
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Luciferase–Rose Bengal conjugates for singlet oxygen generation by bioluminescence resonance energy transfer†
Seonghoon
Kim
 ab,
HyeongChan
Jo
b,
Mijeong
Jeon
b,
Myung-Gyu
Choi
c,
Sei Kwang
Hahn
d and
Seok-Hyun
Yun
*ab
ab,
HyeongChan
Jo
b,
Mijeong
Jeon
b,
Myung-Gyu
Choi
c,
Sei Kwang
Hahn
d and
Seok-Hyun
Yun
*ab
aHarvard Medical School and Wellman Center for Photomedicine, Massachusetts General Hospital, USA. E-mail: syun@hms.harvard.edu
bGraduate School of Nanoscience and Technology, Korea Advanced Institute of Science and Technology, Korea
cDivision of Gastroenterology, Dept. of Internal Medicine, Seoul St. Mary's Hospital, College of Medicine, Catholic University, Korea
dDepartment of Materials Science and Engineering, Pohang University of Science and Technology, Korea
First published on 28th March 2017
Abstract
Conjugates of Rose Bengal and Renilla luciferase generated singlet oxygen upon binding with coelenterazine via bioluminescence resonance energy transfer (BRET). Since the applications of conventional PDT have been limited to superficial lesions due to the limited light penetration in tissue, BRET activated PDT which does not require external light illumination may overcome the limitations of conventional PDT.
Photodynamic therapy (PDT) is used clinically to treat dermatologic lesions, retinal diseases, and epithelial tumors.1,2 PDT employs photosensitizer (PS) molecules and uses light to activate drugs to generate reactive oxygen species (ROS), such as singlet oxygen, and free radicals. These photochemical products can kill target cells and destruct tissues. This toxicity mechanism is different from the cytotoxicity mechanisms of chemotherapy, radiation therapy and immunotherapy. The difference makes PDT an attractive option for stand-alone or combinatorial therapy. Another distinct aspect of PDT comes from the light-induced activation of photosensitizers. This provides an advantage of spatial and temporal controllability of drug activation. However, the need for light also limits the applications of PDT, because of the light's shallow penetration depth in tissues. To date, clinical PDT has been adopted to treat diseases in the skin and retina, which physicians can readily approach with a light source, or in epithelial layers of endoscopically accessible sites such as gastrointestinal tracts. To enhance the therapeutic depth of PDT, considerable efforts have been made in developing PS3 molecules with action spectra in the near-infrared (NIR) range and up-conversion nanoparticles4 that absorb NIR photons and deliver energy to conventional PS drugs. Yet, the limited optical penetration depth (<5 mm) even in the NIR range leaves many diseases out of reach.
To solve this problem, researchers have sought new methods capable of remotely activating PS agents in deep tissues. One such approach is to use Cherenkov radiation produced by beta particles during radioactive decay.5 However, the potential toxicity of radioactive isotopes and inorganic photosensitive nanoparticles, such as TiO2, needs to be addressed for clinical translation.6 Another approach is to use Förster resonance energy transfer from chemiluminescent or bioluminescent molecules. Yuan et al. demonstrated antimicrobial PDT by employing luminol and electrostatically-bound cationic oligo(p-phenylene vinylene).7 Hsu et al. demonstrated cancer therapy by using self-illuminating quantum dots conjugated with mutant Renilla luciferases.8 Kim et al. extended this approach and demonstrated local therapy of cancer cells in draining lymph nodes in mice.9 While these experiments support the feasibility of remotely activated PDT, the long-term toxicity of luminol and quantum dots raise concern about their potential for clinical translation. Furthermore, because donors (luciferases) and acceptors (PSs) are administered separately, bioluminescence resonance energy transfer (BRET) occurs only when they are located close to each other within 5–10 nm. Chemical conjugation of bioluminescent enzymes and PS drugs could solve this problem, but such BRET pairs have not been demonstrated.
Here, we report the conjugation of a luciferase and a PS, for the first time to our knowledge. In this work, we have used mutant Renilla luciferases 8.6 (RLuc8.6)10 and Rose Bengal (RB) dyes to form BRET pairs (Fig. S1, ESI†), where the emission peak of RLuc8.6 at 535 nm is well matched with an RB's absorption peak at 550 nm. RB is an efficient PS with a high quantum efficiency of 0.7–0.8 (measured in aqueous media) in the generation of singlet oxygen.11 We have investigated the efficiency of singlet oxygen generation and the ROS-induced cytotoxicity of the luciferase–RB conjugates for BRET-induced PDT in direct comparison with laser-induced activation used in conventional PDT.
Our initial scheme for conjugating RLuc8.6 and RB via a short linker retained bioluminescence (BL) capability but failed to achieve efficient BRET to RB (Fig. S2, ESI†) presumably due to the quenching of the RB. To solve this problem, we used bovine serum albumin (BSA) as a central piece to which RLuc8.6 and RB were conjugated. The rationales for this design were to provide space between RB and RLuc8.6 to evade quenching and enhance the BRET efficiency by attaching multiple RLuc8.6 molecules in each complex. When the mixing ratio of RB to BSA was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the highest FL intensity was measured from RB–BSA conjugates (Fig. S3a, ESI†). The actual conjugation ratio was estimated to be 2.2:1 by comparing the absorbance of purified RB–BSA conjugates to the absorbance of unpurified simple mixtures of RB–NHS and BSA (Fig. S3b, ESI†). RLuc8.6 was linked to the RB–BSA conjugate by Cu-free click reaction to form an RB–BSA–PEG4–RLuc8.6 conjugate, hereinafter called LucRB (Scheme 1). A polyacrylamide gel electrophoresis of LucRB purified with a 100 kDa filter showed bands near 140, 180, 210 and 250 kDa, respectively, corresponding to 2, 3, 4 and 5 RLuc8.6 molecules per construct (Fig. S3c, ESI†). The LucRB conjugates with heterogeneous molecular weights were used in the experiments without further purification. The hydrodynamic size and zeta potential of LucRB were measured to be 11 nm and −6.6 mV, respectively.
:1, the highest FL intensity was measured from RB–BSA conjugates (Fig. S3a, ESI†). The actual conjugation ratio was estimated to be 2.2:1 by comparing the absorbance of purified RB–BSA conjugates to the absorbance of unpurified simple mixtures of RB–NHS and BSA (Fig. S3b, ESI†). RLuc8.6 was linked to the RB–BSA conjugate by Cu-free click reaction to form an RB–BSA–PEG4–RLuc8.6 conjugate, hereinafter called LucRB (Scheme 1). A polyacrylamide gel electrophoresis of LucRB purified with a 100 kDa filter showed bands near 140, 180, 210 and 250 kDa, respectively, corresponding to 2, 3, 4 and 5 RLuc8.6 molecules per construct (Fig. S3c, ESI†). The LucRB conjugates with heterogeneous molecular weights were used in the experiments without further purification. The hydrodynamic size and zeta potential of LucRB were measured to be 11 nm and −6.6 mV, respectively.
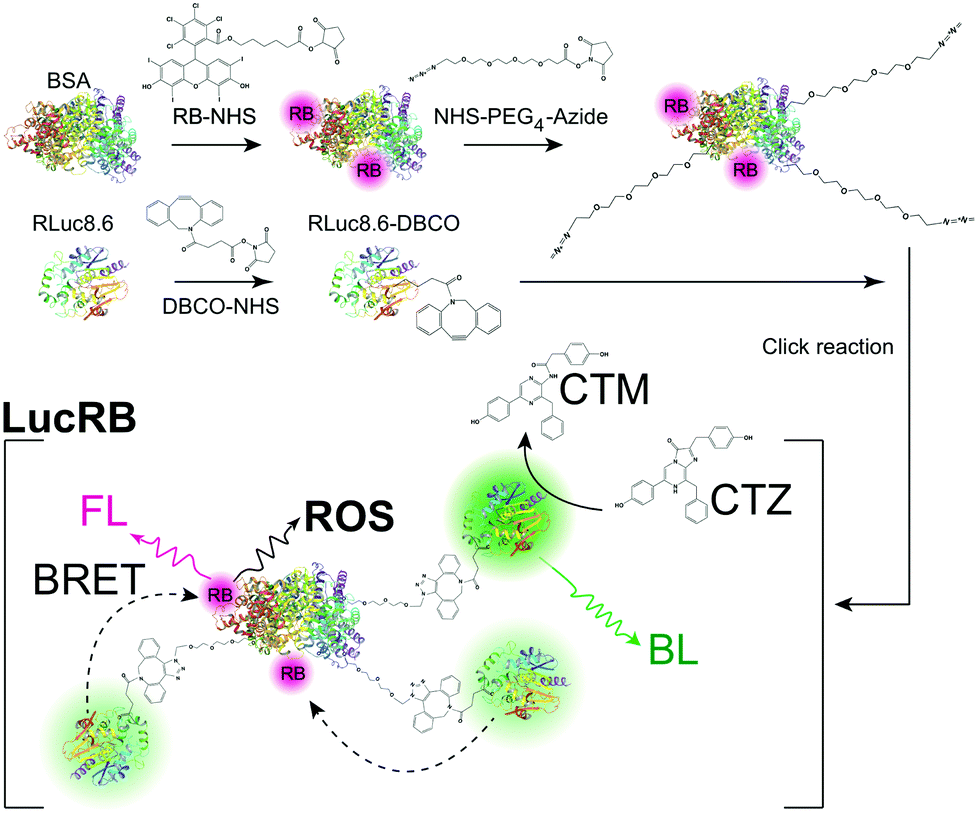 | ||
| Scheme 1 Schematics of the synthesis of a LucRB conjugate and its action for the generation of reactive oxygen species (ROS) via bioluminescence-resonance energy transfer (BRET). RB, Rose Bengal; BSA, bovine serum albumin; CTZ, coelenterazine; CTM, coelecteramide; BL, bioluminescence; FL, fluorescence. | ||
While RLuc8.6 is colorless, LucRB solution has a pink color owing to RB (Fig. 1a). When CTZ was administered, LucRB produced significantly lower BL emission intensity compared to RLuc8.6 solution. This is because of the BRET to RB, and RB has a low fluorescence quantum yield (QY) of ∼5% compared to RLuc8.6's high BL QY of ∼50% (Fig. 1a). To confirm the enzymatic activity of RLuc8.6 in LucRB, we measured the time-lapse curves of BL intensity using a large-area detector (Fig. 1b, inset). We found that the total BL energy integrated over the entire emission time was linearly proportional to the amount of CTZ (up to 10 μg) but independent of sample concentration. The exponential decay rates of BL emission from free RLuc8.6 measured as a function of concentration followed the Michaelis-Mentens kinetic model of the substrate–enzyme reaction (Fig. 1b). The BL decay rate of LucRB at a concentration of 0.4 μM was equivalent to the BL decay rate of RLuc8.6 solution at a concentration of 1.51 μM. This means that the average number of RLuc8.6 in a single LucRB molecule was about 3.8.
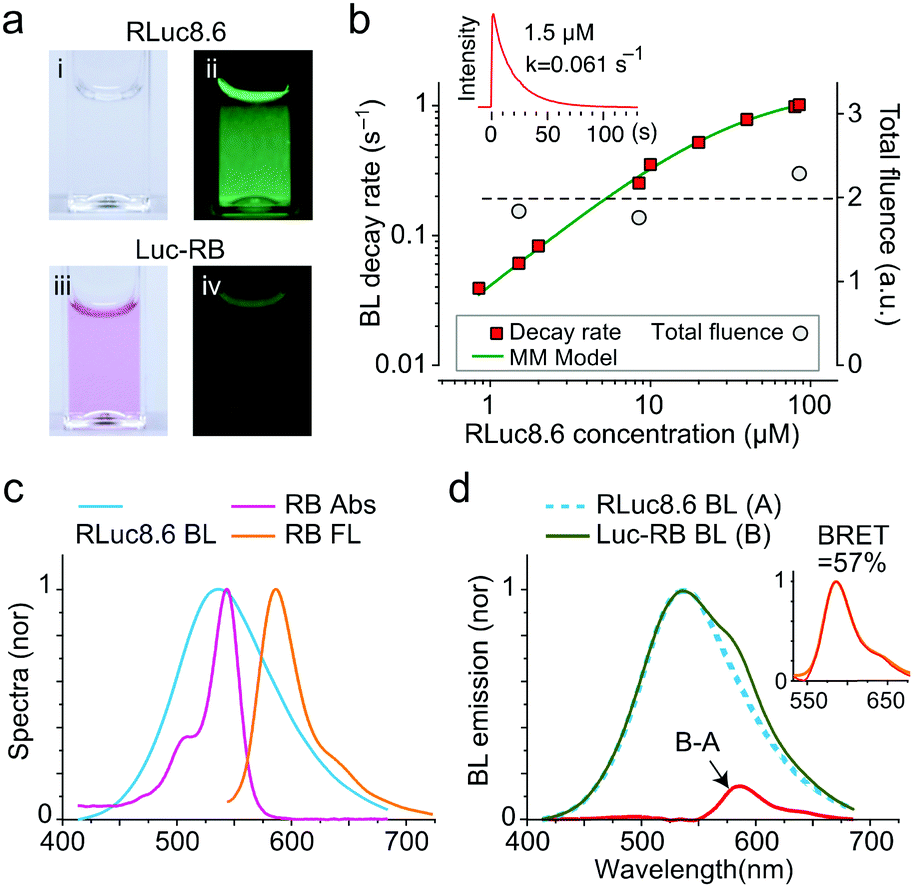 | ||
| Fig. 1 Optical properties of LucRB. (a) Photographs of RLuc8.6 (i, ii) and LucRB (iii, iv) solutions in cuvettes. Left (i, iii): under room light, right (ii, iv): BL in the dark. For 10 μg of CTZ each, the total BL energy emitted from RLuc8.6 (1.5 μM) and LucRB solutions (0.4 μM; same number of RLuc8.6) was estimated to be 50 μJ and 5.8 μJ, respectively. (b) BL emission decay rates and total fluence of LucRB and RLuc8.6 as a function of concentration in PBS buffer (pH 7.4). (c) Normalized absorption (magenta), FL (orange) and BL (cyan) spectra of RB and RLuc8.6. (d) Normalized BL of LucRB (B) and Luc8.6 (A), and the difference (B–A). The inset shows that the difference (magenta dotted line) coincides with the FL spectrum (orange) of LucRB. | ||
RLuc8.6 has a broad BL spectrum that overlaps well with the absorption spectrum of RB (Fig. 1c). By comparison, the BL spectrum of LucRB showed an additional shoulder around 580 nm (Fig. 1d). The difference of the LucRB and RLuc8.6 spectra corresponded to the FL emission spectrum of RB (Fig. 1c and d). The BRET ratio, defined as the FL emission energy from the acceptor (RB) to the BL emission energy of the donor (RLuc8.6), is measured to be 7.5%. The relative QYs of free RB (0.05) and LucRB (0.02) were estimated by comparison with the FL intensity of Rhodamine 6G (QY, 0.95) upon 532 nm excitation in PBS solution. We estimated energy transfer efficiency by comparison with the total BL energy from RLuc8.6 (50 μJ) and transferred energy to RB (23.2 μJ) calculated by dividing RB fluorescence energy (B–A, 0.44 μJ) of LucRB by the LucRB's QY. The calculated BRET efficiency is 46.4% (= 23.2/50 μJ). In summary, 46.4% of the total optical energy is transferred to RB, and 2% of the transferred energy is emitted as FL, while the rest contributes to the generation of singlet oxygen and ROS.
To quantify singlet oxygens generated by the excitation of LucRB, we used a Singlet Oxygen Sensor Green (SOSG) dye whose FL emission is increased by singlet oxygen.12,13 We found that the FL intensity of SOSG was affected when directly exposed to CTZ, but this interference was suppressed by adding BSA proteins to SOSG-containing solution before adding CTZ and LucRB. The premixing with BSA stabilized SOSG so that the FL output was invariant by CTZ up to 100 μg in 2 ml (118 μM) in the absence of LucRB (Fig. 2a, control). By contrast, in the presence of 2 nmol (1 μM) of LucRB, the FL intensity of SOSG increased linearly with increase in the amount of CTZ up to 50 μg (60 μM) until it saturated fully above 65 μg (77 μM) (Fig. 2a). The saturation of SOSG signals at high CTZ concentrations may be in part due to the degradation of LucRB molecules by the generated ROS through repeated enzymatic actions with the substrates. Laser-excited LucRB (532 nm, 1 mW cm−2) without CTZ exhibited a similar linear growth trend as a function of irradiation time (Fig. 2b). This result indicates that for LucRB (1 μM) CTZ (50 μg) can generate as much singlet oxygen as the laser excitation at a fluence of ∼360 mJ cm−2 (6 min at 1 mW cm−2).
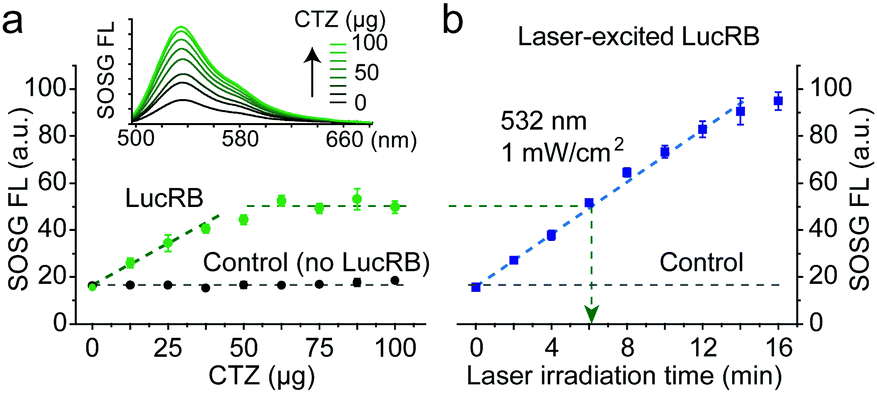 | ||
| Fig. 2 Measurement of ROS generation. (a) FL signal of SOSG increased linearly when CTZ up to 50 μg in 2 ml (47 μM) was added to LucRB (1 μM) solution until it saturated beyond 50 μg, whereas the SOSG signal was invariant in the absence of LucRB (control). (b) Comparison to laser-induced excitation. The SOSG signal from LucRB that was excited by laser light (532 nm, 1 mW cm−2) increased linearly with increasing irradiation time. An optical energy of ∼360 mJ (green arrow) produced an equivalent amount of ROS to the maximum produced by 50 μg of CTZ in (a). | ||
To test LucRB in vitro, we used CT26 cells (murine colon carcinoma cell line) in monolayer culture. Unfortunately, bare LucRB conjugates have a low binding affinity to the cell membrane and thus low internalization efficiency into the cytoplasm. This resulted in nearly no cytotoxicity in both BRET- and laser-excited PDT (532 nm, 1 mW cm−2). To solve this problem, we coupled cell-penetrating peptides (CPP) to LucRB non-covalently. The CPP coupling considerably enhanced the delivery efficiency of LucRB into cells. To quantify the intracellular concentration of LucRB, we measured optical absorption at 550 nm through monolayer cells incubated with 100 μM of CPP-coupled LucRB solution. The measured absorption value (1.6%) was the same as the absorption by monolayer cells incubated with bare-RB solution at a concentration of 12 μM (Fig. 3a). Furthermore, under a laser-excited confocal fluorescence microscope, the intensity of intracellular RB fluorescence from these cells was equivalent to the fluorescence intensity from a bare-RB solution at a concentration of 2.4 μM. These data indicate that the intracellular concentration of RB is ∼5 times lower than the extracellular concentration in the cell medium and that, considering that each LucRB contains average 2.2 RB molecules, the intracellular delivery efficiency of LucRB was 1.1%.
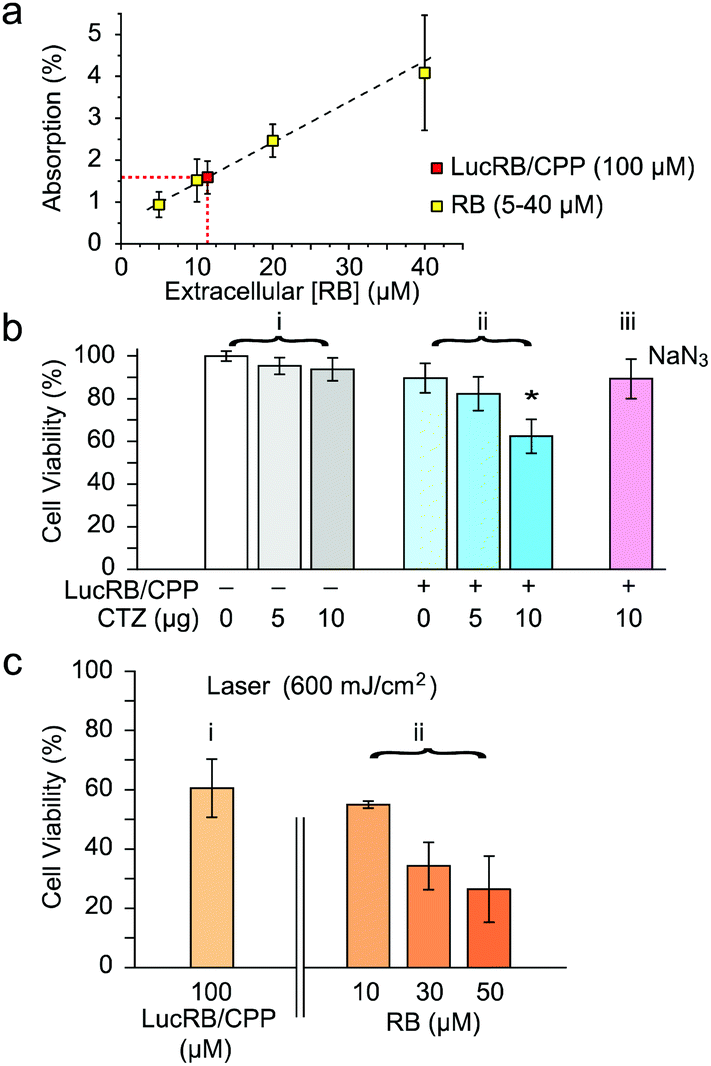 | ||
| Fig. 3 In vitro cytotoxicity assay. (a) Estimated intracellular concentration of LucRB from light absorption by RB ([int. RB]/2.2 = [LucRB], LucRB = ∼5.5 μM). (b) MTT assay of BRET-PDT (LucRB: 100 μM, CTZ: 118 μM (10 μg/200 μl), NaN3: 1 mM). (c) MTT assay of conventional laser-induced PDT. | ||
MTT assays showed no significant cytotoxicity of CTZ at concentrations up to 10 μg in 100 μl of culture medium (232 μM) (Fig. 3b(i)). For CT26 cells incubated at 100 μM LucRB, administration of 10 μg CTZ resulted in cell death in 40% of the cell population (Fig. 3b(ii)). For cells incubated with a ROS scavenger, NaN3, prior to CTZ administration, cell death was negligible (Fig. 3b(iii)), which confirms that the cytotoxicity mechanism is mediated by BRET-induced ROS. For the same LucRB concentration (100 μM) we performed laser-excited PDT at an optical fluence of 600 mJ cm−2 (532 nm, 10 mW cm−2, 1 min) and achieved a similar level (40%) of cell death (Fig. 3c(i)). Laser-induced PDT was performed on cells incubated with bare RB molecules at varying concentrations. The cell death ratio by laser-excited RB at 10 μM (equivalent intracellular RB concentration to 100 μM LucRB) was about 40% (Fig. 3c(ii)), again the same as above. Therefore, these results further support that the cytotoxicity mechanism of LucRB is due to BRET-induced ROS generation. With higher RB concentrations of 30 and 50 μM, the cell death ratio increased to 65 and 75%, respectively. This result is encouraging as it indicates that higher intracellular concentrations of LucRB might produce stronger cytotoxicity.
We performed confocal fluorescence imaging of cells using 2′,7′-dichlorofluorescein diacetate (DCFDA), an ROS indicator. CT26 cells incubated with LucRB show red FL from RB but very low green FL from DCF, indicating low intracellular ROS levels (Fig. 4a). As a positive control, cells incubated with hydrogen peroxide showed strong green FL from oxidized DCF (Fig. 4b). As another positive control, cells incubated with bare RB (10 μM) and treated with laser light (532 nm, 10 mW cm−2, 1 min) generated both red and green FL, confirming ROS generation (Fig. 4c). Cells incubated with LucRB and treated with either laser illumination (10 mW cm−2, 1 min) or CTZ administration (10 μg) exhibited green FL from DCF in 50–70% of the cells (Fig. 4d and e). To quantify DCF FL, flow cytometry was performed subsequently (Fig. 4f–i). Cells incubated with CTZ alone (Fig. 4g) or LucRB prior to CTZ (Fig. 4i) produced slightly higher FL compared to cells without DCFDA (Fig. 4f), but much lower than positive controls incubated with hydrogen peroxide (Fig. 4h). Cells treated with LucRB and CTZ showed a significant increase of DCF signals proportional to the amount of CTZ (5 and 10 μg) (Fig. 4i).
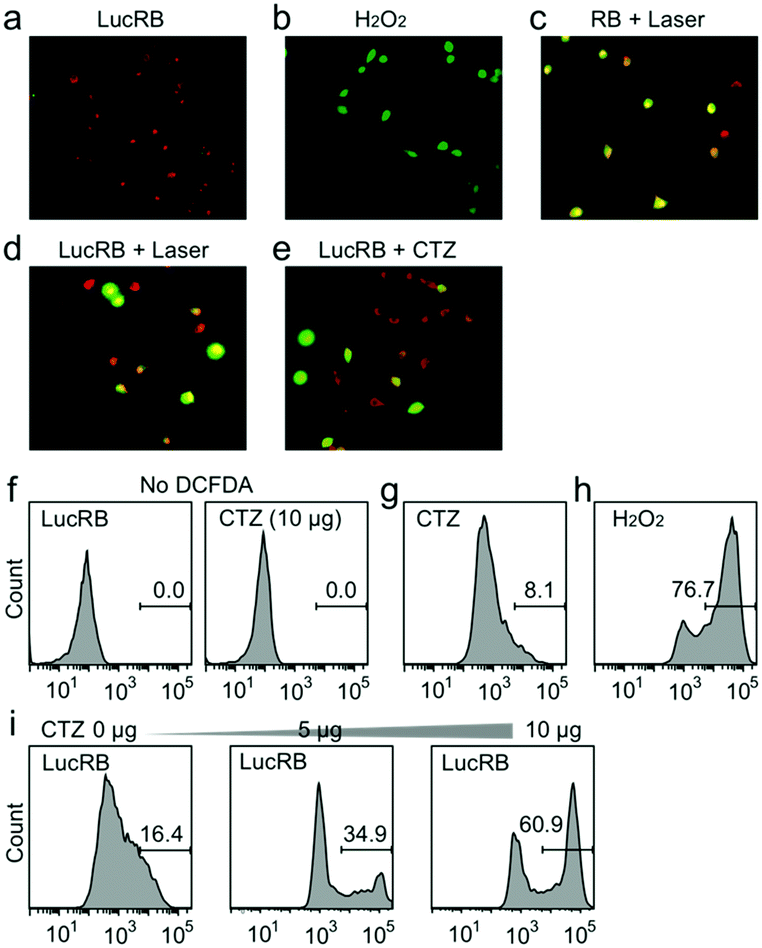 | ||
| Fig. 4 Measurement of oxidative stress in CT26 cells. (a–e) Intracellular ROS generation by BRET-PDT (red: RB or LucRB, green: DCFDA). (f–i) Flow cytometry analysis of DCFDA FL. | ||
The optimization of the conjugation ratio and inter-molecular separation resulted in nearly 50% efficiency in energy transfer from the bioluminescent enzyme–substrate complex to the conjugated PS. The total amount of BL energy produced from 10 μg of CTZ (that is, 1.4 × 1016 molecules) and an excessive amount of free RLuc8 was measured to be ∼50 μJ. 10 μg of CTZ in 100 μl of solution in the cuvettes (Fig. 3) and cytoplasm (Fig. 4) produced the same amount of ROS as that produced from LucRB solution after laser excitation at a dose of ∼600 mJ (10 mW for 1 min). The difference between 600 mJ and 50 μJ may be interpreted as the higher “photon” efficiency of the near-field BRET-induced activation compared to the far-field laser excitation. Given that RB has an extinction coefficient of 90000 M−1 cm−1 at 550 nm and an absorption cross-section of 2.4 × 10−16 cm2, a monolayer of cells (5 μm thick) with 10 μM RB in the cytoplasm would absorb only 0.1% of the topically illuminated light. This accounts for a three orders of magnitude difference in efficiency compared to BRET-induced activation where most of the virtual-photon energy (∼50%) is transferred to RB.
The mechanism of PDT based on ROS generation from LucRB is distinctively different from chemotherapy and radiation therapy. No cross-resistance between BRET-induced PDT and chemotherapy has been known.14 Conventional PDT has been shown to be effective against radio-resistant and chemo-resistant cells15,16 and, also, may sensitize resistant cells to chemotherapy.17 Therefore, BRET-PDT has potential for combination therapy with chemo- and radio-therapy. Unlike conventional chemo agents, LucRB agents are non-toxic on their own until activated by the administration of CTZ. This temporal switch allows BRET PDT to be performed at optimal timing when the most preferred bio-distribution of the drug and maximal therapeutic outcome can be achieved.
The relatively low intracellular uptake efficiency of LucRB limited the amount of cytotoxicity. Other known delivery methods, such as liposomal delivery,18 may increase the intracellular uptake efficiency and thus improve the therapeutic potential. Conjugation of LucRB to antibodies against specific cell-surface biomarkers may enhance targeting and cytotoxicity to tumor cells.3 Finally, other possible combinations of different types of luciferases, photosensitizers, and substrates appear to have potential and are worth investigation for BRET-PDT.
We thank Jae-Myung Park, Yirang Kim, and Tayyaba Hasan for discussion. This work was funded by the U.S. National Institutes of Health (R01CA192878), the National Research Foundation of Korea (NRF-2011-0031644, R31-2008-000-10071-0), and the Human Frontier Science Program (RGP0034/2016).
References
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS PubMed.
- L. Cheng, K. Yang, Y. Li, J. Chen, C. Wang, M. Shao, S. T. Lee and Z. Liu, Angew. Chem., Int. Ed., 2011, 123, 7523–7528 CrossRef.
- N. Kotagiri, G. P. Sudlow, W. J. Akers and S. Achilefu, Nat. Nanotechnol., 2015, 10, 370–379 CrossRef CAS PubMed.
- N. R. Younes, S. Amara, I. Mrad, I. Ben-Slama, M. Jeljeli, K. Omri, J. El Ghoul, L. El Mir, K. B. Rhouma, H. Abdelmelek and M. Sakly, Environ. Sci. Pollut. Res. Int., 2015, 22, 8728–8737 CrossRef PubMed.
- H. Yuan, H. Bai, L. Liu, F. Lv and S. Wang, Chem. Commun., 2015, 51, 722–724 RSC.
- C. Y. Hsu, C. W. Chen, H. P. Yu, Y. F. Lin and P. S. Lai, Biomaterials, 2013, 34, 1204–1212 CrossRef CAS PubMed.
- Y. R. Kim, S. Kim, J. W. Choi, S. Y. Choi, S. H. Lee, H. Kim, S. K. Hahn, G. Y. Koh and S. H. Yun, Theranostics, 2015, 5, 805–817 CrossRef CAS PubMed.
- A. M. Loening, A. M. Wu and S. S. Gambhir, Nat. Methods, 2007, 4, 641–643 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- X. Ragas, A. Jimenez-Banzo, D. Sanchez-Garcia, X. Batllori and S. Nonell, Chem. Commun., 2009, 2920–2922 RSC.
- C. Flors, M. J. Fryer, J. Waring, B. Reeder, U. Bechtold, P. M. Mullineaux, S. Nonell, M. T. Wilson and N. R. Baker, J. Exp. Bot., 2006, 57, 1725–1734 CrossRef CAS PubMed.
- M. G. del Carmen, I. Rizvi, Y. Chang, A. C. Moor, E. Oliva, M. Sherwood, B. Pogue and T. Hasan, J. Natl. Cancer Inst., 2005, 97, 1516–1524 CrossRef CAS PubMed.
- M. C. Luna and C. J. Gomer, Cancer Res., 1991, 51, 4243–4249 CAS.
- A. Casas, G. Di Venosa, T. Hasan and B. Al, Curr. Med. Chem., 2011, 18, 2486–2515 CrossRef CAS PubMed.
- I. Rizvi, T. A. Dinh, W. P. Yu, Y. C. Chang, M. E. Sherwood and T. Hasan, Isr. J. Chem., 2012, 52, 776–787 CrossRef CAS PubMed.
- V. Torchilin, Drug Discovery Today: Technol., 2008, 5, e95–e103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, experimental details, characterization of compounds, and in vitro absorption and bioluminescence spectra measurements. See DOI: 10.1039/c7cc00041c |
| This journal is © The Royal Society of Chemistry 2017 |