
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The fusogenic peptide HA2 impairs selectivity of CXCR4-targeted protein nanoparticles†
L.
Sánchez-García‡
ag,
N.
Serna‡
a,
M.
Mattanovich
ab,
P.
Cazzanelli
ab,
A.
Sánchez-Chardi
c,
O.
Conchillo-Solé
a,
F.
Cortés
d,
X.
Daura
ae,
U.
Unzueta
*fg,
R.
Mangues
fg,
A.
Villaverde
 *ag and
E.
Vázquez
ag
*ag and
E.
Vázquez
ag
aInstitut de Biotecnologia i de Biomedicina and Departament de Genètica i de Microbiologia, Universitat Autònoma de Barcelona, Bellaterra, 08193 Barcelona, Spain. E-mail: antoni.villaverde@uab.es
bDepartment of Biotechnology, University of Natural Resources and Life Sciences (BOKU), Muthgasse 18, 1190 Vienna (BOKU), Austria
cServei de Microscòpia, Universitat Autònoma de Barcelona, Bellaterra, 08193 Barcelona, Spain
dServei de Cultius Cellulars, Producció d’Anticossos i Citometria, (SCAC), Universitat Autònoma de Barcelona, Bellaterra, 08193 Barcelona, Spain
eCatalan Institution for Research and Advanced Studies (ICREA), 08010 Barcelona, Spain
fInstitut d'Investigacions Biomèdiques Sant Pau and Josep Carreras Research Institute, Hospital de la Santa Creu i Sant Pau, 08025 Barcelona, Spain. E-mail: uunzueta@santpau.cat
gCIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Spain
First published on 10th March 2017
Abstract
We demonstrate here that the genetic incorporation of the fusogenic peptide HA2 into a CXCR4-targeted protein nanoparticle dramatically reduces the specificity of the interaction between nanoparticles and cell receptors, a factor to be considered when designing tumor-homing drug vehicles displaying endosomal-escape agents. The loss of specificity is concomitant with enhanced cell penetrability.
Cell-targeted intracellular delivery is a major challenge in innovative medicines, which continuously explore new and more efficient vehicles for conventional and emerging drugs.1 Targeting can be achieved by the incorporation of ligands for cell receptors into the drug vehicle, which direct specific binding and further endosomal-mediated cell uptake. This is particularly convenient when the vehicle itself is produced in a recombinant form, which allows genetic fusion and biological production of the whole polypeptide.2 Unfortunately, endosomal uptake drives the engulfed material to a lysosomal pathway, resulting in acidification and proteolysis. Background endosomal leakage and endosomolytic activities naturally present in the recombinant protein allow a fraction of the complex to reach the cytoplasm. Several natural or modified peptides have been identified as strongly endosomolytic, increasing the fraction of internalized material that escapes from lysosomal degradation. Among them, the N-terminal peptide HA2 from the influenza virus hemagglutinin has been widely explored in a diversity of protein constructs intended as nanoscale intracellular vehicles. In acidic environments, such as the endosome, the anionic amino acids of HA2 get protonated, an alpha helix is formed and the peptide acts as an amphiphilic anionic stretch that destabilizes the cell membrane.3 HA2 alone,4 or in combination with the TAT peptide from the human immunodeficiency virus-1,5,6 promotes endosomal release of fusion proteins or nanoscale constructs, which is observed as a promising activity in vehicles for gene therapy and drug delivery.
So far, the potential use of HA2 in combination with specific cell ligands for receptor-mediated cell-targeted delivery has been neglected. However, efficiently combining the selectivity with endosomal escape would represent a step ahead towards the construction of powerful vehicles for targeted drug delivery. Here we have explored this possibility by the incorporation of peptide HA2 into a CXCR4-targeted protein nanoparticle based on a modular, single chain polypeptide (T22-GFP-H6). T22-GFP-H6 self-assembles as toroid nanoparticles of 12 nm, which penetrate CXCR4+ cells with high selectivity, in cell culture and in vivo.7,8 The cationic peptide T22 acts as both a promoter of protein self-assembly9 and as a specific ligand of CXCR4,10 a chemokine receptor whose overexpression is associated with aggressiveness in several types of human cancers.11 In this context, an efficient HA2 version12 was inserted at two alternative inner positions of T22-GFP-H6 (namely at the amino terminus of the core GFP, or at its carboxy terminus) (Fig. 1A). Both engineered proteins were produced well in E. coli, although the lower yield of soluble T22-GFP-HA2-H6 compared to T22-HA2-GFP-H6 resulted in a higher background in mass spectrometry analysis (Fig. 1B). The purity of both products and the predicted molecular mass were however fully assessed by Western blot (Fig. 1B, inset) and using the purification chromatograms (ESI,† Fig. S1). T22-HA2-GFP-H6 and T22-GFP-HA2-H6 proteins spontaneously self-assembled as stable, supramolecular structures with a toroidal shape, with a diameter of 30 nm and 45 nm, respectively, and similar surface charge (Fig. 1C and D). The untagged parental GFP-H6 protein version remained unassembled (Fig. 1C). Despite their good stability, T22-GFP-HA2-H6 nanoparticles showed a tendency to form aggregates, as observed from a secondary DLS peak at around 200 nm (Fig. 1C). The insertion of the HA2 peptide rendered constructs with significantly lower fluorescence than T22-GFP-H6, especially in the case of T22-GFP-HA2-H6 (Fig. 1A), indicating a conformational impact of the viral peptide on the building block. However, specific fluorescence was in both cases high enough to monitor protein internalization in cultured CXCR4+ HeLa cells.
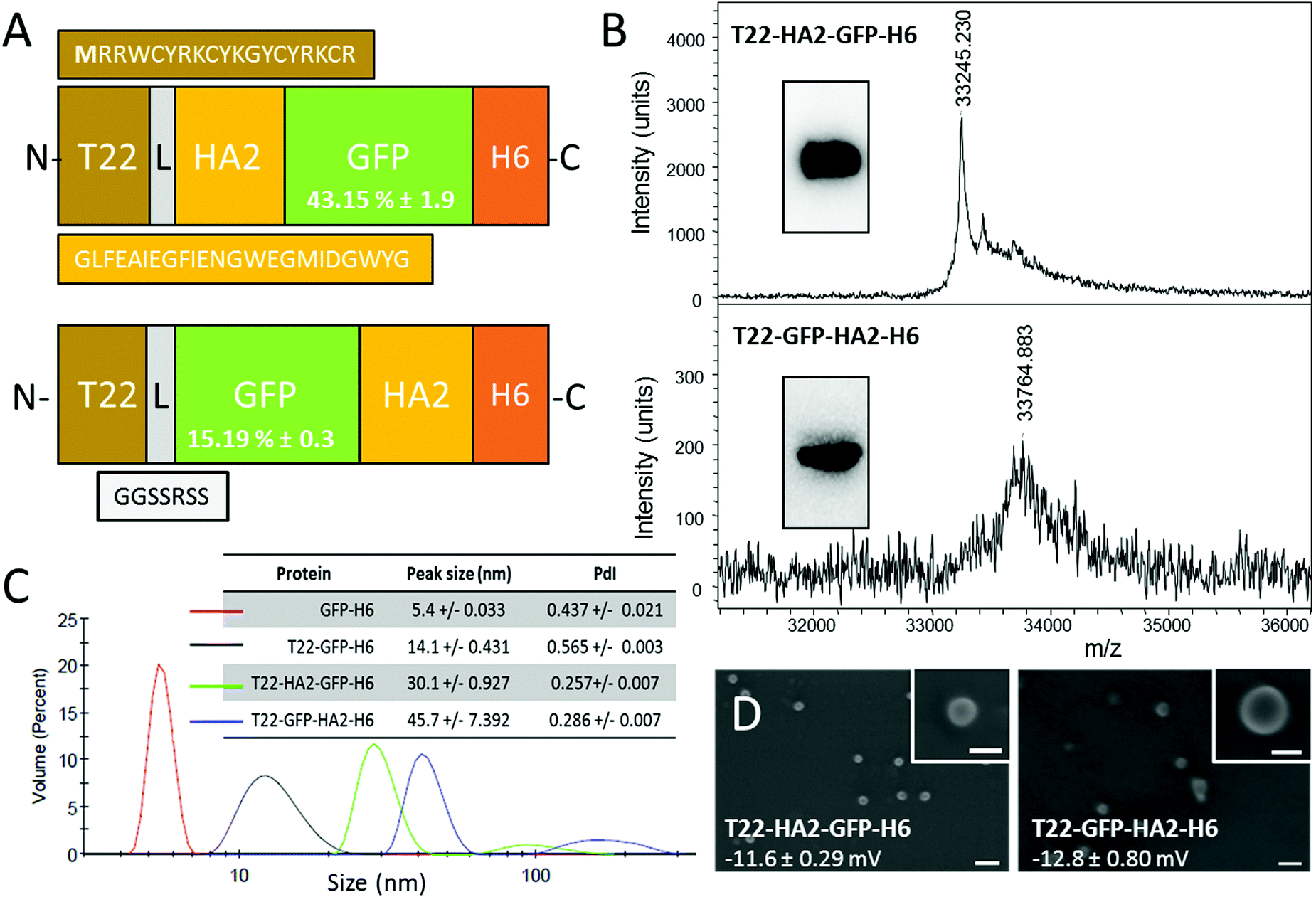 | ||
| Fig. 1 Production and characterization of HA2-empowered protein nanoparticles. (A) Schematic characterization of protein constructs including a HA2 stretch.12 The N-terminal methionine, absent in the original sequence of T22, is shown in bold. L is a ligand that offers molecular flexibility. Figures in GFP boxes represent the specific GFP fluorescence compared to the parental T22-GFP-H6. (B) Mass spectroscopy of HA2 proteins upon IMAC purification. Western blot analyses are shown in the inset. (C) DLS size analysis of the protein materials. The unassembled GFP-H6 is shown as reference. In the inset data, the peak size and the polydispersion index (PdI) are shown. (D) FESEM observations of purified material (size bars are 100 nm, 30 nm in insets). Figures indicate surface charge. Experimental procedures are shown in full in the ESI.† | ||
Cell penetration of both nanoparticles was monitored in the absence and in the presence of a chemical ligand of CXCR4, namely AMD3100, which inhibits binding of T22. As observed (Fig. 2A), the presence of HA2 in the particles enhanced protein penetration in comparison to T22-GFP-H6 nanoparticles, thus confirming the activity of the fusogenic peptide displayed at both accommodation sites. In T22-GFP-HA2-H6, the viral peptide was clearly superior in promoting cell penetration. Even at very low protein concentrations in which the uptake of T22-HA2-GFP-H6 was indistinguishable from that of T22-GFP-H6, its enhancing effect was perceived. Enhanced uptake was not accompanied by cell toxicity (Fig. 2A, inset), an issue that was a matter of concern because of the hemolytic activities of HA2. These effects were mainly observed in HA2 peptide versions with a free amino terminus,12–14 which in the current construct is blocked by T22. On the other hand, the display of HA2 dramatically reduced the specificity of CXCR4-dependent penetration. While AMD3100 inhibited the uptake of T22-GFP-H6 by around 70% it only reduced the penetration of T22-GFP-HA2-H6 by 30% and no inhibition was observed in the case of T22-HA2-GFP-H6 (Fig. 2B). This last protein appears to be internalized in a completely unspecific way (Fig. 2B).
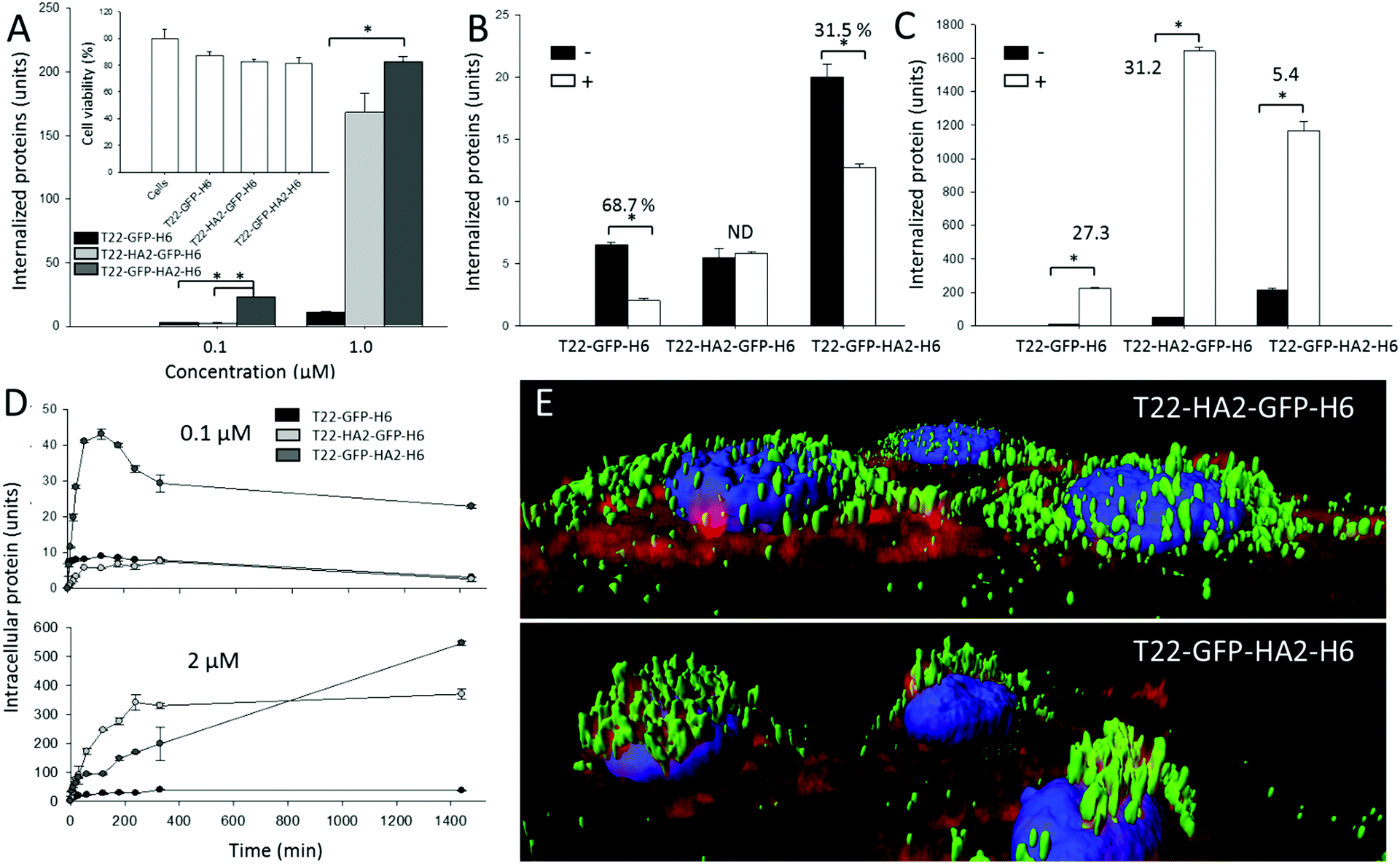 | ||
| Fig. 2 Functional characterization of HA2-containing nanoparticles. (A) Internalization of T22-GFP-H6 and their HA2-containing derivatives in cultured CXCR4+ HeLa cells, after 24 h exposure. Crude fluorescence values were normalized by the specific fluorescence emission of each protein (units) to allow mass-based comparison. In the inset, HeLa cell viability after exposure to 2 μM of modular proteins for 48 h. (B) Analysis of specific CXCR4-mediated internalization in the absence (−) and in the presence (+) of the CXCR4 ligand AMD3100. Data refer to 1 h after exposure. % of inhibition is indicated in each case. ND means not determinable, as the % was 0. (C) Accumulation of HA2-containing nanoparticles in cultured HeLa cells exposed to 1 μM of protein during 24 h in the absence (−) or presence (+) of chloroquine. (D) Intracellular accumulation of proteins, added to two alternative concentrations, in exposed CXCR4+ HeLa cells. Data are presented as arithmetic mean ± standard deviation from two independent experiments. (E) Isosurface representation of HeLa cells within a 3D volumetric z axes stack after incubation for 24 h with nanoparticles (at 0.5 μM). The cell membrane was labeled with CellMask (red signal), cell DNA was labeled with Hoescht 33342 (blue signal) and proteins naturally produced a green signal. | ||
The higher penetrability of HA2-containing nanoparticles respective to the parental T22-GFP-H6 oligomers (Fig. 2A) was supposed to be linked to the enhanced endosomal escape mediated by HA2. To assess this issue, we analyzed the uptake of the HA2-empowered constructs in the presence of chloroquine that inhibits acidification and subsequent lysosomal degradation of the internalized material. This drug equally enhanced the intracellular fluorescence of cells exposed to T22-GFP-H6 and to T22-HA2-GFP-H6 by about 30 fold (Fig. 2C). This indicates that HA2 in T22-HA2-GFP-H6 does not stimulate full endosomal escape of the particles in comparison to the parental T22-GFP-H6. In contrast, chloroquine had much milder enhancing effects on T22-GFP-HA2-H6 (only five-fold increase, Fig. 2C), proving that the viral peptide shows endosomolytic properties in this construct. In this regard, both HA2-displaying nanoparticles are degraded in cells (Fig. 2D) when added to cell cultures at a concentration of 0.1 μM, a dose that has been described below the threshold supporting the endosomolytic properties of HA2.12 However, at 2 μM, over such a transition value, T22-GFP-HA2-H6 but not T22-HA2-GFP-H6 keeps accumulating in cells during prolonged exposure (Fig. 2D). This is again in the line that this protein, but not the related T22-HA2-GFP-H6 (or in a much more moderate way), is able to escape from lysosomal degradation. The enhanced perinuclear accumulation of T22-GFP-HA2-H6 (Fig. 2E) fully supports this hypothesis.
HA2 destabilizes lipid cell membranes at pH 5–5.5.15 However, viral strains with HA2 variants show the ability to replicate in target cells at higher pH values.16–18 Slight variations in charge distribution at the amino terminus of hemagglutinin and in the isoelectric point of the whole protein allow the fusogenic activities of HA2 at less acidic pH. In this regard, the endosomal escape of modular constructs containing histidine-rich peptides, R9, HA2 and cherry, are disrupted by a nuclear localization signal at the carboxy terminus of HA2.12 In the constructs generated here both accommodation sites allow a dramatic enhancement of HA2-mediated cell penetration, although at the expense of a loss of specificity in the interaction with cells. This is particularly deleterious when HA2 is placed in close vicinity to the cationic T22 segment, since T22 is critical for protein–protein contacts.19 In this case, the construct completely missed the ability to specifically interact with CXCR4+ cells, and cell penetration occurs irrespective of endosomal acidification. HA2, in this position, might show enhanced membrane activities influenced by the vicinity of T22. This should produce a differential conformation in the building blocks or the whole nanoparticles already anticipated by differences in the specific fluorescence of the GFP module in both constructs (Fig. 1A). A differential conformational impact of HA2 was confirmed by temperature denaturation followed by Trp emission fluorescence and supported by the molecular modelling of both protein nanoparticles (ESI,† Fig. S2A and B), which resulted in less flexible materials in the case of T22-GFP-HA2-H6. Altogether, while HA2 generically appears as highly appealing for enhancing the integrity of protein-based nanoscale vehicles upon internalization it seems to be poorly appropriate when the efficiency of constructs is based on specific interactions with cell-surface receptors, which might be partially or totally abolished by the viral segment.
The insertion of HA2 in two alternative positions of the tumor-homing nanoparticle T22-GFP-H6 results in a dramatic enhancement of cell penetrability. In the case of T22-GFP-HA2-H6, this is executed through endosomolytic activities and linked to a mild but significant affectation of particle–cell specific interaction. However, regarding T22-HA2-GFP-H6, specificity is completely lost and cell penetration is enhanced in the absence of endosomolytic activity. Taken together, these data indicate that in T22-HA2-GFP-H6, the viral peptide HA2 acts as a cell penetrating peptide rather than as an endosomal escape agent, since it stimulates the penetration of the nanoparticle in a receptor-independent way, probably at the cell surface or at very early endosomal stages. Among the diversity of functional nanoparticles for drug delivery based on biocompatible materials,20 protein nanoparticles offer an unusual structural versatility that allows the precise exploration of functional modular combinations by conventional genetic engineering.
We thank MINECO (BIO2013-41019-P), AGAUR (2014SGR-132) and CIBER de Bioingeniería, Biomateriales y Nanomedicina (project NANOPROTHER) to AV, Marató deTV3 foundation (TV32013-3930) and ISCIII (PI15/00272, co-founding FEDER) to EV and ISCIII (PI15/00378 and PIE15/00028, co-founding FEDER), Marató TV3 (2013-2030) and AGAUR (2014-PROD0005) to RM, for funding our research. Protein production has been partially performed by the ICTS “NANBIOSIS”, more specifically by the Protein Production Platform of CIBER-BBN/IBB (http://www.nanbiosis.es/unit/u1-protein-production-platform-ppp/). PC and MM thank the Erasmus+ for the financial support during this project. LSG was supported by AGAUR (2016FI_B 00034), UU received a Sara Borrell postdoctoral fellowship from ISCIII and AV an ICREA ACADEMIA.
References
- R. Duncan and R. Gaspar, Mol. Pharmaceutics, 2011, 8, 2101–2141 CrossRef CAS PubMed.
- E. Vazquez, R. Mangues and A. Villaverde, Nanomedicine, 2016, 11, 1333–1336 CrossRef CAS PubMed.
- M. Murata, S. Takahashi, S. Kagiwada, A. Suzuki and S. Ohnishi, Biochemistry, 1992, 31, 1986–1992 CrossRef CAS PubMed.
- E. Wagner, C. Plank, K. Zatloukal, M. Cotten and M. L. Birnstiel, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7934–7938 CrossRef CAS.
- S. F. Ye, M. M. Tian, T. X. Wang, L. Ren, D. Wang, L. H. Shen and T. Shang, Nanomedicine, 2012, 8, 833–841 CAS.
- J. S. Wadia, R. V. Stan and S. F. Dowdy, Nat. Med., 2004, 10, 310–315 CrossRef CAS PubMed.
- M. V. Cespedes, et al. , ACS Nano, 2014, 8, 4166–4176 CrossRef CAS PubMed.
- M. V. Cespedes, et al. , Nanomedicine, 2016, 12, 1987–1996 CAS.
- U. Unzueta, et al. , Biomaterials, 2012, 33, 8714–8722 CrossRef CAS PubMed.
- U. Unzueta, M. V. Cespedes, N. Ferrer-Miralles, I. Casanova, J. Cedano, J. L. Corchero, J. Domingo-Espin, A. Villaverde, R. Mangues and E. Vazquez, Int. J. Nanomed., 2012, 7, 4533–4544 CAS.
- F. Balkwill, Nat. Rev. Cancer, 2004, 4, 540–550 CrossRef CAS PubMed.
- J. S. Liou, B. R. Liu, A. L. Martin, Y. W. Huang, H. J. Chiang and H. J. Lee, Peptides, 2012, 37, 273–284 CrossRef CAS PubMed.
- I. Neundorf, R. Rennert, J. Hoyer, F. Schramm, K. Lobner, I. Kitanovic and S. Wolfl, Pharmaceuticals, 2009, 2, 49–65 CrossRef CAS PubMed.
- T. Sugita, T. Yoshikawa, Y. Mukai, N. Yamanada, S. Imai, K. Nagano, Y. Yoshida, H. Shibata, Y. Yoshioka, S. Nakagawa, H. Kamada, S. I. Tsunoda and Y. Tsutsumi, Br. J. Pharmacol., 2008, 153, 1143–1152 CrossRef CAS PubMed.
- J. J. Skehel, K. Cross, D. Steinhauer and D. C. Wiley, Biochem. Soc. Trans., 2001, 29, 623–626 CrossRef CAS PubMed.
- R. S. Daniels, J. C. Downie, A. J. Hay, M. Knossow, J. J. Skehel, M. L. Wang and D. C. Wiley, Cell, 1985, 40, 431–439 CrossRef CAS PubMed.
- C. Scholtissek, Vaccine, 1985, 3, 215–218 CrossRef CAS PubMed.
- E. E. Ooi, J. S. Chew, J. P. Loh and R. C. Chua, Virol. J., 2006, 3, 39 CrossRef PubMed.
- F. Rueda, et al. , Adv. Mater., 2015, 27, 7816–7822 CrossRef CAS PubMed.
- N. Kamaly, B. Yameen, J. Wu and O. C. Farokhzad, Chem. Rev., 2016, 116, 2602–2663 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc09900a |
| ‡ Equally contributed. |
| This journal is © The Royal Society of Chemistry 2017 |