehydes and imines (Scheme 1a).3 In contrast, the reactivity of boronate complexes derived from vinyl boronic esters has been less extensively explored. Upon treatment of a vinyl boronate complex with an electrophile two possible reaction pathways could be envisaged: (1) direct functionalization of an electron rich carbon–boron bond;4 (2) a conjunctive functionalization process, in which the π-system of the vinyl group reacts with the electrophile, triggering a 1,2-metallate rearrangement (Scheme 1a). The latter class of reaction is of particular interest since it represents a three-component coupling between a nucleophile, a vinyl boronic ester and an electrophile. Moreover, the boronic ester is retained in the product, serving as a versatile handle for further functionalization.1,5
 | ||
| Scheme 1 Previous work and strategy for three-component conjunctive coupling. | ||
Although the electrophilic functionalization of vinyl boronate complexes has not received a great deal of attention, several interesting examples have been reported.6–8 Recently, Morken and co-workers disclosed an elegant enantioselective conjunctive variant of the Suzuki–Miyaura reaction (Scheme 1b).9 Mechanistically, this process involves β-functionalization of a vinyl boronate complex by a palladium(II) aryl species, followed by reductive elimination. Another notable example of conjunctive functionalization is the Zweifel olefination (Scheme 1c).10 In this process, a vinyl boronate complex undergoes stereospecific reaction with iodine, resulting in the formation of a β-iodoboronic ester (likely via a transient iodonium ion). This intermediate is unstable, undergoing anti elimination in the presence of a weak base to afford olefin products. Recently we reported a conjunctive functionalization of vinyl boronate complexes with PhSeCl (Scheme 1d).11 The β-selenoboronic ester intermediates were not isolated, but were treated with mCPBA, resulting in syn elimination to form the corresponding alkenes. In this work, we aimed to develop a conjunctive three-component coupling process to obtain boron containing products, which would not undergo β-elimination. The scope with respect to each of the three coupling partners has been evaluated and the factors that influence the efficiency and diastereoselectivity of these reactions are discussed.
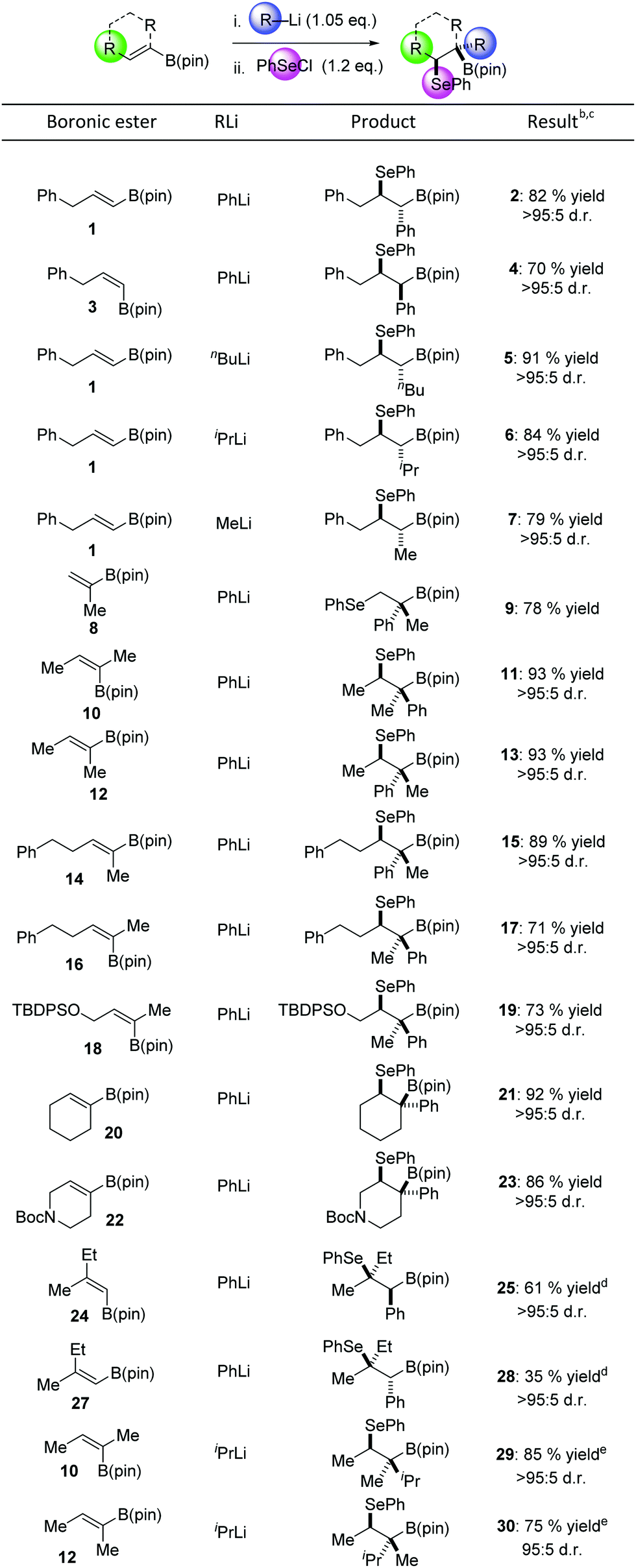
We commenced our study by evaluating the functionalization of E-disubstituted boronic ester 1. Addition of PhLi to a solution of 1 in THF at −78 °C resulted in clean boronate complex formation and upon addition of PhSeCl, we were pleased to find that the product of three-component coupling 2 could be isolated in 82% yield as a single diastereoisomer (Table 1). The anti diastereoselectivity of the reaction was confirmed by X-ray crystallographic analysis of the product. We then carried out an identical sequence beginning with Z-vinyl boronic ester 3. This provided the syn diastereoisomer 4 (>95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 d.r.) in 70% yield, indicating that the process is completely stereospecific. Primary and secondary organolithiums could also be employed in the process, affording alkyl-substituted products 5 and 6 in excellent yields and very high diastereoselectivity. MeLi also served as a successful nucleophile, affording 7 in 79% yield, which is particularly noteworthy given the known poor migratory aptitude of a methyl group.10c A vinyl boronic ester containing an α-substituent was also well tolerated and 9 was formed in 78% yield. When trisubstituted vinyl boronic ester 10 was used as the starting material, 11 was obtained in 93% yield (>95:5 d.r.). The other diastereoisomer could be obtained in 93% yield by employing the corresponding Z-vinyl boronate 12 as the starting material. Homobenzyl substituted boronic esters 14 and 16 also reacted successfully, affording products 15 and 17 in excellent yields and diastereoselectivity. A silyl-ether-containing substrate also reacted cleanly, affording 19 in 73% yield. We were pleased to find that cyclic boronic esters 20 and 22 also efficiently underwent the desired reaction. The latter example demonstrates that high chemoselectivity can be obtained in the presence of a Boc-protected amine.
:5 d.r.) in 70% yield, indicating that the process is completely stereospecific. Primary and secondary organolithiums could also be employed in the process, affording alkyl-substituted products 5 and 6 in excellent yields and very high diastereoselectivity. MeLi also served as a successful nucleophile, affording 7 in 79% yield, which is particularly noteworthy given the known poor migratory aptitude of a methyl group.10c A vinyl boronic ester containing an α-substituent was also well tolerated and 9 was formed in 78% yield. When trisubstituted vinyl boronic ester 10 was used as the starting material, 11 was obtained in 93% yield (>95:5 d.r.). The other diastereoisomer could be obtained in 93% yield by employing the corresponding Z-vinyl boronate 12 as the starting material. Homobenzyl substituted boronic esters 14 and 16 also reacted successfully, affording products 15 and 17 in excellent yields and diastereoselectivity. A silyl-ether-containing substrate also reacted cleanly, affording 19 in 73% yield. We were pleased to find that cyclic boronic esters 20 and 22 also efficiently underwent the desired reaction. The latter example demonstrates that high chemoselectivity can be obtained in the presence of a Boc-protected amine.
|
a Reaction conditions: vinyl boronic ester (1.0 eq.), RLi (1.05 eq.), THF; then PhSeCl (1.2 eq.), THF.
b Yields of isolated products.
c d.r. was determined by integration of crude 1H NMR spectra.
d Reaction with PhSeCl carried out in 3:1 THF/DMF.
e Reaction with PhSeCl carried out in 1:1 THF/CF3CH2OH.
|
|---|
|
We next attempted the coupling of β,β-disubstituted vinyl boronic ester 24. Disappointingly, we found that the desired product 25 was formed in a modest yield of 34% yield, along with 45% yield of vinyl selenide 26. A plausible mechanism accounting for the formation of 26 is outlined in Scheme 2. Initially, the vinyl boronate complex derived from 24 undergoes reaction with PhSeCl to form a zwitterionic seleniranium intermediate. This species can either undergo the desired 1,2-migration, or eliminate to form vinyl selenide 26 along with PhB(pin).12 For the majority of examples, the former pathway predominates, but in the case of boronate complexes derived from β,β-disubstituted boronic esters, the ability to build up partial positive charge at the tertiary β-carbon allows the Cβ–Se bond to lengthen and facilitates the elimination process. We found that the reaction solvent has a significant influence on which pathway is preferred (see ESI† for full details of optimization). Ultimately, we found that in a 3:1 THF/DMF mixture, formation of 26 was suppressed and 25 could be obtained as a single diastereomer in 61% yield. These conditions were also applied to the three-component coupling of isomeric vinyl boronic ester 27 affording the product 28 in 35% yield as a single diastereomer (Table 1).
 | ||
| Scheme 2 Competing pathways in the formation of 25 and the effect of solvent. | ||
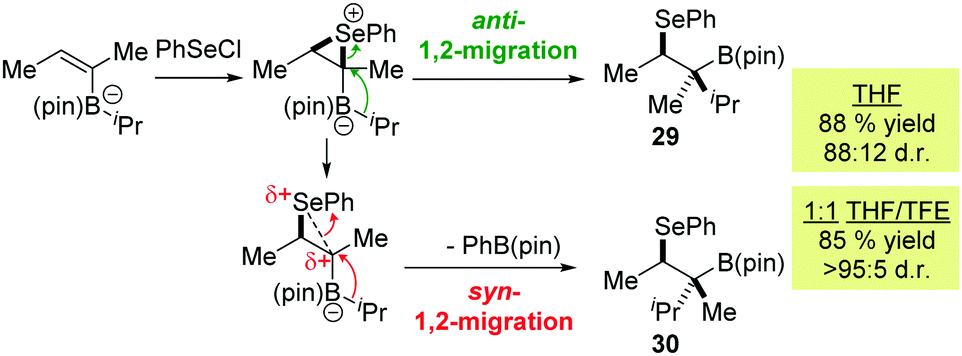
We next carried out a three-component coupling between 10, iPrLi and PhSeCl. Given that this vinyl boronic ester had been reacted with an aryl lithium to afford 11 in very high diastereoselectivity (vide supra) we did not anticipate that this process would be problematic. We were therefore surprised to find that 29 was isolated as an 88:12 mixture of diastereoisomers (Scheme 3). We attribute this reduced diastereoselectivity to the pathway shown in Scheme 3, whereby partial opening of the seleniranium intermediate enables a partial positive charge to develop at the tertiary α-carbon. This allows a syn 1,2-migration process to compete with the usual anti migration, resulting in the formation of both 29 and 30. After solvent optimization (see ESI† for full details) we found that carrying out the reaction in a 1:1 mixture of THF/trifluoroethanol suppressed the undesired pathway, providing 29 in very high diastereoselectivity. We suspect that trifluoroethanol is able to modulate the reactivity of the intermediate seleniranium cation by functioning as a hydrogen bond donor to the basic pinacol oxygen atoms.13 An analogous reaction with isomeric vinyl boronic ester 12 afforded the product 30 in 75% yield and 95:5 d.r.
 | ||
| Scheme 3 Eroded diastereoselectivity in the formation of 29. TFE = CF3CH2OH. | ||
Taking boronate complexes derived from isomeric vinyl boronic esters 10 and 12 as representative examples, coupling with a series of electrophiles was investigated (Table 2; results with PhSeCl are also shown for clarity). With PhSCl the reaction was extremely efficient, producing 31 and 32 in excellent yield. Both products were obtained as a single diastereoisomer indicating that the process is stereospecific. To confirm the relative configuration of 31 and 32, we subjected them to methoxide promoted anti elimination. As expected, this resulted in the formation of isomeric alkenes 39 and 40, respectively (Table 3, entries 3 and 4). We next investigated fluorination of vinyl boronate complexes.2d Employing Selectfluor, β-fluoroboronic esters 33 and 34 were obtained in good yield and high diastereoselectivity (Table 2). Upon exposure of these compounds to sodium methoxide, we were surprised to isolate the unexpected isomers of alkenes 40 and 39 (Table 3, entries 5 and 6). There are two possible explanations for this observation: (1) β-fluoroboronic esters 33 and 34 are formed by a selective syn migration process; (2) elimination proceeds via a syn pathway. To differentiate between these two scenarios, we carried out fluorination of a cyclic boronate complex, obtaining 41 as a single diastereoisomer (Scheme 4). HOESY experiments revealed that an unusual syn migration process had occurred. By analogy, we propose that a similar syn pathway is involved in the formation of 33 and 34. This surprising result suggests that anti diastereoselectivity is only observed in cases where vinyl boronate complexes are activated by electrophiles that form closed three-membered ring intermediates. In line with this hypothesis, electrophilic carbon reagents, such as tropylium tetrafluoroborate and 1,3-benzodithiolylium tetrafluoroborate led to competing syn and anti functionalization, resulting in low levels of diastereoselectivity (Table 2). Finally, we investigated the reaction of vinyl boronate complexes with Umemoto's reagent, a process that is expected to occur by addition of a trifluoromethyl radical followed by single-electron oxidation to form a carbocation intermediate.14 As expected, near identical diastereomeric mixtures of trifluoromethylated products were obtained regardless of which isomer of vinyl boronic ester was employed.
| a Reaction conditions: vinyl boronic ester (1.0 eq.), PhLi (1.05 eq.); then electrophile (1.2 eq.). For details of solvent and reaction conditions see ESI. b Yields of isolated products. c d.r. was determined by integration of crude 1H and 19F NMR spectra. d 19F NMR yield using α,α,α-trifluorotoluene as an internal standard. |
|---|

|
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | d.r. | Major product | E/Z selectivityb |
|
a Reaction conditions: β-substituted boronic ester (1.0 eq.), NaOMe (10 eq.), THF/MeOH (8:1).
b Determined by integration of the crude 1H NMR spectrum.
|
||||
| 1 | 11 (E = SePh) | >95:5 |
39 | >95:5 |
| 2 | 13 (E = SePh) | >95:5 |
40 | <5:95 |
| 3 | 31 (E = SPh) | >95:5 |
39 | 96:4 |
| 4 | 32 (E = SPh) | >95:5 |
40 | 29:71 |
| 5 | 33 (E = F) | 89:11 |
40 | 17:83 |
| 6 | 34 (E = F) | 91:9 |
39 | 92:8 |
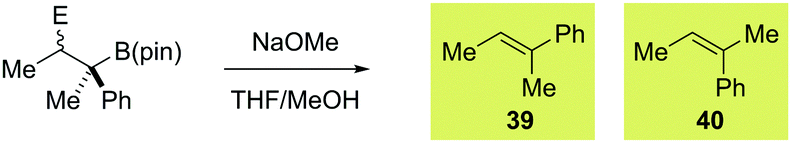
 | ||
| Scheme 4 Probing the diastereoselectivity of fluorination. Reaction conditions: vinyl boronic ester (1.0 eq.), PhLi (1.05 eq.), THF; then Selectfluor I (1.2 eq.), MeCN/THF (5:1). a 19F NMR yield using α,α,α-trifluorotoluene as an internal standard. | ||
In summary, a conjunctive three-component coupling between vinyl boronic esters, carbon nucleophiles and electrophiles has been developed. The diastereoselectivity of the process is strongly dependent upon the nature of the electrophile. Reactions that proceed via closed three-membered cyclic intermediates exhibit very high anti diastereoselectivity. In the case of more reactive, charged electrophiles, the increasingly asynchronous nature of the bond formation enables syn migration to compete, in some cases becoming the preferred pathway. This work will be of use for the synthesis of boron-containing materials and will inform the development of other conjunctive functionalization processes.
We thank EPSRC (EP/I038071/1) and Bristol University for financial support. C. G.-R. thanks the Ramón Areces Foundation for a postdoctoral fellowship. We thank Dr Eddie Myers for helpful discussions, Dr Hazel A. Sparkes and Dr Natalie E. Pridmore for assistance with X-ray crystallography and Dr E. Kriemen for carrying out preliminary studies.
Notes and references
- Synthesis and application of organoboron compounds, ed. E. Fernández and A. Whiting, Springer, Heidelberg, 2015 Search PubMed.
- (a) H. C. Brown, N. R. De Lue, G. W. Kabalka and H. C. Hedgecock, J. Am. Chem. Soc., 1976, 98, 1290–1291 CrossRef CAS; (b) H. C. Brown and B. Singaram, Acc. Chem. Res., 1988, 21, 287–293 CrossRef CAS; (c) R. Larouche-Gauthier, T. G. Elford and V. K. Aggarwal, J. Am. Chem. Soc., 2011, 133, 16794–16797 CrossRef CAS PubMed; (d) C. Sandford, R. Rasappan and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10100–10103 CrossRef CAS PubMed.
- (a) Organic Reactions: Allylboration of Carbonyl Compounds, ed. H. Lachance and D. G. Hall, Wiley, Weinheim, 2009, vol. 73 Search PubMed; (b) J. W. J. Kennedy and D. G. Hall, Angew. Chem., Int. Ed., 2003, 42, 4732–4739 CrossRef CAS PubMed; (c) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763–2794 CrossRef CAS PubMed.
- (a) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169–6193 CrossRef CAS PubMed; (b) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (c) H. C. Brown, T. Hamaoka and N. Ravindran, J. Am. Chem. Soc., 1973, 95, 5786–5788 CrossRef CAS; (d) S. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 15438–15439 CrossRef CAS PubMed; (e) A. Mitchell and J. W. Bode, J. Am. Chem. Soc., 2009, 131, 18057–18059 CrossRef PubMed.
- (a) H. K. Scott and V. K. Aggarwal, Chem. – Eur. J., 2011, 17, 13124–13132 CrossRef CAS PubMed; (b) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed; (c) C.-Y. Wang, J. Derosa and M. R. Biscoe, Chem. Sci., 2015, 6, 5105–5113 RSC; (d) C. Sandford and V. K. Aggarwal, Chem. Commun., 2017, 53 10.1039/C7CC01254C.
- For selected examples with alkynyl boronate complexes, see: (a) M. Naruse, K. Utimoto and H. Nozaki, Tetrahedron Lett., 1973, 14, 2741–2744 CrossRef; (b) N. Miyaura, T. Yoshinari, M. Itoh and A. Suzuki, Tetrahedron Lett., 1974, 15, 2961–2964 CrossRef; (c) K. K. Wang and S. Dhumrongvaraporn, Tetrahedron Lett., 1987, 28, 1007–1010 CrossRef CAS; (d) N. Ishida, W. Ikemoto, M. Narumi and M. Murakami, Org. Lett., 2011, 13, 3008–3011 CrossRef CAS PubMed.
- For selected examples with aromatic boronate complexes, see: (a) G. M. Davies, P. S. Davies, W. E. Paget and J. M. Wardleworth, Tetrahedron Lett., 1976, 17, 795–798 CrossRef; (b) I. Akimoto and A. Suzuki, Synthesis, 1979, 146–147 CrossRef CAS; (c) M. Ishikura and H. Kato, Tetrahedron, 2002, 58, 9827–9838 CrossRef CAS; (d) A. Bonet, M. Odachowski, D. Leonori, S. Essafi and V. K. Aggarwal, Nat. Chem., 2014, 6, 584–589 CrossRef CAS PubMed; (e) M. Odachowski, A. Bonet, S. Essafi, P. Conti-Ramsden, J. N. Harvey, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2016, 138, 9521–9532 CrossRef CAS PubMed.
- For conjunctive intramolecular alkylation, see: Y. Kobayashi, M. Asano and Y. Kiyotsuka, Heterocycles, 2009, 77, 787–791 CrossRef CAS.
- (a) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef CAS PubMed; (b) G. J. Lovinger, M. D. Aparece and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 3153–3160 CrossRef CAS PubMed.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652–3653 CrossRef CAS; (b) D. A. Evans, T. C. Crawford, R. C. Thomas and J. A. Walker, J. Org. Chem., 1976, 41, 3947–3953 CrossRef CAS PubMed; (c) H. C. Brown and N. G. Bhat, J. Org. Chem., 1988, 53, 6009–6013 CrossRef CAS; (d) S. Xu, C.-T. Lee, H. Rao and E. Negishi, Adv. Synth. Catal., 2011, 353, 2981–2987 CrossRef CAS PubMed.
- R. J. Armstrong, C. García-Ruiz, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 786–790 CrossRef CAS PubMed.
- In analogy with related reactions of vinyl silanes, this process proceeds with stereochemical retention: T. A. Blumenkopf and L. E. Overman, Chem. Rev., 1986, 86, 857–873 CrossRef CAS.
- I. Shuklov, N. Dubrovina and A. Börner, Synthesis, 2007, 2925–2943 CrossRef CAS.
- (a) Y. Wang, A. Noble, C. Sandford and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 1810–1814 CrossRef CAS PubMed; (b) M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Science, 2017, 355, 936–938 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1534237. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc01671a |
| This journal is © The Royal Society of Chemistry 2017 |