
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Properties and emerging applications of mechanically interlocked ligands
James E. M.
Lewis
,
Marzia
Galli
and
Stephen M.
Goldup
*
Chemistry, University of Southampton, University Road, Southampton, SO17 1BJ, UK. E-mail: s.goldup@soton.ac.uk
First published on 7th November 2016
Abstract
Mechanically interlocked molecules have a long and rich history as ligands thanks to the key role coordination chemistry has played in the development of high yielding passive template syntheses of rotaxanes and catenanes. In this Feature Article, we highlight the effect of the mechanical bond on the properties of metal ions bound within the sterically hindered environment of the macrocycle cavity, and discuss the emerging applications of interlocked ligands in catalysis, sensing and supramolecular materials.
Introduction
Species in which two or more covalent sub-components are entangled and held together not through direct covalent interactions but through the inability of bonds and atoms to pass through one another are referred to as mechanically interlocked molecules (MIMs).1 The archetypal examples of MIMs are catenanes, consisting of two or more mechanically interlocked macrocycles, and rotaxanes which, in their simplest form, consist of a macrocyclic component wrapped around a linear axle, with dissociation of the subcomponents prevented by bulky stopper units at the axle termini.Initially an esoteric curiosity, the exploration of MIMs for a variety of applications has increased dramatically over the last three decades, a trend inextricably associated with the development of template-directed methods for their synthesis.1 The first such high yielding method was reported by Sauvage and co-workers in 1983 and relied on the predictable tetrahedral coordination geometry of a CuI ion with phenanthroline ligands to arrange macrocycle 1 and macrocycle precursor 2 perpendicular to one another in such an orientation that cyclisation of 2 gave relatively facile and high yielding access to [2]catenane 3 (Fig. 1).2 Soon after, Gibson and co-workers demonstrated that the introduction of bulky stoppering units to 2 rather than cyclisation, using the same CuI–phenanthroline passive template,3 gave the corresponding [2]rotaxane species.4
 | ||
| Fig. 1 Sauvage's seminal passive CuI-template directed synthesis of [2]catenane 3.2 | ||
Sauvage's seminal work on the CuI–phenanthroline template inspired the development of many other template-directed methods, including a great many examples based on metal ions. MIMs prepared by these methods inherently possess the ability to bind metal ions between the two components in a mechanically chelating manner where the donor atoms are linked via the mechanical bond. Perhaps most famously, this has been further capitalised on in the development of molecular switches where the relative position of the covalent sub-components (their co-conformation) is controlled by metal–ligand interactions.1
The synthesis and operation of interlocked molecular machines using such passive metal templates has been comprehensively reviewed elsewhere and will not be covered here.1 Instead, in this Feature Article, we shall discuss the effect of the mechanical bond on the chemistry of coordinated metal ions and emerging applications of mechanically interlocked ligand (MIL) scaffolds, focussing on MIMs as ligands for catalysis, MILs as sensors, and finally MIMs as structural components within metallo-supramolecular architectures.
The effect of the mechanical bond on the chemistry of coordinated metal ions
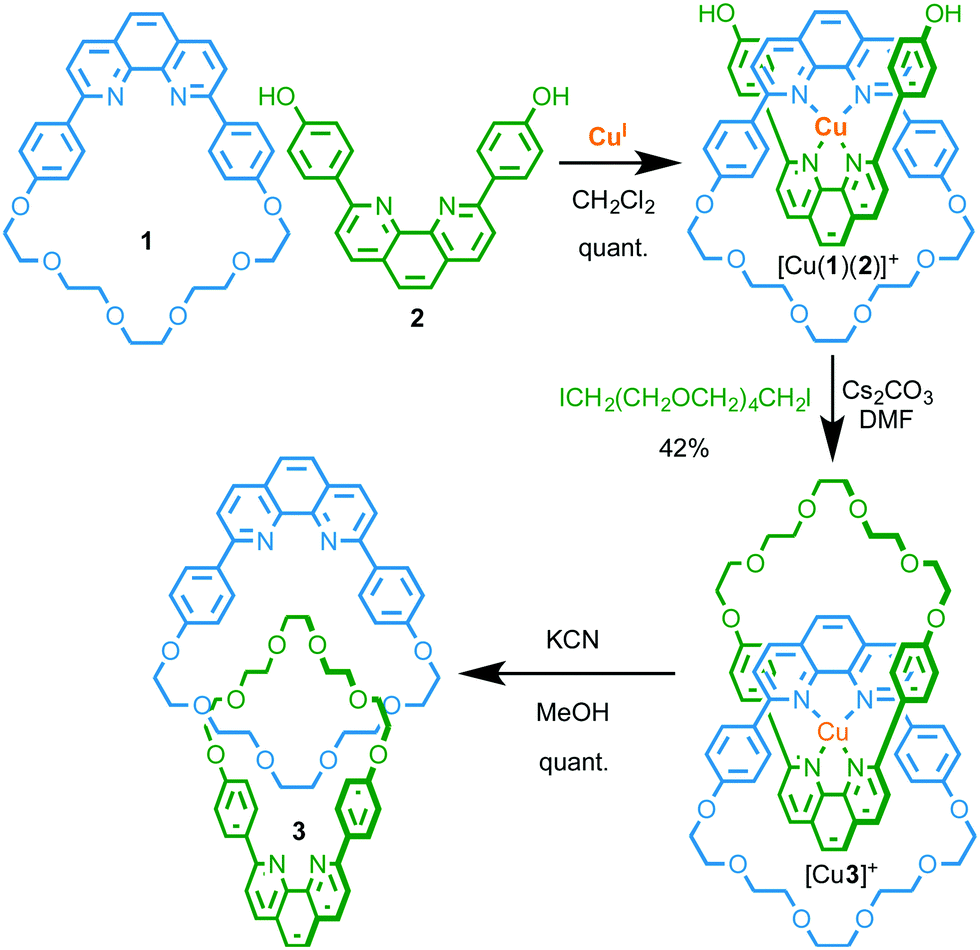
Given the preponderance of metal ion templated syntheses of MIMs and the inherent ability of the products to bind metal ions strongly in an endohedral manner between the two covalent components, this is unsurprisingly the most prominent mode of coordination by MIM-based ligands. Furthermore, these “mechanically chelating” ligands inherently place metal ions in an unusual sterically shielded and conformationally restricted environment, and this can have significant consequences for their properties.Firstly, the interlocked product of a passive metal template synthesis typically forms more kinetically stable complexes with the metal ion template than the precursor ligands. Sauvage observed this phenomenon early in the development of his CuI–phenanthroline template; the templating CuI ion remained bound to catenane 3 following column chromatography, requiring the use of −CN to achieve demetallation.2 Subsequent studies demonstrated that the rate of CuI removal by KCN was lower in the case of catenane 3 than analogous non-interlocked phenanthroline complexes (Fig. 2).5 Sauvage termed this increased kinetic stability the “catenand”6 effect by analogy with the cryptate effect,7 and attributed the observed stabilisation to the pre-organisation of the ligand donor atoms in the catenane architecture, combined with steric protection of the metal centre, afforded by the organic foliage of the MIM itself.
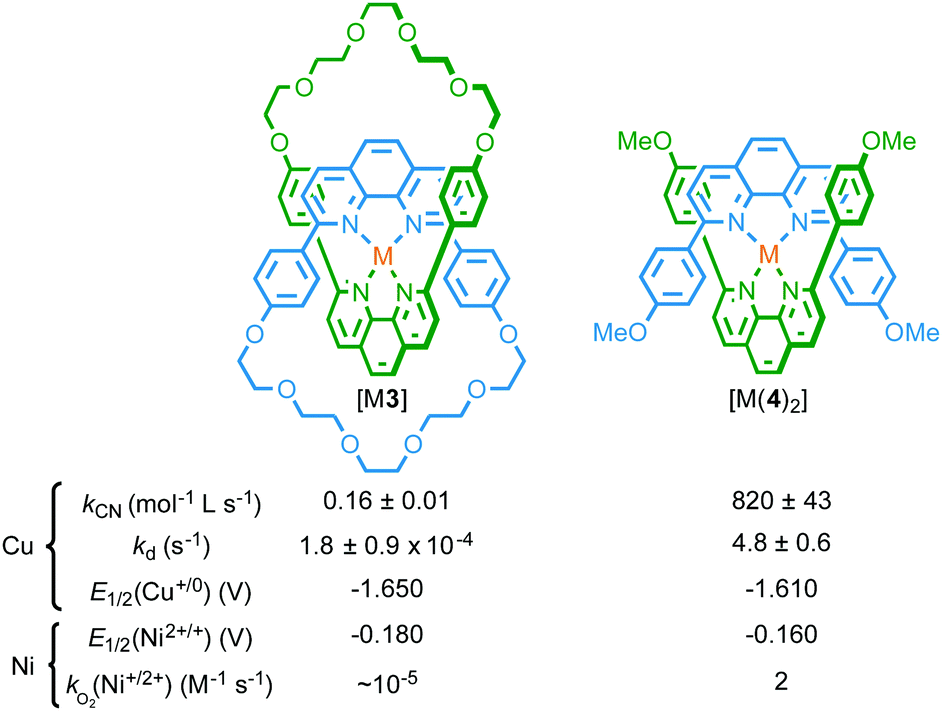 | ||
| Fig. 2 Effect of the mechanical bond on the stability and electrochemistry of metal complexes of catenane [M(3)] compared with non-interlocked analogue [M(4)2].5,8 | ||
Secondly, endotopic coordination within the binding pocket of catenane 3 was shown to alter the redox chemistry of the bound metal ion. Sauvage and co-workers found that catenane 3 is capable of binding a range of metal cations (LiI, AgI, ZnII, CdII, CoII, NiII, FeII, PdII) other than CuI, demonstrating that, although the ligand pocket is restricted, it is flexible enough to accommodate ions of different radii; indeed, the enforced association of the phenanthroline ligands in catenane 3 allowed tetracoordinate complexes of LiI and FeII to be isolated, whereas complex formation was not observed with non-interlocked ligand 4.8,9 However, the mechanical bond of catenane 3 significantly altered the accessible oxidation states of the bound metal ions by disfavouring higher oxidation states; FeIII and CoIII proved inaccessible under the experimental conditions used.8 The inability to access higher oxidation states was attributed to the catenane structure preventing coordination of additional ligands to provide the higher coordination numbers required to stabilise the high charge density of the trivalent cations. Conversely, reduction potentials to access lower oxidation states (CuI/0 and NiII/I) were not significantly altered in the interlocked structure.
Finally, the mechanical bond also modifies the kinetics of chemical oxidation. Thus, although [NiII(3)] and non-interlocked analogue [NiII(4)2] were reduced at comparable potentials to give spectroscopically similar NiI species, with EPR studies confirming the formally d9 electronic state of the NiI ion in both cases, the rate constant for oxidation of catenane complex [NiI(3)] by dissolved O2 in CH2Cl2 solution was found to be 105 times smaller than the non-interlocked analogue.10
Thus the mechanical bond in catenanes has been shown to significantly alter the coordination chemistry, stability and redox properties of the interlocked metal complex. The observation of similar effects in the case of rotaxanes is much less common, at least in part because the environment of the mechanical bond is typically less constrained than in catenanes. However, similarly to Sauvage's report in the case of catenane 3, Leigh and co-workers have demonstrated that the enforced association of convergent binding sites within a rotaxane can give rise to complexes that are not accessible in the case of the corresponding non-interlocked ligands.11 More recently, we have demonstrated that the active template Cu-mediated alkyne–azide cycloaddition (AT-CuAAC)12 approach to rotaxanes can be used to generate very hindered interlocked molecules13 to the point where this unusual environment can stabilise a reactive CuI–organometallic species; when NiPr2Et is employed to accelerate the AT-CuAAC reaction, the isolated product is not the expected rotaxane, but interlocked CuI–triazolide [Cu(8)] intermediate of the CuAAC reaction (Fig. 3).14 Given the well-established protolytic instability of CuI organometallic species, the stability of [Cu(8)] is remarkable; not only does it survive aqueous work up with NH3-EDTA, it even displays an appreciable stability in the presence of a variety of carboxylic acids. Tellingly, the time taken for protonation of the Cu–C bond is strongly correlated with the steric bulk of the acid.
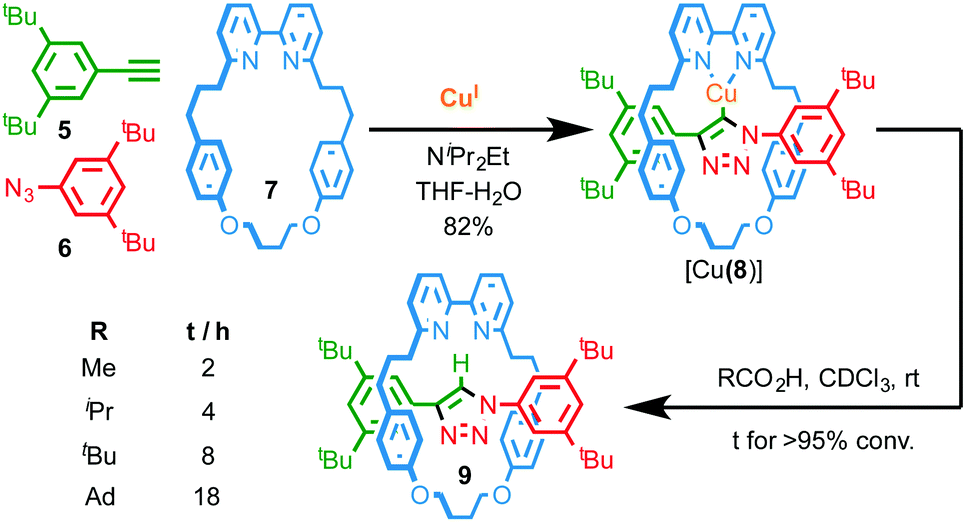 | ||
| Fig. 3 Synthesis of CuI triazolide [Cu(8)] under aqueous conditions and the effect of carboxylic acid size on the rate of protodemetallation.14 Ad = 1-adamantyl. | ||
It is perhaps surprising that, although the catenand effect and the modification of redox behaviour of metal ions were two of the first concrete examples of the effect of mechanical bonding on the chemistry of metal complexes, both remain largely unused as a method for generating metal complexes with tailored properties. Indeed, although a great many interlocked ligands with endohedrally bound metals have been reported, in many cases detailed comparisons with the corresponding non-interlocked metal complex are not discussed. Given the many potential applications of metal ions, for instance in bioimaging through radiochemical, optical and magnetic modes, the key role complex stability plays in their biocompatibility, and the flexibility provided by mechanically chelating ligands,15 this is clearly an area that warrants further investigation.
Mechanically interlocked ligands for catalysis
Despite the large number of interlocked metal complexes reported to date, the majority of these are poorly suited to catalytic applications; mechanically chelating ligands produced through metal-based passive template methods are typically coordinatively saturated and sterically hindered, limiting their catalytic potential. For this reason, relatively few metal complexes of mechanically interlocked ligands have been reported as catalysts. However, the unusual properties of the mechanical bond, principally the restricted relative motion of the covalent sub-components and the ability to generate a crowded but flexible reaction environment, have been shown to impart unusual properties on catalysts based on interlocked molecules.Intramolecular transformations of metal-ion containing MIMs
Architectures in which a catalytically active metal ion is bound to the ring component of a rotaxane bear comparison with information processing enzymes such as DNA polymerase III that operate by threading their polymeric template through a toroidal clamp bearing the catalytic subunit.16 This arrangement of catalyst and substrate results in high processivity – the number of catalytic cycles before the catalyst and substrate separate. Inspired by these natural systems, in 2003 Rowan and Nolte reported a rotaxane in which an endotopic MnIII centre in the macrocycle was demonstrated to epoxidise a threaded polybutadiene substrate in a highly processive manner (Fig. 4a).17 The interlocked catalyst–substrate architecture is assembled through π–π stacking interactions between a viologen unit in the axle and the electron rich aromatic units of a glycoluril-clip macrocycle. Subsequent addition of a bulky pyridine ligand that binds to the MnIII centre blocks the outer face of the Mn-porphyrin catalytic unit, ensuring that catalysis can only take place in the macrocycle cavity. Addition of an oxidant (PhIO) results in the rapid epoxidation of the threaded substrate with catalysis taking place inside the cavity of the macrocycle. In addition to ensuring extremely high processivity, this unusual catalyst–substrate arrangement leads to high selectivity for the trans epoxide product (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 trans–cis) whereas the corresponding intermolecular reaction produces the cis epoxide (20:80). Subsequent studies of the threading and sliding process indicate that the enzyme mimic operates by randomly sliding along its macromolecular substrate rather than transforming each individual alkene in the order they are encountered. More recently, the same authors reported the extension of this approach to the site selective oxidation of DNA by tethering a similar MnIII porphyrin to the toroidal sliding clamp protein of bacterio-phage T4, to produce a biohybrid mechanically interlocked catalyst (Fig. 4b).18
:20 trans–cis) whereas the corresponding intermolecular reaction produces the cis epoxide (20:80). Subsequent studies of the threading and sliding process indicate that the enzyme mimic operates by randomly sliding along its macromolecular substrate rather than transforming each individual alkene in the order they are encountered. More recently, the same authors reported the extension of this approach to the site selective oxidation of DNA by tethering a similar MnIII porphyrin to the toroidal sliding clamp protein of bacterio-phage T4, to produce a biohybrid mechanically interlocked catalyst (Fig. 4b).18
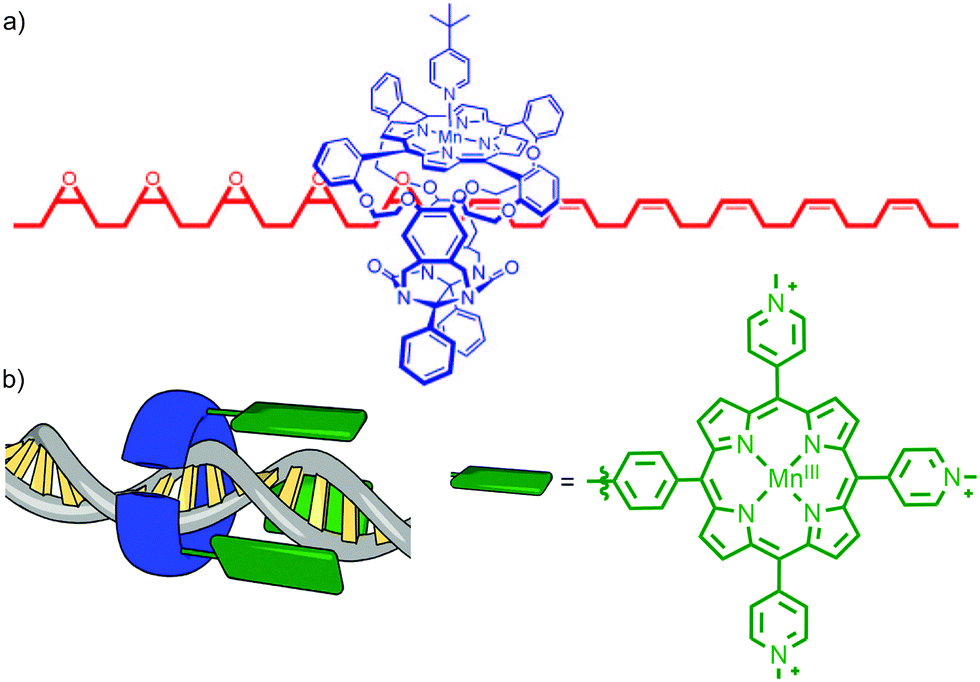 | ||
| Fig. 4 (a) Partial structure of Rowan and Nolte's DNA polymerase mimic showing the macrocycle containing the Mn porphyrin catalyst and the partially oxidised polybutadiene axle.17 Adapted from ref. 17c. Copyright 2007 National Academy of Sciences. (b) a cartoon of the recently reported biohybrid mechanically interlocked catalyst.18 Adapted by permission from ref. 18. Copyright 2013 Nature Publishing Group. | ||
In Rowan and Nolte's systems, the catalytic unit of the macrocycle component was not employed directly in the synthesis of the interlocked structure. More recently, Takata and co-workers reported examples of rotaxanes assembled using a PdII passive template in which the metal ion can then be used to catalyse the conversion of allylic carbamate units in the axle to produce oxazolidinone rings (Fig. 5).19 Coordination of pyridine 11 to Pd complex [Pd(10)(MeCN)] produced threaded complex [Pd(10)(11)] which was subsequently trapped by reaction with an isocyanate to give rotaxane Pd complex [Pd(12)]. Heating [Pd(12)] in the presence of Mg(OH)2 resulted in the cyclisation of the urethane units to produce [Pd(13)]. Test reactions demonstrated that both the PdII complex and the base were required for the reaction to take place efficiently. Furthermore, the reaction of the non-interlocked axle with the same macrocyclic PdII complex led to no conversion demonstrating that the reaction takes place within the cavity of the rotaxane.20
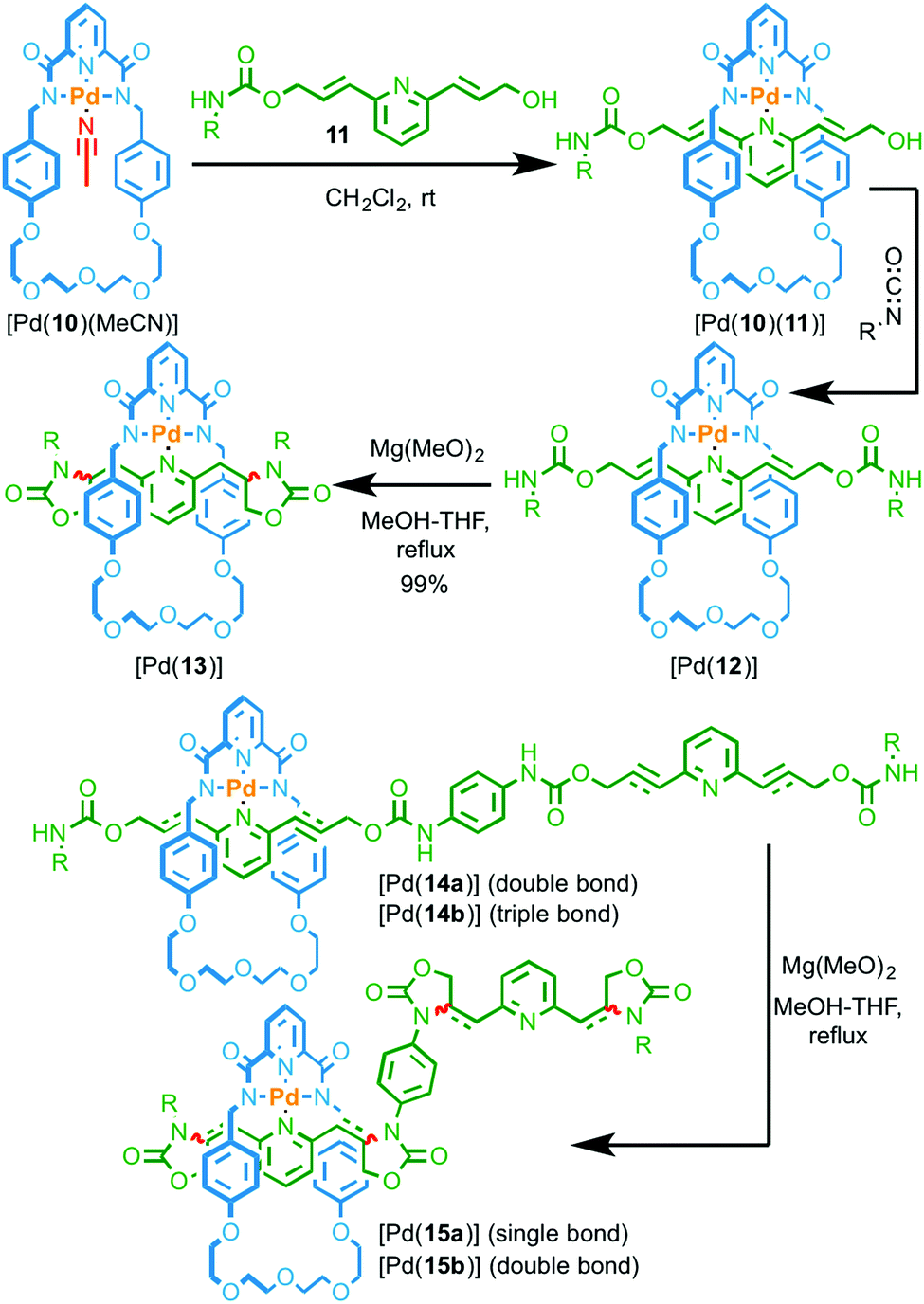 | ||
| Fig. 5 Intramolecular cyclisation of allylic and propargylic carbamate groups by a threaded macrocyclic Pd catalyst. R = 4-C6H4(C(4-tBuC6H4)3).19 | ||
The reaction could be extended to rotaxane [Pd(14a)] containing four allyl groups and an excellent yield of tetra-cyclised product [Pd(15a)] was maintained (99%). However, when propargylic rotaxane [Pd(14b)] was reacted under the same conditions, a much lower yield of the corresponding tetra-oxazolone product [Pd(15b)] was obtained (16%). Although the oxazolone unit of [Pd(15b)] is unstable under the reaction conditions, inherently reducing the yield of this reaction, the difference in yield between [Pd(15a)] and [Pd(15b)] was, in part, attributed to the inability of the macrocycle to move freely along the axle in intermediates leading to [Pd(15b)]. Conversely, the macrocycle can move freely in the case of more flexible [Pd(15a)]. This suggests that the modification of the axle is once again not sequence-processive, with the cyclisation reaction taking place by a random walk pathway.
Recently, Saito and co-workers reported the transformation of rotaxane 16, whose axle contains a diyne unit, into pyrrole-containing rotaxane 17 by addition of aniline to the diyne unit mediated by a CuI ion coordinated in the macrocyclic cavity (Fig. 6).21 The rate of the double hydroamination reaction was found to be significantly higher in the case of the interlocked structure than the non-interlocked axle with a range of CuI sources, confirming that the reaction is significantly accelerated by the endotopically bound CuI ion in rotaxane 16. By varying the size of the aniline nucleophile employed the authors were able to control the rate of macrocycle shuttling in the product.
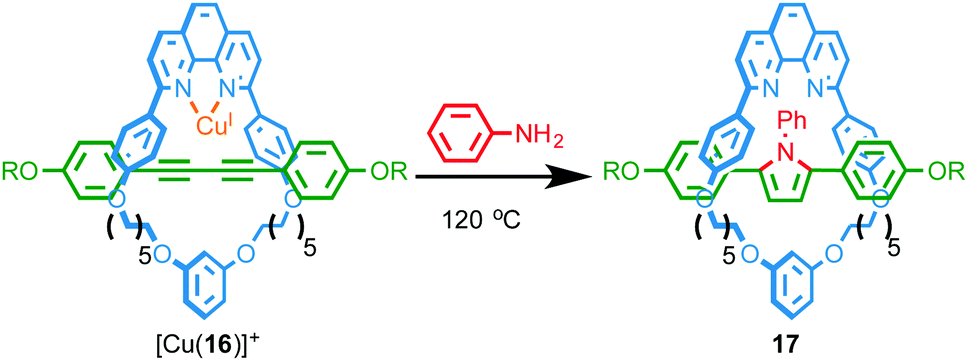 | ||
| Fig. 6 Double hydroamination of rotaxane 16 mediated by an endotopically coordinationed CuI ion.21 | ||
These examples demonstrate that metal ions bound within the macrocyclic cavity of rotaxanes can enable reactions of the axle that are hard to perform otherwise and, in some cases, lead to high processivity when transforming multiple functional groups, suggesting potential applications in the post-synthetic modification of polymers. However, unlike the enzymes they set out to mimic, in the systems disclosed to date the macrocyclic catalyst explores the axle and randomly transforms the functional groups. If these systems are to deliver sequence controlled post-synthetic polymer modification, methods are needed to match the rate of macrocycle motion to the rate of the catalytic process, or the development of other strategies to control the relative position of the macrocycle and axle, as has been demonstrated in related systems relying on organocatalysis.22
MIM complex-based catalysts with exogenous substrates
The sterically crowded nature of the mechanical bond can profoundly affect the reactivity of functional groups on the interlocked components23 including bound metal ions (cf. the catenand effect). Logically, this effect should extend to metal ions capable of catalysing reactions between exogenous substrates and thus interlocked ligands present an interesting opportunity for the development of new catalysts with unusual properties. However, only limited examples of ligands that take advantage of this effect have been reported to date.In 2008 Hagiwara and co-workers described catenane 18, which is functionalised by two phosphine moieties, and synthesised using a hydrogen bonding passive template (Fig. 7).24 This ligand has the potential to act as an unusual bidentate phosphine in which the bite angle is determined by the mechanical bond. Catenane 18 was investigated in the Pd-catalysed Suzuki–Miyaura cross-coupling reaction between bromobenzene and phenylboronic acid. Although the reaction was successful, producing biphenyl in 66% yield, no comparison was made with the non-interlocked ligand making it hard to assess the effect of the mechanical bond on the reaction. Furthermore, the ratio between the metal ion (1.5 mol%) and the potentially bidentate ligand 18 (2.8 mol%) suggests that only one of the two phosphine moieties is involved in coordination to Pd.
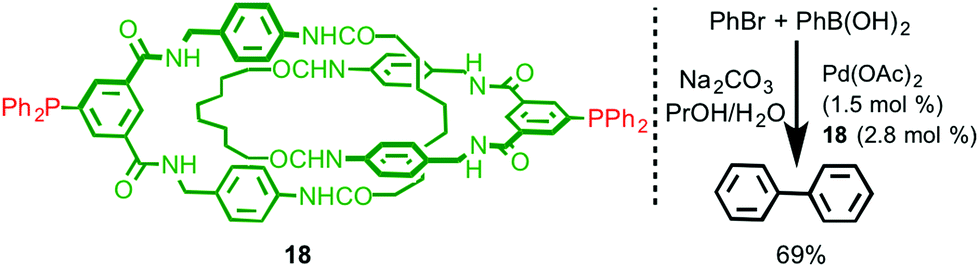 | ||
| Fig. 7 Suzuki–Miyaura coupling reaction with catenane 18 as a ligand.24 | ||
As in the case of Rowan and Nolte, and Takata's catalysts above, one of the advantages of interlocked molecules is the ability of the covalent components to move relative to one another. Osakada and co-workers investigated the potential advantages of this effect for transformation of exogenous substrates by synthesising [3]rotaxane [Pd2(19)(Cl)4] in which both macrocycles contain an exotopically ligated PdII ion (Fig. 8).25 They then investigated this binuclear PdII complex as a catalyst for the Heck cyclisation of bis-alkene 20 and bis-iodobenzene 21. The reaction was successful, producing target macrocycle 22 in higher yield and with greater selectivity than the same reaction mediated by Pd(OAc)2 in the presence of four equivalents of PPh3. The authors proposed that this might be due to the rotaxane architecture allowing both metal centres to react with the substrates simultaneously to promote macrocyclisation due to the ability of the two macrocycles to move relative to one another. This is, to date, the only example of using the mechanical bond to provide a flexible environment for bimetallic catalysis, an exciting potential application of MIM-based catalysts.26
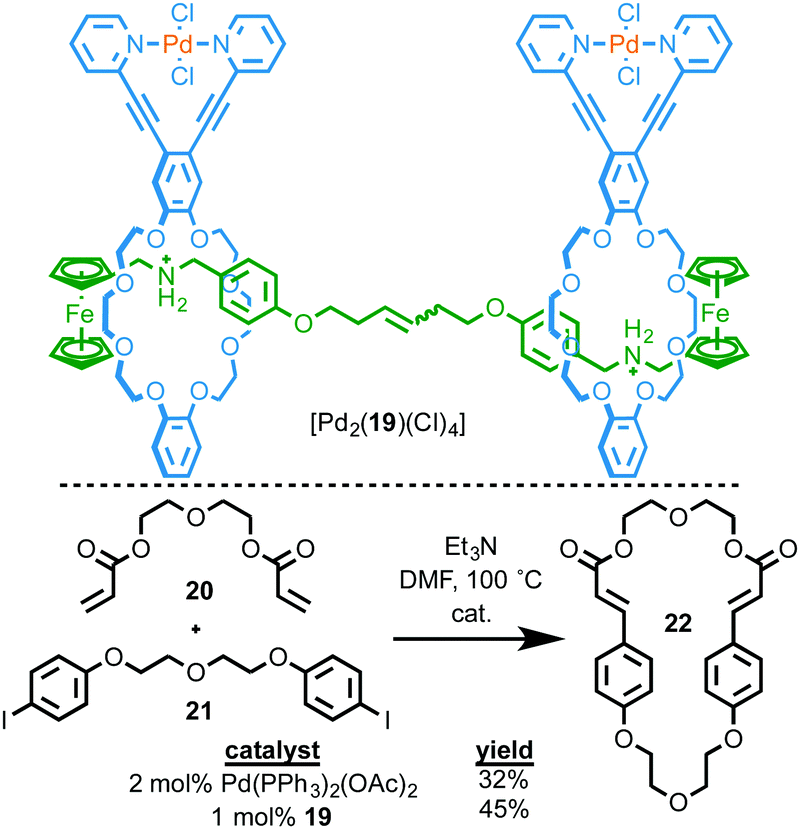 | ||
| Fig. 8 Ring-closing Mizoroki–Heck reaction by [3]rotaxane Pd complex [Pd2(19)(Cl)4].25 | ||
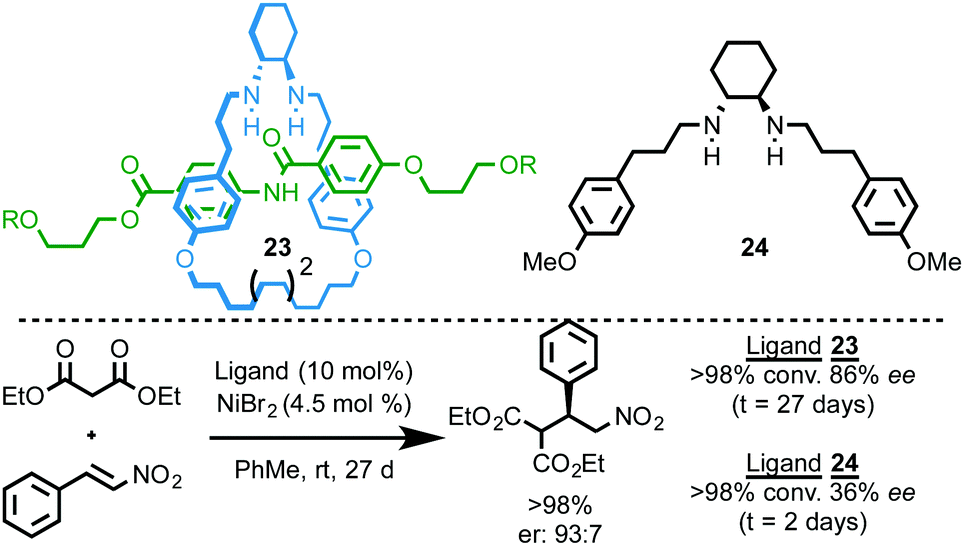
Leigh and co-workers reported a chiral [2]rotaxane ligand for a Nickel-catalysed enantioselective Michael addition (Fig. 9).27 Rotaxane 23, which is synthesised using an active template methodology, is unusual in that it can form coordinatively unsaturated endotopically bound complexes. The NiBr2 complex of rotaxane 23 mediates the Michael addition between diethyl malonate and trans-β-nitrostyrene to give the product in 86% ee. Rotaxane 23 delivers the product in higher ee than corresponding acyclic amine ligand 24 (86% and 36% ee respectively), although the rotaxane-mediated reaction is an order of magnitude slower (27 days vs. 48 h), presumably due to the hindered nature of the metal centre.
 | ||
| Fig. 9 An enantioselective Michael-addition reaction mediated by a rotaxane ligand.27 | ||
Recently we reported an interlocked gold catalyst with a number of unusual properties.28 Rotaxane AuI–phosphine complex [Au(25)(Cl)] was synthesised using an AT-CuAAC approach and its performance in the cyclopropanation reaction between styrene and propargyl ester 26 was compared with the corresponding non-interlocked catalyst. Surprisingly, although the non-interlocked catalyst mediates the cyclopropanation of styrene in moderate diastereoselectivity, rotaxane catalyst [Au(25)(Cl)] fails to mediate the reaction at all (Fig. 10).
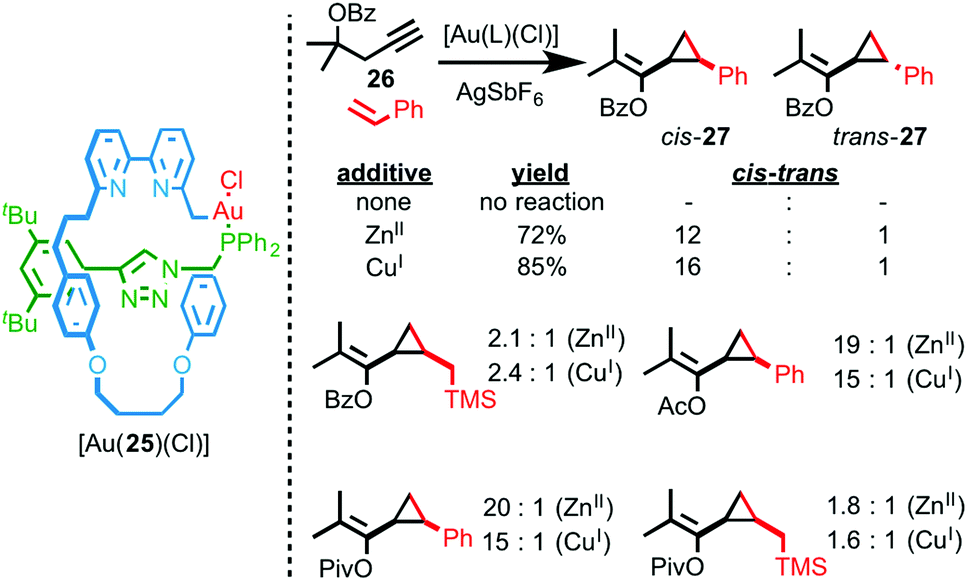 | ||
| Fig. 10 Sterically hindered, stimuli responsive rotaxane catalyst [Au(25)(Cl)] was studied in the Au-mediated cyclopropanation reaction shown and the effect of catalytically innocent cofactors on the diastereoselectivity of the reaction demonstrated.28 | ||
Ultimately, 1H NMR analysis suggested that binding of the bipyridine donors to the AuI centre inhibited the interlocked catalyst. Accordingly, addition of a catalytically innocent guest (CuI, ZnII), that binds inside the macrocycle cavity, reverses the inhibition and switches the catalyst on, leading to higher yields and diastereoselectivity compared with the non-interlocked axle. The enhanced diastereoselectivity of [Au(25)(Cl)] was rationalised by considering the buried volume (%Vbur – a measure of the space occupied by ligands in the coordination sphere of metal ions)29 values of the rotaxane and non-interlocked Au complexes, which demonstrated the enhanced steric hindrance provided by the mechanical bond.30 Intriguingly, not only does the identity of the additive influence the yield and diastereoselectivity of the reaction catalysed by [Au(25)(Cl)], the “best” additive varies depending on the substrate, with aryl esters favouring CuI as the co-factor and alkyl esters giving the highest selectivity in the presence of ZnII. This variation in dr with additive was rationalised by the effect of additive binding on the shape of the catalyst and thus the steric hindrance around the reaction site.
The development of rotaxanes as catalysts is still in its early stages. In order to demonstrate the key advantages of the mechanical bond in catalysis, in particular the effect of the crowded, flexible environment of the mechanical bond, it is important that proper controls are provided for comparison and this is not always trivial. For instance, the behaviour of [3]rotaxane [Pd2(19)(Cl)4] and rotaxane ligand 23, in both of which the catalytic metal ion is bound by the macrocyclic component, were compared with acyclic non-interlocked catalysts which makes it hard to tease apart the potential influences of ligand topology31 and the mechanical bond. This point notwithstanding, it is clear that interlocked ligands for catalysis bear further investigation as the studies above suggest the potential for the mechanical bond to influence the activity, chemo- and stereo-selectivity of catalysed reactions.
Mechanically interlocked ligands as sensors
Rotaxanes and catenanes have the proven potential to act as hosts for ions and small molecules by binding the guest in the crowded environment created by the mechanical bond.32 The extension of this property to sensors requires the development of systems that display a “readable” signal (typically optical or electrochemical) that selectively distinguish between different potential guests.MILs that report the binding of metal ions
A simple way to design a metal responsive MIM is by employing the mechanically chelating ligand produced in a metal ion mediated passive template synthesis to bind the analyte. Swager and co-workers demonstrated this approach by employing Sauvage's phenanthroline–CuI template to produce rotaxane 28 that displays exciplex emission in the metal-free state (Fig. 11).33a Addition of ZnII to a solution of 28 leads to quenching of the longer wavelength emission as the energy levels of the coordinated phenanthroline moiety and relative orientation of the chromophores in metal complex [Zn(28)]2+ disfavour exciplex formation.34 Other metal ions also quenched the exciplex emission. The authors later extended this system to the solid state by electropolymerisation of an interlocked precursor and demonstrated that ZnII or CuI binding alters the electrical conductivity and optical properties of the conjugated polymer.33b,c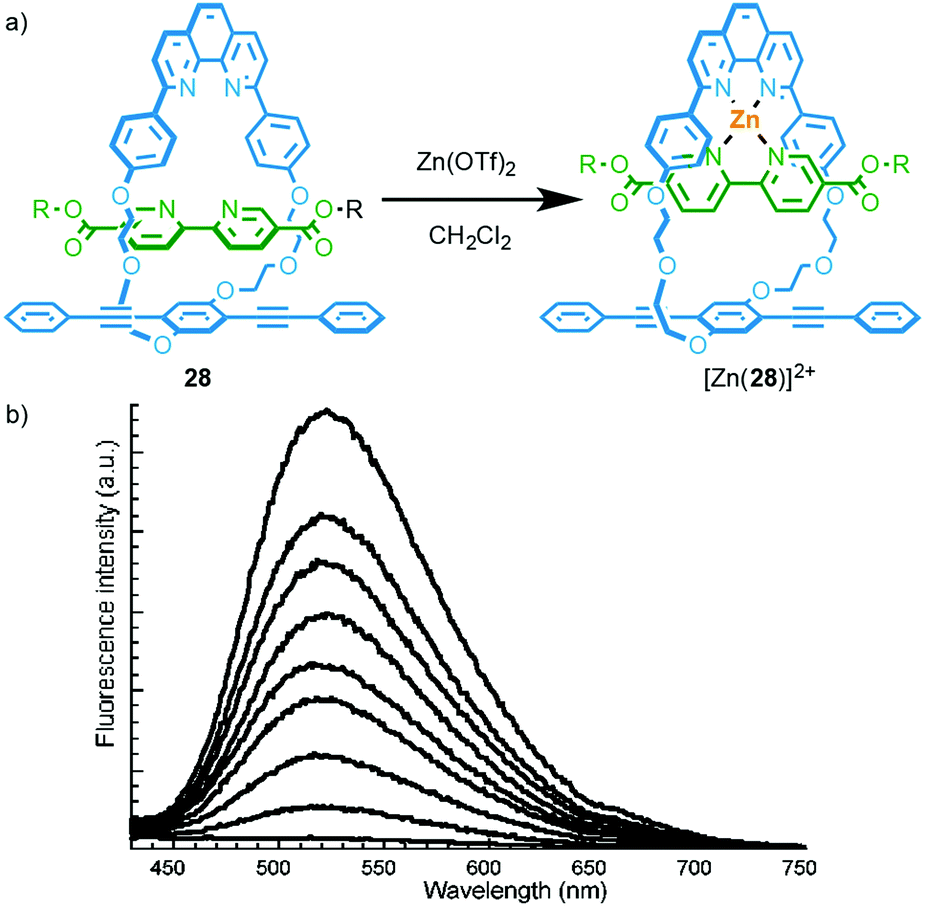 | ||
| Fig. 11 (a) Emissive rotaxane 28; (b) the response of 28 to ZnII.33a Adapted with permission from ref. 33a. Copyright 2001 American Chemical Society. | ||
Rotaxane 28 relies on metal binding in the mechanically chelating ligand produced in the passive template synthesis to induce electronic and conformational changes that result in optical and electrochemical read out. It is also possible to use metal binding in systems where the metal binding unit is not residual from the method of synthesis. Catenane 29, which is synthesised through a hydrogen bond directed strategy, exhibits a switch on fluorescent response in the presence of ZnII (Fig. 12).35 The origin of this effect is not entirely clear but it appears that the binding of ZnII interrupts the salt bridge between the amidine and carboxylate units,36 leading to enhanced relative motion of the two macrocycles, as shown by the loss of CD signal on complexation. Addition of a cryptand to sequester the ZnII ion regenerates the free ligand.
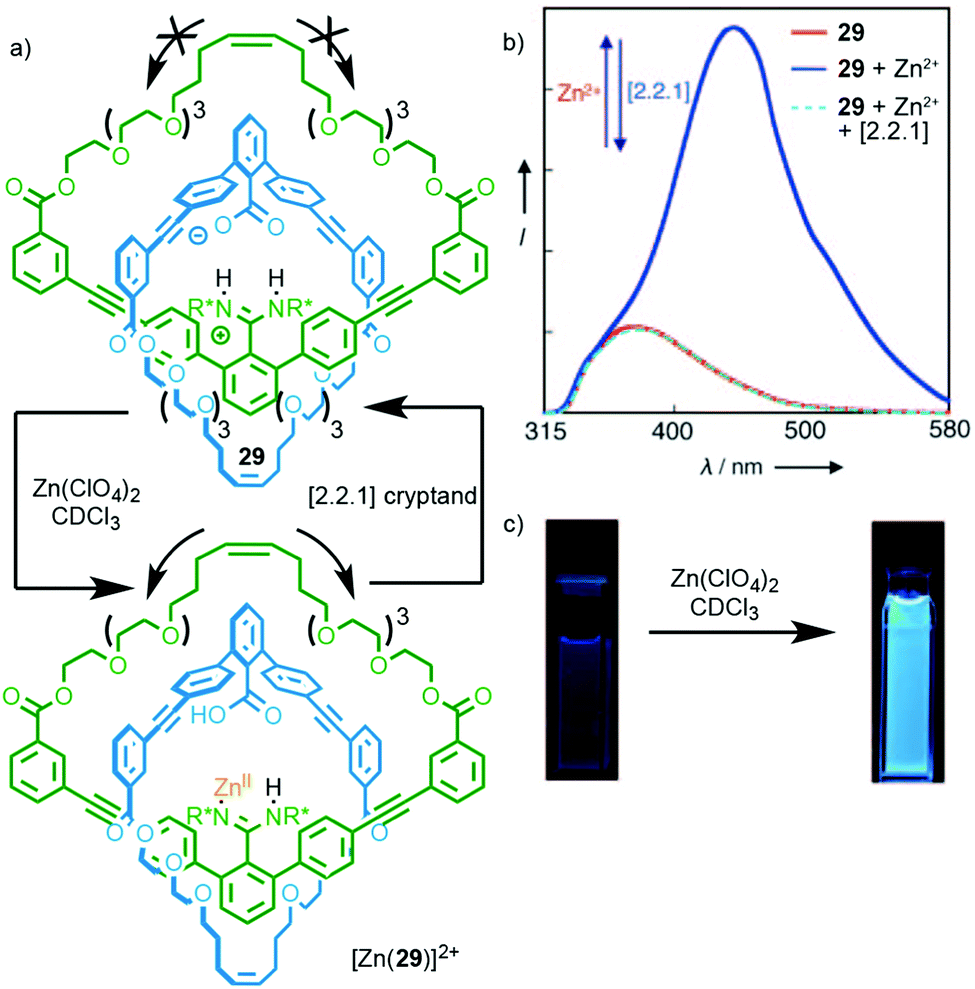 | ||
| Fig. 12 (a) Catenane 29 synthesised using a salt-bridge template; (b) addition of ZnII interrupts the salt bridge leading to visibly enhanced fluorescence (c).35 Adapted with permission from ref. 35. Copyright © 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In 2004 Hiratani and co-workers reported [1]rotaxane3730 that selectively binds LiI over other group I metal ions (Fig. 13);381H NMR studies demonstrated that LiI was able to bind in the cavity formed between the crown ether macrocycle and the amide axle component, whereas NaI and KI did not bind.39 Furthermore, binding of LiI was proposed to lead to a (co)conformational change that brings the macrocycle and anthracene moiety into closer proximity. This in turn enhanced energy transfer between the naphthol moiety of the macrocycle and the anthracene moiety of the axle, resulting in an enhanced emission. The same authors later extended this approach to a [3]rotaxane that shows different coordination behaviour with LiI and the larger CsI ion;40 while LiI is coordinated within the macrocycle cavity and forms a 2:1 complex, the larger CsI ion is coordinated between the macrocycles in a 1:1 complex. Coordination of CsI is reported to result in an increase in fluorescence intensity.41
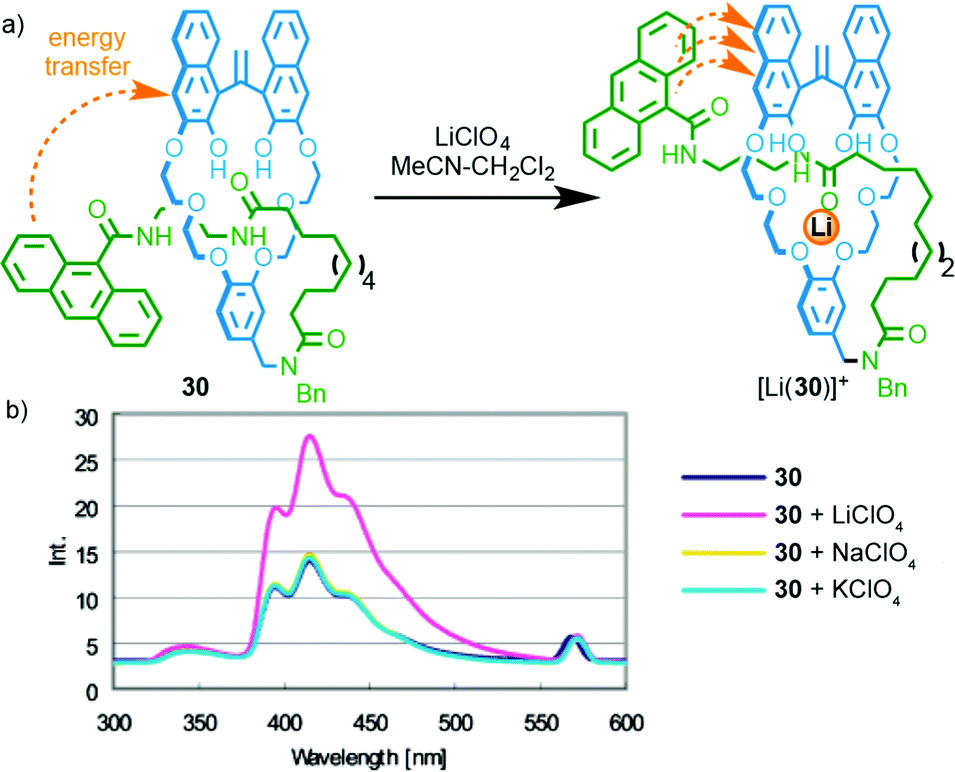 | ||
| Fig. 13 (a) Li-selective rotaxane based host 30 and (b) its fluorescence response in the presence of group I metal ions. Adapted with permission from ref. 38. Copyright 2004 American Chemical Society. | ||
Perhaps the best-known role of metal ions in the development of stimuli responsive MIMs is as molecular shuttles where addition or removal of metal ions leads to changes in the relative position of the interlocked components.1 Most of these systems are studied either by NMR or cyclic voltammetry. However, a small number of examples have been reported in which metal driven changes in the co-conformation leads to an optical output.
In 2008 Li and co-workers demonstrated metal-responsive shuttle 31 that displays changes in optical output depending on its coordination state (Fig. 14).42 In the neutral state, the macrocycle occupies the amide station and photo-induced electron transfer from the amine moieties of the axle and macrocycle quenches the anthracene emission. Addition of LiI leads to an increase in emission that the authors propose is due to a change in orientation of the macrocycle on the amide station reducing the efficiency of the quenching process. Addition of ZnII to rotaxane 31 led to shuttling of the macrocycle from the amide station to the amine to provide the Zn2+ ion with a tetra-aza binding pocket. This led to a much larger increase in the fluorescence output from the anthracene moiety as the amine in the axle and the macrocycle, now coordinated to ZnII, are no longer able to quench the excited state by electron transfer.
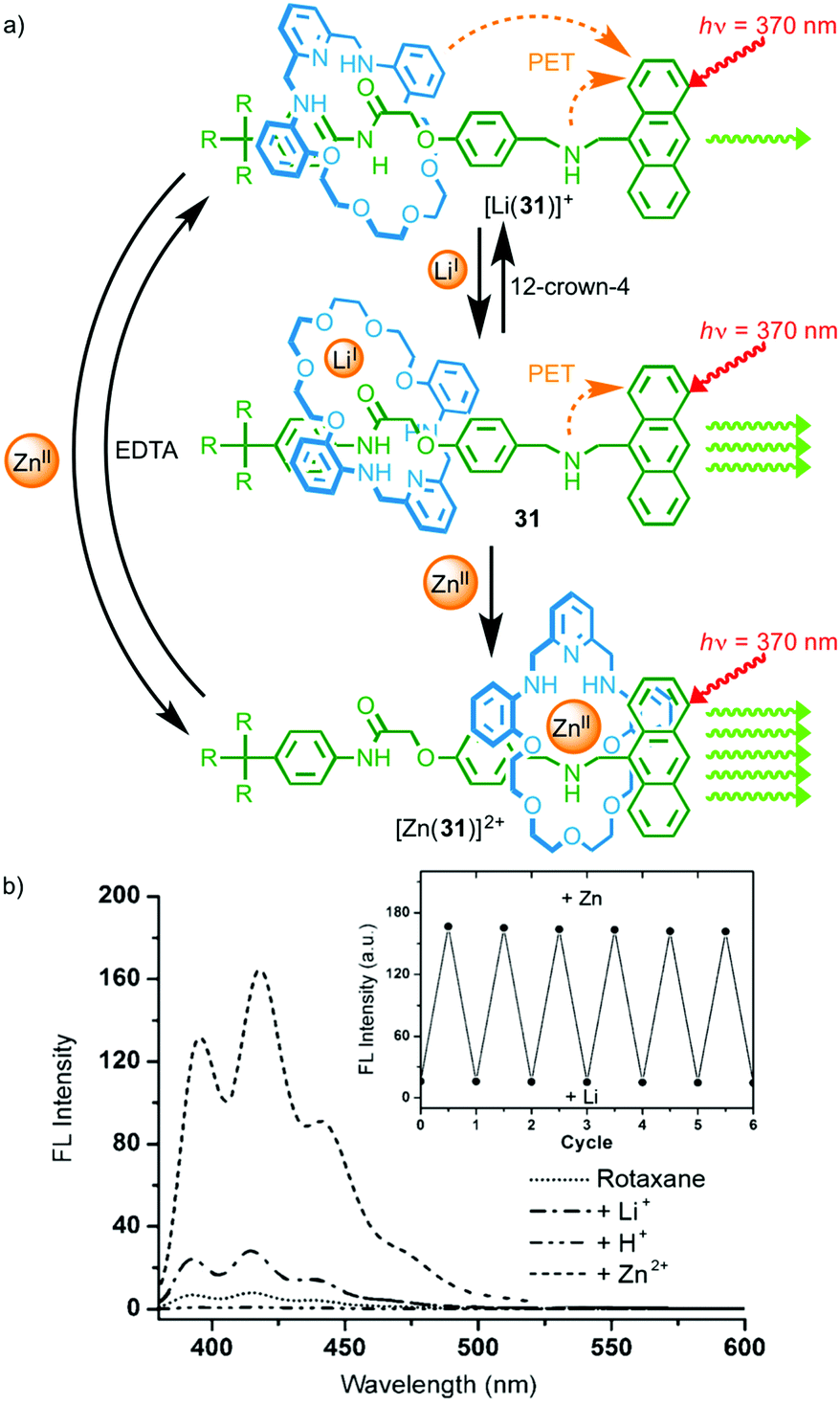 | ||
| Fig. 14 (a) Metal responsive rotaxane 31 and (b) the observed changes in emission intensity depending on the coordination state.42 The exchange of ZnII for LiI and vice versa was reversible over a number of cycles (inset). Adapted with permission from ref. 42. Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Chiu and co-workers took a different approach in the development of rotaxane molecular shuttle 32, which also shows optical read out of metal ion binding (Fig. 15).43 In this case, the macrocycle occupies the bipyridinium station in the absence of metal ions. This exposes the squarine dye to the polar solvent (MeCN) and results in quenching of the emission. Addition of NaI leads to shuttling of the macrocycle to the squarine station due to ion–dipole interactions between the crown ether windows of the macrocycle and the alkoxide moieties of the dye, shielding the dye from its environment and increasing the quantum yield of the squarine unit.
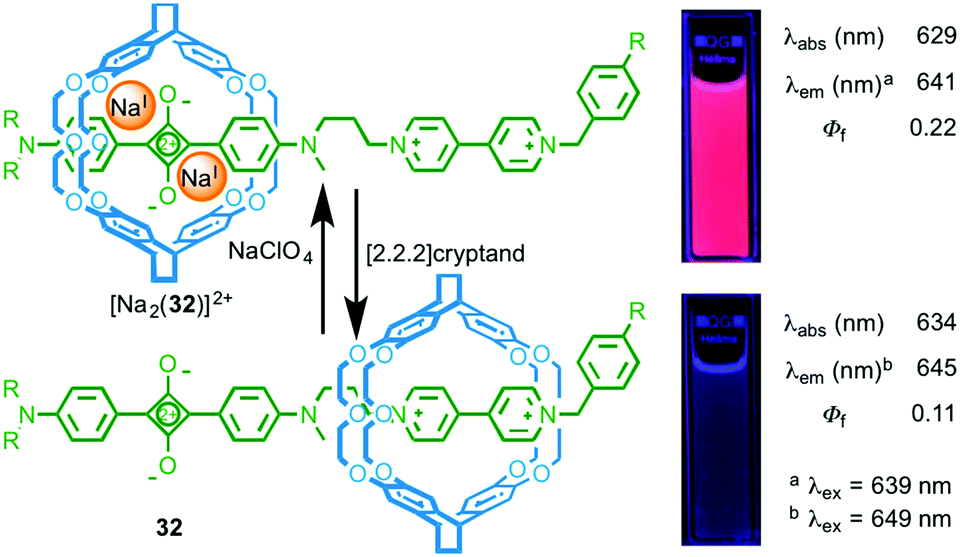 | ||
| Fig. 15 Molecular shuttle 32 and its response to NaI binding.43 Adapted with permission from ref. 43. Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Relatively few examples of MIMs that display a “readable” signal on metal binding have been reported, despite the extremely large number of reported MIMs with metal binding units. Furthermore, although these examples produce a useful optical read-out, the majority do not report selectivity in the response; only in the case of [1]rotaxane 30 and shuttle 31 are different responses reported for different metal ions. Thus, although all of the above examples demonstrate the potential of MIMs to bind metal ions and report that binding event, only Hiratani's system (Fig. 13) demonstrates the potential of this approach in the development of sensors by using the confined cavity of the rotaxane to develop a selective three dimensional binding site for a specific analyte. Given the proven potential of rotaxanes as sensors for small molecules and ions (see below), this seems to be an area with significant potential.
Metal ions as reporter units in rotaxane-based sensors
In contrast to the limited examples of selectivity in metal ion binding with MILs, interlocked hosts have been reported to bind to a range of anionic and small molecule analytes thanks to the restrictions imposed by the mechanical bond.32 Work from Beer and co-workers demonstrates both the potential of MIMs as selective hosts and an alternative application of metal–ligand interactions in sensor development; using the bound metal ion as a reporter unit.As with metal ion templated MIMs, MIMs prepared via anion templation methods tend to have high affinities for anions. Importantly, the shape and size of the cavity generated between the MIM components often leads to high selectivity between anions, which in some cases contradicts those obtained with the non-interlocked components. For example, although the corresponding non-interlocked macrocycle alone binds most strongly to ClI, interlocked hydrogen bonding host 33, which contains a ReI centre bound to the exocyclic bipyridine unit, shows a greater than 10-fold higher binding constant for HSO4− (Fig. 16). The presence of the emissive Re(CO)3Cl complex allowed the binding of anions to be monitored by fluorescence spectroscopy (Fig. 16b).44,45 Recently Beer and co-workers have extended this approach to halogen bonding systems that display selectivity for iodide under aqueous conditions.46 The same approach has been extended to other emissive metal ion reporters including MILs for EuIII,47 and RuII.48
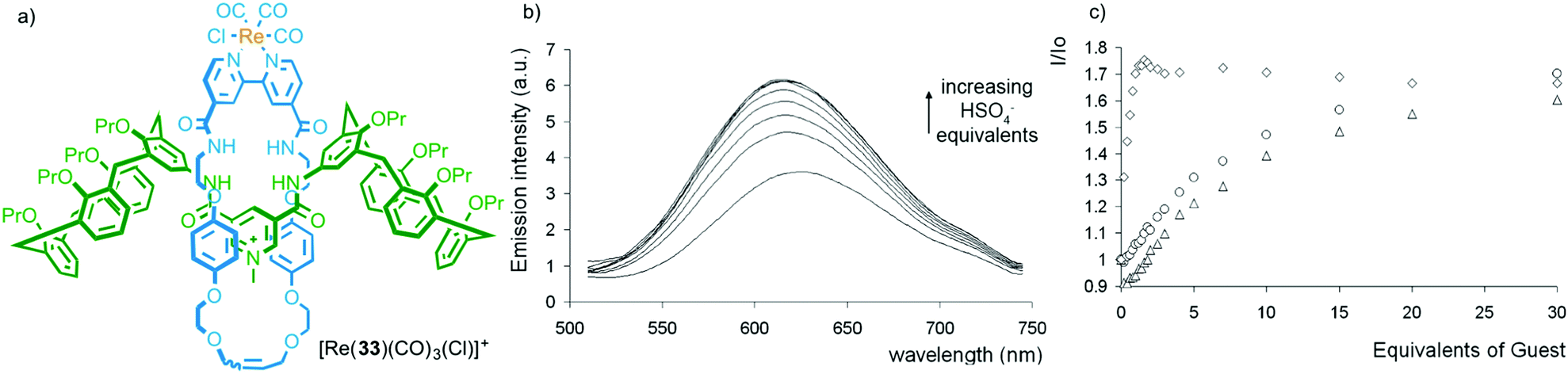 | ||
| Fig. 16 (a) Beer's hydrogen bonding host [Re(33)(CO)3(Cl)]+, (b) its optical read out on HSO4− addition and (c) a comparison with the response of the rotaxane host (diamonds), the corresponding non-interlocked macrocycle (triangles) and an acyclic analogue (circles).44 Reproduced from ref. 44 with permission from the Royal Society of Chemistry. | ||
In addition to their emissive properties, metal ions also offer the possibility of electrochemical read out. Beer and co-workers have demonstrated this using ferrocene substituted MIMs in which anion binding perturbs the FeII/III couple leading to a cathodic shift (Fig. 17).49 Extending this to OsIII, as in rotaxane 34, and mounting the host on a gold surface allowed the anion binding event to be detected both optically and electrochemically.50
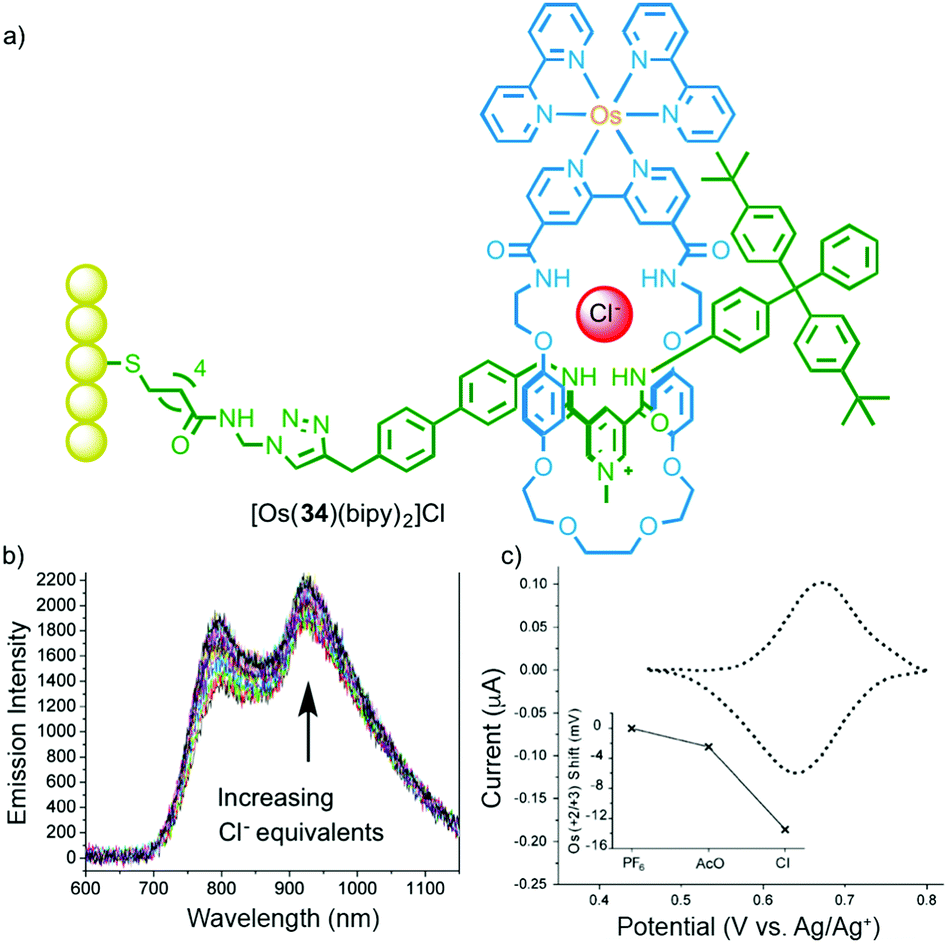 | ||
| Fig. 17 (a) Surface appended electrochemical anion sensor [Os(34)(bipy)2] with bound chloride anion and (b) its optical and (c) electrochemical response to Cl.50 Adapted with permission from ref. 50. Copyright 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The examples presented above clearly show that metal ions bound within the MIM structure can be used to report changes upon guest binding which, combined with the selectivity imparted by the mechanical bond, provide an excellent platform for sensor development.
Mechanically interlocked ligands in metallo-supramolecular chemistry
There is currently a growing interest in metallo-supramolecular materials in which metal–ligand interactions are used to direct the assembly of complex 1D, 2D and 3D architectures.51 In the final section of this Feature Article we will discuss systems that attempt to combine MILs with metallo-supramolecular assembly. We have chosen to focus on robust, permanently interlocked building blocks rather than metallo-supramolecular systems based on pseudorotaxane units in which the metal–ligand interactions are integral to the stability of the interlocked structure.52Discrete metallo-supramolecular assemblies
As a first step towards metallo-supramolecular assemblies, in 2003 Loeb and co-workers demonstrated the homo-dimerisation of rotaxane ligand 353+, which is functionalised by a terpyridine stopper unit, to give a simple MIM-based metal complex. Treatment with FeII led to a homoleptic low-spin FeII complex in which two rotaxane ligands bind to the Fe centre and the resulting complex was characterised by 1H NMR, MS and UV-vis spectroscopy (Fig. 18).53 What is perhaps noteworthy about this relatively simple coordination complex is that, by associating the axle units using metal–ligand interactions, the macrocycle components are also brought into proximity. Crowley and co-workers demonstrated the inverse of this effect by substituting the macrocycle component of a [2]rotaxane with terpyridine moieties.54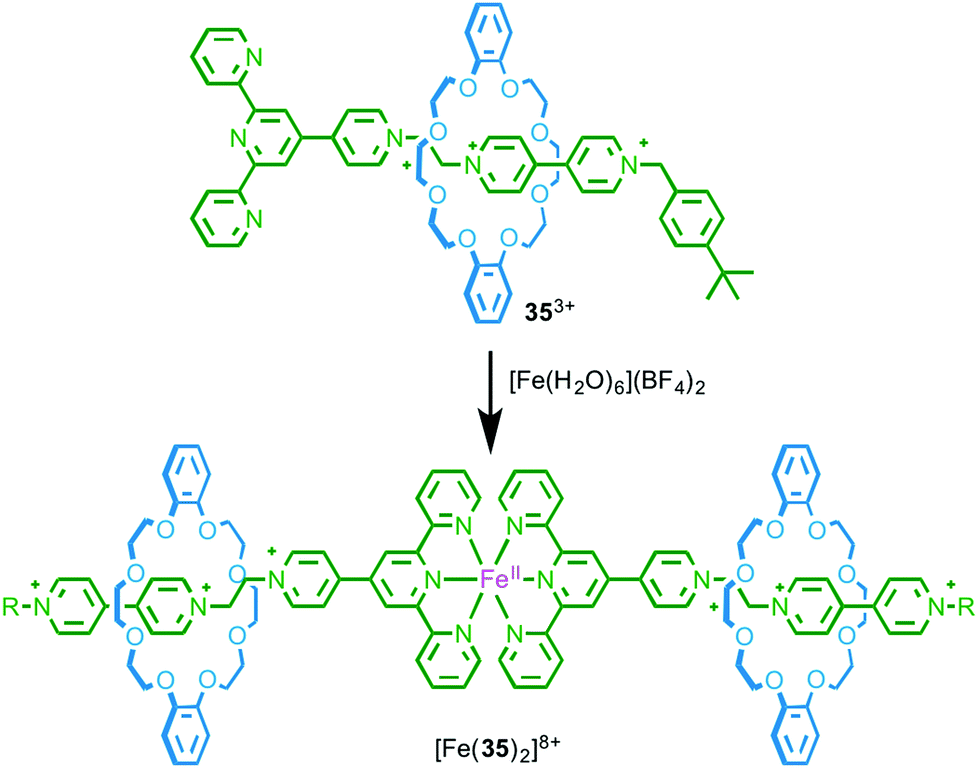 | ||
| Fig. 18 Dimerisation of rotaxane ligand 353+ in the presence of FeII.53 | ||
Wang and co-workers took advantage of such a coordination-driven dimerisation of [2]rotaxane ligands to construct supramolecular polymers using hierarchical self-assembly.55 Monomer unit [2]rotaxane 36 is stoppered by a terpyridine moiety and the macrocycle is substituted with a Hamilton receptor (Fig. 19). Addition of ZnII leads to the efficient dimerisation of the rotaxane ligand, which produces an assembly containing two Hamilton receptors. At concentrations above ∼7 mM, this dimer self-assembles through H-bonding interactions in the presence of bis-cyanuric acid derivative 37 to produce a supramolecular polymer, and the polymerisation process was monitored by DOSY NMR and viscometry. Importantly, neither ZnII nor bis-cyanuric acid derivative 37 alone are sufficient for supramolecular polymerisation to take place. The product polymer forms flexible, transparent free-standing films.
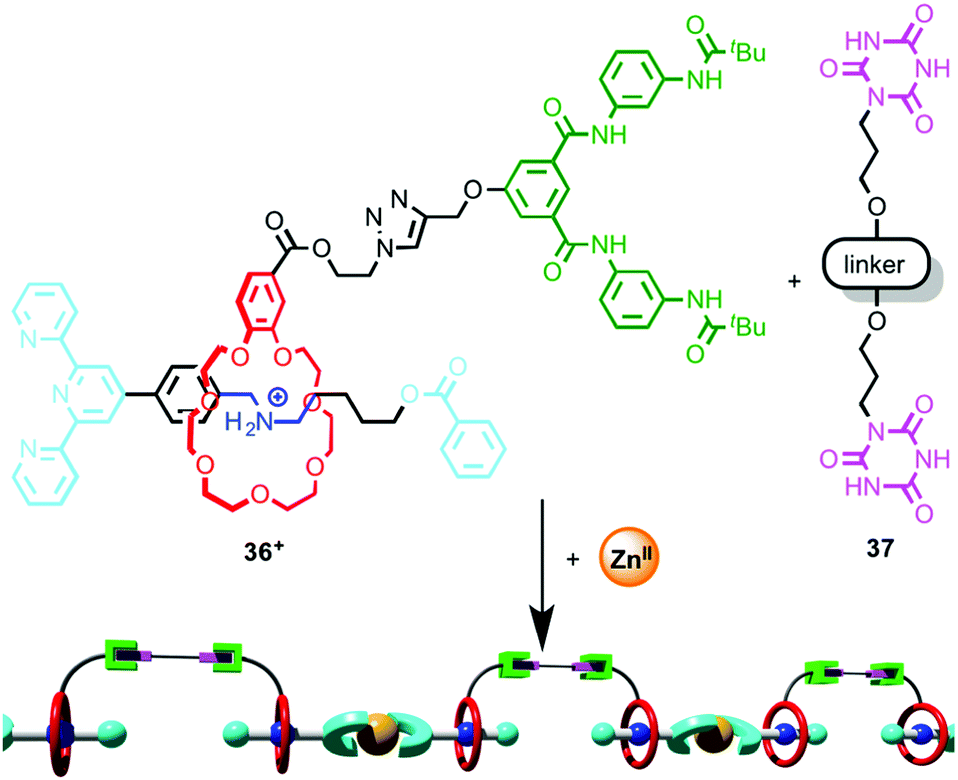 | ||
| Fig. 19 Hierarchical self-assembly of a metallo-supramolecular dimer.55 | ||
In the case of the examples above either the axle or macrocycle component provide the ligand used in the assembly of the metallo-dimer and the only effect of the mechanical bond is to associate the second covalent component with the assembly. In 2014 Crowley and co-workers observed a dimeric complex in which both the macrocycle and thread provide donor atoms (Fig. 20). In the presence of AgI ions, [2]rotaxane 38 dimerised to form an M2L2 assembly; in the solid state the silver ions adopted a distorted trigonal planar geometry, with the 1,2,3-triazole units of the thread acting as bridging ligands between two AgI ions (forming a six membered metallocycle), and the pyridines of the macrocycle completing the coordination sphere (Fig. 20b).56
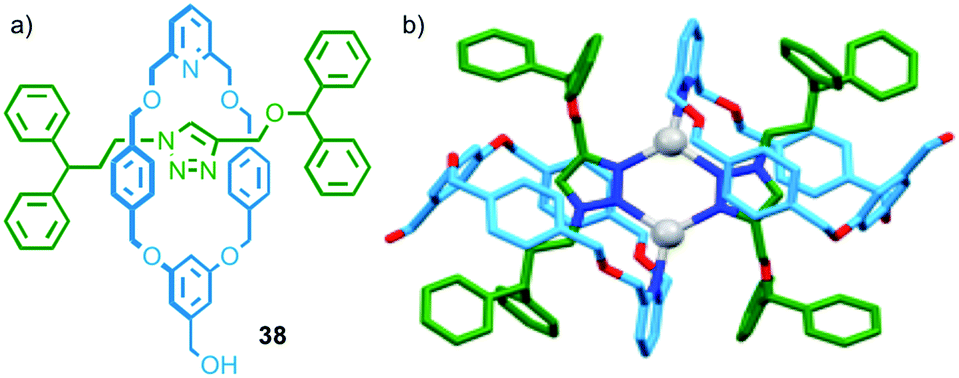 | ||
| Fig. 20 (a) Interlocked rotaxane ligands 38 and (b) the solid state structure of the corresponding dimer formed in the presence of AgI.56 | ||
Schalley and co-workers recently extended this approach to surface modification by using a PdII ion to link rotaxane 39 to a gold surface and study the ordering of the interlocked molecule at the interface (Fig. 21a).57 The terpyridine ligand of rotaxane 39 was first coordinated to PdII to give a stable complex with a labile coordination site. A monolayer was prepared functionalised by a linear mono-pyridine ligand. Immersion of this monolayer in a solution of [Pd(39)(MeCN)] resulted in coordination to the monopyridine ligands, generating an ordered array of rotaxanes bound to the gold surface. X-ray photoelectron spectroscopy (XPS) confirmed the presence of the iodine-labeled rotaxane axle on the surface, with near-edge X-ray absorption fine structure (NEXAFS) spectroscopy revealing nitrogen resonances indicative of the macrocycle, thus leading to the conclusion that the rotaxanes had been successfully bound to the surface. A linear dichroism effect was observed in the NEXAFS C K-edge, suggesting the formation of ordered arrays of the surface-bound rotaxanes, and a packing density of 15–20% was estimated from XPS data.
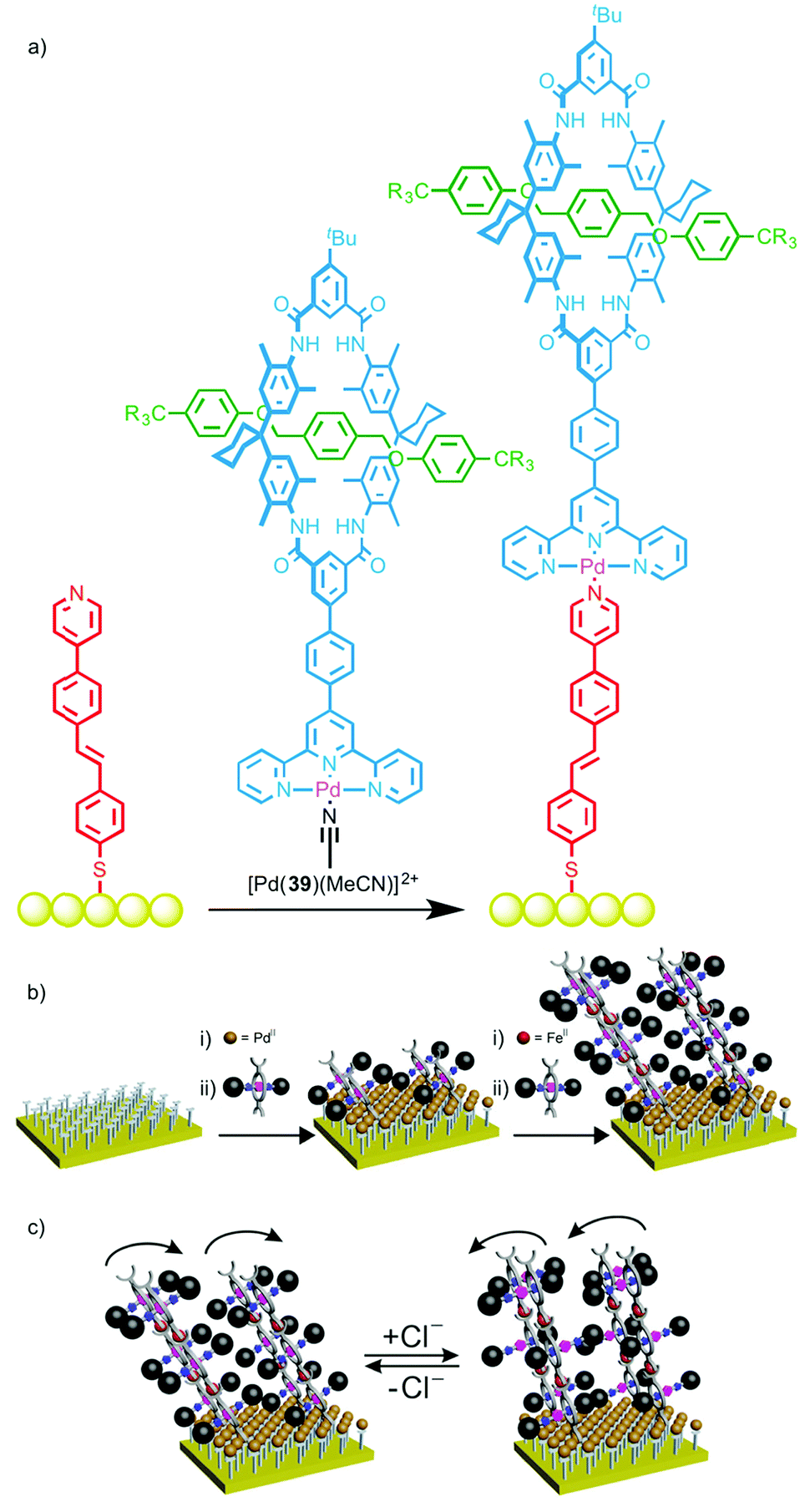 | ||
| Fig. 21 (a) Schalley's rotaxane monolayer assembled using metallosupramolecular chemistry;57 (b) a cartoon representation of the layer-by-layer deposition of Cl-responsive rotaxanes and (c) their proposed switching in the presence of Cl anions.58 Adapted with permission from ref. 58. Copyright 2015 American Chemical Society. | ||
This self-assembly technique was subsequently employed to deposit multilayers of a chloride-responsive rotaxane in which the macrocycle was substituted with two terpyridine moieties (Fig. 21b).58 This allowed the layer-by-layer deposition by sequentially washing the surface with solutions of Fe(BF4)2·6H2O and the rotaxane, with addition of up to 20 layers reported. Most impressively, the surface-bound rotaxane multilayer retained its ability to bind chloride ions; reversible changes in contact angle measurements and angle-resolved NEXAFS spectra upon addition and removal of chloride ions suggested on-surface switching took place (Fig. 21c).
Although there is increasing interest in the use of MIMs in metallo-supramolecular assembly, relatively little attention has been paid to the effect of the mechanical bond on the outcome of the assembly process. Loeb has recently found that bis-pyridine ligand 40 and its interlocked analogue 41 form different architectures in the presence of PtII(dppp) (Fig. 22);59 non-interlocked axle 40 formed a simple M2L2 metallo-rectangle whereas [2]rotaxane 41 formed a M3L3 molecular necklace and higher order MnLn complexes. The difference in behaviour between rotaxane 41 and axle 40 was attributed to the higher rigidity of the interlocked ligand induced by the encircling macrocycle, combined with the increased steric demand provided by the macrocycle; the macrocycles completely fill the cavity of the triangle.
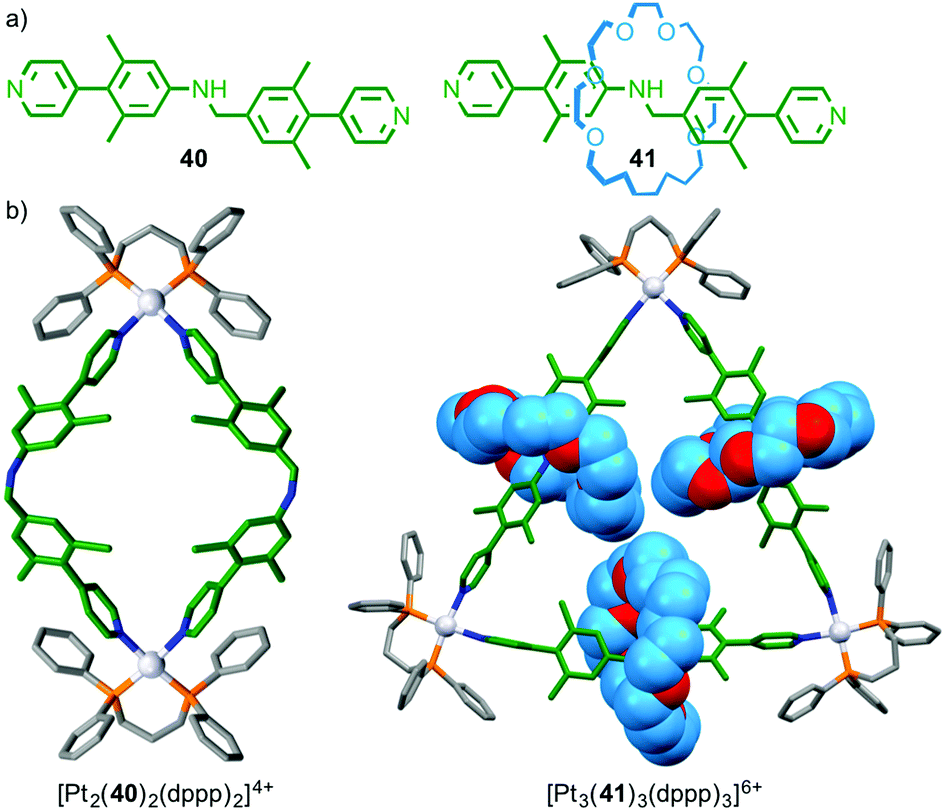 | ||
| Fig. 22 (a) Rotaxane 41 and corresponding non-interlocked axle 40 and (b) the products of their self-assembly in the presence of PtII.59 | ||
Interlocked metallo-supramolecular polymers
The discrete assemblies above, in particular Loeb's molecular triangle, suggest the possibility of larger metallo-polymers.60 The first such structure based on a permanently interlocked [2]rotaxane linker relied on ligands not in the axle component but in the macrocycle.61 In 2011 Loeb and co-workers reported a series of rotaxanes in which the macrocycle component was appended with pyridine-based ligands. When rotaxane 42, in which each macrocycle bears four 3-substituted pyridine units (Fig. 23a), was combined with CdII a coordination polymer was obtained in which the pyridine ligands of each macrocycle coordinate to a different CdII ion (Fig. 23b). The authors later extended the approach by replacing the pyridine ligands with thio-ether units to create coordination polymers using AgI–S interactions.62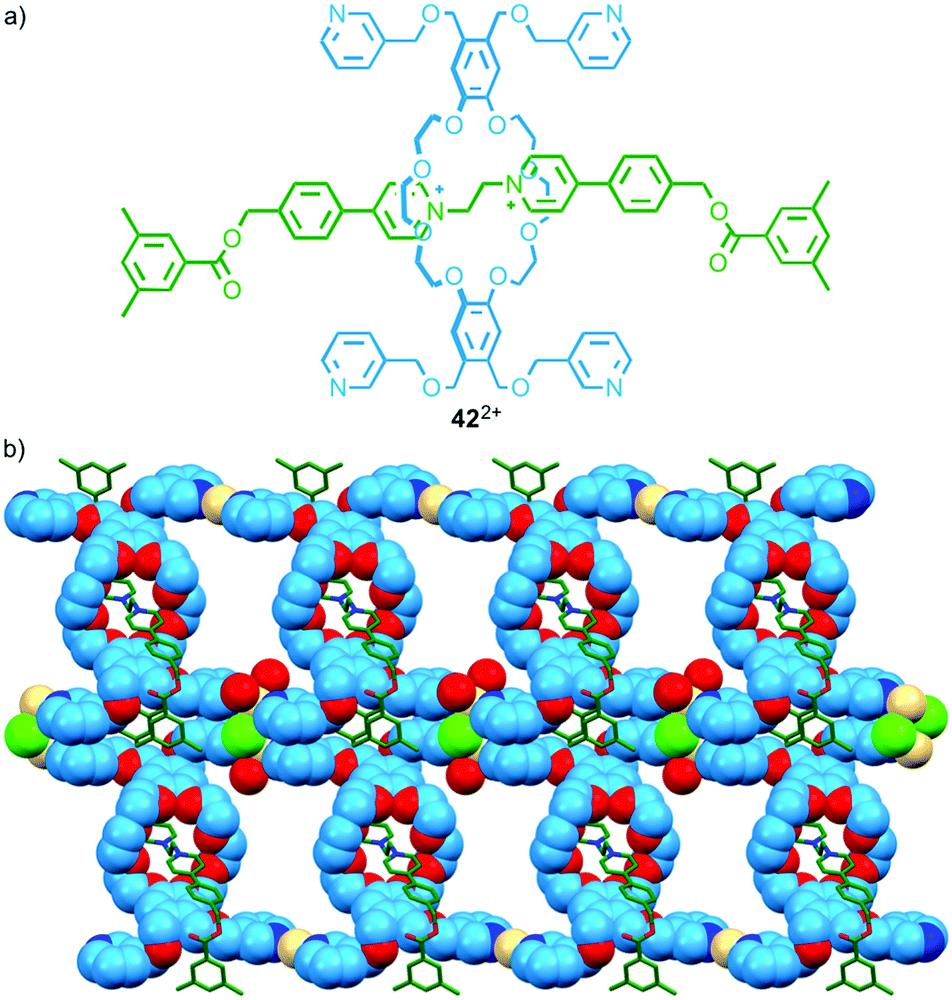 | ||
| Fig. 23 (a) Tetradentate interlocked ligand 422+ and (b) the product of its self-assembly in the presence of CdII.61 | ||
Through post-synthetic modification of a [2]rotaxane, Crowley and co-workers were able to install terpyridine units on both the macrocycle and thread components (Fig. 24).54 Addition of [Fe(H2O)6](BF4)2 to a dilute solution of functionalised rotaxane 43 resulted in formation of a simple ML complex, with coordination of the macrocyclic and thread terpyridine units from the same rotaxane to an FeII ion. Conversely, addition of FeCl2 to a concentrated solution of the rotaxane yielded metallo-oligomeric species, underlining the important role that the reaction conditions play in determining the outcome of coordination-based polymerisation reactions. Using both DOSY NMR and GPC the metallo-oligomers were estimated to be formed from between 11 and 13 rotaxane units.
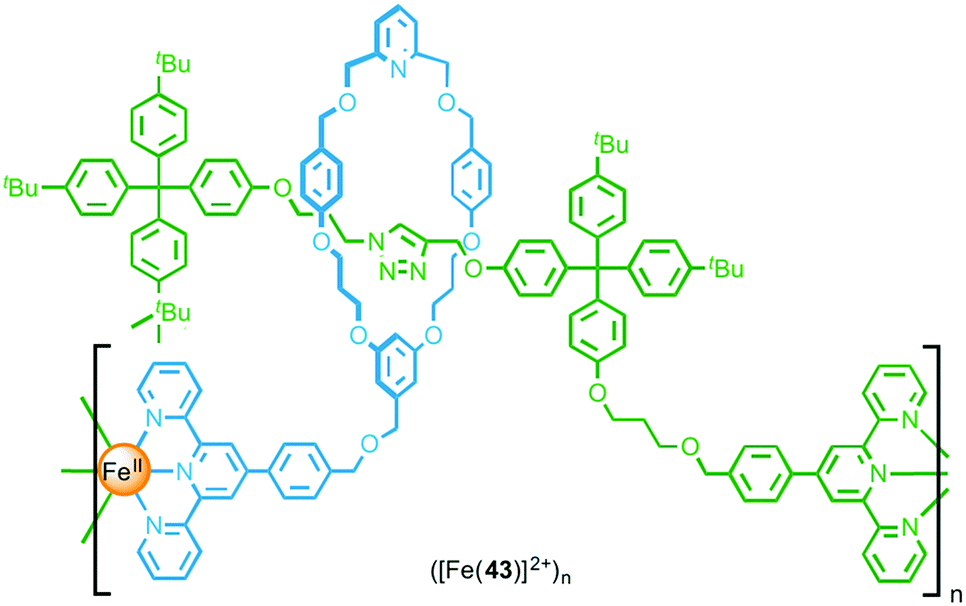 | ||
| Fig. 24 A metallo-supramolecular polymer formed by coordination of FeII to both the macrocycle and the axle. | ||
In 2014 Huang and co-workers reported a rotaxane molecular machine-based metallo-supramolecular polymer.63 Rotaxane 44 (Fig. 25) is based on their previously reported “molecular spring” in which changing the solvent environment changes the preferred positions of the macrocycles on the axle, resulting in a change in the average length of the assembly.64 In the presence of FeCl2, 44 assembles into a 1D supramolecular polymer and the resulting fibres were visualised by TEM and SEM. As with the monomeric building block 44 (Fig. 25), variation in the solvent environment changes the length of the assembled metallo-supramolecular polymer and these changes were visualised using DLS.
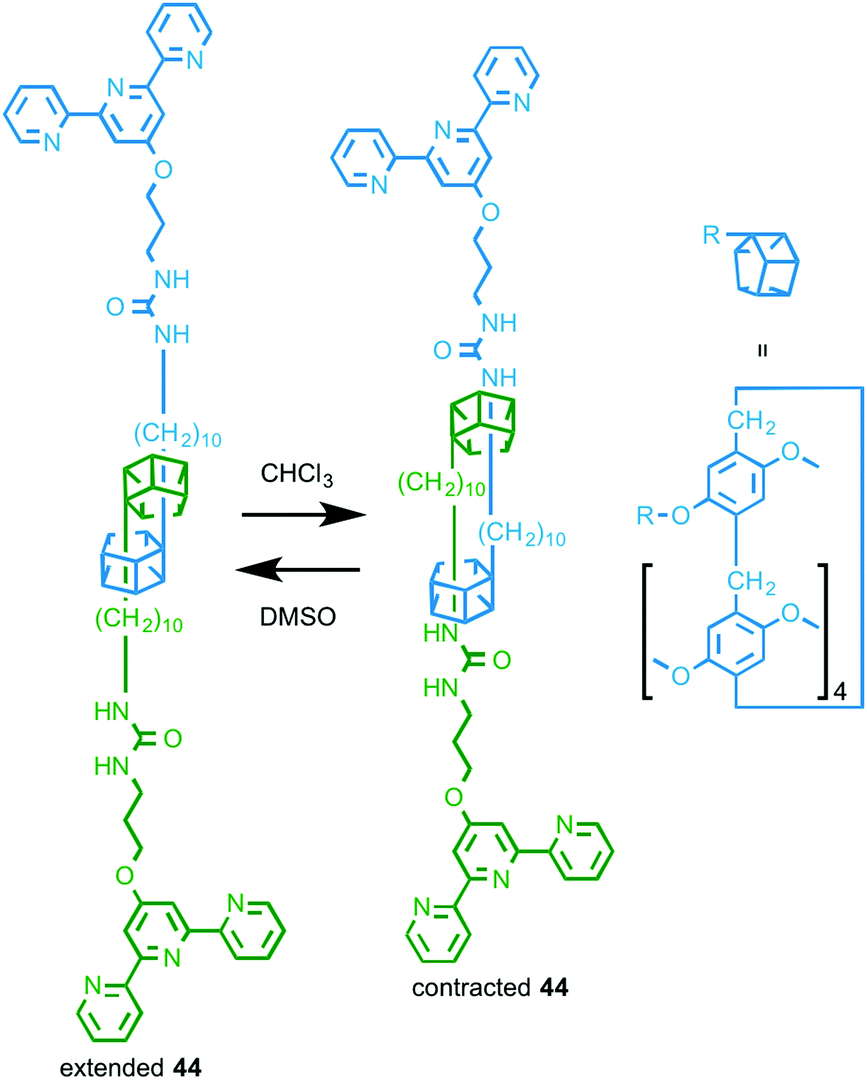 | ||
| Fig. 25 Solvent responsive monomer 44 that assembles in the presence of FeII to give a stimuli-responsive metallo-supramolecular polymer.63 | ||
Metal–organic frameworks (MOFs)
Crystalline coordination polymers in which the network contains appreciable void space – commonly referred to as metal organic frameworks (MOFs) – have become an intense area of research for applications as diverse as drug delivery, gas sorption, sorting of chemical mixtures,65 and recently as an analytic tool in X-ray crystallography.66 In recent years several research groups have sought to combine MOFs and MIMs to generate networks with interlocked ligand components.Loeb and co-workers have demonstrated a number of crown ether–ammonium templated rotaxanes as MOF building blocks. In 2012 they reported that tetra-carboxylic acid rotaxane 45 formed a crystalline MOF when heated with Cu(NO3)2·3H2O in DMF–EtOH–H2O and allowed to cool slowly (Fig. 26).67 Single crystal X-ray analysis confirmed the porous nature of UWDM-1 and revealed that the crown ether macrocycle is hydrogen bonded to a water molecule bound to a CuII paddle-wheel unit (Fig. 26b). This interaction, combined with steric interactions with further molecules of H2O occupying void space in the lattice, lock the macrocycle in place, preventing free rotation of the macrocycle relative to the lattice.
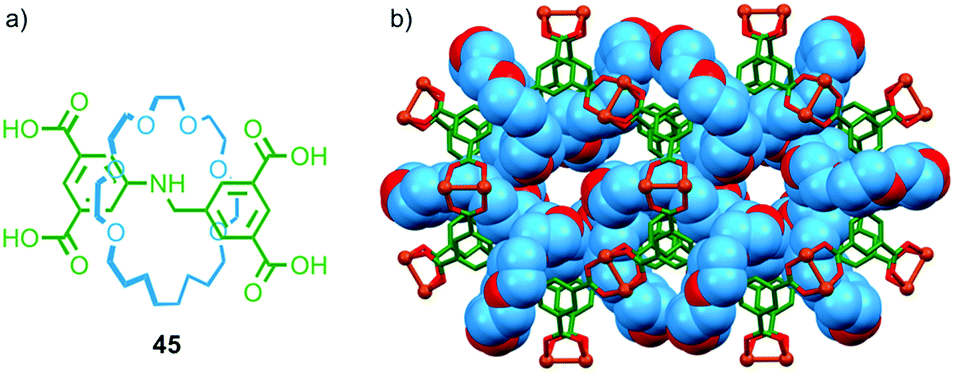 | ||
| Fig. 26 (a) Tetracarboxylic acid ligand rotaxane 45 and (b) the MOF formed by 45 in the presence of CuII.67 | ||
Heating the green, as-synthesised material to 150 °C in vacuo removed these water molecules. In the new lattice the macrocycle components are significantly less restricted and a number of dynamic modes were observed including pirouetting of the ring around the axle. This activation was reversible and the material could be switched between rotating and locked states repeatedly. The rotation of the macrocycles around the struts of the framework could be monitored by variable temperature solid-state 13C and 2H NMR (of a labelled analogue), which showed the barrier to be around 50 kJ mol−1.
In later work Loeb and co-workers investigated the effect of macrocycle structure on the solid-state dynamics of a family of related MOFs.68 Replacing the 24 membered crown ether with a 22-membered analogue resulted in a framework that does not display macrocycle rotation in the solid state, which was attributed to the enhanced hydrogen bonding interaction between the smaller ring and the aniline proton. Similarly, when a bulkier 24-membered crown ether was investigated, free rotation was not observed, presumably due to the increased steric barrier. This study suggests that the solid-state dynamics of such systems can be tuned rationally.
Having demonstrated the pirouetting of the macrocycle of a rotaxane within a MOF framework Loeb and co-workers turned their attention to the issue of macrocycle shuttling in the solid state.69 Molecular shuttle 46 with two degenerate benzimidazole stations was prepared, with 50% of the stations labelled using 13C at the central imidazole carbon (Fig. 27a). Solution phase studies demonstrated that the chemical shift of the 13C labelled imidazole station varies depending on whether it is encircled by the macrocycle or not (46-13C-occupied or 46-12C-occupied respectively). Ligand 46 assembled in the presence of ZnII to form a porous MOF (Fig. 27b). Using line shape analysis, solid state 13C NMR allowed rate constants to be determined for the shuttling motion in the MOF framework and the macrocycle was shown to be in dynamic exchange even at room temperature. Unsurprisingly given the crowded nature of the solid-state structure, the rate of shuttling in the rotaxane MOF framework was five orders of magnitude lower than in solution.
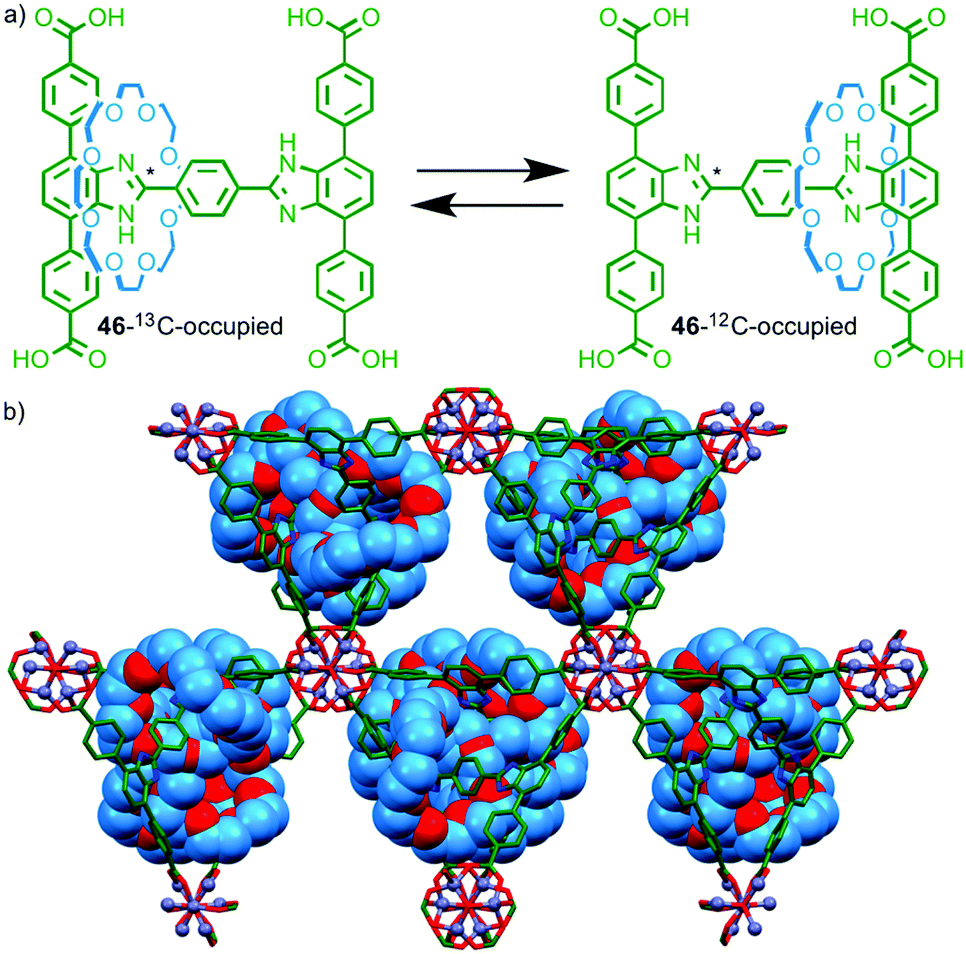 | ||
| Fig. 27 (a) Pseudo-degenerate molecular shuttle 46 and (b) the MOF formed by reaction of 46 with ZnII. Solid state 13C NMR line shape analysis revealed that the macrocycle continues to shuttle even in the solid state.64 | ||
Stoddart, Yaghi and co-workers have investigated the synthesis of catenane-based coordination polymers. Catenanes 474+, which were assembled using donor–acceptor π–π interactions, contain a rigid strut appended by carboxylate groups, common ligands in the assembly of metallo-supramolecular polymers (Fig. 28a). The length of the strut was found to be crucial to the metallo-supramolecular structure formed in the presence of CuI; both ligands gave the same coordination mode about the metal ions but the smaller (19.3 Å) strut length (47a4+) gave a 2D coordination network,70 whilst the larger (32.9 Å) strut (47b4+) resulted in a 3D MOF (Fig. 28b).71
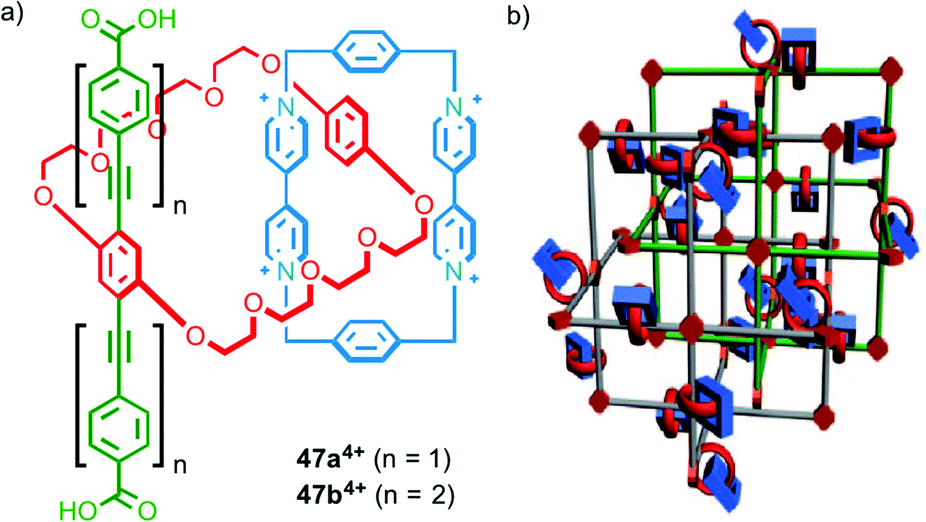 | ||
| Fig. 28 (a) Catenanes 474+ with different length strut units and (b) a cartoon representation of the MOF formed by 47b4+ in the presence of CuI.71 Adapted with permission from ref. 71. Copyright 2010 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Combining metallo-supramolecular chemistry with MIMs is a relatively new area that holds promise in the development of functional materials by providing new structural motifs, combining the benefits of supramolecular polymerisation and stimuli responsive behaviour and, particularly in the case of MOFs, offering the possibility of dynamic behaviour in systems that are typically required to be rigid to maintain porosity and stability; Loeb's recent contributions regarding large amplitude motion of the macrocycle component in the solid state clearly demonstrate this principle of “robust dynamics”.52c By providing a new approach to interfacing the behaviour of molecular machines with the macroscopic world, metallo-supramolecular chemistry brings real-world applications of these intriguing molecules one step closer.
Conclusions and outlook
The exploitation of MIMs as ligands has a long history given the central role metal–ligand interactions have played in the synthesis of rotaxanes and catenanes. However, the application of such metal complexes, beyond the operation of molecular machines, is a relatively recent phenomenon. In this Feature Article we have highlighted some of the under-exploited areas that we believe MIMs have the potential to have an impact in. In particular, the ability of the interlocked structure to augment the properties of the bound metal ion in terms of the kinetic and thermodynamic stability of the metal complex and its catalytic activity have only received relatively cursory attention to date. For the potential of these systems to be realised it is important that, wherever possible, relevant comparisons with a corresponding non-interlocked ligand are performed. Similarly, the use of interlocked molecules as scaffolds for metal ion sensing is currently underdeveloped, despite the success of interlocked molecules for sensing other charged species. Again, simple tests of metal ion selectivity need to be performed routinely.In both sensing and catalysis, systems that take advantage of the potential for unusual stereoisomerism in interlocked structures have yet to be reported.72 This arguably represents a significant and untapped area for study; the ability to rapidly generate chiral, crowded but flexible chemical fields for sensing or catalysis could bring benefits in sensing of chiral analytes and enantioselective catalysts.73
The area that has shown the greatest development recently is that of metallo-supramolecular materials but it is clear that there is still much to discover; the first rotaxane shuttle in a metal organic framework was only reported very recently. The potential to combine metallo-supramolecular assembly with some of the other areas we have highlighted to produce, for example, frameworks that selectively sequester metal ions for extraction, metallo-supramolecular supports for heterogeneous interlocked catalysts and sensors, and stimuli responsive frameworks with dynamic properties, present exciting opportunities for further study.
As more details emerge of the potential benefits of ligand frameworks based on rotaxanes and catenanes in a variety of areas, it seems likely that the field will expand rapidly. Based on the results presented in this Feature article we have no doubt that mechanically interlocked molecules have a bright future as ligands for a range of applications.
Acknowledgements
We thank the University of Southampton, EPSRC (EP/L016621/1) and Royal Society for financial support. J. E. M. L. is an EU Marie Skłodowska-Curie Fellow, receiving financial support from the European Union's Horizon 2020 research and innovation programme under Marie Sklodowska-Curie grant agreement No. 660731. M. G. thanks the EPRSC for a Doctoral Prize. S. M. G. is a Royal Society Research Fellow.Notes and references
- For selected recent reviews on the synthesis and applications of MIMs see: (a) J. E. Beves, B. A. Blight, C. J. Campbell, D. A. Leigh and R. T. McBurney, Angew. Chem., Int. Ed., 2011, 50, 9260 CrossRef CAS PubMed; (b) A. Coskun, M. Banaszak, R. D. Astumian, J. F. Stoddart and B. A. Grzybowski, Chem. Soc. Rev., 2012, 41, 19 RSC; (c) N. H. Evans and P. D. Beer, Chem. Soc. Rev., 2014, 43, 4658 RSC; (d) E. A. Neal and S. M. Goldup, Chem. Commun., 2014, 50, 5128 RSC; (e) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081 CrossRef CAS PubMed; (f) M. Xue, Y. Yang, X. Chi, X. Yan and F. Huang, Chem. Rev., 2015, 115, 7398 CrossRef CAS PubMed.
- C. O. Dietrich-Buchecker, J.-P. Sauvage and J. P. Kintzinger, Tetrahedron Lett., 1983, 24, 5095 CrossRef CAS; C. O. Dietrich-Buchecker, J.-P. Sauvage and J. M. Kern, J. Am. Chem. Soc., 1984, 106, 3043 CrossRef.
- We refer to methods in which metal-ligand interactions simply assemble the MIM precursors and those in which the metal ion also mediates the reaction that captures the interlocked structure as “passive” and “active” respectively: J. D. Crowley, S. M. Goldup, A.-L. Lee, D. A. Leigh and R. T. McBurney, Chem. Soc. Rev., 2009, 38, 1530 RSC.
- C. Wu, P. R. Lecavalier, Y. X. Shen and H. W. Gibson, Chem. Mater., 1991, 3, 569 CrossRef CAS.
- A. M. Albrecht-Gary, Z. Saad, C. O. Dietrich-Buchecker and J. P. Sauvage, J. Am. Chem. Soc., 1985, 107, 3205 CrossRef CAS.
- The kinetic stabilisation of chelated metal complexes and complexes of cryptand ligands are known as the chelate and cryptate effects respectively (Pure Appl. Chem. 1977, 49, 857). However, when Sauvage and co-workers observed the kinetic stabilisation of metal complexes of catenane ligands (also known as catenands) they named this the “catenand effect”, presumably to avoid confusion with the existing verb “to catenate”, meaning to arrange atoms in a chain.
- D. H. Busch, Chem. Rev., 1993, 93, 847 CrossRef CAS.
- C. Dietrich-Buchecker, J.-P. Sauvage and J.-M. Kern, J. Am. Chem. Soc., 1989, 111, 7791 CrossRef CAS.
- A. J. Blake, C. O. Dietrich-Buchecker, T. I. Hyde, J.-P. Sauvage and M. Schröder, J. Chem. Soc., Chem. Commun., 1989, 1663 RSC.
- C. O. Dietrich-Buchecker, J.-M. Kern and J.-P. Sauvage, J. Chem. Soc., Chem. Commun., 1985, 760 RSC.
- D. A. Leigh, P. J. Lusby, A. M. Z. Slawin and D. B. Walker, Angew. Chem., Int. Ed., 2005, 44, 4557 CrossRef CAS PubMed.
- V. Aucagne, K. D. Hänni, D. A. Leigh, P. J. Lusby and D. B. Walker, J. Am. Chem. Soc., 2006, 128, 2186 CrossRef CAS PubMed.
- H. Lahlali, K. Jobe, M. Watkinson and S. M. Goldup, Angew. Chem., Int. Ed., 2011, 50, 4151 CrossRef CAS PubMed.
- J. Winn, A. Pinczewska and S. M. Goldup, J. Am. Chem. Soc., 2013, 135, 13318 CrossRef CAS PubMed.
- For a recent example that showcases the flexibility of MILs: G. Baggi and S. J. Loeb, Angew. Chem., Int. Ed., 2016, 55, 12533 CrossRef CAS PubMed.
- Z. Kelman and M. O’Donnell, Annu. Rev. Biochem., 1996, 64, 171 CrossRef PubMed.
- (a) P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915 CrossRef CAS PubMed; (b) P. H. Ramos, R. G. E. Coumans, A. B. C. Deutman, J. M. M. Smits, R. de Gelder, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, J. Am. Chem. Soc., 2007, 129, 5699 CrossRef PubMed; (c) R. G. E. Coumans, J. A. A. W. Elemans, R. J. M. Nolte and A. E. Rowan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19647 CrossRef CAS PubMed.
- S. F. M. van Dongen, J. Clerx, K. Nørgaard, T. G. Bloemberg, J. J. L. M. Cornelissen, M. a. Trakselis, S. W. Nelson, S. J. Benkovic, A. E. Rowan and R. J. M. Nolte, Nat. Chem., 2013, 5, 945 CrossRef CAS PubMed.
- N. Miyagawa, M. Watanabe, T. Matsuyama, Y. Koyama, T. Moriuchi, T. Hirao, Y. Furusho and T. Takata, Chem. Commun., 2010, 46, 1920 RSC.
- For a related study on the effect of macrocycle size on the efficiency of such macrocyclic Pd complexes see: M. Ogawa, M. Nagashima, H. Sogawa, S. Kuwata and T. Takata, Org. Lett., 2015, 17, 1664 CrossRef CAS PubMed.
- Y. Matsuoka, Y. Mutoh, I. Azumaya, S. Kikkawa, T. Kasama and S. Saito, J. Org. Chem., 2016, 81, 3479 CrossRef CAS PubMed.
- B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D’Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189 CrossRef CAS PubMed; G. De Bo, S. Kuschel, D. A. Leigh, B. Lewandowski, M. Papmeyer and J. W. Ward, J. Am. Chem. Soc., 2014, 136, 5811 CrossRef PubMed.
- For examples in non-metal based systems see: (a) A. H. Parham, B. Windisch and F. Vögtle, Eur. J. Org. Chem., 1999, 1233 CrossRef CAS; (b) N. Kihara, Y. Tachibana, H. Kawasaki and T. Takata, Chem. Lett., 2000, 506 CrossRef CAS; (c) T. Oku, Y. Furusho and T. Takata, Org. Lett., 2003, 5, 12384 CrossRef PubMed; (d) C. B. Caputo, K. Zhu, V. N. Vukotic, S. J. Loeb and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 960 CrossRef CAS PubMed; (e) A. Martinez-Cuezva, C. Lopez-Leonardo, D. Bautista, M. Alajarin and J. Berna, J. Am. Chem. Soc., 2016, 138, 8726 CrossRef CAS PubMed.
- M. Yamazaki, T. Hagiwara, M. Sekiguchi, T. Sawaguchi and S. Yano, Synth. Commun., 2008, 38, 553 CrossRef CAS.
- Y. Suzaki, K. Shimada, E. Chihara, T. Saito, Y. Tsuchido and K. Osakada, Org. Lett., 2011, 13, 3774 CrossRef CAS PubMed.
- For an example of a related approach to developing bimetallic hosts for flexible guests see: J. Frey, C. Tock, J. P. Collin, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 2008, 130, 4592 CrossRef CAS PubMed.
- S. Hoekman, M. O. Kitching, D. A. Leigh, M. Papmeyer and D. Roke, J. Am. Chem. Soc., 2015, 137, 7656 CrossRef CAS PubMed.
- M. Galli, J. E. M. Lewis and S. M. Goldup, Angew. Chem., Int. Ed., 2015, 54, 13545 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759 CrossRef CAS.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759 CrossRef CAS.
- Macrocyclic ligands have been shown to display enhanced stereoselectivity compared with their acyclic analogues: T. Portada, M. Roje, Z. Raza, V. Čaplar, M. Žinić and V. Šunjić, Eur. J. Org. Chem., 2007, 838 CrossRef CAS.
- For selected examples and reviews of sensors based on interlocked molecules see: (a) N. Kameta, Y. Nagawa, M. Karikomi and K. Hiratani, Chem. Commun., 2006, 3714 RSC; (b) J. J. Gassensmith, S. Matthys, J. J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem. – Eur. J., 2010, 16, 2916 CrossRef CAS PubMed; (c) W. Wong, K. C.-F. Leung and J. F. Stoddart, Org. Biomol. Chem., 2010, 8, 2332 RSC; (d) J. J. Gassensmith, S. Matthys, J. J. Lee, A. Wojcik, P. V. Kamat and B. D. Smith, Chem. – Eur. J., 2010, 16, 2916 CrossRef CAS PubMed; (e) X. Ma and H. Tian, Chem. Soc. Rev., 2010, 39, 70 RSC; (f) M. J. Langton and P. D. Beer, Acc. Chem. Res., 2014, 47, 1935 CrossRef CAS PubMed; (g) M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem. – Eur. J., 2016, 22, 185 CrossRef CAS PubMed; (h) R. Mitra, M. Thiele, F. Octa-Smolin, M. C. Letzel and J. Niemeyer, Chem. Commun., 2016, 52, 5977 RSC.
- (a) M. J. MacLachlan, A. Rose and T. M. Swager, J. Am. Chem. Soc., 2001, 123, 9180 CrossRef CAS PubMed; (b) S. S. Zhu, P. J. Carroll and T. M. Swager, J. Am. Chem. Soc., 1996, 118, 8713 CrossRef CAS; (c) P. H. Kwan and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 5902 CrossRef CAS PubMed.
- For a related system in which metal binding controls electron transfer between Zn and Au porphyrins see: J. C. Chambron, A. Harriman, V. Heitz and J. P. Sauvage, J. Am. Chem. Soc., 1993, 115, 7419 CrossRef CAS.
- Y. Nakatani, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2010, 49, 5463 CrossRef CAS PubMed.
- The role of the Zn2+ ion may be to coordinate to the carboxylate or the amidine moiety. However, the authors provide evidence that amidine coordination is preferred and we have adopted their representation here.35.
- Arguably, structures such as 30 are not mechanically interlocked because the macrocycle and axle components are covalently linked. However, they are commonly referred to as [1]rotaxanes, where “[1]” refers to the single covalent component present, and we follow this convention here. For more information on nomenclature see: A. Yerin, E. S. Wilks, G. P. Moss and A. Harada, Pure Appl. Chem., 2008, 80, 2041 CrossRef CAS.
- K. Hiratani, M. Kaneyama, Y. Nagawa, E. Koyama and M. Kanesato, J. Am. Chem. Soc., 2004, 126, 13568 CrossRef CAS PubMed.
- For an example of the selective discrimination of alkali metal ions by a rotaxane host using 1H NMR see: N.-C. Chen, P.-Y. Huang, C.-C. Lai, Y.-H. Liu, Y. Wang, S.-M. Peng and S.-H. Chiu, Chem. Commun., 2007, 4122 RSC.
- Y. Nagawa, J. Suga, K. Hiratani, E. Koyama and M. Kanesato, Chem. Commun., 2005, 749 RSC.
- Unfortunately the photophysical properties of the LiI complex have not, to our knowledge, been reported.
- W. Zhou, J. Li, X. He, C. Li, J. Lv, Y. Li, S. Wang, H. Liu and D. Zhu, Chem. – Eur. J., 2008, 14, 754 CrossRef CAS PubMed.
- S. Y. Hsueh, C. C. Lai and S. H. Chiu, Chem. – Eur. J., 2010, 16, 2997 CrossRef CAS PubMed.
- D. Curiel and P. D. Beer, Chem. Commun., 2005, 1909 RSC.
- For selected examples of a related approach where metal ion-containing molecular shuttles are monitored by a change in luminescence see: (a) H. Zhang, J. Hu and D. H. Qu, Org. Lett., 2012, 14, 2334 CrossRef CAS PubMed; (b) H. Zhang, B. Zhou, H. Li, D.-H. Qu and H. Tian, J. Org. Chem., 2013, 78, 2091 CrossRef CAS PubMed; (c) H. Li, X. Li, Z.-Q. Cao, D.-H. Qu, H. Ågren and H. Tian, ACS Appl. Mater. Interfaces, 2014, 6, 18921 CrossRef CAS PubMed; (d) X. Ma, J. Zhang, J. Cao, X. Yao, T. Cao, Y. Gong, C. Zhao and H. Tian, Chem. Sci., 2016, 7, 4582 RSC.
- B. R. Mullaney, A. L. Thompson and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11458 CrossRef CAS PubMed.
- C. Allain, P. D. Beer, S. Faulkner, M. W. Jones, A. M. Kenwright, N. L. Kilah, R. C. Knighton, T. J. Sørensen and M. Tropiano, Chem. Sci., 2013, 4, 489 RSC; M. J. Langton, O. A. Blackburn, T. Lang, S. Faulkner and P. D. Beer, Angew. Chem., Int. Ed., 2014, 53, 11463 CrossRef CAS PubMed.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem. – Eur. J., 2016, 22, 185 CrossRef CAS PubMed; L. M. Hancock, E. Marchi, P. Ceroni and P. D. Beer, Chem. – Eur. J., 2012, 18, 11277 CrossRef PubMed.
- (a) N. H. Evans and P. D. Beer, Org. Biomol. Chem., 2011, 9, 92 RSC; (b) J. Y. C. Lim, M. J. Cunningham, J. J. Davis and P. D. Beer, Dalton Trans., 2014, 43, 17274 RSC; (c) N. H. Evans, C. J. Serpell and P. D. Beer, Chem. Commun., 2011, 47, 8775 RSC; (d) N. H. Evans, C. J. Serpell, N. G. White and P. D. Beer, Chem. – Eur. J., 2011, 17, 12347 CrossRef CAS PubMed; (e) N. H. Evans, H. Rahman, A. V. Leontiev, N. D. Greenham, G. A. Orlowski, Q. Zeng, R. M. J. Jacobs, C. J. Serpell, N. L. Kilah, J. J. Davis and P. D. Beer, Chem. Sci., 2012, 3, 1080 RSC.
- J. Lehr, T. Lang, O. A. Blackburn, T. A. Barendt, S. Faulkner, J. J. Davis and P. D. Beer, Chem. – Eur. J., 2013, 19, 15898 CrossRef CAS PubMed.
- For recent reviews on the principles and applications of metallo-supramolecular chemistry see: (a) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (b) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354 RSC; (c) M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860 RSC; (d) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (e) L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201 CrossRef CAS PubMed.
- For selected reviews of interlocked metallo-supramolecular systems including pseudorotaxanes see: (a) S. J. Loeb, Chem. Commun., 2005, 1511 RSC; (b) S. J. Loeb, Chem. Soc. Rev., 2007, 36, 226 RSC; (c) H. Deng, M. A. Olson, J. F. Stoddart and O. M. Yaghi, Nat. Chem., 2010, 2, 439 CrossRef CAS PubMed; (d) V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896 RSC; (e) J. Yang, J.-F. Ma and S. R. Batten, Chem. Commun., 2012, 48, 7899 RSC; (f) P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815 RSC.
- G. J. E. Davidson and S. J. Loeb, Dalton Trans., 2003, 4319 RSC.
- A. Noor, S. C. Moratti and J. D. Crowley, Chem. Sci., 2014, 5, 4283 RSC.
- Y. Shi, Z. Yang, H. Liu, Z. Li, Y. Tian and F. Wang, ACS Macro Lett., 2015, 4, 6 CrossRef CAS.
- A. Noor, W. K. C. Lo, S. C. Moratti and J. D. Crowley, Chem. Commun., 2014, 50, 7044 RSC.
- S. Richter, J. Poppenberg, C. H. H. Traulsen, E. Darlatt, A. Sokolowski, D. Sattler, W. E. S. Unger and C. A. Schalley, J. Am. Chem. Soc., 2012, 134, 16289 CrossRef CAS PubMed.
- T. Heinrich, C. H.-H. Traulsen, M. Holzweber, S. Richter, V. Kunz, S. K. Kastner, S. O. Krabbenborg, J. Huskens, W. E. S. Unger and C. A. Schalley, J. Am. Chem. Soc., 2015, 137, 4382 CrossRef CAS PubMed.
- E. Viljoen, K. Zhu and S. J. Loeb, Chem. – Eur. J., 2016, 22, 7479 CrossRef CAS PubMed.
- V. N. Vukotic and S. J. Loeb, Chem. Soc. Rev., 2012, 41, 5896 RSC.
- D. J. Mercer, V. N. Vukotic and S. J. Loeb, Chem. Commun., 2011, 47, 896 RSC.
- N. C. Frank, D. J. Mercer and S. J. Loeb, Chem. – Eur. J., 2013, 19, 14076 CrossRef CAS PubMed.
- L. Gao, Z. Zhang, B. Zheng and F. Huang, Polym. Chem., 2014, 5, 5734 RSC.
- Z. B. Zhang, C. Y. Han, G. C. Yu and F. H. Huang, Chem. Sci., 2012, 3, 3026 RSC.
- For recent reviews on the synthesis and applications of MOFs see: (a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed; (b) H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed; (c) O. K. Farha, Chem. Commun., 2015, 51, 3501 RSC; (d) A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804 RSC; (e) A. Dhakshinamoorthy, A. M. Asiri and H. García, Angew. Chem., Int. Ed., 2016, 55, 5414 CrossRef CAS PubMed; (f) Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483 CrossRef CAS PubMed; (g) I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127 RSC; (h) Q. Wang, J. Bai, Z. Lu, Y. Pan and X. You, Chem. Commun., 2016, 52, 443 RSC.
- (a) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461 CrossRef CAS PubMed; (b) M. Hoshino, A. Khutia, H. Xing, Y. Inokuma and M. Fujita, IUCrJ, 2016, 3, 139 CAS.
- V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456 CrossRef CAS PubMed.
- (a) V. N. Vukotic, C. A. O’Keefe, K. Zhu, K. J. Harris, C. To, R. W. Schurko and S. J. Loeb, J. Am. Chem. Soc., 2015, 137, 9643 CrossRef CAS PubMed; (b) N. Farahani, K. Zhu, C. A. O’Keefe, R. W. Schurko and S. J. Loeb, ChemPlusChem, 2016, 81, 836 CrossRef CAS.
- K. Zhu, C. A. O’Keefe, V. N. Vukotic, R. W. Schurko and S. J. Loeb, Nat. Chem., 2015, 7, 514 CrossRef CAS PubMed.
- Q. Li, W. Zhang, O. Š. Miljanić, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Chem. Commun., 2010, 46, 380 RSC.
- Q. Li, C.-H. Sue, S. Basu, A. K. Shveyd, W. Zhang, G. Barin, L. Fang, A. A. Sarjeant, J. F. Stoddart and O. M. Yaghi, Angew. Chem., Int. Ed., 2010, 49, 6751 CrossRef CAS PubMed.
- For selected examples of unusual stereoisomerism as a result of the mechanical bond see: (a) C. Yamamoto, Y. Okamoto, T. Schmidt, R. Jäger and F. Vögtle, J. Am. Chem. Soc., 1997, 119, 10547 CrossRef CAS; (b) S. Meskers, H. Dekkers, G. Rapenne and J.-P. Sauvage, Chem. – Eur. J., 2000, 6, 2129 CrossRef CAS PubMed; (c) S. A. Vignon, J. Wong, H. Tseng and J. F. Stoddart, Org. Lett., 2004, 6, 1095 CrossRef CAS PubMed; (d) Y. Makita, N. Kihara, N. Nakakoji, T. Takata, S. Inagaki, C. Yamamoto and Y. Okamoto, Chem. Lett., 2007, 36, 162 CrossRef CAS; (e) M. Alvarez-Pérez, S. M. Goldup, D. A. Leigh and A. M. Z. Slawin, J. Am. Chem. Soc., 2008, 130, 1836 CrossRef PubMed; (f) A.-M. L. Fuller, D. A. Leigh and P. J. Lusby, J. Am. Chem. Soc., 2010, 132, 4954 CrossRef CAS PubMed; (g) R. J. Bordoli and S. M. Goldup, J. Am. Chem. Soc., 2014, 136, 4817 CrossRef CAS PubMed; (h) T. Prakasam, M. Lusi, E. Nauha, J.-C. Olsen, M. Sy, C. Platas-Iglesias, L. J. Charbonnière and A. Trabolsi, Chem. Commun., 2015, 51, 5840 RSC; (i) E. A. Neal and S. M. Goldup, Angew. Chem., Int. Ed., 2016, 55, 12488 CrossRef CAS PubMed.
- For a recent example of a mechanically point chiral organocatalyst see: Y. Cakmak, S. Erbas-Cakmak and D. A. Leigh, J. Am. Chem. Soc., 2016, 138, 1749 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |