
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(I)-catalysed transfer hydrogenations with ammonia borane†
Eva
Korytiaková
,
Niklas O.
Thiel
,
Felix
Pape
and
Johannes F.
Teichert
 *
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: johannes.teichert@chem.tu-berlin.de; Fax: +49 30 314 28829; Tel: +49 30 314 22791
First published on 19th December 2016
Abstract
Highly Z-selective alkyne transfer semihydrogenations and conjugate transfer hydrogenations of enoates can be effected by employing a readily available and air-stable copper(I)/N-heterocyclic carbene (NHC) complex, [IPrCuOH]. As an easy to handle and potentially recyclable H2 source, ammonia borane (H3NBH3) is used.
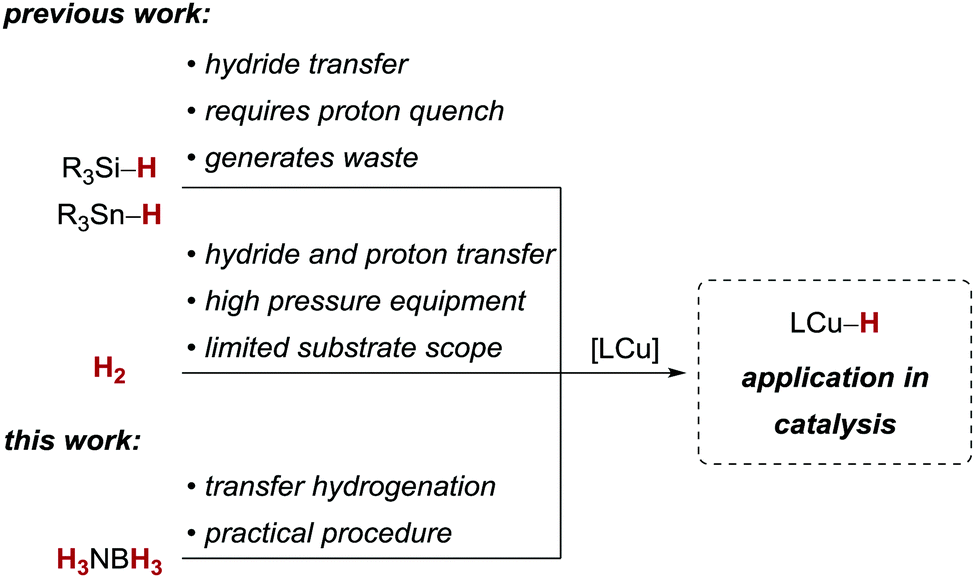
Ammonia borane (H3NBH3), an easy to prepare and manipulate solid, has attracted interest as a potential dihydrogen (H2) storage material.1–6 Based on the fact that ammonia borane dehydrogenation products (e.g., borazines and polyiminoboranes) can potentially be recycled,7 and its exceptionally high H2 content, many studies have been directed toward controlled catalytic release of H2 (dehydrocoupling) from ammonia borane.1–6,8 Notably, to a much lesser extent has H3NBH3 been employed as a reagent for synthesis, for example in the reduction of functional groups or transfer hydrogenations.9–11
Copper(I) hydride complexes have emerged as powerful catalysts for a wide variety of reductive transformations, generally relying on silanes12–15 or, to a much lesser extent, on tin hydrides16 or dihydrogen17,18 (Scheme 1). These terminal reducing agents differ conceptually: Si- and Sn-based compounds will deliver a hydride equivalent and the reaction products remain silylated or stannylated until hydrolysis or protonation. On the other hand, reactions with H2 can directly deliver the hydride and proton equivalent, circumventing the need for an additional proton source and reducing the waste generated in these processes. As an example, copper(I)-catalysed alkyne semihydrogenations have recently demonstrated the potential replacement of silanes with H2.18–20 However, most of these procedures require elevated H2 pressure and/or have a limited substrate scope.
 | ||
| Scheme 1 Hydride and dihydrogen sources employed for copper(I) hydride catalysis. | ||
On the other hand, homogeneous transfer hydrogenations with copper catalysts are scarce,21,22 but circumvent the need for high pressure equipment. Given the synthetic utility of copper hydride complexes, we decided to investigate ammonia borane as a reagent for copper-catalysed transfer hydrogenations.
We decided to optimise the copper(I)-catalysed transfer hydrogenation with H3NBH3 employing alkyne transfer semihydrogenation of pentynol-derived alkyne 1 as a test reaction (Table 1).23 Due to the reported reactivity with H2,18–20 we chose well-defined copper(I)/NHC complexes bearing a copper–oxygen bond as the starting point for our studies. With a copper(I) tert-butanolate complex, generated in situ from [IMesCuCl] and NaOtBu, we observed no turnover to the corresponding alkene Z-2 at room temperature, but full conversion at 50 °C (Table 1, entries 1 and 2). A similar trend was observed with a copper(I) hydroxide/NHC complex, [IPrCuOH],18b,24,25 which also underwent full conversion to Z-2 at 50 °C (Table 1, entries 3–5). Increasing the temperature further to 60 °C led to diminished conversion of 60% (Table 1, entry 6). The use of [IPrCuOH] is particularly attractive, as it does not require the generally common in situ preactivation (to give activated copper(I) alkoxide complexes) and is air-stable for months. It should be noted that in all cases excellent Z-selectivity and negligible overreduction to the corresponding alkane 3 were maintained. This is synthetically appealing, as alkyne semihydrogenations are commonly carried out with heterogeneous palladium catalysts which generally suffer from these particular limitations.26 Furthermore, another report of alkyne transfer semihydrogenation with copper catalysts does not give turnover with internal alkynes,21c which underscores the orthogonal reactivity of the present catalyst.27
|
|
|||
|---|---|---|---|
| Entry | Conditions | Conv.a [%] | Z-2/E-2/3a |
| a Determined by 1H NMR and GC analysis. b Slow addition of H3NBH3 in THF solution over 3 h. c Isolated yield: 96%. | |||
| 1 | [IMesCuCl], 7.5 mol% NaOtBu, 3 equiv. of H3NBH3, THF, rt, 16 h | — | — |
| 2 | [IMesCuCl], 7.5 mol% NaOtBu, 3 equiv. of H3NBH3, THF, 50 °C, 16 h | >99 | >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0:0 :0:0 |
| 3 | [IPrCuOH], 3 equiv. of H3NBH3, THF, rt, 16 h | 9 | >99:0:0 |
| 4 | As entry 3, 40 °C | 68 | >99:0:0 |
| 5 | As entry 3, 50 °C | >99 | >99:0:0 |
| 6 | As entry 3, 60 °C | 60 | >99:0:0 |
| 7 | As entry 5, in C6H6 | 18 | >99:0:0 |
| 8 | As entry 5, in CH2Cl2 | 6 | n.d. |
| 9b | [IPrCuOH], 3 equiv. of H3NBH3, THF, 50 °C, 4 h | >99c | >99:0:0 |
| 10b | [IPrCuOH], 2 equiv. of H3NBH3, THF, 50 °C, 4 h | 88 | >99:0:0 |
| 11 | [IPrCuOH], 20 mol% H3NBH3, 1 bar H2, THF, 50 °C, 16 h | 20 | >99:0:0 |
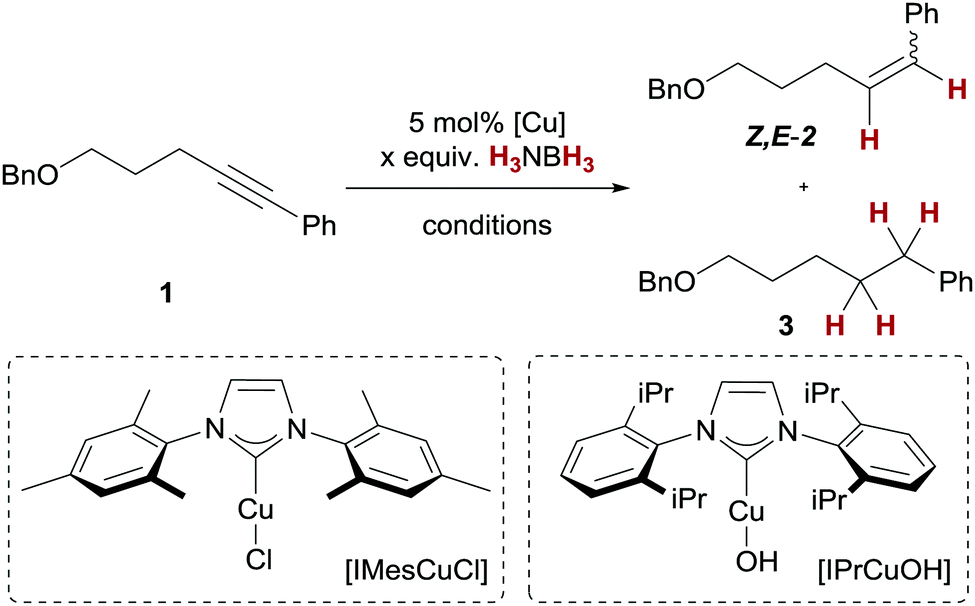
The use of THF as a solvent proved to be optimal, as the reactions in benzene and dichloromethane under otherwise similar reaction conditions gave lower conversion of 1 (Table 1, entries 7 and 8). At the onset of the described reactions in THF, gas evolution (most likely H2) was observed, accompanied by a relatively slow reaction rate (>99% conversion was reached after ∼12 h reaction time). This hinted at the loss of H2 equivalents for the desired transfer semihydrogenation through concomitant dehydrocoupling. Consequently, when an H3NBH3 solution in THF was added slowly (3 h) to the reaction mixture, a generally faster conversion and higher yield of Z-2 were observed (Table 1, entry 9, 96%). Nevertheless, a total of three equivalents of H3NBH3 were required for >99% conversion since slow addition of two equivalents of ammonia borane did not lead to complete consumption of 1 (Table 1, entry 10).28 Based on these results, we decided to probe whether a direct transfer of a hydride equivalent to copper or H2 activation after dehydrocoupling of ammonia borane was present. Therefore, we carried out the alkyne transfer semihydrogenation with a catalytic amount of H3NBH3 under a H2 atmosphere. These reactions gave only 20% conversion of 1 (Table 1, entry 11); hence, we conclude that a direct hydride transfer mechanism is present.
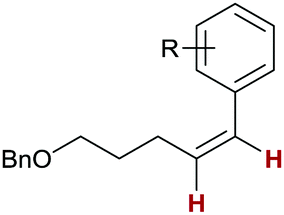
With the optimised reaction conditions in hand, we explored the substrate scope of the copper-catalysed alkyne transfer semihydrogenation. A variety of phenylacetylene derivatives with both electron-donating and electron-withdrawing functional groups 4a–4g could successfully be transformed into alkenes 5a–5g with good yields and consistently high Z-selectivity (Table 2, entries 1–7). Of note is the low conversion achieved with 3-anisole derivative 4d (30%, Table 2, entry 4); we suspect substrate coordination detrimental to the catalyst, which is absent with the other two regioisomers 4c and 4e (Table 2, entries 3 and 5). With strongly electron-withdrawing substituents (as in 5h–5j, Table 2, entries 8–10), we observed partial overreduction to the corresponding alkanes, although the Z/E-ratios remained high. The same is true for the acetophenone derivative 4k, where overreduction to the benzyl alcohol 5k was detected (Table 2, entry 11).29 A variety of Z-stilbenes 5l–5o can be synthesised from the corresponding tolane derivatives 4l–4o with good yields (Table 2, entries 12–15). Here, a similarly detrimental influence of the electron-withdrawing trifluoromethyl substituent in 5o was observed as 8% alkane was detected (Table 2, entry 15). The alkyne transfer semihydrogenation can also be applied to dialkylalkynes, as demonstrated by the formation of Z-alkenes 5p and 5q (Table 2, entries 16 and 17). For full consumption of the former, one more equivalent of H3NBH3 was added. Finally, protected allyl alcohols 5r and 5s can be obtained with high Z-selectivity from the corresponding propargylic silyl ethers 4r and 4s (Table 2, entries 18 and 19). Generally, the obtained yields are good; in some cases we observed undesired hydroboration/oxidation of the alkene products.23 We attribute the somewhat diminished yields to this side reaction.
|
|
|||
|---|---|---|---|
| Entry | Product | Yield [%] | Z/E/alkanea |
| a Determined by 1H NMR and GC analysis. b 4 equiv. of H3NBH3 was used. | |||
| 1 |
5a: R = 4-Me
|
63 | >99:0:0 |
| 2 | 5b: R = 4-tBu | 50 | >99:0:0 |
| 3 | 5c: R = 4-OMe | 69 | 95:5:0 |
| 4 | 5d: R = 3-OMe | 30 conv. | >99:0:0 |
| 5 | 5e: R = 2-OMe | 71 | >99:0:0 |
| 6 | 5f: R = Br | 80 | >99:0:0 |
| 7 | 5g: R = Cl | 63 | >99:0:0 |
| 8 | 5h: R = CO2Me | 65 | 80:0:20 |
| 9 | 5i: R = CN | 51 | 89:4:7 |
| 10 | 5j: R = CF3 | 71 | 81:0:19 |
| 11 |
5k:
|
55 | 84:1:15 |
| 12 |
5l: R = H
|
57 | >99:0:0 |
| 13 | 5m: R = 4-OMe | 66 | >99:0:0 |
| 14 | 5n: R = 4-Cl | 77 | >99:0:0 |
| 15 | 5o: R = 4-CF3 | 50 | 92:0:8 |
| 16b |
5p:
|
74 | >99:0:0 |
| 17 |
5q:
|
80 conv. | >99:0:0 |
| 18 |
5r:
|
74 | >99:0:0 |
| 19 |
5s:
|
63 | >99:0:0 |
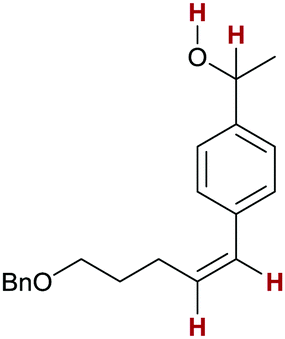
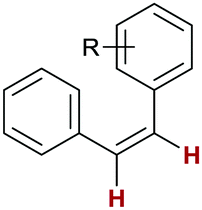
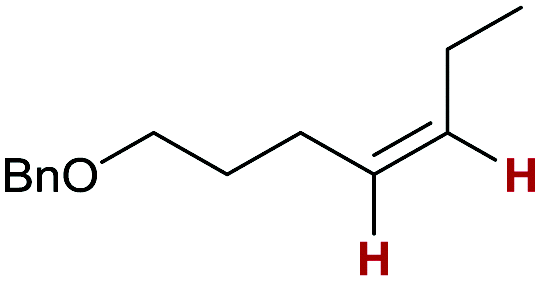
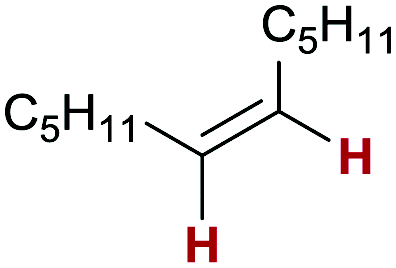
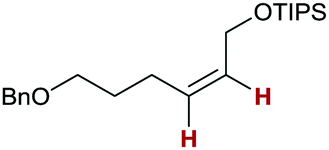
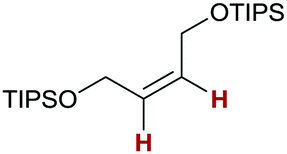
Finally, we investigated the transfer semihydrogenation of alkyl and aryl propiolates 6 (Scheme 2). In both cases the desired Z-acrylates 7a and 7b were found, however, in the presence of overreduced esters 8a and 8b. This indicated the viability of a conjugate transfer hydrogenation of the initially formed acrylates 7. We therefore decided to investigate the conjugate transfer hydrogenation of α,β-unsaturated esters with ammonia borane next.
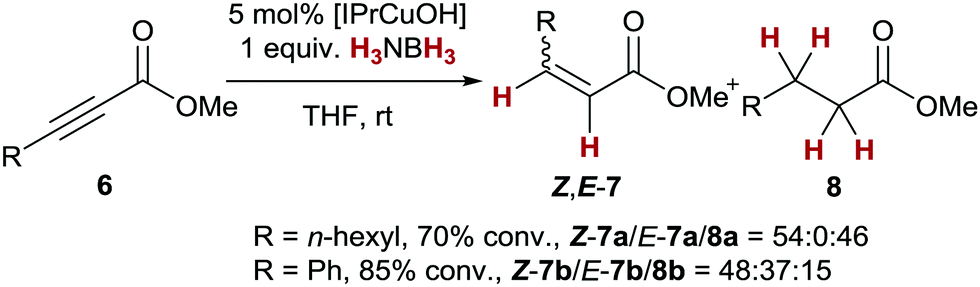 | ||
| Scheme 2 Transfer semihydrogenation of propiolates. | ||
After initial optimisation we also found that conjugate transfer hydrogenation of enoates 9 with ammonia borane was feasible under similar reaction conditions (Table 3). Conjugate transfer hydrogenation of disubstituted ethyl esters 9a–9i could be realised with generally good isolated yields when the reaction was carried out under previously optimised conditions (Table 3, entries 1–9). Isolated yields for esters 10 generally are higher than in the alkyne transfer semihydrogenation. This is possibly due to the absence of a reactive double bond in the products, which could lead to further reactions. Only thiophene derivative 10f was obtained in a somewhat lower yield (Table 3, entry 6). Of note is the different reactivity of the catalyst toward double bond isomers: while the E-enoate 9d underwent smooth conversion to the desired saturated product 10d (Table 3, entry 4), the reaction with Z-enoate 9e proceeded sluggishly (46% conversion to 10e after 16 h) under otherwise similar conditions (Table 3, entry 5). Higher substituted acrylates 10j–10l proved to be more difficult to obtain, most probably due to steric hindrance (Table 3, entries 10–12). However, by addition of 6 equivalents of H3NBH3, esters 10j–10l could be isolated in good yields.30
|
|
||
|---|---|---|
| Entry | Product | Yield [%] |
| a These reactions reached >99% conversion with 2 equiv. of H3NBH3. b The methyl ester of 9e was employed. c 6 equiv. of H3NBH3 was used. d Contains 14% of the starting material. | ||
| 1a | 10a: R1 = Ph, R2 = H | 82 |
| 2a | 10b: R1 = 2-naphthyl, R2 = H | 83 |
| 3a | 10c: R1 = 4-OMe-C6H4, R2 = H | 83 |
| 4 | 10d: R1 = 4-OBn-C6H4, R2 = H | 87 |
| 5b | 10e: R1 = H, R2 = 4-OBn-C6H4 | 46 conv. |
| 6 | 10f: R1 = 2-thiophenyl, R2 = H | 66 |
| 7a | 10g: R1 = cyclohexyl, R2 = H | 88 |
| 8 | 10h: R1 = 4-Br-C6H4, R2 = H | 82 |
| 9 | 10i: R1 = 4-CF3-C6H4, R2 = H | 89 |
| 10c | 10j: R1 = 2-naphthyl, R2 = Me | 93 |
| 11c | 10k: R1 = Ph, R2 = Me | 76 |
| 12c | 10l: R1 = Ph, R2 = Ph | 73d |
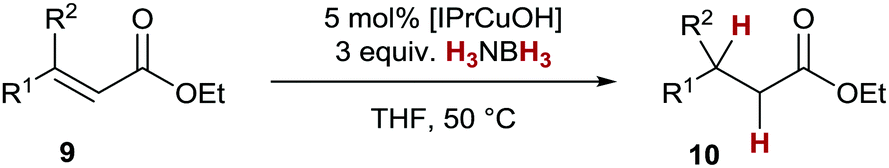
In summary, we have developed copper(I) hydride-mediated transfer hydrogenation reactions employing ammonia borane (H3NBH3) as a H2 equivalent. Alkynes can be converted into the corresponding alkenes with high Z-selectivities in transfer semihydrogenations. Also, the protocol enables a conjugate transfer hydrogenation of enoates. An air-stable and preactivated copper(I) hydroxide/NHC complex is used, circumventing the need for generally common in situ activation of the copper catalyst. We think that these results could be of importance with foresight to the use of readily available transition metal catalysts (such as copper) for transfer hydrogenations.
This research was supported by the Fonds der Chemischen Industrie (Liebig-Stipendium for J. F. T.) and the German Academic Exchange Service (DAAD) through a Leistungsstipendium (for E. K.). F. P. is supported by a predoctoral scholarship of the Berlin International Graduate School of Natural Sciences and Engineering (BIG-NSE). We kindly thank Prof. Dr. Martin Oestreich (TU Berlin) for generous support. Prof. Dr. Gerhard Erker (WWU Münster) is thanked for fruitful discussion.
Notes and references
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed.
- H.-L. Jiang and Q. Xu, Catal. Today, 2011, 170, 56–63 CrossRef CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC.
- P. Wang and X.-D. Kang, Dalton Trans., 2008, 5400–5413 RSC.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- (a) Y. Tan and X. Yu, RSC Adv., 2013, 3, 23879–23894 RSC; (b) N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS.
- (a) A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed; (b) R. Waterman, Chem. Soc. Rev., 2013, 42, 5629–5641 RSC; (c) R. J. Less, R. L. Melen and D. S. Wright, RSC Adv., 2012, 2, 2191–2199 RSC; (d) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 7170–7179 CrossRef CAS PubMed.
- X. Yang, Z. Xie, J. He and L. Yu, Chin. J. Org. Chem., 2015, 35, 603–609 CrossRef CAS.
- For selected examples, see: (a) Z. M. Heiden and T. B. Rauchfuss, J. Am. Chem. Soc., 2007, 129, 14303–14310 CrossRef CAS PubMed; (b) N. Blaquiere, S. Diallo-Garcia, S. I. Gorelsky, D. A. Black and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 14034–14035 CrossRef CAS PubMed; (c) Y. Jiang, O. Blacque, T. Fox, C. M. Frech and H. Berke, Organometallics, 2009, 28, 5493–5504 CrossRef CAS; (d) R. Barrios-Francisco and J. J. García, Appl. Catal., A, 2010, 385, 108–113 CrossRef CAS; (e) H. Dong and H. Berke, J. Organomet. Chem., 2011, 696, 1803–1808 CrossRef CAS; (f) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 15310–15313 CrossRef CAS PubMed; (g) J. K. Pagano, J. P. W. Stelmach and R. Waterman, Dalton Trans., 2015, 44, 12074–12077 RSC.
- For the use of ammonia borane as the H2 donor in transfer hydrogenations with alkynes and/or alkenes, see for example: (a) R. Barrios-Francisco and J. J. García, Appl. Catal., A, 2010, 385, 108–113 CrossRef CAS; (b) M. E. Sloan, A. Staubitz, K. Lee and I. Manners, Eur. J. Org. Chem., 2011, 672–675 CrossRef CAS; (c) C. E. Hartmann, V. Jurčík, O. Songis and C. S. J. Cazin, Chem. Commun., 2013, 49, 1005–1007 RSC; (d) S. Fu, N.-Y. Chen, X. Liu, Z. Shao, S.-P. Luo and Q. Liu, J. Am. Chem. Soc., 2016, 138, 8588–8594 CrossRef CAS PubMed.
- A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed.
- C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- S. Rendler and M. Oestreich, Angew. Chem., Int. Ed., 2007, 46, 498–504 CrossRef CAS PubMed.
- B. H. Lipshutz, in Copper(I)-mediated 1,2- and 1,4-Reductions. Modern Organocopper Chemistry, ed. N. Krause, Wiley-VCH, Weinheim, 2002, pp. 167–187 Search PubMed.
- (a) B. H. Lipshutz, C. Ung and S. Sengupta, Synlett, 1989, 64–66 CAS; (b) L. T. Leung, S. K. Leung and P. Chiu, Org. Lett., 2005, 7, 5249–5252 CrossRef CAS PubMed.
- (a) W. S. Mahoney and J. M. Stryker, J. Am. Chem. Soc., 1989, 111, 8818–8823 CrossRef CAS; (b) H. Shimizu, D. Igarashi, W. Kuriyama, Y. Yusa, N. Sayo and T. Saito, Org. Lett., 2007, 9, 1655–1657 CrossRef CAS PubMed; (c) K. Junge, B. Wendt, D. Addis, S. Zhou, S. Das, S. Fleischer and M. Beller, Chem. – Eur. J., 2011, 17, 101–105 CrossRef CAS PubMed.
- (a) F. Pape, N. O. Thiel and J. F. Teichert, Chem. – Eur. J., 2015, 21, 15934–15938 CrossRef CAS PubMed; (b) N. O. Thiel and J. F. Teichert, Org. Biomol. Chem., 2016, 14, 10660–10666 RSC.
- K. Semba, R. Kameyama and Y. Nakao, Synlett, 2015, 318–322 CrossRef CAS.
- T. Wakamatsu, K. Nagao, H. Ohmiya and M. Sawamura, Organometallics, 2016, 35, 1354–1357 CrossRef CAS.
- For examples of homogeneous copper-catalysed transfer hydrogenations, see: (a) J. W. Yang and B. List, Org. Lett., 2006, 8, 5653–5655 CrossRef CAS PubMed; (b) K. Saito, Y. Kajiwara and T. Akiyama, Angew. Chem., Int. Ed., 2013, 52, 13284–13288 CrossRef CAS PubMed; (c) H. Cao, T. Chen, Y. Zhou, D. Han, S.-F. Yin and L.-B. Han, Adv. Synth. Catal., 2014, 356, 765–769 CrossRef CAS.
- For alkyne transfer semihydrogenations with ammonia borane and heterogeneous catalysts, see: (a) D. van der Waals, A. Pettman and J. M. J. Williams, RSC Adv., 2014, 4, 51845–51849 RSC; (b) E. Vasilikogiannaki, I. Titilas, G. Vassilikogiannakis and M. Stratakis, Chem. Commun., 2015, 51, 2384–2387 RSC.
- See the ESI† for details and additional optimisation (reaction parameters, Cu catalysts, addition mode) data.
- G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3966–3972 CrossRef CAS.
- For the use of IPrCuOH as the catalyst in carboxylation reactions, see: (a) I. I. F. Boogaerts, G. C. Fortman, M. R. L. Furst, C. S. J. Cazin and S. P. Nolan, Angew. Chem., Int. Ed., 2010, 49, 8674–8677 CrossRef CAS PubMed; (b) O. Santoro, F. Lazreg, Y. Minenkov, L. Cavallo and C. S. J. Cazin, Dalton Trans., 2015, 44, 18138–18144 RSC.
- (a) C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313–1350 CrossRef CAS PubMed; (b) M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz and L. Kiwi-Minsker, ACS Catal., 2012, 2, 1773–1786 CrossRef CAS; (c) H. Lindlar, Helv. Chim. Acta, 1952, 35, 446–450 CrossRef CAS.
- The transfer semihydrogenation of terminal alkynes with the present catalyst leads to mixtures of products with the terminal alkene and a dimerised 1,3-diene as the major products. See the ESI† for details.
- Reaction analysis by 11B NMR shows the formation of polyaminoboranes (see the ESI† for details), which indicates the transfer of 1 equiv. of H2 per molecule of ammonia borane. This could account for the high loadings of ammonia borane required.
- The reduction of ketones with H3NBH3 is most likely an uncatalysed process, see: (a) G. C. Andrews and T. C. Crawford, Tetrahedron Lett., 1980, 21, 693–696 CrossRef CAS; (b) G. C. Andrews, Tetrahedron Lett., 1980, 21, 697–700 CrossRef CAS; (c) L. Shi, Y. Liu, Q. Liu, B. Wei and G. Zhang, Green Chem., 2012, 14, 1372–1375 RSC.
- Even though varying amounts of H3NBH3 are necessary in these transformations, it should be noted that from a practical perspective, the exact amount required can be determined by in situ analysis of the reaction: later addition of further equivalents of H3NBH3 is possible, as the catalyst remains active in solution.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation and characterisation data as well as 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c6cc09067b |
| This journal is © The Royal Society of Chemistry 2017 |