
A planar model of the vessel wall from cellularized-collagen scaffolds: focus on cell–matrix interactions in mono-, bi- and tri-culture models
Caroline
Loy
a,
Sébastien
Meghezi
a,
Lucie
Lévesque
a,
Daniele
Pezzoli
a,
Heena
Kumra
b,
Dieter
Reinhardt
b,
Jayachandran N.
Kizhakkedathu
c and
Diego
Mantovani
*a
aLaboratory for Biomaterials and Bioengineering, Canada Research Chair I in Biomaterials and Bioengineering for the Innovation in Surgery, Department of Min-Met-Materials Engineering, Research Center of CHU de Quebec, Division of Regenerative Medicine, Laval University, Quebec, QC, Canada G1V 0A6. E-mail: diego.mantovani@gmn.ulaval.ca; Web: http://www.lbb.ulaval.ca Fax: +418 656 5343; Tel: +418 656 2131 # 6270
bFaculty of Medicine, Department of Anatomy and Cell Biology, and Faculty of Dentistry McGill University, Montréal, QC, Canada H3A 0C7
cCentre for Blood Research, Department of Pathology and Laboratory Medicine, Department of Chemistry, University of British Columbia, Vancouver, BC, Canada V6T 1Z3
First published on 5th December 2016
Abstract
The acquisition of new thorough knowledge on the interactions existing between vascular cells would represent a step forward in the engineering of vascular tissues. In this light, herein we designed a physiological-like tri-culture in vitro vascular wall model using a planar cellularized collagen gel as the scaffold. The model can be obtained in 24 h and features multi-layered hierarchical organization composed of a fibroblast-containing adventitia-like layer, a media-like layer populated by smooth muscle cells and an intima-like endothelial cell monolayer. After 7 days of static culture, the compaction of the collagen matrix by the vascular cells was achieved, and the deposition of the vascular extracellular matrix components fibronectin, fibrillin-1 and tropoelastin was observed. The blood-compatible functionality of the endothelial cell monolayer was demonstrated by a blood clotting assay: after 7 days of maturation, clotting was prevented on the endothelialized constructs (more than 80% free hemoglobin maintained after 60 min of blood contact) but not at all on non-endothelialized ones (less than 20% free hemoglobin). In addition, western blotting results suggested that in the tri-culture model the loss of smooth muscle cell phenotype was delayed compared to what was observed in the mono-culture model, finally resulting in a behaviour more similar to the in vivo conditions. Overall, our findings indicate that this in vitro model has the potential to be used as an advanced system to examine vascular cell behavioural interactions, as well as for drug testing and the investigation of physiological and pathological processes.
1. Introduction
Engineered blood vessels can be instantly impactful as in vitro models to test drugs, assess functional devices, and even study the physiology of vascular tissues and the etiology of cardiovascular diseases.1 Despite numerous attempts to produce reliable vascular tissue models, significant challenges remain, such as (1) the distribution, organization, and retention of living cells inside the constructs; (2) the obtainment of a functional extracellular matrix (ECM); and (3) the development of a non-thrombogenic endothelium.2Vascular tissues are hierarchically organized into three layers, namely, the tunica intima, the media, and the adventitia. These layers are respectively populated by endothelial cells (ECs), smooth muscle cells (SMCs) and fibroblasts (FBs) immersed in a highly organized ECM which is composed primarily of type I and III collagen and elastin, the principal components of elastic fibers. The natural formation and maintenance of functional blood vessels are cell-driven, active processes that involve cell growth, death, and migration, as well as tissue structural modifications due to the production and degradation of the ECM by the cells themselves.3–5 In fact, in vivo, the cells intimately interact with the surrounding ECM and with other cells to influence cellular behavior, in addition to the tissue structure and functionality. These factors contribute to the increased interest of researchers to develop reliable co-culture systems to delve further into the close interactions between vascular cells in blood vessels.6,7
Several co-culture models have been developed in the past few years to investigate the interactions of various vascular cell types, notably between ECs and SMCs.8–10 One study on the co-culture of ECs and SMCs has demonstrated the effect of the EC proliferative state on SMC activity, phenotype, and morphology under both static and dynamic culture conditions.11 The influence of FBs on SMCs has also been investigated,12,13 showing that FBs interact with SMCs, thereby affecting their growth, synthetic activity, and ECM deposition. More recently, the addition of FBs in a model of microvessel development composed of polymer fibers embedded in a fibrin gel resulted in mature microvessels containing more endothelial cell–cell connections.14 These studies highlight the key role of vascular cell interactions in the regulation of their activity as well as the biological processes occurring in blood vessels. Moreover, they point out the need to favorably exploit these relationships when engineering vascular tissue models.
Thus far, only a few examples of vascular tissue tri-culture models have been reported in the literature. Gulbins et al. seeded ECs on pre-formed layers of SMCs and FBs cultured on polyurethane vascular prostheses, demonstrating improved EC adhesion and resistance to shear stress,15 while L'Heureux and colleagues were the first to develop a completely biological tissue-engineered human blood vessel using a self-assembly method, the preparation of which takes close to three months.16 Although these approaches are promising for vascular tissue engineering, either they involve synthetic scaffolds which do not mimic the natural 3D matrix or they require weeks of maturation to obtain the final tissue.
In this context, the goal of this study was to design and develop an easy-to-prepare 3D tri-culture vascular wall model and to investigate how the interactions between vascular cells could guide the generation of functional arteries. To the best of our knowledge, no other study has incorporated the layered culture of three vascular cell types into an ECM-mimicking matrix using planar type I collagen gels as scaffolds for the investigation of vascular cell interactions. More specifically, based on a previously reported original protocol for the processing of collagen and SMCs,17 tri-layered planar cellularized scaffolds were developed. The first (adventitia-like) and the second (media-like) layers were composed of a collagen gel matrix cellularized with a combination of FBs and SMCs and SMCs alone, respectively. The third (intima-like) layer was composed of ECs seeded as a monolayer on the upper surface of the media-like layer. Of importance is that the resulting model finely reproduced the structural properties of native blood vessels, with preparation times of ca. two hours and the final construct was ready in 24 h.
2. Materials and methods
2.1. Cell isolation and culture
Human umbilical artery SMCs (HUASMCs) and human umbilical vein ECs (HUVECs) were isolated from human umbilical cord samples. Written informed consent was obtained from all mother donors according to the Declaration of Helsinki. All experiments were performed in compliance with the Canadian Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans and institutional CHU de Quebec – Laval University guidelines. The protocol was approved by the Ethics Committee of the CHU de Quebec Research Centre (CER #S11-03-168). FBs from human aortic adventitia were purchased from Lonza (AoAF cc-7014, Walkersville, MD, USA). ECs and SMCs were cultured at 37 °C and with 5% CO2 respectively in M199 (Gibco, Life Technologies, Grand Island, NY, USA) and Dulbecco's Modified Eagle's Medium (DMEM, Gibco), and both were supplemented with 10% fetal bovine serum (Gibco), 1% penicillin/streptomycin (Gibco), 2 ng mL−1 of basic fibroblast growth factor (FGFb, Gibco), 0.5 ng mL−1 of epidermal growth factor (EGF, Invitrogen, Life Technologies), 1 μg ml−1 of L-ascorbic acid (Sigma, St Louis, MO, USA), 5 μg mL−1 of human insulin solution (Santa Cruz Biotechnology, Dallas, TX, USA), 1 μg mL−1 of hydrocortisone (Sigma), 90 μg mL−1 of porcine heparin sodium salt (Grade I-A, Sigma), and 1 μg mL−1 of endothelial cell growth supplement (ECGF, Becton Dickinson, Oakville, ON, Canada). The complete media is hereafter referred to as M199+ and DMEM+, respectively. Finally, the FB culture medium used was stromal cell growth medium (SCGM BulletKit, CC-3204 & CC-4181, Lonza).For the cell isolation studies, umbilical cord samples approximately 15 cm in length were obtained from normal-term pregnancies with the consent of the donor mothers. The umbilical cord samples were then collected in phosphate-buffered saline solution (PBS, Fisher Scientific, Fair Lawn, NJ, USA) supplemented with 5% penicillin/streptomycin solution to avoid any contamination, and maintained at 4 °C until processing. For HUVEC isolation, the vein was rinsed with PBS, filled with 10× trypsin–EDTA solution (Gibco), and incubated for 15 min at 37 °C, after which the trypsin–EDTA solution containing the cells was collected. PBS was added to wash the lumen, collected along with the previous solution, and centrifuged at 1000 rpm for 5 min. Thereafter, the supernatant was removed and the cells were resuspended in M199+ and seeded in a 75 cm2 flask (Corning, Oneonta, NY, USA). The culture medium was changed after 24 h and then every 48 h until confluence. ECs were characterized using a mouse antibody against CD31 (Dako, M0623, dilution 1/50, Burlington, ON, Canada) and a rabbit antibody against von Willebrand factor (VWF, Abcam, Ab6994, dilution 1/100, Toronto, ON, Canada). For HUASMC isolation, Wharton's jelly and all of the associated connective tissues surrounding the umbilical arteries were carefully removed, the arteries were opened longitudinally, and the EC layer was removed by scraping the intima. The arteries were subsequently cut into small pieces by means of a scalpel blade and carefully placed in a Petri dish containing DMEM+. After two weeks, SMCs had migrated from the explants to colonize the surface of the cell-culture dish. The pieces of artery were removed thereafter and the cells were expanded in DMEM+ medium. The SMCs were identified by staining their markers, namely, smooth muscle-α-actin (SM-α-actin) and calponin, using the appropriate antibodies (Abcam, Ab7817, dilution 1/200 and Ab46794, dilution 1/200, respectively). In the reported experiments, all of the cells were used up to passage 6.
2.2. Cellularized collagen-based construct preparation
Type I collagen was extracted from rat tails, solubilized in acetic acid solution (0.02 N) at a concentration of 4 g L−1, and processed as previously reported.18 For the cellularized gel preparation, SMCs or a mixture of SMCs and FBs were suspended in either DMEM+ or SCGM, and then mixed with the collagen solution supplemented with NaOH (15 mM) and Hepes (20 mM) to a final collagen concentration of 2 g L−1 and pH 7.2. The resulting collagen–cell suspension was poured into either 12- or 6-well culture plates (Corning) or 4-well culture slides (Becton Dickinson), where a sterile gauze was previously deposited and glued to anchor the forming gels to the wells for the duration of the culture (Fig. 1A).19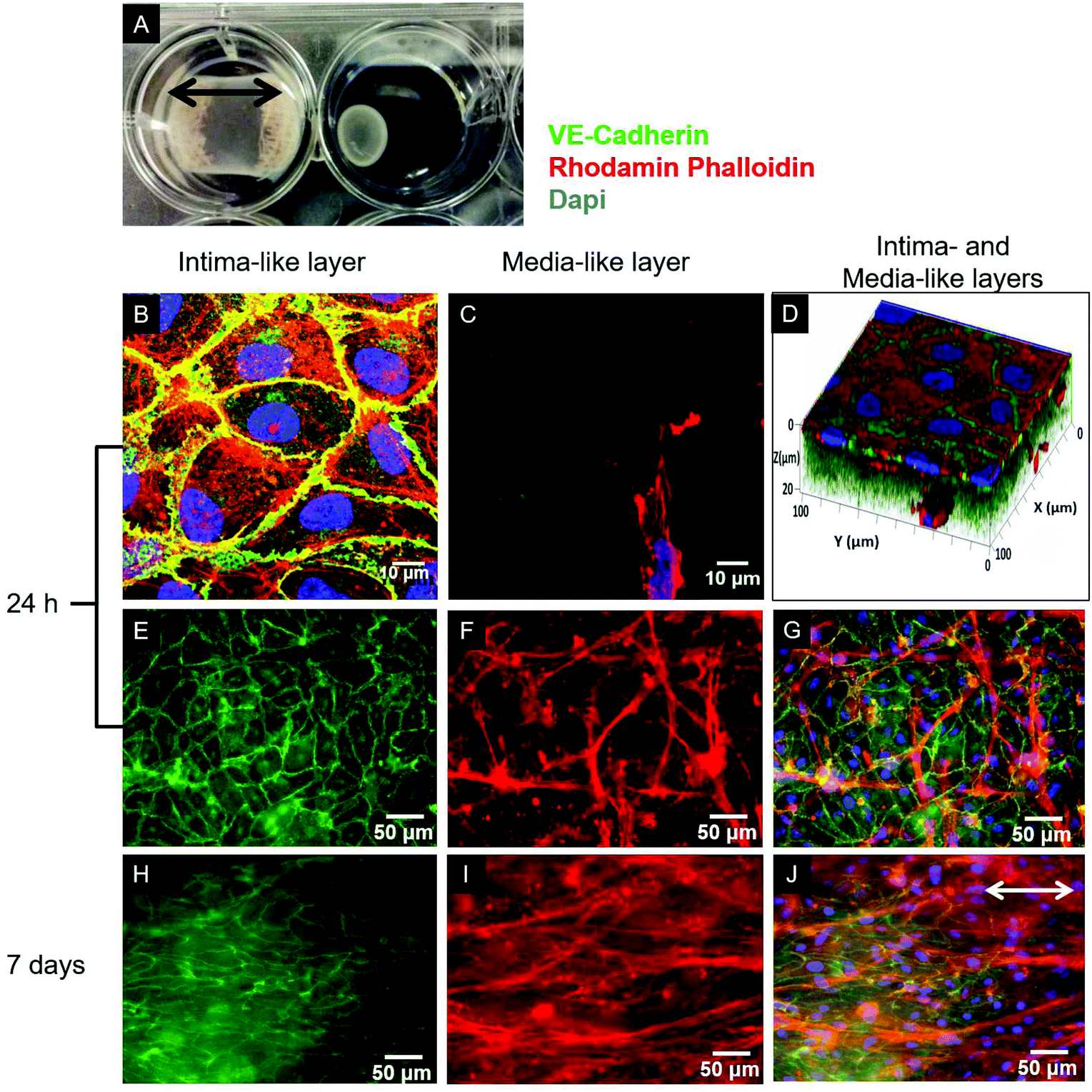 | ||
| Fig. 1 (A) Representative photograph of SMC-cellularized collagen gel-based constructs anchored (left) or not (right) to the culture well with a gauze after 3 days of static culture. Cell-mediated gel compaction is evident, demonstrating the necessity to anchor the gel to the well to retain the compaction in the longitudinal (indicated by the black arrows) and lateral directions. (B–J) Characterization by immunofluorescence of the tri-culture model. VE-Cadherin (green), F-actin (rhodamine-phalloidin, red) and nuclei (DAPI, blue) were stained. (B, C) 2D sequential confocal images at different Z stacks from the surface (1 μm and 18 μm depth, respectively) and (D) 3D rendering of confocal Z-stack images of the tri-culture construct after 24 h of culture confirmed the formation of an EC monolayer and the presence of underlying SMCs inside the collagen gel. (E, H) Superficial EC monolayer (VE-cadherin, green) and (F, I) first layers of SMCs (F-actin, red) embedded in the collagen gel after (E, F) 24 h and (H, I) 7 days of culture, as visualized by widefield fluorescence microscopy by focusing at different depths. (G, J) Merged images representing the structure of the very first cellular layers of the construct. The progressive alignment of SMCs in the direction of the retaining compaction (arrow) can be observed. | ||
The tri-culture collagen-based models were prepared as follows: an adventitia-like layer containing 5 × 105 SMCs per mL and 5 × 105 FBs per mL was prepared by adding 0.11 mL cm−2 of the collagen–cell suspension. After 5 min of incubation at room temperature (r.t.), a media-like layer containing 106 SMCs per mL was molded over the adventitia-like layer (0.22 mL cm−2, with a final volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 with respect to the total collagen gel volume), and the obtained construct was incubated for 30 min at 37 °C. A superficial EC monolayer (intima-like layer) was then prepared by seeding cells on the surface of the media-like layer at a density of 80000 cells per cm2 in M199+. The resulting constructs were then incubated at 37 °C in a cell culture incubator. After 24 h, the culture medium was replaced with DMEM+ and was subsequently changed every 48 h for either 3 or 7 days. The mono- and bi-culture models were respectively composed of only the media-like and the media- and intima-like layers, and were prepared as described above for the tri-culture model. For blood clotting studies another model was used, composed of a non-cellularized collagen gel covered by an intima-like layer.
:3 with respect to the total collagen gel volume), and the obtained construct was incubated for 30 min at 37 °C. A superficial EC monolayer (intima-like layer) was then prepared by seeding cells on the surface of the media-like layer at a density of 80000 cells per cm2 in M199+. The resulting constructs were then incubated at 37 °C in a cell culture incubator. After 24 h, the culture medium was replaced with DMEM+ and was subsequently changed every 48 h for either 3 or 7 days. The mono- and bi-culture models were respectively composed of only the media-like and the media- and intima-like layers, and were prepared as described above for the tri-culture model. For blood clotting studies another model was used, composed of a non-cellularized collagen gel covered by an intima-like layer.
2.3. Evaluation of EC adhesion by shear exposure experiments
To assess EC adhesion to the underlying collagen gels, the bi- and tri-culture constructs were placed in a flow chamber (Sticky-Slide I 0.4 Luer, Ibidi, Madison, WI, USA) adapted to the gel geometry. Continuous flow of culture medium was generated by using a peristaltic pump (Masterflex, Cole-Parmer, Montreal, QC, Canada), thereby eliciting a shear stress of 15 dynes per cm2 on the ECs of the intima-like layers, which was representative of physiological arterial hemodynamic shear stress.20 Following flow-exposure for 24 h at 37 °C, EC adhesion and morphology were determined by immunofluorescence (IF). The other experiments were performed without flow exposure.2.4. Histological analysis
Histological analyses were performed on the developed models to evaluate cell distribution and cell/matrix organization. After 3 or 7 days of static culture, the collagen gel constructs were first rinsed with PBS and fixed in 3.7% formaldehyde (Sigma) for 20 min and embedded in paraffin, after which 5 μm sections were cut. For the histochemical analyses, the sections were deparaffinized with toluene, rehydrated through graded washes of ethanol in water, refixed in Bouin solution overnight, and finally stained using a modified Masson's trichrome procedure. Three different dyes were used to differentiate the cells from the ECM: Weigert's iron hematoxylin solution for nuclear staining (dark brown); acid fuchsin and xylidine ponceau solution for cell cytoplasm (red); and light green SF yellowish solution for collagen (green, no species specificity). For the immunohistochemical analyses, deparaffinized and rehydrated sections were stained for SM-α-actin and von Willebrand factor using the appropriate antibodies (Abcam, Ab7817, dilution 1/200 and Ab6994, dilution 1/100, respectively), while nuclei were stained with DAPI (1/3000, Life Technologies). Images were captured under an Olympus BX51 fluorescence microscope. Staining of the ECM proteins was performed on deparaffinized and rehydrated tissue sections using the Dako Envision + System-HRP (AEC) immunohistochemistry kit (Dako, K4008), while heat-induced and proteinase K-mediated methods were used for antigen retrieval. The sections were stained using anti-human fibronectin (Sigma, F3648), anti-human tropoelastin (Elastin Products Company, PR398) with anti-human fibrillin-1 C-terminal21 antibodies and hematoxylin as the counterstain. Images were recorded under a Zeiss Axio Imager M2 microscope.2.5. Immunofluorescence analysis
After 1, 3, or 7 days of culture, the constructs were washed twice with PBS, fixed with 3.7% formaldehyde (Sigma) for 20 min, and treated with 0.5% Triton X-100 in PBS for 30 min at r.t. to permeabilize the cells. The constructs were incubated thereafter for 1 h at r.t. with the mouse monoclonal antibody against VE-cadherin (Abcam, Ab7047, dilution 1/50), washed twice with 0.05% Tween 20 in PBS, and finally incubated at r.t. for 1 h with the anti-mouse Alexa Fluor 488 antibody (Life Technologies). Rhodamine-phalloidin (Sigma) and DAPI were used to stain F-actin and nuclei, respectively. Images were obtained under an Olympus BX51 fluorescence microscope and an LSM 700 confocal laser scanning microscope (Zeiss) controlled by ZEN 2009 software for image acquisition and further analysis.2.6. Western blot analysis
The expression of SMC phenotypic markers SM-α-actin and calponin was determined by western blot analysis. The constructs were frozen in liquid nitrogen and then ground in a mortar. Thereafter, 0.1 g of the obtained powder were mixed with radio immunoprecipitation (RIPA) lysis buffer (25 mM of Tris, 75 mM of NaCl, 5 mM of EDTA, 0.25% Triton X-100, and 0.1% SDS), supplemented with a mini EDTA-free proteinase inhibitor cocktail (1:10 mL, Roche, Mississauga, ON, Canada). The protein concentration was determined by using the standard Bradford method. Samples containing equal amounts of proteins were then boiled for 5 min in 1X Laemmli buffer (BioRad, Mississauga, ON, Canada) containing 2% β-mercaptoethanol (Sigma). Equal amounts of proteins were then loaded in the lanes of 4–15% Mini-PROTEAN TGX stain-free protein gels (BioRad) and subsequently run at 50 mA (maximum voltage 200 V) using a Mini-PROTEAN electrophoresis cell kit (Bio-Rad). The proteins were then transferred to nitrocellulose membranes by means of a Trans-Blot Turbo™ transfer system (BioRad), with the blots incubated thereafter for 1 h in a blocking solution of 5% non-fat dry milk in Tris-buffered saline (TBS) containing 0.2% Tween 20 (TBS-Tween). The membranes were then incubated with either mouse anti-SM-α-actin (Sigma, dilution 1/400) or rabbit anti-calponin (Abcam, dilution 1/2000) antibodies for 2 h in TBS-Tween containing 0.1% non-fat dry milk and were then rinsed 3 times for 10 min in TBS-Tween. The membranes were then incubated in either goat anti-mouse or anti-rabbit IgG (H + L)-HRP conjugate (BioRad) and anti-GAPDH-HRP conjugate antibodies (Abcam) for 1 h and subsequently rinsed 3 times in TBS-Tween. An enhanced chemiluminescence kit (ECL, BioRad) was used to visualize the protein bands. The SM-α-actin and calponin band intensities were quantified by densitometry using ImageJ22 software and were then normalized over the signal of the corresponding bands of GAPDH (loading control).
2.7. Blood clotting studies
The coagulation of whole blood in contact with the surfaces of the developed models was evaluated by monitoring the hemoglobin (Hb) concentration (%) using the Drabkin method.23,24 All experiments were performed in compliance with the Canadian Tri-Council Policy Statement: Ethical Conduct for Research Involving Humans and the Centre for Blood Research of the University of British Columbia guidelines. The protocol for blood collection was approved by the Clinical Research Ethics Board (CREB; certificate number H10-01896) of the University of British Columbia, and written consent was obtained from donors in accordance with the Declaration of Helsinki. Briefly, 250 μl of human citrated blood were mixed with 50 μl of 0.1 M CaCl2 (Sigma). The constructs were covered with 10 μl of this solution and incubated thereafter at 37 °C. After 10, 20, 30, 40, 50, and 60 min, 700 μl of PBS were added to each surface and 75 μl of the resulting supernatant were mixed with 75 μl of Drabkin's reagent (Sigma). After 30 min of incubation at r.t., the absorbance at 540 nm was measured by means of a spectrophotometer. Drabkin's reagent alone was used as a reference and blood in contact with the surfaces for 0 min was used as a control. The experiments were performed in triplicate using blood from three different donors.2.8. Statistical analysis
The experiments were performed at least in triplicate, while the data were expressed as means ± SD and the statistical analyses were conducted using GraphPad InStat 3 software (GraphPad, La Jolla, CA, USA). All of the data were analyzed by one-way ANOVA (multiple comparisons) along with a post-hoc Tukey test to indicate significant differences between the experimental groups. The significance was retained when p < 0.05.3. Results
In this study, a collagen gel-based 3D model of vascular tissues containing ECs, SMCs, and FBs was produced, developed, and characterized. The final construct was composed of three highly organized layers: (1) an EC monolayer mimicking the natural tunica intima and providing blood compatibility properties to the construct; (2) an SMC-enhanced collagen gel layer modeling the tunica media; and (3) a collagen gel layer cellularized with FBs and SMCs, corresponding to the tunica adventitia. To compare the properties of the developed tri-culture construct, three additional models were prepared: a bi-culture model composed of media-like and intima-like layers, a mono-culture model composed of only the media-like layer, and finally a monolayer of ECs seeded over a collagen gel (EC-collagen model).3.1. Formation of a stable EC monolayer on collagen gel-based 3D models
As evidenced in the IF images reported in Fig. 1B, D, E, and G, after 24 h of culture in the tri-culture model, ECs stained for EC marker VE-cadherin appeared confluent, with the typical polygonal shape, suggesting that they were able to adhere and proliferate on the underlying collagen surface. No ECs could be observed 20 μm below the superficial EC monolayer, indicating that ECs are confined to the gel surface (Fig. 1C). The presence of SMCs inside the gel immediately below the surface was confirmed by the elongated cells stained for actin filaments (rhodamine phalloidin) and not for VE-cadherin visible beyond the first layer of the ECs, as observed by confocal microscopy images in Fig. 1C, and D, and by widefield fluorescence microscopy images in Fig. 1F, and G. To confirm that the ECs had well adhered, the monolayer was exposed to mechanical stimulation. Following exposure for 24 h to a shear stress of 15 dyne per cm2, comparable to the average shear stress in healthy arteries,20 the ECs did not detach from the surface and were elongated and aligned in the direction of the flow, as observed in Fig. 2.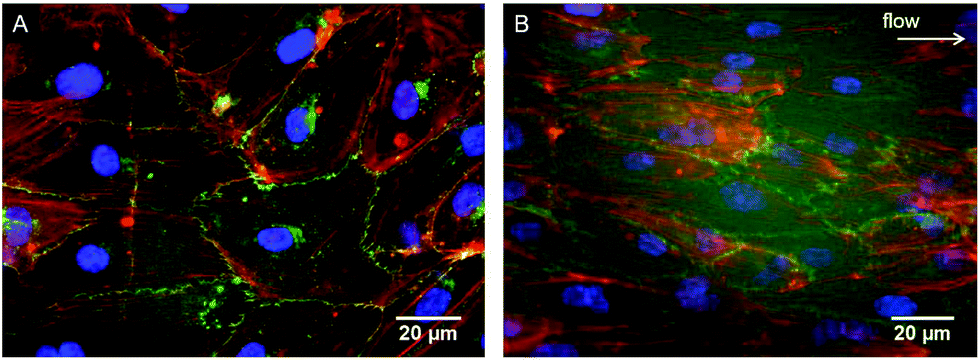 | ||
| Fig. 2 Immunofluorescence staining of ECs on the surface of the tri-culture model cultured for 24 h under (A) static or (B) dynamic conditions (shear stress of 15 dynes per cm2). Cell alignment along the flow direction was observed (arrow). VE-Cadherin (green), F-actin (rhodamine-phalloidin, red) and nuclei (DAPI, blue) were stained. | ||
3.2. Assessment of cellular organization and cell-mediated matrix remodeling and deposition
After 7 days of growth under static culture conditions, the SMCs in the tri-, bi- and mono-culture models were elongated and aligned in the direction of the anchors due to cell-mediated gel compaction, as observed in Fig. 1I and J, with the ECs remaining attached and the monolayer intact (Fig. 1H). In contrast, when ECs were seeded on uncellularized collagen models, no compaction was observed (data not shown). Staining of SM-α-actin confirmed that the cells embedded in the collagen matrix immediately beneath the EC monolayer are SMCs.Histological staining of transverse sections of the tri-culture models (Fig. 3) confirmed the presence of the three layers and the alignment of SMCs and FBs in the direction of the anchors which prevented compaction in the longitudinal direction; this was particularly evident after 7 days of culture (Fig. 3B and E). Initially, the thickness of the bottom layer (adventitia-like layer) was lower than that of the second layer (media-like layer) because of the different starting volumes (ratio 1:2). After 7 days of maturation, the adventitia/media-like layer thickness ratio inverted to approximately 2:1, owing to the higher compaction ability of the SMCs involved; however, no delamination between the two layers was observed. A comparable gel compaction with the associated increase in cell density was also observed in the bi- and mono-culture constructs. Immunohistochemical staining with an antibody specific to ECs (vWF) showed intact and continuous EC monolayers and confirmed that no migration of ECs occurred inside the collagen gel in either the tri-culture model (as shown in Fig. 3C and D) or the bi-culture and EC-collagen constructs (not shown).
 | ||
| Fig. 3 Histological characterization of transverse sections of the tri-culture model. Sections were stained after (A) 3 days and (B, E) 7 days with Masson's trichrome to visualize collagen (green, no species specificity), cell cytoplasm (red), and cell nuclei (dark purple). SMCs were homogenously dispersed within the collagen gel, with a visible alignment in the direction of the anchors (arrow), notably after 7 days of maturation. Immunostaining of von Willebrand factor (red), specific for ECs, at (C) 3 days and (D) 7 days of culture, confirming that ECs did not migrate inside the underlying collagen gel populated by SMCs, as supported by the SM-α-actin (green) and nuclear (DAPI, blue) staining. | ||
In order to investigate the deposition of critical vascular ECM proteins by the vascular cells, immunohistochemical staining of human fibronectin, fibrillin-1, and tropoelastin was performed on transverse sections after 7 days of maturation (Fig. 4). The tri-layered organization of the tri-culture construct was again evident and the deposition of newly synthesized fibronectin, fibrillin-1, and tropoelastin was observed on each of the tested constructs. However, contrary to the relatively modest deposition of the three proteins in the mono- and bi-culture models (Fig. 4A, C, D, and F), a strong upregulation was observed in the tri-culture model (Fig. 4G–I).
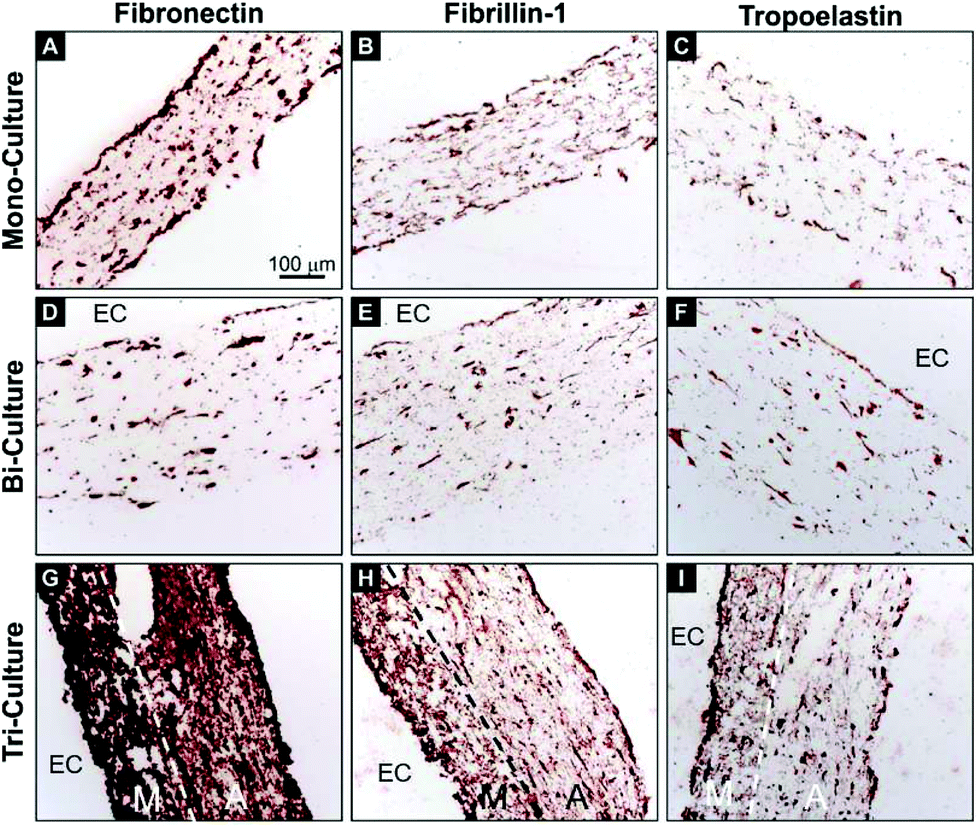 | ||
| Fig. 4 Immunohistochemistry images of transverse sections of the (A–C) mono-, (D–F) bi-, and (G–I) tri-culture models after 7 days of static maturation. Sections were stained using antibodies against (A, D, G) fibronectin, (B, E, H) fibrillin-1, and (C, F, I) tropoelastin (red-brown staining). Hematoxylin was used as the counterstain (blue). Maximum staining for each antibody was observed in the tri-culture model. The endothelialized side of the construct, when present, was indicated by “EC”, the dashed lines indicate the interface between the media-like layer (M) and the adventitia-like layer (A). | ||
3.3. Evaluation of the SMC phenotype in 3D models
SMCs are capable of phenotypically switching from a synthetic to a contractile state. The expression of two specific proteins, namely, early development marker SM-α-actin and mid-stage development marker calponin, which indicates phenotypic differentiation from a synthetic to a contractile state, was investigated by western blot analysis after 0, 3, and 7 days of culture in the tri-, bi-, and mono-culture models (Fig. 5). As expected, SMCs expressed SM-α-actin and calponin regardless of the model, and although the expression kinetics of both markers were markedly different in each model, they did follow a similar trend. In the mono-culture model after 7 days, the contractile phenotype marker expression decreased by approximately 70 and 75% compared to day 0 for SM-α-actin and calponin, respectively (p < 0.05). In the bi-culture model, a bell-shaped trend was observed, with an increase in expression of approximately 50% after 3 days (p < 0.05 for SM-α-actin) and a return to values comparable to day 0 after 7 days for calponin (p > 0.05) and slightly lower for SM-α-actin (20% reduction, p < 0.05). The observed behaviour of the tri-culture model was halfway between that of the mono- and bi-cultures; indeed, no important differences were observed after 3 days, whereas after 7 days, expression levels decreased by 37 and 23% with respect to day 0 for SM-α-actin (p < 0.05) and calponin, respectively.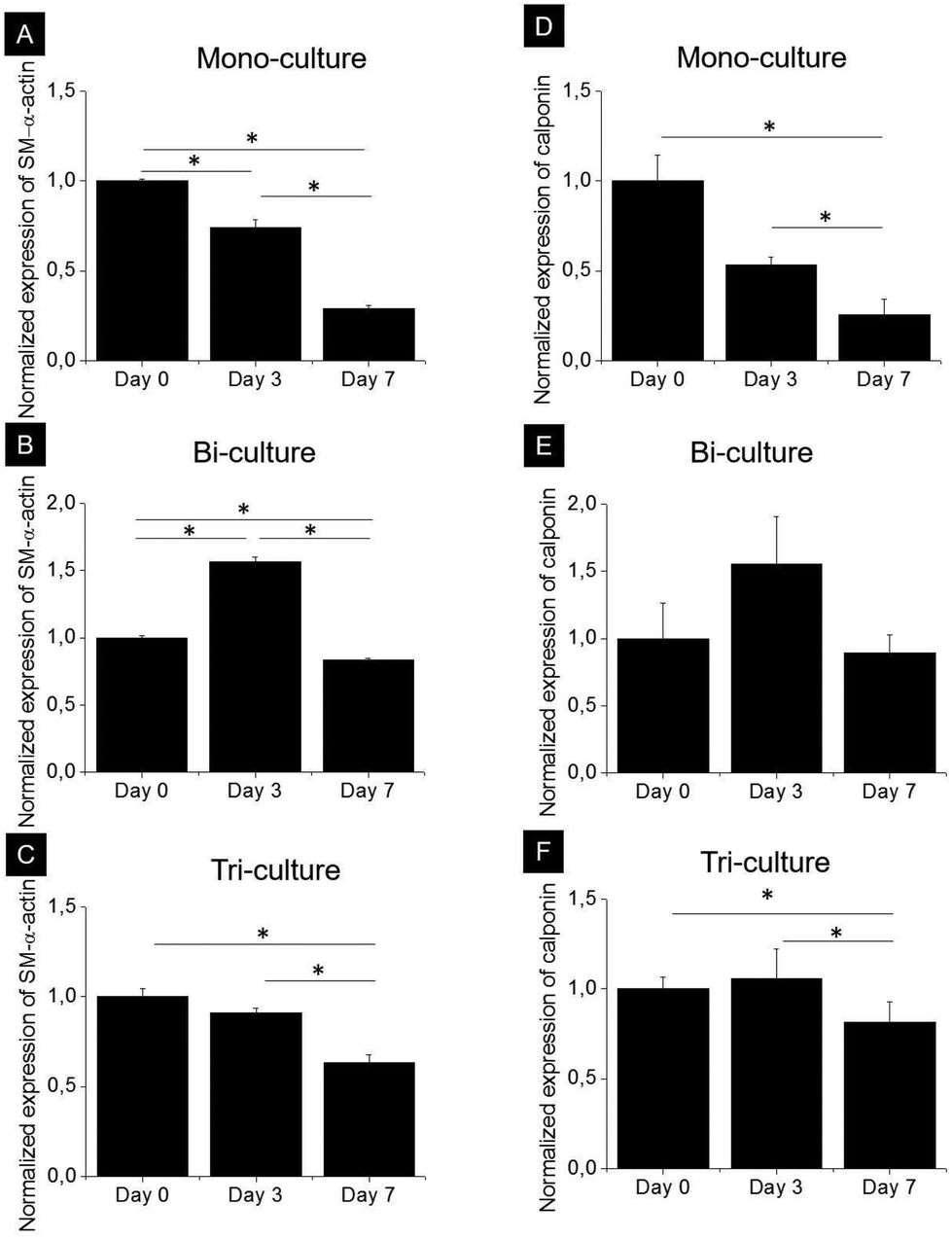 | ||
| Fig. 5 Analysis of the expression of (A, B, C) SM-α-actin and (D, E, F) calponin by SMCs in the (A, D) mono-, (B, E) bi-, and (C, F) tri-culture models after 0, 3, and 7 days of culture. Protein levels were normalized to GAPDH as the loading control and to the corresponding expression at day 0. Data are expressed as means ± SD; * indicates p < 0.05. | ||
3.4. Blood clotting studies
Recalcified citrated whole blood was used to perform blood clotting studies to assess the thrombogenicity of the collagen gel models. In this assay, blood was deposited on the surface of collagen gel constructs and allowed to clot. As coagulation occurred, the hemoglobin was retained in the clot, with the size of the clot being inversely proportional to the hemoglobin concentration in the supernatant, as measured by the absorbance. Fig. 6A shows the kinetics of the blood clotting on the surface of different collagen gel models after 3 days of culture. The blood was considered completely clotted when the percentage of free hemoglobin in solution was lower than 20%.25 On the non-cellularized collagen gel and mono-culture models (SMC-cellularized collagen gel), the blood completely clotted within 30 min of incubation, a time comparable to the blood clotting time under normal conditions on thrombogenic surfaces. On each endothelialized model, the blood did not completely clot after 60 min, however the amount of free hemoglobin decreased over time up to values close to 50%. It is of significance that after 30 min, the clotting on the tri-culture model was significantly lower than that observed on both the bi-culture model and the EC-seeded plain collagen (% of free hemoglobin: 85 ± 6% vs. 68 ± 1% and 51 ± 3%, p < 0.05), showing that the EC monolayer grown over the tri-culture model slowed down the clot formation, thus reflecting its superior antithrombogenic properties. In each endothelialized model after 7 days of maturation (Fig. 6B), no significant clotting was observed within the first 60 min, with a higher than 80% concentration of free hemoglobin.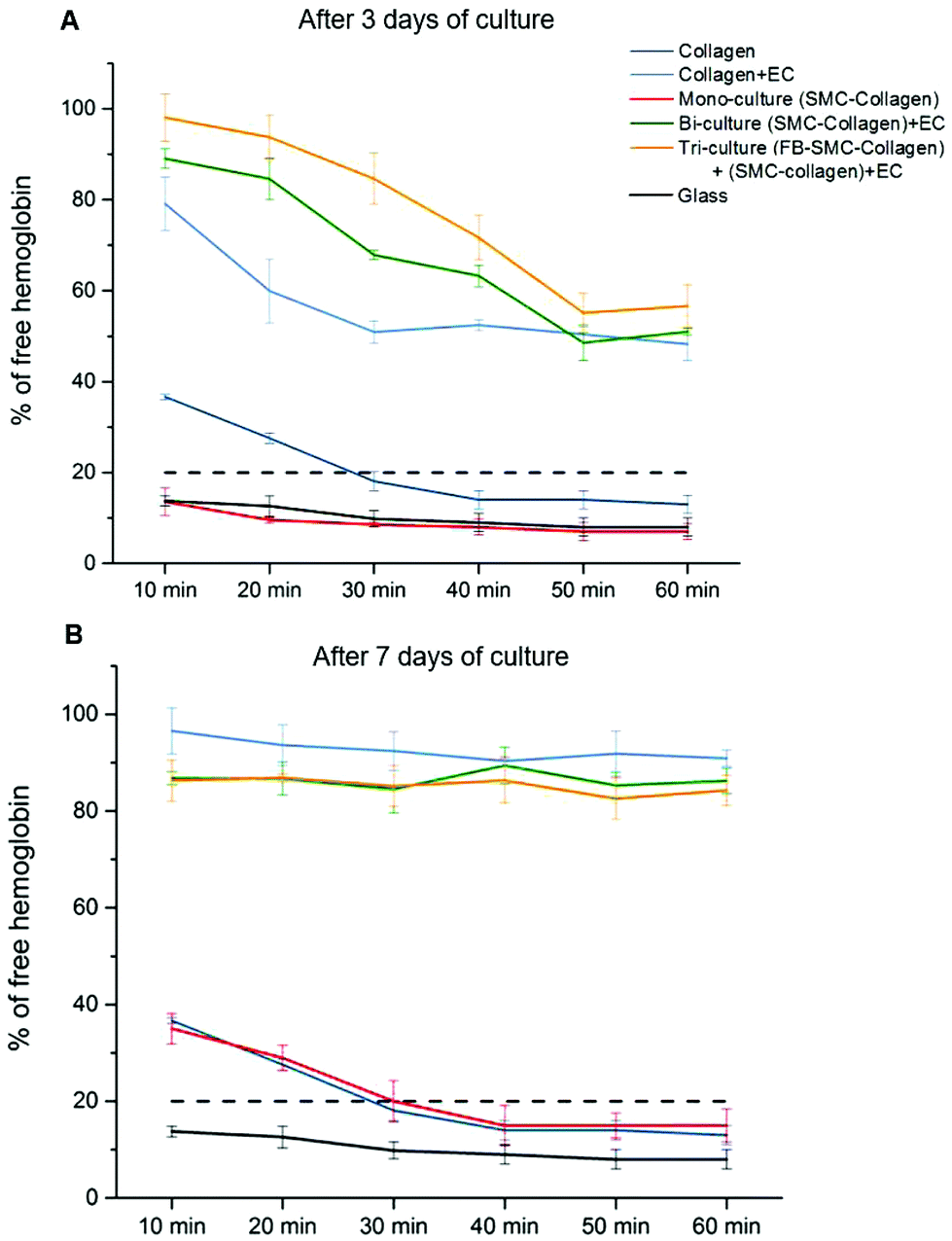 | ||
| Fig. 6 Kinetics of whole blood clotting for different collagen gel models after (A) 3 or (B) 7 days of culture. Positive blood clotting controls were performed on glass slides. The size of the clot was inversely proportional to the hemoglobin concentration in the supernatant. Blood was considered completely clotted when free hemoglobin was lower than 20% (dotted lines). The experiments were performed in triplicate with blood from three different donors. | ||
4. Discussion
In recent years, the design of systems for the co-culture of vascular cells has attracted considerable attention because of the increasing demand of reliable in vitro models to investigate the physiological and pathological processes of vascular tissues in vivo. Initially, 2D systems were proposed;9,10,26 ultimately, however, 2D cell cultures cannot adequately reproduce the complex physiology of native arteries. Scaffold materials with suitable properties have thus been introduced to bridge the gap between classical cell cultures and native tissues27,28 and a few 3D co-culture systems have been developed29–35 which mainly use only two cell types.In the present study, based on these premises, a planar cellularized collagen gel-based 3D model of the arterial wall containing ECs, SMCs, and FBs was designed and characterized. Substantial efforts were devoted to considerably reduce the time required for model preparation, with the protocol developed in this work resulting in preparation times of ca. two hours and the obtainment of a confluent EC monolayer on the surface of the vascular wall model already 24 h after preparation. Collagen-based hydrogels were chosen as the scaffold material, with the following unique combination of benefits: (1) collagen is one of the main components of the ECM of native blood vessels; (2) it is abundantly available and can be easily purified;18 (3) it is non-antigenic and biocompatible; (4) collagen gel can be directly and uniformly seeded with cells during the fabrication process;17 (5) the collagen network can be actively remodeled by cells36 and (6) can also be used as a tissue regeneration scaffold.37,38
Because SMCs were capable of compacting the collagen gel matrix, a sterile gauze was previously glued inside the Petri dishes to anchor the gels to the underlying substrate and retain compaction in the longitudinal (indicated by the arrows in Fig. 1A) and lateral directions. Consequently, the subsequent compaction occurred mainly in terms of thickness of the constructs (vertical direction). It is of interest that in addition to the classical intima- and media-like layers, an engineered adventitia was introduced in this model, which made it possible to mimic the highly organized tri-layered structure of native blood vessels (Fig. 3). Traditionally, the adventitia layer was considered to provide only structural support to blood vessels, but it is now emerging as an important player in both vessel remodeling and the pathogenesis of cardiovascular diseases.5 As the ability of adventitial fibroblasts to contract collagen is much lower than that of SMCs, the latter were also added in the engineered adventitia to obtain the same compaction rate in the two collagen layers and to avoid delamination. We acknowledge that the presence of SMCs in the adventitia layer is less physiological than a pure FB layer; however, this in no way hindered our investigation on how FBs influenced the behavior of the other cell types and tissue maturation in general. Moreover, it is worth mentioning that despite the strong compaction of the collagen matrix, the SMC and FB cell density remained quite low after 7 days of maturation (Fig. 3), particularly when compared to that of native vessels. That said, in the proposed system, cell numbers may be easily increased thanks to our simple collagen gel cellularization process.
4.1. Endothelialization
A major issue limiting current vascular tissue model engineering is the lack of a functional (antithrombogenic), adherent, and shear-resistant EC layer.39 Therefore, to initially optimize endothelialization of the upper surface of the constructs, EC seeding was performed using M199+ culture medium, which was shown to perform better than DMEM in promoting better EC growth and proliferation to form a monolayer.30 After 24 h of culture, the ECs completely covered the surface of the collagen constructs (Fig. 1B, D, E and G) and their adhesion to the collagen substrate was confirmed by the resistance of the endothelial layer upon exposure to physiological-like shear stress for 24 h. It is of relevance that the typical EC cobblestone pavement structure and the intercellular VE-cadherin-mediated junctions between the ECs were completely preserved, with evident cell alignment in the direction of flow (Fig. 2). These findings demonstrate that the seeding and culture conditions chosen for the endothelialization phase were appropriate to obtain a stable and confluent intima-like layer within 24 h. Furthermore, our blood clotting experiment results show that each model featuring an engineered intima was able to slow down blood coagulation after 3 days of culture and inhibited the process (in our experimental setup) after 7 days, thereby confirming the functionality of the obtained EC monolayers (Fig. 6). However, the different coagulation rates observed after 3 days of culture in the endothelialized models suggest that both SMCs and FBs can influence the behavior, activity, and functionality of ECs, particularly in terms of the more rapid formation of an antithrombogenic EC layer.4.2. Expression of SMC contractile phenotype markers
In order to favor the cell-mediated remodeling of the collagen matrix 24 h after construct preparation, the medium was changed to DMEM+ medium, a widely used basal medium supplemented with specific vitamins and growth factors recommended to increase matrix deposition and remodeling by SMCs and FBs.40,41 As expected, SMC protein expression was also significantly influenced by the presence of other vascular cell types in co-culture. It is known that immediately after isolation from arterial walls, SMCs primarily display a contractile phenotype.9 However, in vitro static culture with serum-containing medium42 is known to induce a synthetic phenotype. While several studies have shown that both ECs and FBs are capable of regulating the SMC phenotype,8,12 contrasting results have been reported, due to differences among the 2D models employed. For example, in a 2D model of co-cultured ECs seeded directly on contractile SMCs, Wallace et al. reported increased calponin expression by the SMCs,43 whereas Sakamoto et al., in an indirect 2D bi-culture model, showed a decreased expression of SM-α-actin.9 To the best of our knowledge, our study is the first to combine the effects of both ECs and FBs on SMCs in a 3D model, thus profoundly mimicking the physiology of native tissues. Due to the presence of ECs, the study was carried out in serum-supplemented medium. In the mono-culture constructs, the expression of contractile phenotype markers by SMCs was progressively downregulated after 3 and 7 days of culture (Fig. 5), as already observed in 3D type I collagen gels.42In contrast, in the bi-culture constructs, contractile phenotype marker expression was upregulated at day 3 but not at day 7 whereas the tri-culture models showed an intermediate behavior. These observations suggest that the presence of ECs could delay the loss of the contractile phenotype of SMCs, while FBs mitigated this effect, most likely by promoting ECM synthesis.4.3. ECM synthesis
It is an established fact that FBs are capable of inducing ECM synthesis by SMCs12 and that ECs can influence the composition of the synthesized ECM.44 With this in mind, the deposition of ECM proteins was investigated in these models after 7 days of maturation. We focused on fibronectin, fibrillin-1, and tropoelastin for several reasons. Fibronectin is a key player in the biogenesis and organization of the vascular ECM and is crucial for the biogenesis of a number of critical ECM proteins, including fibrillin-1,45 latent TGF-β binding protein-1,46 collagen I and III,47 and others. In turn, assembled fibrillin-1 microfibrils provide a template for the deposition of soluble tropoelastin in the form of small amorphous aggregates, representing the first steps in elastic fiber formation. Moreover, promoting in vitro elastogenesis remains among the principal challenges in vascular tissue engineering.48 Here, a significant improvement in ECM deposition was observed in the tri-culture model but not in the bi- and mono-culture systems (Fig. 4), thus demonstrating the key role of FBs in modulating ECM synthesis. It is of particular relevance that the significant deposition of fibronectin and fibrillin-1 in the tri-culture constructs, and more so in the media-like layers, indicates that the process of elastogenesis was underway, as confirmed by the superior expression of tropoelastin. These results are of importance, as a significant presence of tropoelastin was already evident after 7 days, whereas elastin deposition usually requires at least 3–4 weeks of culture to be observed in classical 3D vascular constructs containing only SMCs.495. Conclusion
By partially lifting the veil on the complex cell–cell and cell–matrix interactions and focusing on the mutual influence of different vascular cell types, we designed and developed an original, easy-to-prepare tri-layered model that mimics natural arteries by combining three types of vascular cells in a 3D collagen gel matrix scaffold. Indeed, this innovative tri-culture arterial wall model not only promoted cell–matrix remodeling and the early expression of elastic fiber-related proteins but also improved the antithrombogenic properties of the engineered endothelium. Furthermore, our findings highlight the importance of co-culturing SMCs, FBs, and ECs to obtain engineered vascular tissues with adequate physiological-like properties to appropriately investigate vascular cell behaviors such as vascular cell phenotype, functionality and contractility, which will be the subject of future studies. By capturing the complex 3D structure, organization, and cellular composition of native blood vessels, our results indicate that this new in vitro model has the potential to shed light on the processes underlying the intricate interactions between vascular cells. Furthermore, this advanced in vitro system may be deployed in drug testing applications as well as for the investigation of physiological and pathological processes occurring in native vessels, such as functional endothelial layer formation, SMC phenotypic switching, and elastic fiber deposition and maturation.Acknowledgements
CL and DP were awarded a doctoral and a post-doctoral scholarship, respectively, from the NSERC CREATE Program in Regenerative Medicine. This work was partially funded by NSERC-Canada, CIHR-Canada, FRQ-NT-Quebec, and CFI-Canada. JNK is the recipient of a Michael Smith Foundation for Health Research Career Scholar Award.References
- M. C. Gibbons, M. A. Foley and K. O. Cardinal, Tissue Eng., Part B, 2013, 19, 14–30 CrossRef CAS PubMed.
- D. G. Seifu, A. Purnama, K. Mequanint and D. Mantovani, Nat. Rev. Cardiol., 2013, 10, 410–421 CrossRef CAS PubMed.
- R. M. Touyz, Exp. Physiol., 2005, 90, 449–455 CrossRef CAS PubMed.
- G. H. Gibbons and V. J. Dzau, N. Engl. J. Med., 1994, 330, 1431–1438 CrossRef CAS PubMed.
- J. C. McGrath, C. Deighan, A. M. Briones, M. M. Shafaroudi, M. McBride, J. Adler, S. M. Arribas, E. Vila and C. J. Daly, Exp. Physiol., 2005, 90, 469–475 CrossRef PubMed.
- C. J. Kirkpatrick, S. Fuchs and R. E. Unger, Adv. Drug Delivery Rev., 2011, 63, 291–299 CrossRef CAS PubMed.
- J. C. Kirkpatrick, S. Fuchs, M. Iris Hermanns, K. Peters and R. E. Unger, Biomaterials, 2007, 28, 5193–5198 CrossRef PubMed.
- J.-J. Chiu, L.-J. Chen, C.-N. Chen, P.-L. Lee and C.-I. Lee, J. Biomech., 2004, 37, 531–539 CrossRef PubMed.
- N. Sakamoto, T. Kiuchi, M. Sato, N. A. S. Akamoto, T. A. K. Iuchi and M. A. S. Ato, Ann. Biomed. Eng., 2011, 39, 2750–2758 CrossRef PubMed.
- M. D. Lavender, Z. Pang, C. S. Wallace, L. E. Niklason and G. a. Truskey, Biomaterials, 2005, 26, 4642–4653 CrossRef CAS PubMed.
- X. Zhang, X. Wang, V. Keshav, X. Wang, J. T. Johanas, G. G. Leisk and D. L. Kaplan, Biomaterials, 2009, 30, 3213–3223 CrossRef CAS PubMed.
- S. Javerzat, P. Auguste and A. Bikfalvi, Trends Mol. Med., 2002, 8, 483–489 CrossRef CAS PubMed.
- D. Seliktar, R. M. Nerem and Z. S. Galis, Tissue Eng., 2003, 9, 657–666 CrossRef CAS PubMed.
- I. Sukmana and P. Vermette, Biomaterials, 2010, 31, 5091–5099 CrossRef CAS PubMed.
- H. Gulbins, M. Dauner, R. Petzold, A. Goldemund, I. Anderson, M. Doser, B. Meiser and B. Reichart, J. Thorac. Cardiovasc. Surg., 2004, 128, 372–377 CrossRef PubMed.
- N. L'Heureux, N. Dusserre, G. Konig, B. Victor, P. Keire, T. N. Wight, N. A. F. Chronos, A. E. Kyles, C. R. Gregory, G. Hoyt, R. C. Robbins and T. N. McAllister, Nat. Med., 2006, 12, 361–365 CrossRef PubMed.
- F. Boccafoschi, N. Rajan, J. Habermehl and D. Mantovani, Macromol. Biosci., 2007, 7, 719–726 CrossRef CAS PubMed.
- N. Rajan, J. Habermehl, M.-F. Coté, C. J. Doillon and D. Mantovani, Nat. Protoc., 2006, 1, 2753–2758 CrossRef CAS PubMed.
- N. Bono, D. Pezzoli, L. Levesque, C. Loy, G. Candiani, G. B. Fiore and D. Mantovani, Biotechnol. Bioeng., 2016, 113(10), 2254–2263 CrossRef CAS PubMed.
- K. B. Chandran, S. E. Rittgers, A. P. Yoganathan, K. B. Manning, K. B. Chandran, S. E. Rittgers and A. P. Yoganathan, Cardiovascular Engineering and Technology, CRC Press, 2012, vol. 3, pp. 351–352 Search PubMed.
- K. Tiedemann, B. Bätge, P. K. Müller and D. P. Reinhardt, J. Biol. Chem., 2001, 276, 36035–36042 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- N. A. A. Rossi, I. Mustafa, J. K. Jackson, H. M. Burt, S. a. Horte, M. D. Scott and J. N. Kizhakkedathu, Biomaterials, 2009, 30, 638–648 CrossRef CAS PubMed.
- N. A. A. Rossi, I. Constantinescu, R. K. Kainthan, D. E. Brooks, M. D. Scott and J. N. Kizhakkedathu, Biomaterials, 2010, 31, 4167–4178 CrossRef CAS PubMed.
- A. Abdulmajeed, A. K. Kokkari, J. Käpylä, J. Massera, L. Hupa, P. K. Vallittu and T. O. Närhi, J. Mater. Sci.: Mater. Med., 2014, 25, 151–162 CrossRef CAS PubMed.
- J. H. Campbell and G. R. Campbell, Annu. Rev. Physiol., 1986, 48, 295–306 CrossRef CAS PubMed.
- F. Pampaloni, E. G. Reynaud and E. H. K. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS PubMed.
- J. W. Haycock, Methods Mol. Biol., 2011, 695, 1–15 CAS.
- C. Williams and T. M. Wick, Ann. Biomed. Eng., 2005, 33, 920–928 CrossRef PubMed.
- J. J. Ross and R. T. Tranquillo, Matrix Biol., 2003, 22, 477–490 CrossRef CAS PubMed.
- C. B. Weinberg and E. Bell, Science, 1986, 231, 397–400 CAS.
- S. Villaschi and R. F. Nicosia, Lab. Invest., 1994, 71, 291–299 CAS.
- Z. Shi, K. G. Neoh, E.-T. Kang, C. Poh and W. Wang, Tissue Eng., Part A, 2009, 15, 417–426 CrossRef CAS PubMed.
- N. L'Heureux, L. Germain, R. Labbé and F. A. Auger, J. Vasc. Surg., 1993, 17, 499–509 CrossRef.
- F. Dietrich and P. I. Lelkes, Angiogenesis, 2006, 9, 111–125 CrossRef CAS PubMed.
- F. Couet, S. Meghezi and D. Mantovani, Med. Eng. Phys., 2012, 34, 269–278 CrossRef PubMed.
- I. U. Allan, B. A. Tolhurst, R. V. Shevchenko, M. B. Dainiak, M. Illsley, A. Ivanov, H. Jungvid, I. Y. Galaev, S. L. James, S. V. Mikhalovsky and S. E. James, Biomater. Sci., 2016, 4, 1007–1014 RSC.
- R. L. DiMarco, R. E. Dewi, G. Bernal, C. Kuo and S. C. Heilshorn, Biomater. Sci., 2015, 3, 1376–1385 RSC.
- T. Welman, S. Michel, N. Segaren and K. Shanmugarajah, Bioengineered, 2015, 6, 257–261 CrossRef CAS PubMed.
- J. M. Davidson, P. A. LuValle, O. Zoia, D. Quaglino and M. Giro, J. Biol. Chem., 1997, 272, 345–352 CrossRef CAS PubMed.
- J. Shi, A. Wang, S. Sen, Y. Wang, H. J. Kim, T. F. Mitts and A. Hinek, Am. J. Pathol., 2012, 180, 715–726 CrossRef CAS PubMed.
- J. P. Stegemann, H. Hong and R. M. Nerem, J. Appl. Physiol., 2005, 98, 2321–2327 CrossRef PubMed.
- C. S. Wallace, J. C. Champion and G. A. Truskey, Ann. Biomed. Eng., 2007, 35, 375–386 CrossRef PubMed.
- R. A. A. Pullens, M. Stekelenburg, F. P. T. Baaijens and M. J. Post, J. Tissue Eng. Regener. Med., 2009, 3, 11–18 CrossRef CAS PubMed.
- L. Sabatier, D. Chen, C. Fagotto-Kaufmann, D. Hubmacher, M. D. McKee, D. S. Annis, D. F. Mosher and D. P. Reinhardt, Mol. Biol. Cell, 2009, 20, 846–858 CrossRef CAS PubMed.
- S. L. Dallas, P. Sivakumar, C. J. P. Jones, Q. Chen, D. M. Peters, D. F. Mosher, M. J. Humphries and C. M. Kielty, J. Biol. Chem., 2005, 280, 18871–18880 CrossRef CAS PubMed.
- T. Velling, J. Biol. Chem., 2002, 277, 37377–37381 CrossRef CAS PubMed.
- A. Patel, B. Fine, M. Sandig and K. Mequanint, Cardiovasc. Res., 2006, 71, 40–49 CrossRef CAS PubMed.
- L. Venkataraman and A. Ramamurthi, Tissue Eng., Part A, 2011, 17, 2879–2889 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |