
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monodentate coordination of the normally chelating chiral diamine (R,R)-TMCDA†
Ana I.
Ojeda-Amador
,
Antonio J.
Martínez-Martínez
,
Alan. R.
Kennedy
,
David R.
Armstrong
and
Charles T.
O'Hara
 *
*
WestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: charlie.ohara@strath.ac.uk
First published on 7th December 2016
Abstract
After isolating an unusual binuclear, but monosolvated NaHMDS complex [{(R,R)-TMCDA}·(NaHMDS)2]∞ which polymerises via intermolecular electrostatic Na⋯MeHMDS interactions, further (R,R)-TMCDA was added to produce the discrete binuclear amide [{κ2-(R,R)-TMCDA}·(NaHMDS)2{κ1-(R,R)-TMCDA}], whose salient feature is the unique monodentate coordination of one of the chiral diamine ligands.
Chiral diamine ligands, for example (−)-sparteine, its (+)-sparteine surrogate and N,N,N′,N′-(1R,2R)-tetramethylcyclohexane-1,2-diamine [(R,R)-TMCDA] have attracted considerable attention in asymmetric synthesis in a whole host of transition metal catalysed methodologies.1 From an s-block perspective, when paired with an organolithium reagent it can be envisaged that ‘chiral carbanions’ are created, which can be used in subsequent enantioselective syntheses.2 Focusing particularly on the C2-symmetric ligand (R,R)-TMCDA, it has come to prominence recently as the availability of the historically more widely utilised diamine (−)-sparteine, has been unreliable over the past few years.3 In terms of its coordination chemistry, (R,R)-TMCDA has worldwide interest and has been well studied. Over 50 metal complexes containing its ligated form have been reported, spanning both the s- (Li,4 Na,4e K,4e and Mg,5) and d-block metals (Cu,6 Zn,7 Ru,8 Pd,9 Pt10 and Hg11). Within s-block chemistry and germane to this work, Strohmann has comprehensively studied (R,R)-TMCDA complexes of synthetically important organolithium reagents (such as tBuLi,4a MeLi,4biPrLi,4bsBuLi,4bnBuLi,4c BH3P(Ph)(Me)CH2Li,4d MeLi,4g PhLi,4h (allyl)Li4h and (benzyl)Li4i derivatives). An all-encompassing feature of all known structures is that the chiral diamine ligand adopts exclusively a κ2-bidentate chelating mode. Due to the less flexible, fixed bite angle in (R,R)-TMCDA, with respect to that of N,N,N′,N′-tetramethylethylenediamine (TMEDA),12 it is a stronger chelating ligand than the latter,13 with a recent study noting that it ‘displays no tendency to bind as a monodentate ligand.’14 This has been attributed to the κ1 (or by implication η1) form of (R,R)-TMCDA inducing severe steric strain due to the juxtaposition of the metal–NMe2 with the uncoordinated NMe2 group. The structural chemistry of alkali metal amide complexes continues to be an important topic of research.15 We have recently discovered that lithium and sodium 1,1,1,3,3,3-hexamethyldisilazide (LiHMDS and NaHMDS) can capture alkali metal halide salts in the presence of donor ligands to form ion pair metal anionic crown (MAC) complexes, for example [Li{(R,R)-TMCDA}2]+[Li5HMDS5Cl]−.4f,16 A key starting material which remained hitherto elusive in our studies involving sodium is the (R,R)-TMCDA–solvated NaHMDS complex. Crystallisation of other donor ligated [e.g., Me6TREN17 and (−)-sparteine18] NaHMDS complexes has proven difficult, although the polymeric TMEDA [(μ-TMEDA)·(NaHMDS)2]∞19 and N,N,N′,N′-tetramethylpropanediamine (TMPDA) [(μ-TMPDA)·(NaHMDS)2]∞20 complexes, which propagate via the non-chelating diamine ligand, are known (Fig. 1). These have similar structural motifs to Williard's lithium diisopropylamide (LDA) complex [(μ-TMEDA)·(LDA)2]∞.19
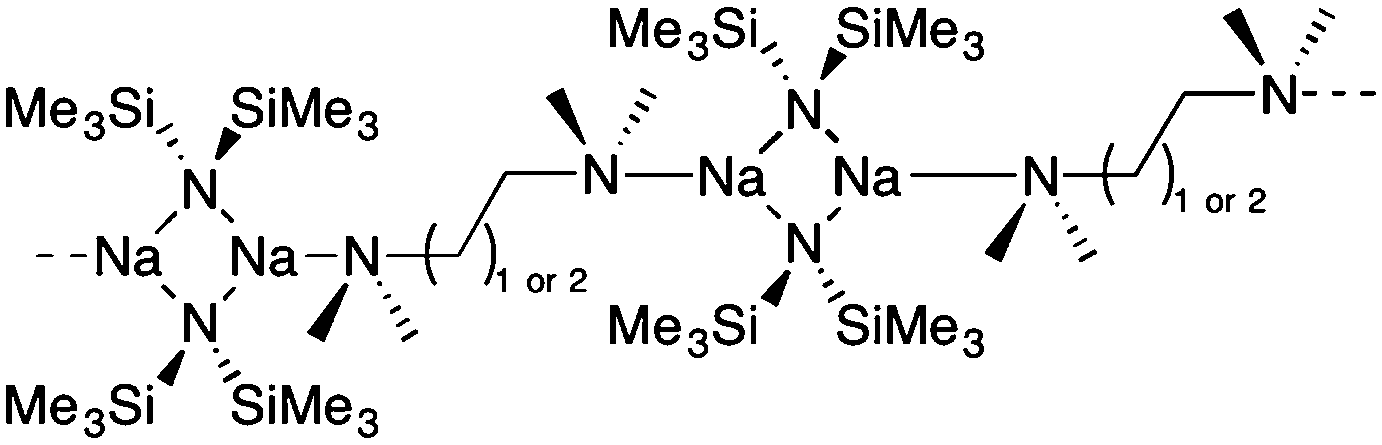 | ||
| Fig. 1 Structures of previously known polymeric [(μ-TMEDA)·(NaHMDS)2]∞ and [(μ-TMPDA)·(NaHMDS)2]∞. | ||
In an effort to prepare the (R,R)-TMCDA complex of NaHMDS, an equimolar mixture of NaHMDS and (R,R)-TMCDA was combined in n-hexane medium and left to stir at ambient temperature for 1 hour (Scheme 1). The reaction mixture was then cooled to −33 °C and crystals suitable for X-ray crystallographic analysis deposited after 48 hours (27% non-optimised, crystalline yield; maximum yield 50% based on (R,R)-TMCDA consumption). X-ray data reveal the mono-(R,R)-TMCDA, binuclear [{(R,R)-TMCDA}·(NaHMDS)2]∞1 (Fig. 2a). There are six crystallographically distinct but essentially chemically equivalent molecules of [{(R,R)-TMCDA}·(NaHMDS)2] in the structure of 1, thus for brevity only one is discussed here. Interestingly, the empirical formula of 1, i.e., [(donor)·(NaHMDS)2] is identical to that for the aforementioned TMEDA and TMPDA derivatives; however, in keeping with previously known (R,R)-TMCDA complexes, the diamine adopts a chelating bonding mode, and with respect to the N donor atoms, renders one Na metal centre (Na1) four-coordinate in a distorted tetrahedral arrangement (bond angles range from 68.70(9) to 151.55(10)°, see ESI† for full details). Additionally, Na1 has two long Na⋯Me interactions with a methyl group from each HMDS ligand [Na1⋯C12 2.987(4) and Na1⋯C22 2.987(4) Å]. The second Na metal centre (Na2) remains only two-coordinate with respect to the bridging amido N atoms. To satisfy this electron deficiency, Na2 engages a solitary intermolecular Na⋯Me(SiMe2) [Na2⋯C65 distance, 2.818(4) Å] electrostatic interaction (Fig. 2b), which is short in comparison to known literature examples [range Na⋯Me(SiMe2) 2.947–3.138 Å].21 This sole intermolecular Na⋯Me interaction induces propagation of binuclear units in a zigzag polymer chain. This change in the coordination chemistry of (R,R)-TMCDA in 1 with respect to the bridging TMEDA and TMPDA ligands in the aforementioned polymeric sodium amides emphasises the propensity for the chiral 1,2-diamine to remain as a chelating ligand rather than binding in a monodentate fashion. As a consequence of this coordination mismatch, significantly shorter Na2–NHMDS bonds (mean distance, 2.356 Å) are observed when compared with Na1–NHMDS bonds (mean distance, 2.530 Å). Despite utilising a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of NaHMDS:(R,R)-TMCDA in this synthesis, it is clearly evident that the ultimate ratio in 1 is 2:1. When this optimised ratio is used in the synthesis, 1 was again the sole product isolated (36% crystalline yield).
:1 ratio of NaHMDS:(R,R)-TMCDA in this synthesis, it is clearly evident that the ultimate ratio in 1 is 2:1. When this optimised ratio is used in the synthesis, 1 was again the sole product isolated (36% crystalline yield).
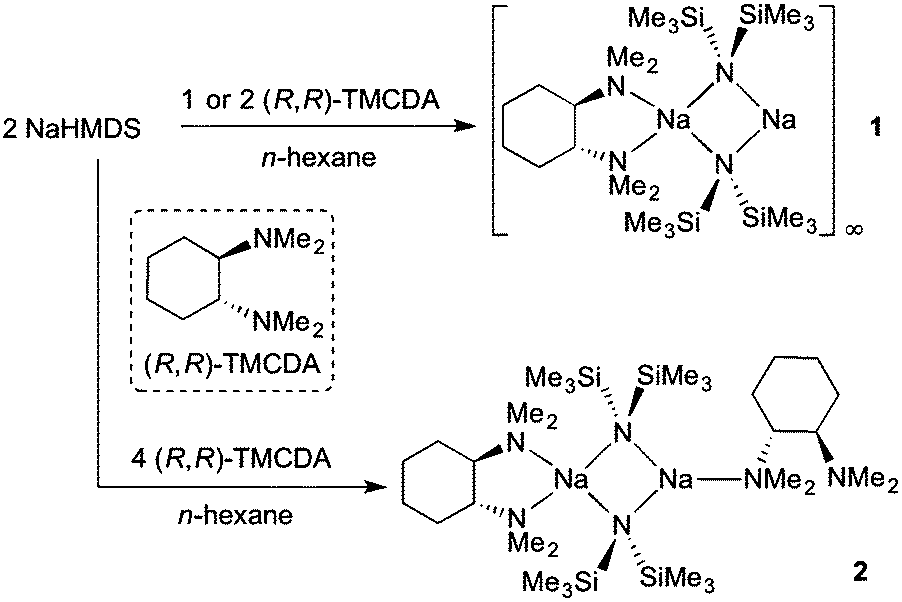 | ||
| Scheme 1 Syntheses of [{(R,R)-TMCDA}·(NaHMDS)2]∞1 and [{κ2-(R,R)-TMCDA}·(NaHMDS)2{κ1-(R,R)-TMCDA}] 2. | ||
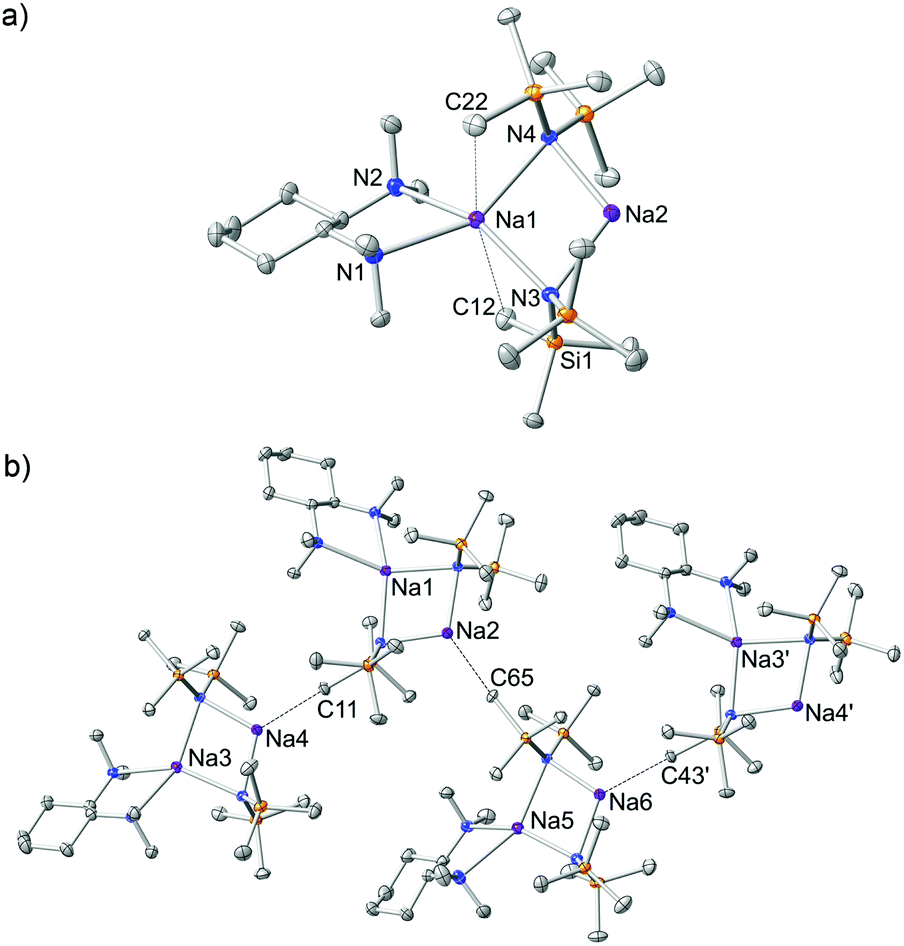 | ||
| Fig. 2 (a) Molecular structure of [{(R,R)-TMCDA}·(NaHMDS)2]∞1 showing one molecule from the asymmetric unit. Hydrogen atoms omitted for simplicity and thermal ellipsoids are displayed at 35% probability. (b) Section of the zigzag polymeric chain of 1. The dashed lines illustrate Na⋯Me(SiMe2) interactions. The symmetry operation used to generate the atoms labelled with ′ is −x + 1, y + 1/2, −z + 1. | ||
Complex 1 is a rare example of a solvated sodium amide which contains an unsolvated Na site. Bochmann revealed the mono(tetrahydrofuran), mono(THF), complex [(THF)·(NaHMDS)2] where one Na atom is two coordinate whilst the other binds to the ether to render it three coordinate.22 Interestingly, seven years prior to this report Dehnicke published the bis(THF) analogue [(THF)2·(NaHMDS)2] where both Na atoms are three coordinate.23 This begged the question: ‘could the coordinatively unsaturated (Lewis acidic) Na atom in1, act as a host for another Lewis base?’
A logical route to address this question would be to utilise monodentate donors such as THF and diethylether, in an attempt to saturate the deficient metal centre; but, it is highly likely that these strong σ-donors would also displace the chelating (R,R)-TMCDA ligand. Therefore to maintain synthetic simplicity, we repeated the preparation of 1 but employing an excess (two molar equivalents) of (R,R)-TMCDA with respect to NaHMDS in an attempt to coordinate a second molecule of the Lewis base ligand to the donor-free metal centre. High quality crystals (39% crystalline yield) were obtained by storing the resultant solution at −33 °C for 24 h, which were analysed by X-ray crystallography and were pleasingly found to be the target bis(solvated) derivative [{κ2-(R,R)-TMCDA}·(NaHMDS)2{κ1-(R,R)-TMCDA}] 2 (Fig. 3). The distorted tetrahedral coordination sphere of Na1 in 2 (bond angles around Na1 range from 66.90(6) to 151.05(8), see ESI†) is essentially identical to that found in 1, exhibiting additional long contacts with a methyl group from each HMDS amido ligand [Na1⋯C27 2.968(3) and Na⋯C24 2.976(3) Å]. However, the second sodium metal centre, Na2, is additionally coordinated to an extra molecule of (R,R)-TMCDA, giving rise to a distorted trigonal planar geometry. As such there are two distinct coordinated diamine ligands within the structure of 2. Undoubtedly, the most eye-catching feature is that one (R,R)-TMCDA ligand adopts a previously unseen κ1-coordination mode. To change from a κ2- to a κ1-coordination mode, it appears that inversion of the N1 atom of the (R,R)-TMCDA has occurred, no longer allowing the ligand to chelate to Na2 (Fig. 3).
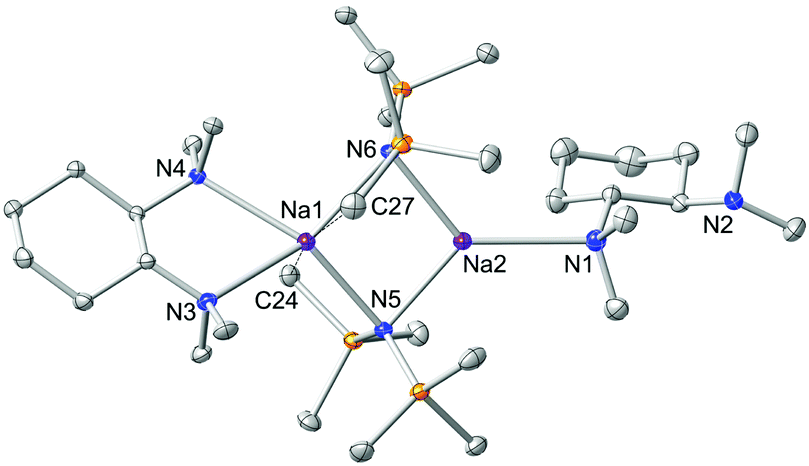 | ||
| Fig. 3 Molecular structure of [{κ2-(R,R)-TMCDA}·(NaHMDS)2{κ1-(R,R)-TMCDA}] 2. Hydrogen atoms and one disordered component of the mono-dentate (R,R)-TMCDA ligand are omitted for simplicity. Thermal ellipsoids are displayed at 35% probability. | ||
Complex 2 is a discrete dimeric entity, despite the potential availability for N2 to coordinate further. In theory, this could be achieved if this N atom could also invert thus allowing an additional exo-coordination site; however, it is unlikely that this would occur due to high steric strain (buttressing).14 The κ1-coordinated (R,R)-TMCDA is disordered over two domains, but its atomic connectivity and geometry are unequivocal. The κ2- and the hitherto unseen κ1-coordination mode (R,R)-TMCDA observed in 2 can be compared with DFT calculations (at the B3P86/6-311+G* level) performed for its diamine relative (−)-sparteine (Fig. 4).24 It has been shown that when (−)-sparteine binds to a metal complex, it always adopts a chelating ‘cis’ configuration. However, in the absence of a metal complex, it is actually slightly more stable (by 3.4 kcal mol−1) in a ring-flipped ‘trans’ configuration [akin to our κ1-coordinated (R,R)-TMCDA] where the lone pairs of electron present on the N atoms are not adjacent to each other. We have performed similar DFT studies (ESI†) on (R,R)-TMCDA and have shown that there is negligible difference (less than 1 kcal mol−1) between the potentially κ1- and κ2-coordination modes.
 | ||
| Fig. 4 Relative stabilities of cis and trans isomers of uncoordinated (−)-sparteine.24 | ||
As 1 and 2 are both highly soluble in non-polar hydrocarbon and arene solutions, solutions of these compounds were studied by NMR spectroscopy. Using 1H NMR spectroscopy, it was evident that the expected 1:2 and 2:2 (R,R)-TMCDA:HMDS ratios were observed respectively. For 1, a single amido resonance (at δ 0.25) was observed and the (R,R)-TMCDA resonances (at δ 2.01, 1.90, 1.47 and 0.74) in C6D6 solution appeared to correspond to a metallo-coordinated ligand (see ESI† for full details). For 2, the amido resonance appears at δ 0.31 in the same solvent. If the solid state structure of 2 was to be retained in solution, two unique sets of (R,R)-TMCDA resonances would be expected. In reality a single set of resonances (at δ 2.06, 1.99, 1.51 and 0.80 in C6D6 solution) is observed. This indicates that a single (R,R)-TMCDA environment exists at 300 K in arene solution, indeed, a variable temperature NMR spectroscopic study of 2 in [D8]-toluene solution unveiled that this situation was maintained even at low temperature (down to 206 K, see ESI†). In addition, 1H and 13C NMR spectra obtained in non-polar [D12]-cyclohexane also reveal this situation (see ESI†). Therefore due to the steric bulk of the HMDS ligands within the molecule [thus precluding a dual κ2-situation for the (R,R)-TMCDA ligands], it is likely that the spectra show a time-averaged situation between dynamic κ1- and κ2-coordinated (R,R)-TMCDA ligands.
In closing, we have shown that counter to previous studies, (R,R)-TMCDA can indeed bind to an alkali metal in a non-chelating κ1-manner.
This work was supported by the EPSRC (through a Career Acceleration Fellowship to CTOH, EP/J001872/1 and EP/L001497/1). The authors would like to thank Professors Mulvey and Hevia, and Dr Robertson for useful discussions. We would also like to thank Dr Claire McGoldrick for help with artwork design. The research data associated with this paper is openly available at http://dx.doi.org/10.15129/103ad89d-550d-44ad-8aa8-62d54c47e3ac.
Notes and references
- (a) K. Mikami and M. Yamanaka, Chem. Rev., 2003, 103, 3369 CrossRef CAS PubMed; (b) R. Noyori, Adv. Synth. Catal., 2003, 345, 15 CrossRef CAS; (c) V. Bette, A. Mortreux, F. Ferioli, G. Martelli, D. Savoia and J.-F. Carpentier, Eur. J. Org. Chem., 2004, 3040 CrossRef CAS.
- (a) D. Hoppe and T. Hense, Angew. Chem., Int. Ed. Engl., 1997, 2282 CrossRef CAS; (b) J. Clayden, Organolithiums: Selectivity for Synthesis, Pergamon, New York, 2002 Search PubMed; (c) O. Chuzel and O. Riant, Top. Organomet. Chem., 2005, 15, 59 CrossRef CAS; (d) C.-A. B. Ferber, H. B. Kagan, O. Lafon and P. Lesot, Tetrahedron: Asymmetry, 2008, 19, 2666 CrossRef; (e) P. O'Brien, Chem. Commun., 2008, 655 RSC; (f) Q. Perron, J. Praz and A. Alexakis, Tetrahedron: Asymmetry, 2009, 20, 1004 CrossRef CAS; (g) J. Praz, J. Graff, L. Egger, L. Guénée, S. Wagschal, E. P. Kündig and A. Alexakis, Chem. Commun., 2015, 51, 16912 RSC.
- J. D. Firth, P. O'Brien and L. Ferris, Org. Biomol. Chem., 2014, 12, 9357 CAS.
- (a) C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 8281 CrossRef CAS PubMed; (b) C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2007, 129, 8952 CrossRef CAS PubMed; (c) C. Strohmann and V. H. Gessner, J. Am. Chem. Soc., 2008, 130, 11719 CrossRef CAS PubMed; (d) V. H. Gessner, S. Dilsky and C. Strohmann, Chem. Commun., 2010, 46, 4719 RSC; (e) P. García-Álvarez, A. R. Kennedy, C. T. O'Hara, K. Reilly and G. M. Robertson, Dalton Trans., 2011, 40, 5332 RSC; (f) A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, G. M. Robertson and S. D. Robertson, Angew. Chem., Int. Ed., 2011, 50, 8375 CrossRef CAS PubMed; (g) K. Götz, V. H. Gessner, C. Unkelbach, M. Kaupp and C. Strohmann, Z. Anorg. Allg. Chem., 2013, 639, 2077 CrossRef; (h) P. K. Eckert, B. Schnura and C. Strohmann, Chem. Commun., 2014, 46, 4719 Search PubMed; (i) S. G. Koller, U. Kroesen and C. Strohmann, Chem. – Eur. J., 2015, 21, 641 CrossRef CAS PubMed.
- J. Francos, B. J. Fleming, P. García-Álvarez, A. R. Kennedy, K. Reilly, G. M. Robertson, S. D. Robertson and C. T. O'Hara, Dalton Trans., 2014, 43, 14424 RSC.
- (a) A. P. Cole, D. E. Root, P. Mukherjee, E. I. Solomon and T. D. P. Stack, Science, 1996, 273, 1848 CrossRef PubMed; (b) E. C. Brown, J. T. York, W. E. Antholine, E. Ruiz, S. Álvarez and W. B. Tolman, J. Am. Chem. Soc., 2005, 127, 13752 CrossRef CAS PubMed; (c) A. P. Cole, V. Mahadevan, L. Mirica, X. Ottenwaelder and T. D. P. Stack, Inorg. Chem., 2005, 44, 7345 CrossRef CAS PubMed; (d) J. T. York, I. Bar-Nahum and W. B. Tolman, Inorg. Chem., 2007, 46, 8105 CrossRef CAS PubMed; (e) J. E. Bercaw, G. S. Chen, J. A. Labinger and B.-L. Lin, J. Am. Chem. Soc., 2008, 130, 17654 CrossRef CAS PubMed; (f) P. Verma, J. Weir, L. Mirica and T. D. P. Stack, Inorg. Chem., 2011, 50, 9816 CrossRef CAS PubMed.
- (a) H. Y. Lee, J. U. Yoon and J. H. Jeong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2471 Search PubMed; (b) P. K. Eckert, I. d. S. Vieira, V. H. Gessner, J. Borner, C. Strohmann and S. Herres-Pawlis, Polyhedron, 2013, 49, 151 CrossRef CAS.
- W.-C. Cheng, W.-Y. Yu, J. Zhu, K.-K. Cheung, S.-M. Peng, C.-K. Poon and C.-M. Che, Inorg. Chim. Acta, 1996, 242, 105 CrossRef CAS.
- (a) S. V. Pavlova, Y.-S. Wen and S. I. Chan, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m792 CAS; (b) G. Lu and H. C. Malinakova, J. Org. Chem., 2004, 69, 4701 CrossRef CAS PubMed; (c) N. Miklasova, E. Fischer-Fodor, R. Miklas, L. Kuckova, J. Kozisek, T. Liptaj, O. Soritau, J. Valentova and F. Devinsky, Inorg. Chem. Commun., 2014, 46, 229 CrossRef CAS.
- M. Benedetti, G. Tamasi, R. Cini and G. Natile, Chem. – Eur. J., 2003, 9, 6122 CrossRef CAS PubMed.
- P. K. Eckert, V. H. Gessner, M. Knorr and C. Strohmann, Inorg. Chem., 2012, 51, 8516 CrossRef CAS PubMed.
- (a) D. Barr, W. Clegg, R. E. Mulvey, R. Snaith and D. S. Wright, J. Chem. Soc., Chem. Commun., 1987, 716 RSC; (b) M. Westerhausen, M. Wieneke, W. Ponikwar, H. Nöth and W. Schwarz, Organometallics, 1998, 17, 1438 CrossRef CAS; (c) M. A. Beswick, C. N. Hamer, P. R. Raithby, A. Steiner, M. Tombil and D. S. Wright, J. Organomet. Chem., 1999, 573, 267 CrossRef CAS; (d) E. Hevia, D. J. Gallagher, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and C. Talmard, Chem. Commun., 2004, 2422 RSC; (e) M. M. Meinholz and D. Stalke, Eur. J. Inorg. Chem., 2011, 4578 CrossRef CAS.
- (a) B. L. Lucht, M. P. Bernstein, J. F. Remenar and D. B. Collum, J. Am. Chem. Soc., 1996, 10707 CrossRef CAS; (b) J. F. Remenar, B. L. Lucht and D. B. Collum, J. Am. Chem. Soc., 1997, 119, 5567 CrossRef CAS; (c) D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 1998, 120, 5810 CrossRef CAS; (d) J. L. Rutherford, D. Hoffmann and D. B. Collum, J. Am. Chem. Soc., 2002, 124, 264 CrossRef CAS PubMed.
- T. S. De Vries, A. M. Bruneau, L. R. Liou, H. Subramanian and D. B. Collum, J. Am. Chem. Soc., 2013, 135, 4103 CrossRef CAS PubMed.
- (a) R. Michel, T. Nack, R. Neufeld, J. M. Dieterich, R. A. Mata and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 734 CrossRef CAS PubMed; (b) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef CAS PubMed; (c) R. Neufeld, R. Michel, R. Herbst-Irmer, R. Schöne and D. Stalke, Chem. – Eur. J., 2016, 22, 12340–12346 CrossRef CAS PubMed.
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O'Hara, Inorg. Chem., 2015, 54, 9833 CrossRef CAS PubMed.
- D. M. Cousins, M. G. Davidson, C. J. Frankis, D. García-Vivo and M. F. Mahon, Dalton Trans., 2010, 39, 8278 RSC.
- N. M. Clark, P. García-Álvarez, A. R. Kennedy, C. T. O'Hara and G. M. Robertson, Chem. Commun., 2009, 5835 RSC.
- M. P. Bernstein, F. E. Romesberg, D. J. Fuller, A. T. Harrison, D. B. Collum, Q. Y. Liu and P. G. Williard, J. Am. Chem. Soc., 1992, 114, 5100 CrossRef CAS.
- K. W. Henderson, A. E. Dorigo, Q. Y. Liu and P. G. Williard, J. Am. Chem. Soc., 1997, 119, 11855 CrossRef CAS.
- A recent CCDC search ( C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed ) reveals a selection of examples comprising Na⋯Me(Si) interactions, for example see: F. Antolini, P. B. Hitchcock, M. F. Lappert and P. Merle, Chem. Commun., 2000, 1301 RSC; O. Bénaud, J.-C. Berthet, P. Thuéry and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117 CrossRef PubMed.
- Y. Sarazin, S. J. Coles, D. L. Hughes, M. B. Hursthouse and M. Bochmann, Eur. J. Inorg. Chem., 2006, 3211 CrossRef CAS.
- M. Karl, G. Seybert, W. Massa, K. Harms, S. Agarwal, R. Maleika, W. Stelter, A. Greiner, W. Heitz, B. Neümuller and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 1301 CrossRef CAS.
- K. B. Wiberg and W. F. Bailey, J. Mol. Struct., 2000, 556, 239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General synthetic procedures, crystal structure determinations and NMR spectroscopic data. CCDC 1501992 and 1501993. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc07190b |
| This journal is © The Royal Society of Chemistry 2017 |