
Synthesis, properties and biomedical applications of hydrolytically degradable materials based on aliphatic polyesters and polycarbonates
Ruairí P.
Brannigan
and
Andrew P.
Dove
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk
First published on 14th November 2016
Abstract
Polyester-based polymers represent excellent candidates in synthetic biodegradable and bioabsorbable materials for medical applications owing to their tailorable properties. The use of synthetic polyesters as biomaterials offers a unique control of morphology, mechanical properties and degradation profile through monomer selection, polymer composition (i.e. copolymer vs. homopolymer, stereocomplexation etc.) and molecular weight. Within this review, the synthetic routes, degradation modes and application of aliphatic polyester- and polycarbonate-based biomaterials are discussed.
Introduction to biomaterials and degradable polymers
One of the most rapidly evolving area in materials science is the development of biodegradable polymers for medical applications.1,2 A wide range of naturally occurring polymeric materials have been employed as biomaterials such as proteins (collagen,3 elastin,4 silk fibroin5), polysaccharides (chitosan,6 hyaluronate7), polyhydroxyalkanoates8 (PHAs) (poly(hydroxybutyrate)9 (PHB), poly(hydroxybutyrate-co-valerate)10 (PHBV)) (Fig. 1) etc. While the utilisation of natural polymers offers advantages based on the ease of access, low cost, lower barriers to gaining regulatory approval and well established biocompatibility, the range of applications and criteria for which biomaterials are required has become too vast for natural polymers to endure (e.g. controlled/sustained pharmaceutical delivery in varying environments, degradable textiles, wound dressings, site-specific regenerative medicine/tissue engineering etc.).11,12 | ||
| Fig. 1 Naturally occurring polymeric materials used as biomaterials. | ||
The development of novel synthetic materials, which undergo bio-degradation, have been at the forefront of biomaterials research as a consequence of the issues surrounding common ‘biostable’ materials e.g. long-term incompatibility and invasive revision surgeries.13,14 While the use of synthetic polymers as biomaterials can be limited as a consequence of their often slow degradation profiles and undesirable degradation products, their application not only allows for the development of modulus-matched polymeric materials with ‘site-specific’ degradation profiles but also negates certain disadvantages found with their natural counterparts such as immunogenic responses, difficult purification, inhomogeneity and poor processability.15
In general terms, there are 4 prolific mechanisms of biomaterial degradation; hydrolytic degradation (scission of susceptible bonds by water), enzymatic degradation (enzyme catalysed scission of bonds), oxidative degradation (radical attack supported by peroxide producing inflammatory reactions) and physical degradation (i.e. mechanical loading and wear, swelling–deswelling etc.). Natural polymeric materials tend to be highly susceptible to enzymatic and oxidative degradation, and as a consequence of varying enzymatic and macrophage levels from patient-to-patient, uniform degradation profiles may be unachievable. Conversely, the uniformity of degradation and thermo-mechanical properties achieved with synthetic biodegradable biomaterials, which generally undergo hydrolytic degradation, makes these approaches more attractive.16–18
Hydrolytic degradation
Hydrolysis is defined as the reaction of water with susceptible bonds of a compound to form two or more products (Schemes 1 and 2).19 When discussing bulk polymer degradation there are several factors that determine the rate of hydrolysis-induced mass loss of a material; susceptibility of bonds and reactivity of hydrolysable bonds, diffusion rates of hydrolysis-contributing chemicals (i.e. water, ions, degradation products/small polymer chains etc.) and thermodynamics of polymer–water interactions. In some studies, definition of the rate of hydrolysis of polymer bonds has been undertaken in solution in order to negate the effects of polymer–water interactions and diffusion rates.20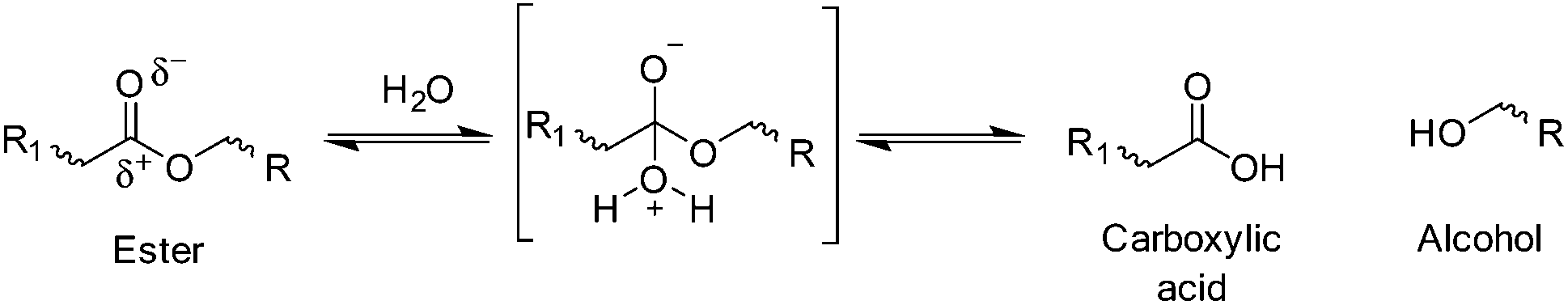 | ||
| Scheme 1 Simplified example of cleavage of hydrolysable ester bond and products. | ||
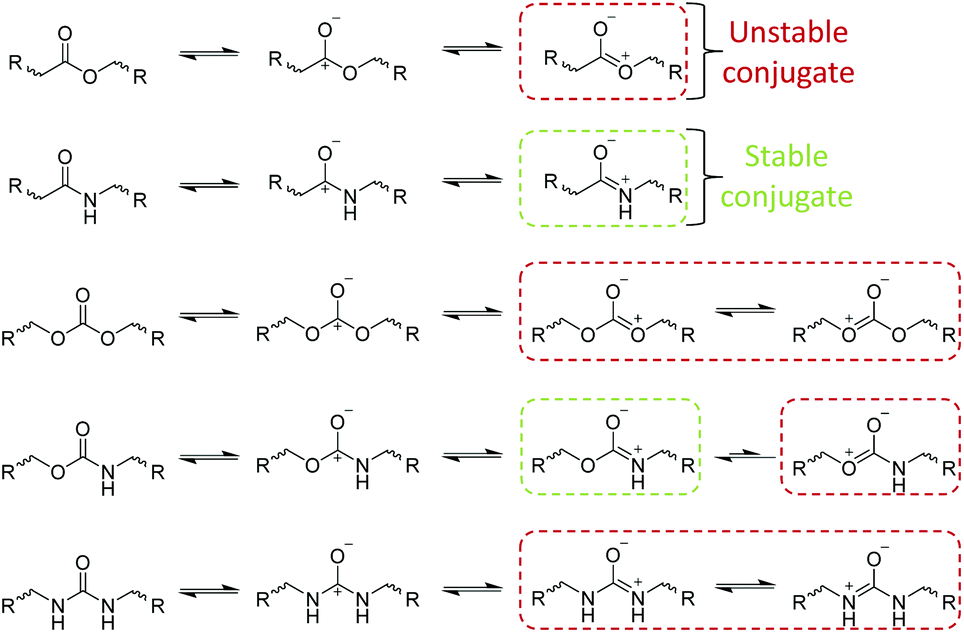 | ||
| Scheme 2 Example conjugate structures of hydrolysable bonds. | ||
Hydrolytic degradation of polymers in solution
In solution, the hydrolysis of susceptible polymeric bonds proceeds via 2nd order reaction kinetics, meaning the rate of hydrolysis is proportional to the concentration of water and hydrolysable bonds. The susceptibility of a chemical bond to undergo hydrolysis is primarily dependant on the charge value of the reacting carbon atoms (Scheme 1). Extensively demonstrated in literature, chemical functionalities with charge values >0.3 generally exhibit a higher susceptibility to hydrolysis (i.e. esters, amides, carbonates, carbamates, ureas, anhydrides, orthoesters etc.).21 Secondary to charge effects, steric effects and conjugate structures have a substantial effect on the rate of hydrolysis of chemically susceptible bonds.Although exhibiting high charge values (0.45–0.72), amide, carbonate, carbamate, urea and aromatic ester-based polymers exhibit lower rates of hydrolysis owing to stable conjugate structures (Scheme 2) which reduce the susceptibility of hydrolytic attack at the carbonyl site through delocalisation of the positive charge. Furthermore, steric effects exhibited by substituted reactants can drastically reduce the rate of hydrolysis as a consequence of bond inaccessibility. An excellent example of steric effects can be seen in the reduced rate of hydrolysis of polydimethylsiloxane (PDMS), where the two methyl substituents significantly protect the silicon atoms from hydrolysis irrespective of its high charge value (0.62). Importantly, it has been shown that the hydrolytic degradation of polymers, both in solution and in bulk, proceed by random scission of hydrolysable bonds e.g. all chemically identical hydrolysable bonds in a polymer are equally reactive irrespective of chain position (i.e. chain end vs. mid-block).20
Hydrolytic degradation of polymers in bulk
As may be obvious, the most significant difference between polymers in solution and polymers in bulk is the molecular mobility. The restrictions in molecular mobility mean, when in bulk, diffusion of water and reactants are limited. Therefore, hydrolytic degradation in polymeric systems in bulk can be defined as a combination of the diffusion of water into a polymer network and subsequent random cleavage of hydrolysable bonds.12,18 The diffusion of water into a bulk polymer network of a given composition (i.e. poly(lactic acid) (PLA)) is constant and is relatively independent of morphology or type i.e. amorphous poly(D,L-lactic acid-co-L-lactic acid) (PDLLA-co-PLLA). Although this diffusion coefficient may affect the macroscopic degradation of a material, degradation kinetics are still determined by the hydrolysis reaction on a molecular scale. In simplistic terms, hydrolytic degradation of synthetic materials can be split into two modes; bulk degradation and surface erosion (Fig. 2), however, both mechanisms are not necessarily independent of each other.22,23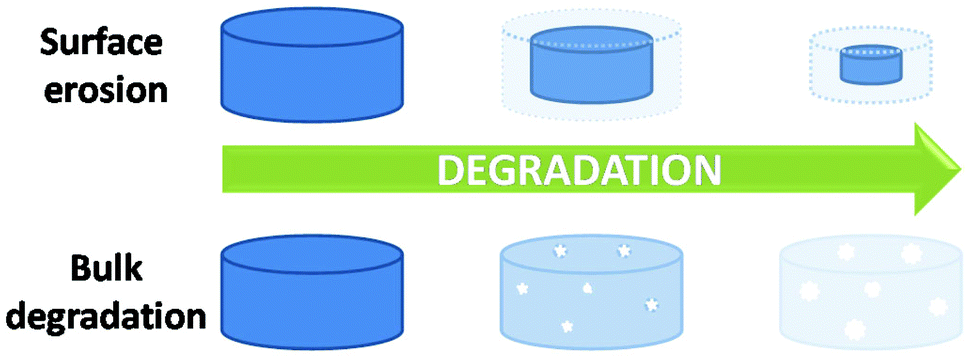 | ||
| Fig. 2 Degradation mechanisms; surface erosion versus bulk degradation. | ||
The dominant mode of degradation may be predicted by examining the relationship between the time taken for hydrolysis of surface polymer chains into small soluble components (Te), dependent on quantum chemical effects mentioned earlier, and water diffusion into a polymer within this given time (DT), an intrinsic property of a polymer defined by the physico-chemical properties of the material (i.e. polymer density, hydrophobicity, phase behaviour etc.).24
Bulk degradation is characterised by a higher rate of water diffusion to hydrolysis (DT ≫ Te), leading to sample saturation by the degradation media, and a non-linear mass loss over time (Fig. 3(a)). Bulk degradation is the dominant mechanism in most polyester-based polymers i.e. polylactide (PLA), polyglycolide (PGA), poly(ε-caprolactone) (PCL) etc.18,23 A further important characteristic of bulk degradation is the presence of autocatalytic effects as a consequence of the accumulation of degradation products.20
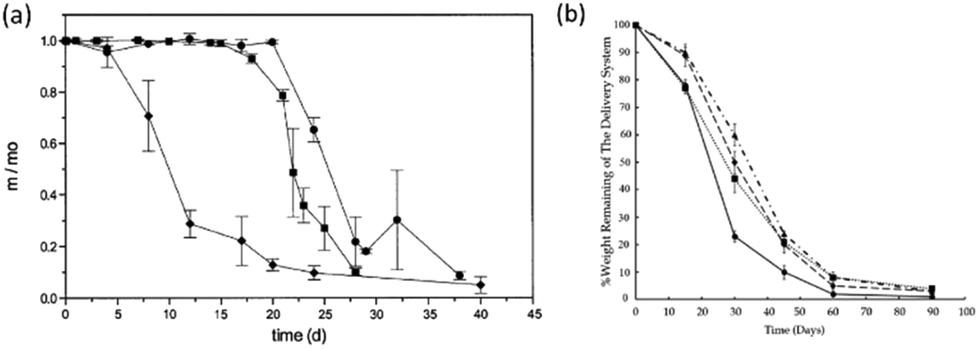 | ||
| Fig. 3 Example of (a) bulk degradation of polyester copolymers,24 (b) surface of erosion of a polyester-based block copolymer25 at 37 °C in pH 7.4 phosphate buffer solution. | ||
In bulk degradation, degradation products which may partake in the hydrolysis reaction (i.e. hydroxyl and carboxylic acid groups formed during ester cleavage) have reduced diffusivity through the polymer matrix leading to accumulation.20 This accumulation results in areas of accelerated degradation and leads to the loss of structural integrity and mechanical stability and ultimately leads to the collapse and disintegration of the polymer matrix. As a result, autocatalysis is an important consideration when designing medical devices which undergo bulk degradation. Some functionalities such as polyorthoesters and certain polyanhydrides and polycarbonates (poly(trimethylene carbonate) (PTMC)), however, undergo mainly surface erosion. Surface erosion is characterised by a faster rate of hydrolysis than water diffusion (DT ≤ Te) and generally expresses a linear loss in mass over time (Fig. 3(b)).26–28 In the case of surface degradation, degradation products are free to diffuse away from the polymer matrix and therefore have no catalytic effect on the degradation.
Other mechanisms of degradation
Enzymatic degradation
Enzymatic degradation is defined as the enzyme catalysed scission of bonds of a compound to form two or more products and is typically affected by 4 mechanistic steps; (1) diffusion of the enzyme through solution to the substrate interface, (2) adsorption and complexation of the enzyme to the substrate, (3) catalysis of the scission reaction and (4) diffusion of soluble degradation products away from the substrate surface.19,29 Owing to the versatility of nature, a wide range of enzyme-catalysed degradation reactions are available, however, the only known mechanism of enzyme-catalysed synthetic polymer degradation is hydrolysis.20 As a consequence of hydrolysis being the only mechanism of enzymatic degradation, hydrolase enzymes such as lipases, esterases, proteases/peptideases etc. are typically employed to demonstrate the enzymatic degradability of a synthetic biomaterial.30 The in vitro enzymatic degradation of synthetic biomaterials are typically conducted under conditions imitative of the implantation site (e.g. 37 °C, phosphate buffer pH 7.2 when using cholesterol esterase; a broad acting enzyme released during inflammatory responses), however, the mimetic capabilities of these systems are severely limited owing to the constant fluctuation of numerous enzymatic concentrations in vivo.29Unlike standard hydrolytic degradation, enzymatic degradation is not common in synthetic polymers as a consequence of the substrate specific nature of many enzymes.29 It is important to note that hydrolase substrate specificity can greatly affect the rate hydrolysis observed.31 For example, cholesterol esterase has been shown to have a higher affinity for polyesters which contain longer methylene chains in the repeat unit (e.g. poly(ε-caprolactone) > polypropiolactone).32 Contrarily, R. delemar lipase, a microbial hydrolase, exhibits a substrate specificity for polyesters with shorter methylene chains (e.g. polypropiolactone > poly(ε-caprolactone)).33 Furthermore, akin to passive hydrolytic degradation, the physical properties of the polymer (i.e. crystallinity, molecular weight etc.) can also affect the rate of degradation.17,29
Oxidative degradation
As mentioned previously, oxidative degradation occurs as a result of the scission of susceptible bonds by radical attack supported by peroxide-producing inflammatory reactions.12 Consequent to implantation and acceptance of a synthetic or natural material, owing to acute inflammatory responses, implants are encapsulated in a matrix chiefly consisting of fibroblasts, collagens, foreign body giant cells (FBGCs) and varying levels of macrophages.20 In order to facilitate the clearing of degradable implanted devices, FBGCs and macrophages precipitate peroxides at the surface of the implant to illicit degradation.34 These latent peroxides are readily degraded by naturally occurring or process induced free radicals which in turn proliferate and partake in oxidative degradation of susceptible bonds of the implanted device. Although the vast majority of polymer bonds are susceptible to oxidative reactions, structures which can facilitate the proliferation of free radicals with adequate lifetime, such as polyolefins, polyethers vinyl polymers, polyurethanes and polyamines, are generally more susceptible.20,35The oxidative degradation of implanted materials occurs through a free radical process of initiation (production of free radicals), proliferation, radical transfer/chain break-up and termination of radicals. As previously mentioned, FBGCs and macrophages produced during inflammatory responses are a potential sources of free radicals, however, the production of free radicals may also occur during processing of devices before implantation (i.e. thermally induced, photoinduced and radiation induced initiation upon sterilization) (Scheme 3).36–38
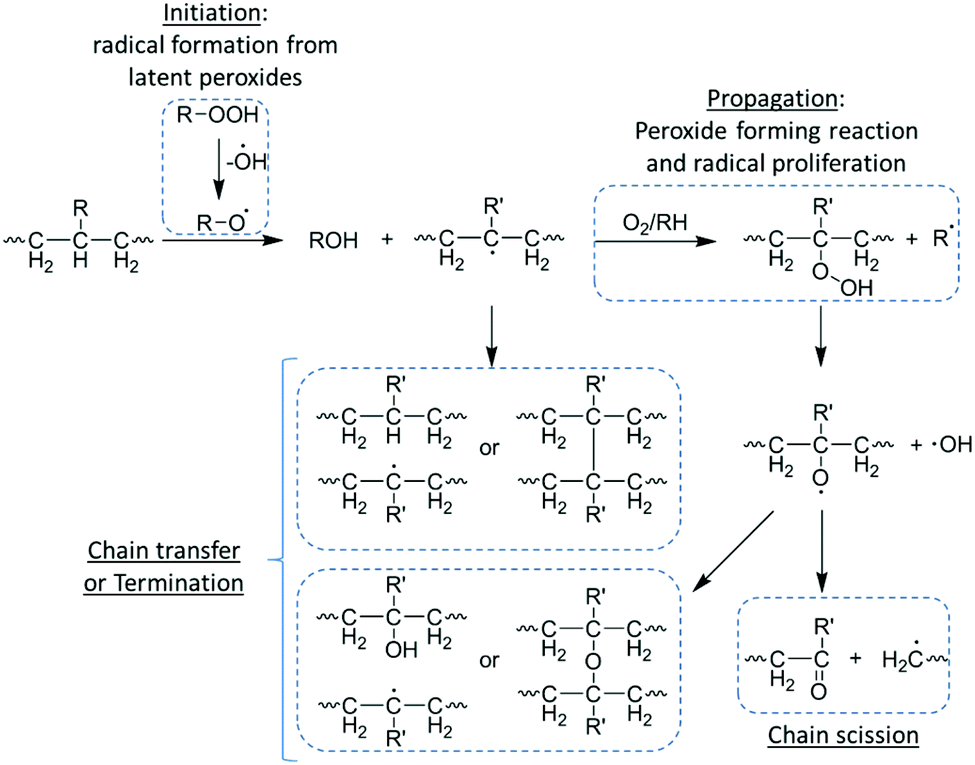 | ||
| Scheme 3 Oxidative degradation of a polyolefin. | ||
Once initiated, free radicals can proliferate by a series of peroxide-forming reactions with solubilised oxygen within the polymer matrix producing more free radicals, which in turn can proliferate further in an auto-accelerated process. Alternately, free radicals can undergo chain transfer in which free radicals are imparted onto the polymer chain in a radical net neutral process (i.e. no new radical species are formed), leading to the formation of small molecule side products such as water.39 The process of chain transfer can facilitate an increase in radical mobility as it passes along the polymer chain, however, and more importantly, it can facilitate chain break-up leading to polymer degradation.20 The oxidative degradation of the polymer chain occurs at the radical site and results in the formation of an alkene terminated chain and a radical chain end, which can further facilitate chain transfer and degradation. Owing to their highly reactive nature, termination can occur via coupling of two radical species, leading to a reduction of free radicals and consequently oxidative degradation. It is important to note that as termination increases with radical concentration (i.e. initiation and proliferation), oxidative degradation is generally localised to an ‘infected zone’ until the diffusion of soluble degradation products from the polymer surface.40
Physical degradation
Physical degradation is considered as the scission of bonds, both physical and chemical, within a polymer chain as a consequence of the application of mechanical stresses upon the material.41 As with other forms of degradation, physical or mechanical degradation is dependent on a number of physical properties such as molecular weight, molecular composition and viscosity, however, external factors such as applied shear stress, temperature and water-induced plasticisation also need to be considered, especially when considering modulus-matched degradable biomaterials.42 Typically, high molecular weight polymers with high viscosities exhibit a higher mechanical deterioration when appropriate shear stresses are applied to them. This is a consequence of reduced chain mobility allowing for the polymer chains to experience the full extent of the shear force applied.43 Conversely, polymers with low viscosities are able to dissipate shear stress through enhanced chain mobility and multilateral translations. Furthermore, application of shear stress on low viscosity polymers may induce increased amounts of chain entanglement and recombination negating or ‘masking’ the effects brought on by mechanical degradation (i.e. reduction in mechanical strength, permeability etc.).42 Although low viscosity polymers exhibit a better resistance to mechanical stresses, they are subject to other adverse characteristics such as reduced creep resistance and viscous deformation.43Another key factor in the mechanical degradation of biomaterials is the effect of plasticisation and delamination induced by the uptake of water (Fig. 4). Water-induced plasticisation of polymers manifests itself by a reduction in the glass transition (Tg) leading to increased chain mobility.20 As with inherently low viscosity polymers, the reduction in the Tg leads to a reduction in creep resistance and possible mechanical deformation (i.e. unrecoverable plastic deformation, viscous flow etc.) and structural failure (i.e. micro-fractures, reduced load-bearing capabilities etc.). In the case of phase separated polymers, which often require synergistic interactions of each component phase, absorption of water may leads to disruption or delamination of the phases with loss of synergistic interactions, and ultimately, mechanical failure. It is important to note that delamination of phases tends only to occur in materials in which each component phase are relatively hydrophilic.44,45
 | ||
| Fig. 4 SEM of delamination of a polyurethane composite reinforced with glass fibres.20 | ||
Polyesters for use in biomaterials
Owed to their synthetic versatility and diverse properties in conjunction with their historic and regulatory compliance, polyesters have been the most widely investigated synthetic polymer for biomedical applications. Although all polyesters may undergo degradation, aliphatic polyesters with relatively short chain lengths possess suitable degradation profiles for biomedical applications.23 As mentioned earlier, the degradation of the majority of polyesters proceeds by bulk degradation which, characteristically, results in the accumulation of acidic degradation products. The accumulation of leachable acidic by-products, and low molecular weight degradants, in degradable medical devices has been found to be a key contributor in inflammatory responses in tissue.47,48 As such, implantation sites which exhibit low metabolic activity or vascularity (corneal tissue,49 articular cartilage,50 epithelial tissue51etc.), and therefore low ability to eliminate by-products, may be unsuitable for polyester-based materials and should be met with great consideration.47,52 Aliphatic polyesters may be synthesised by the condensation of diacids/diacid chlorides with diols (A + B),53,54 self-condensation of hydroxyacids (AB monomer),55 ester interchange reaction (transesterification)56 or, more recently, by dehydrogenation polymerisation of diols.57 Although these methods offer a simplistic route to the synthesis of aliphatic polyesters, molecular weight control and low dispersities are often unachievable.Ring opening polymerisation (ROP) of cyclic lactones, carbonates and O-carboxyanhydrides (OCAs) (Fig. 6) allow for the synthesis of aliphatic polyesters and polycarbonates with excellent control over molecular weight, with high molecular weights achievable, while maintaining low dispersities (<1.1–1.2).58–60 Furthermore, the ROP of these cyclic monomers require milder reaction conditions and shorter reaction times while negating the production of unwanted condensation by-products (i.e. HCl, H2etc.). Although ROP of lactones had previously been limited to relatively small cyclic species (C4–C7), recent reports have successfully shown the controlled ‘immortal’ ROP (iROP) of macrolactones (C8–C17), however, their use as biomaterials has been limited as a consequence of their slow degradation profiles.61–63 Owing to these distinct advantages, the ROP of lactones and cyclic carbonates has become the most industrially relevant methodology in the synthesis of aliphatic polyesters and polycarbonates for biomedical applications.
Polyglycolide (PGA)
Polyglycolide (PGA) is the simplest of aliphatic polyester and was used in the development of the first biodegradable synthetic polymer (DEXON®) investigated for medical applications by Frazza et al. (American Cyanamid Company).64 PGA is synthesised via the ROP of glycolide, a cyclic diester comprising of two units glycolic acid, and is a highly crystalline polymer (Xc = 46–52%).65 PGA displays a glass transition temperature (Tg) between 35–40 °C and a melting temperature (Tm) range of 225–230 °C, with low solubility in most organic solvents.66 As a consequence of its thermal and morphological characteristics, PGA has a high Young's modulus (E) (E = 12.8 GPa) and is an excellent candidate for bone internal fixation devices (Fig. 5) (i.e. Biofix®).12 However, PGA undergoes mass loss by hydrolytic degradation within 12 months with loss of tensile strength after just 1–2 months. This, in combination with its poor solubility, limits the applications of PGA homopolymers for soft tissue implants. As a result, several PGA containing copolymers have been developed (i.e. poly(glycolide/lactide) (Vicry®), poly(glycolide/caprolactone) (MONOCRYL®), poly(glycolide/trimethylene carbonate) (MAXON®) etc.). In order to improve the range of biomedical applications of PGA, a number of copolymers have been synthesised. The most extensively researched of these materials are copolymers with polylactide (PLA), which will be discussed later (Scheme 4).67–69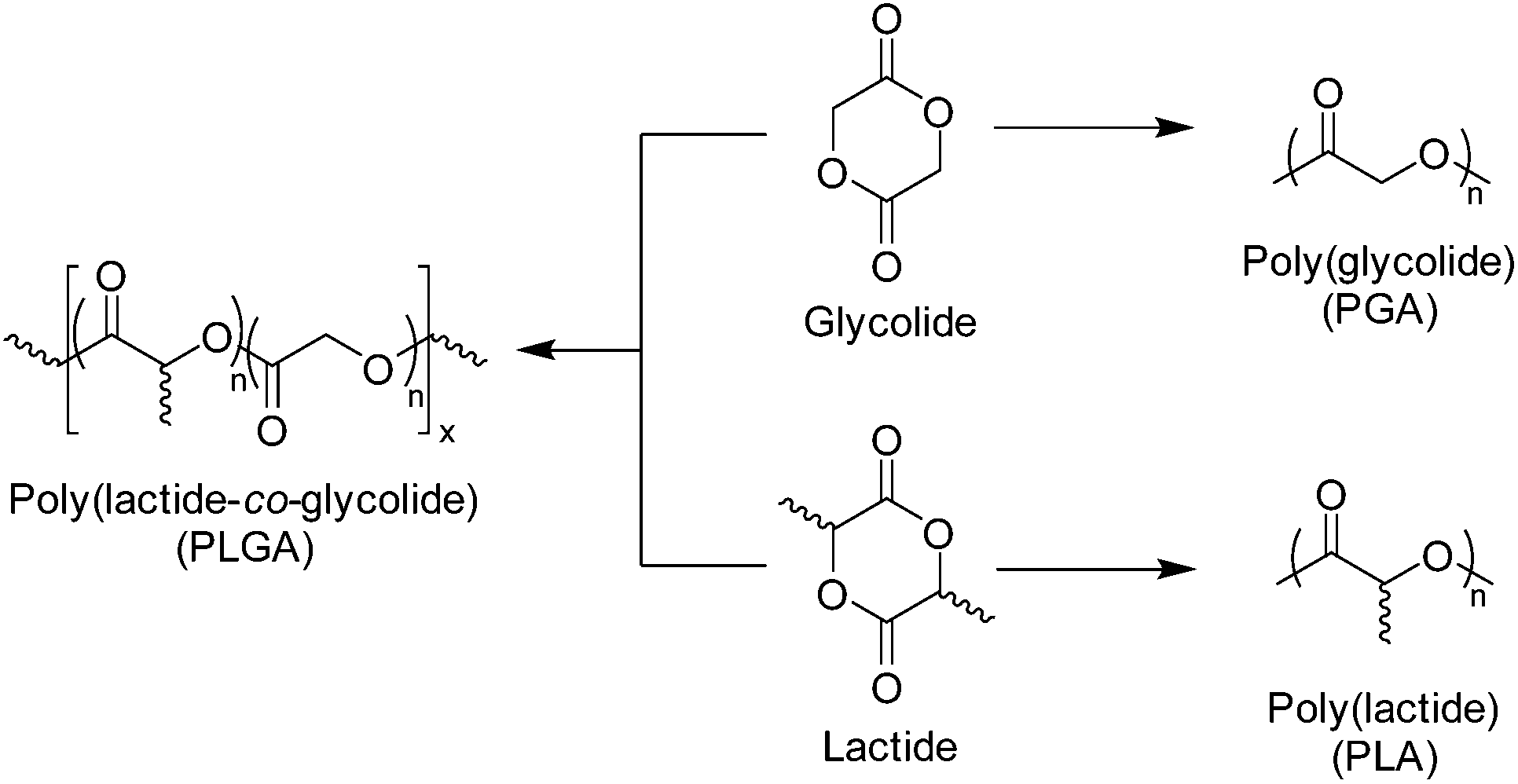 | ||
| Scheme 4 Structure of glycolide and lactide monomers and corresponding homopolymers and copolymer, poly(lactide-co-glycolide) (PLGA). | ||
 | ||
| Fig. 5 Commercially developed polyester-based all-inside meniscal repair devices. (Left to right) Lactosorb® Meniscal Staple, Lactosorb® Meniscal Screw, Clearfix® Meniscal Screw, Biostinger®, Meniscal Dart®, Contour Meniscus Arrow®.46 | ||
Polylactide (PLA)
Like glycolide, lactide is a cyclic diester, however comprises of two units of lactic acid, which differs from glycolic acid by a methyl group As a consequence of the chiral centre of lactic acid, lactide is present as three isomers, SS (L-lactide), RR (D-lactide) and RS (meso-lactide), with L-lactide being the naturally occurring isomer (Fig. 7). Stereo-pure poly(L-lactide) (PLLA) homopolymers are considered crystalline polymers (30–35% dependant on molecular weight and processing) with relatively high Tg's (57–60 °C), a high E (∼4.8 GPa) and low strains before mechanical failure (∼6%).66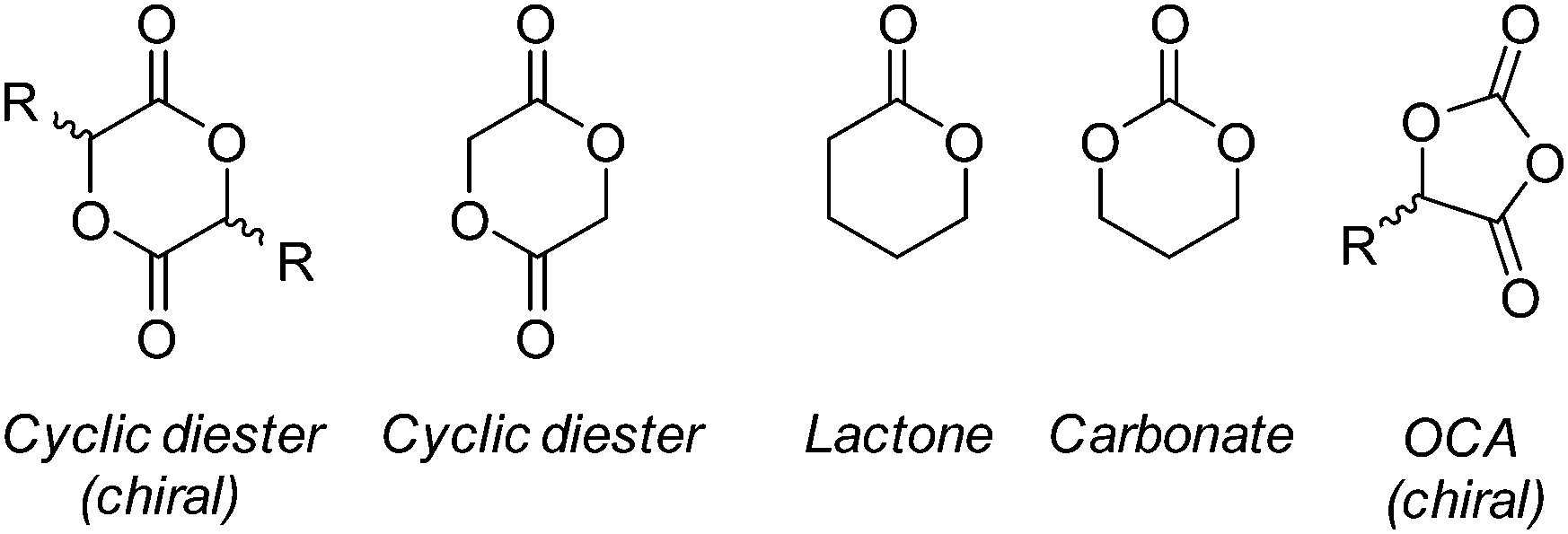 | ||
| Fig. 6 Cyclic monomers for the synthesis of polyesters by ROP. | ||
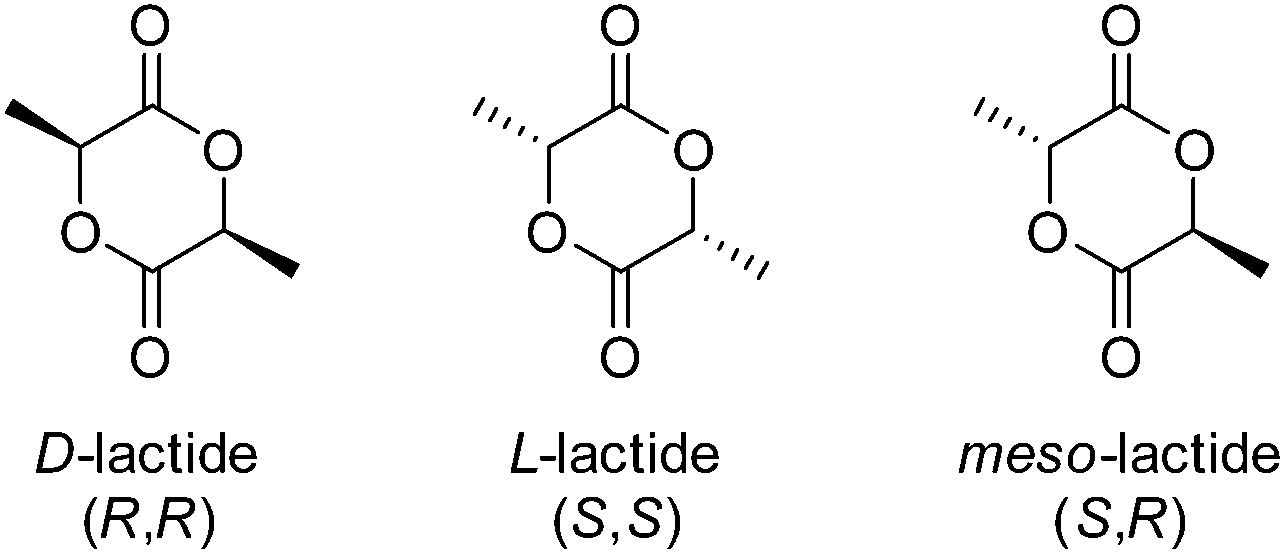 | ||
| Fig. 7 Isomers of lactide. | ||
As a consequence of steric factors, mentioned earlier, PLA exhibits a reduction in hydrolytic activity, compared to PGA, which manifests in reabsorption times ranging from months to decades dependant on polymer processing (i.e. crystallinity, orientation, specimen type etc.), polymer purity (e.g. tacticity), molar mass and distributions.67,70 PLLA degradation products (e.g.L-lactic acid) is easily eliminated from the body by the Cori cycle (later by the Krebs cycle) as glucose (via pyruvate). Owing to this enhanced hydrolytic durability and mechanical strength, PLLA has been successfully employed in the development of several commercially available medical devices for meniscal repair (Lactosorb® Meniscal Staple, Bionx Meniscal Arrow®, Linvatec BioStinger™ etc.). Furthermore, copolymerisation of PLLA and PGA has resulted in numerous medical devices with adjustable reabsorption time dependent on the incorporation of PGA such as the multifilament sutures Vicryl® and Vicryl Rapid®.68
Owing to stereo-irregularities in the polymer microstructure, the non-selective ROP of racemic lactide (rac-lactide) results in atactic polymers which are amorphous with a reduced Tg (50–53 °C). Owing to its amorphous nature, poly(rac-lactide) exhibits a higher susceptibility to hydrolytic degradation (<5 months) as a consequence of enhance permeability.71 More recently, the effects of stereocomplexation of PLLA/PDLA blends on the physical, mechanical and degradation properties have been investigated. As discussed by Tsuji, the stereocomplexation of PLLA and PDLA result in materials with superior mechanical durability, E ≈ 8.6 GPa and elongation at break of 30%. Furthermore, stereocomplexed materials exhibited a drastic change in Tg (PLLA = 57–60 °C blend = 65–72 °C), Tm (PLLA = 170–190 °C, blend = 220–230 °C), and percentage crystallinity, (PLLA = 35%, blend = 70%) (Scheme 5).72
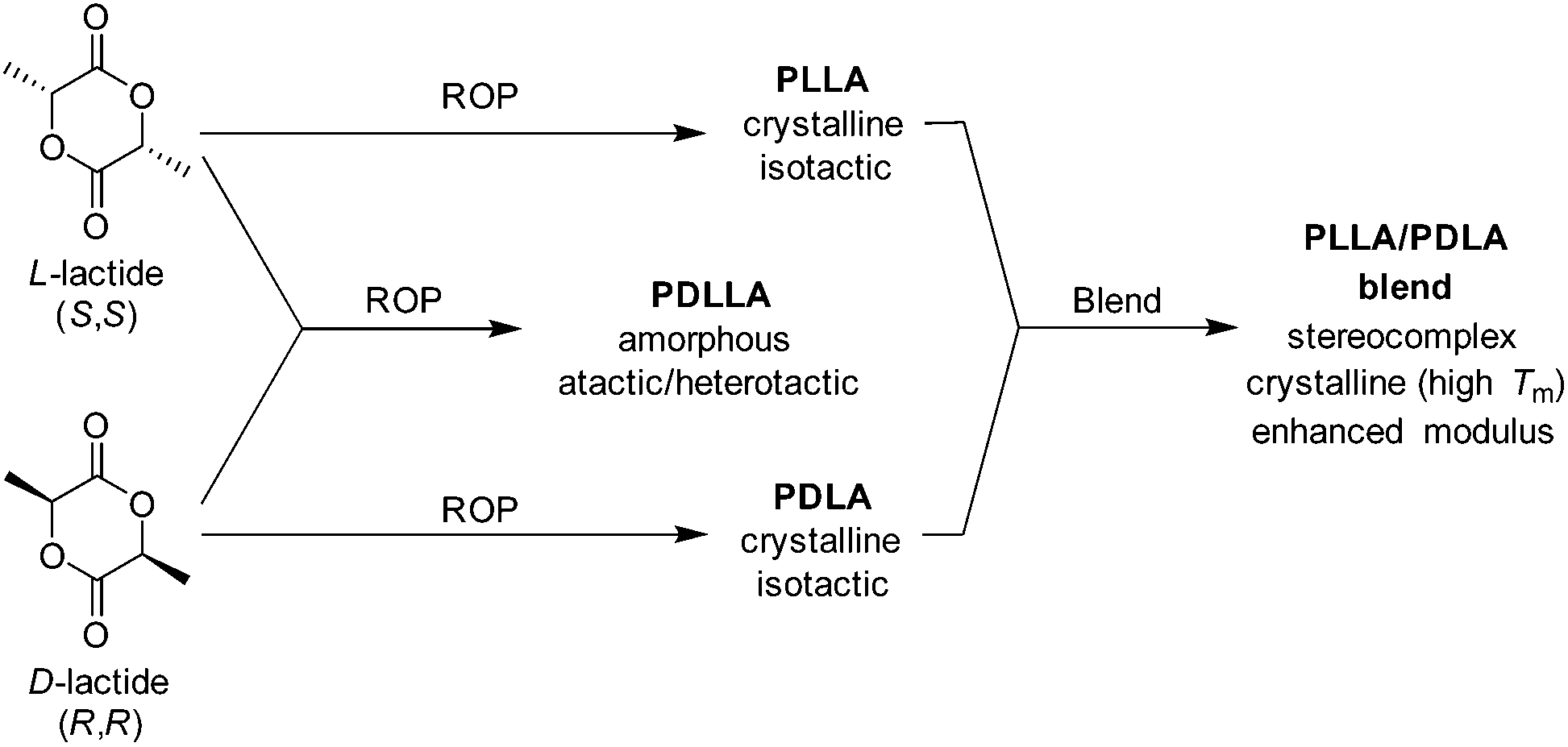 | ||
| Scheme 5 Resultant polymer properties from homo- and copolymerisation of D- and L-lactide and isotactic stereocomplexation of homopolymers. | ||
As a consequence of the high crystallinity, and therefore low permeability/mobility, stereocomplexed PLLA/PDLA blends exhibit a lower susceptibility to hydrolytic degradation.71–73 Furthermore, Tsuji et al. demonstrated that stereocomplexed blends were found to retain their tensile strength at extended periods of degradation (16 months) compared to PLLA homopolymers under the same conditions (4 months) (Fig. 8).72,73 Although stereocomplexation allows for a vast improvement in the mechanical strength PLA materials, PLLA/PDLA blends still exhibit relatively low extension before mechanical failure is observed. As a result, PLLA-based copolyesters have been developed (i.e. poly(LLA-co-caprolactone), poly(LLA-trimethylene carbonate-LLA)).
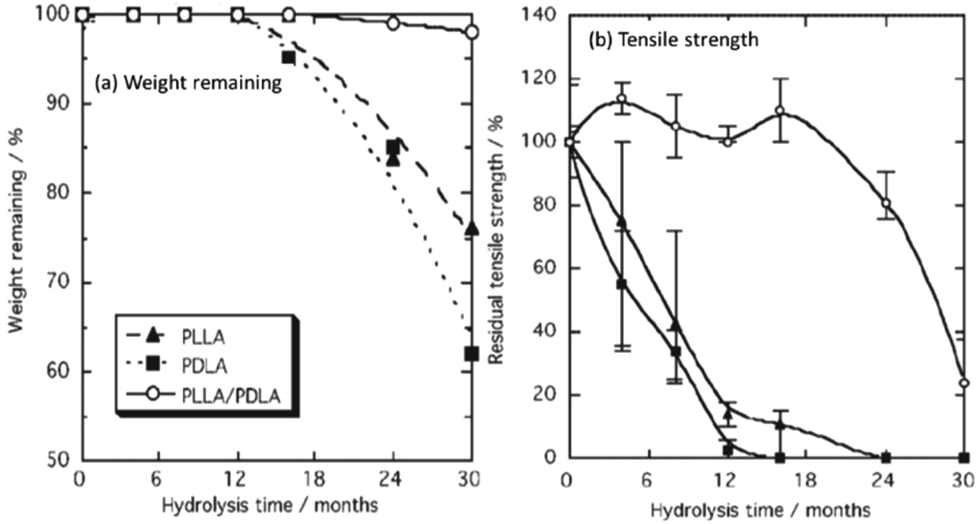 | ||
| Fig. 8 Percentage weight remaining (a), and residual tensile strength (b) of PLLA, PDLA and PLLA–PDLA stereocomplexed blends during hydrolytic degradation. | ||
Poly(lactide-co-glycolide) (PLGA)
In order to expand the range of biomedical applications of PGA and PLA-based materials, copolymers of glycolide (GA) and lactide (LA), both L- and DL-, have been researched.74–77 As has been extensively reported, owing to the differing hydrolytic stability of PGA and PLA homopolymers, the degradation PLGA copolymers can be tailored based on the copolymer composition. In an early study, Reed and Gilding demonstrated that PLGA copolymers with compositions between 25–70 mol% glycolide, for poly(L-LA-co-GA), and 0–70 mol% glycolide, for poly(DL-LA-co-GA), resulted in polymers which were completely amorphous and exhibited an increased susceptibility to hydrolytic degradation.65 Furthermore, studies found that PLGA with the copolymer composition of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mol% LA:GA, demonstrates the highest susceptibility to hydrolytic degradation (∼1–2 months) and that resistance to hydrolytic degradation was enhanced when approaching the extremes of the copolymer composition range (75:25 ∼ 4–5 months, 85:15 ∼ 5–6 months) (Fig. 9).78,79
:50 mol% LA:GA, demonstrates the highest susceptibility to hydrolytic degradation (∼1–2 months) and that resistance to hydrolytic degradation was enhanced when approaching the extremes of the copolymer composition range (75:25 ∼ 4–5 months, 85:15 ∼ 5–6 months) (Fig. 9).78,79
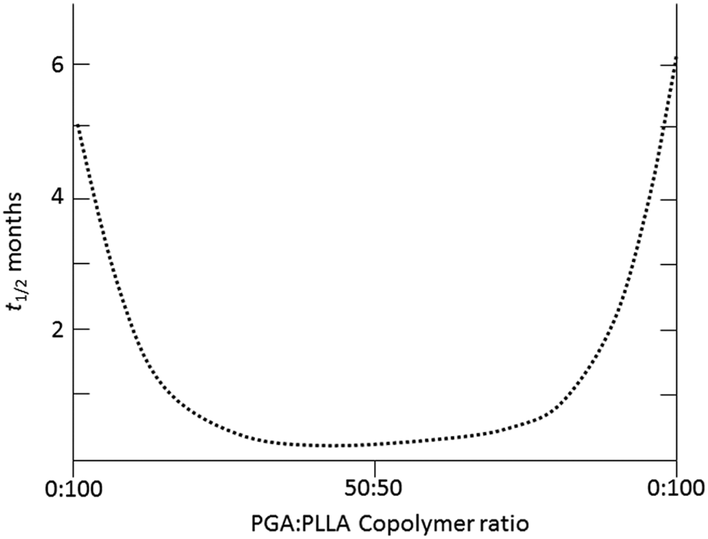 | ||
| Fig. 9 Exemplar half life (t1/2, months) of implanted materials with varying copolymer ratios of PLA and PGA exhibiting hydrolytic stability (i.e. 100:0) or susceptibility (i.e. 50:50).79 | ||
Owing to this control of degradation rate, PLGA has been commercially developed for a range of biomedical applications such as degradable multifilament sutures and suture reinforcements (Vicryl®, Vicryl Rapid®), degradable scaffolds for tissue engineering (Dermagraft®, Vicryl Mesh®), guided tissue regeneration (CYTOPLAST resorb®) drug delivery (LUPRON DEPOT®, Suprecur® MP). Furthermore, CorbionPURAC® have commercialised semi-crystalline PLGA pellets (PuraSorb®PLG, LA:GA molar ratio = 80:20) as a thermoplastic material for use in biodegradable and reabsorbable devices.
Poly(ε-caprolactone) (PCL)
Poly(ε-caprolactone) (PCL) is a semi-crystalline linear aliphatic polyester comprising of an ester-linked C5 hydrocarbon repeat unit. Traditionally, PCL is synthesised by the alcohol initiated ROP of ε-caprolactone (CL), however, more recently PCL has been prepared through the free radical or xanthate-mediated radical addition–fragmentation chain-transfer (RAFT) polymerisation by the ring-opening of 2-methylene-1,3-dioxepane (MDO) (Scheme 6).80,81 It is important to note, however, although the controlled radical ROP of MDO has seen great advancements in recent times, adverse side-reactions such as branching and ring-retention result in polyesters with poorly defined molecular weights and physical properties.82 Furthermore, well-defined high molecular weight PCL is only achievable through the ROP of ε-caprolactone.81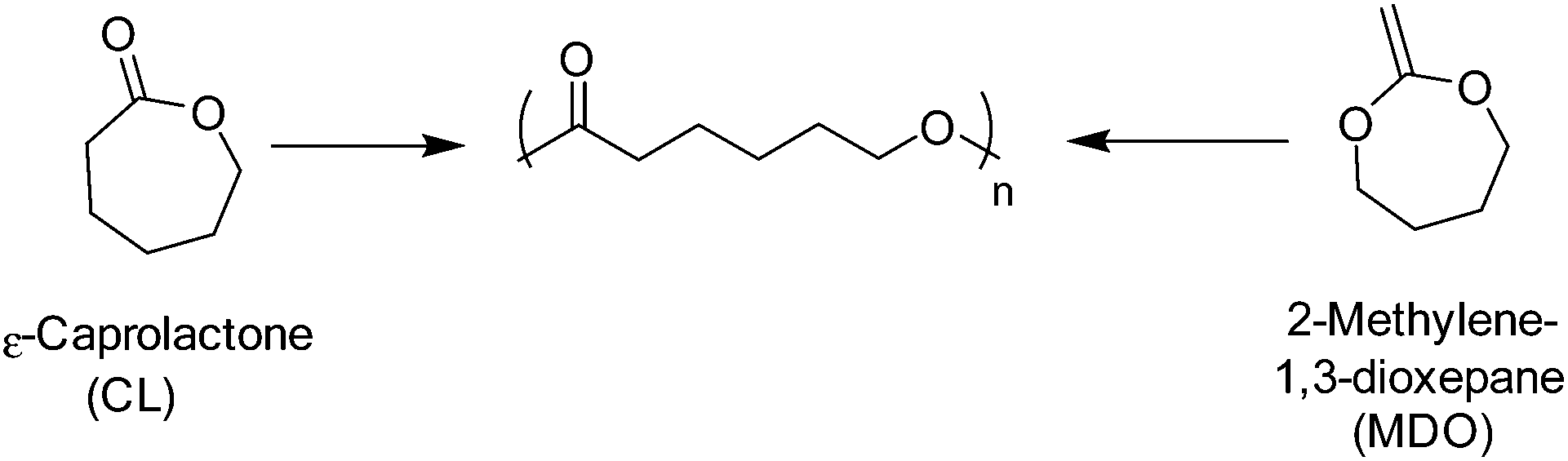 | ||
| Scheme 6 ROP of ε-caprolactone (left) and the radical ring-opening polymerisation of 2-methylene-1,3-dioxepane (right). | ||
In comparison to PLA and PGA-based materials, PCL exhibits quite low mechanical strength (E = 0.4 GPa), however, owing to relatively good elongation at break (εbreak = 300–500%), in conjunction with its reasonable processing temperatures (Tm = 59–60 °C, Tg ≈ −60 °C), solubility, permeability and blend compatibility, PCL has gained much attention for its potential use in biomedical applications (i.e. active pharmaceutical ingredient (API) blended composites, elastomeric copolymers, bioactive scaffolds etc.).66,83 Furthermore, PCLs slow degradation time (∼2 years), compared to PGA and PDLLA, owed to increased hydrophobicity, and ability to be resorbed and eliminated through the citric acid cycle as 6-hydroxyl caproic acid/acetyl coenzyme A, offers a further point of interest for its application as a degradable biomaterial.84
As a consequence of the poor load bearing capabilities of PCL homopolymers its use in commercially viable biomedical applications has been hindered, however recent advancements in biomedical and processing technologies has allowed for a resurgence in PCL-centred materials research.84 In more commercial areas, PCL copolymers, composites and blends were investigated as long term (>1 year) drug delivery sutures (Capronor, Ethicon Inc.: MONOCRYL®) owing to their tailorable degradation times. However, PCL-based materials have found applications in nerve regeneration (NEUROLAC®, PDLLA-co-PCL),85 oral surgery (Resilon™, bioactive glass/bismuth oxychloride/barium sulphate/PCL composite),86 tissue reinforcement/torn tendon replacement (Fig. 10) (Artelon® Sportmesh™, PCL-based polyester-urethane-urea),87 anti-adhesives (Mesofol®, PCL-co-PLLA)88 and wound dressings.89,90 Arguably the most prolific area of poly(ε-caprolactone) copolymers is in the synthesis of degradable segmented thermoplastic polyester-urethanes (TPEUs) which will be discussed later.
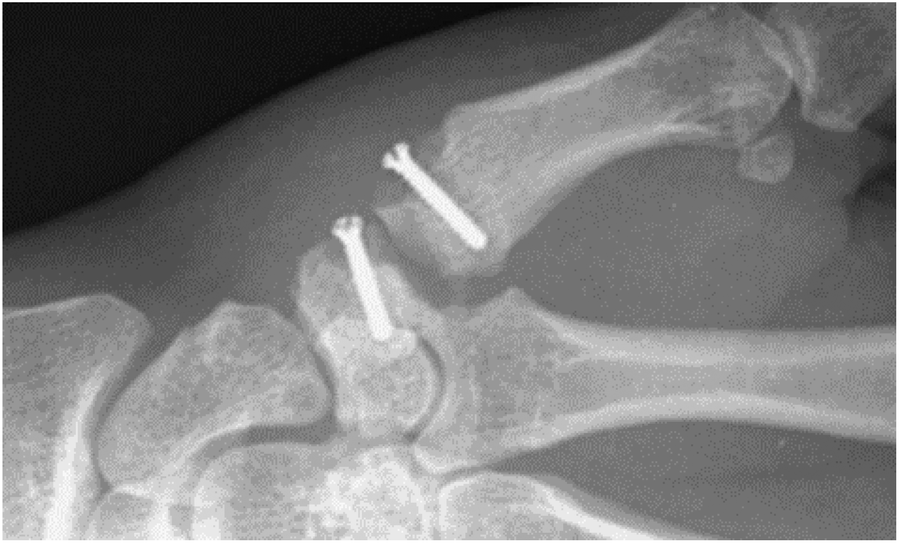 | ||
| Fig. 10 Implanted PCL-based Artelon® spacer and reinforcement screw fixation.125 | ||
Poly(trimethylene carbonate) (PTMC)
First reported by Carothers and Van Natta in 1930, poly(trimethylene carbonate) (PTMC), similar to PCL, remained relatively unused in biomedical applicable research until the 1970s.91,92 PTMC is an amorphous polymer with a low Tg (−17 °C), low Young's modulus (∼3 MPa) and relatively good strain at break (∼160%).66 Commercially, as a consequence of their low Young's modulus, PTMC-based devices have been limited to copolymers, generally with glycolides, for applications as flexible sutures (Biosyn™) tissue fixation devices (Suretac™) and orthopaedic screws (Acufex®). However, owing to its elastic properties as a consequence of increased molecular mobility, high molecular weight (HMW) PTMC is an excellent candidate, and has been extensively researched, in the fabrication of currently non-commercial devices for soft tissue regeneration.93,94 Furthermore, low molecular weight (LMW) PTMC/poly(ethylene glycol) (PEG) triblock copolymers have been used for the sustained release of corticosteroids (Scheme 7).95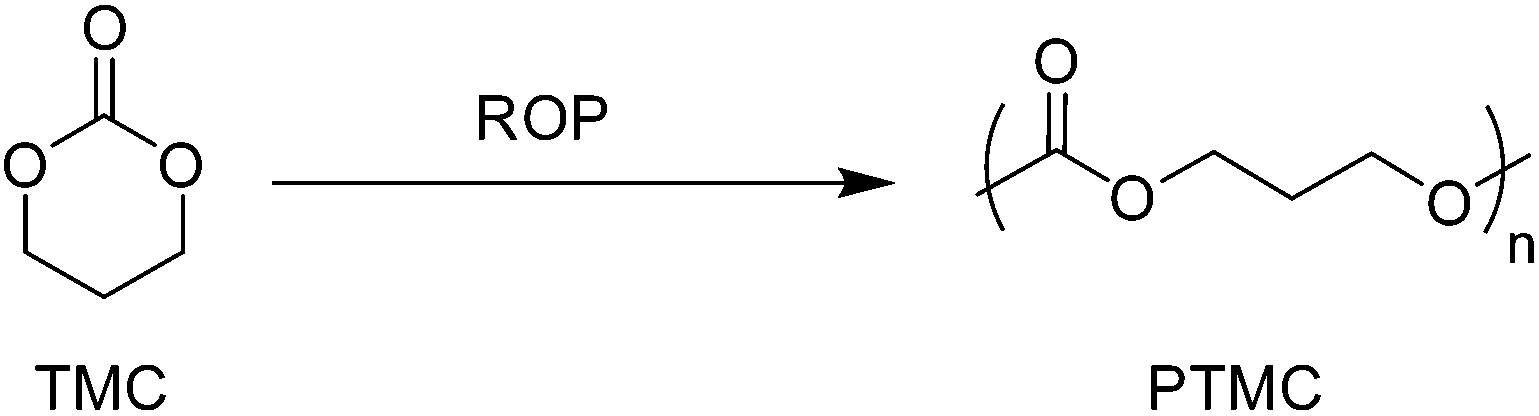 | ||
| Scheme 7 ROP of trimethylene carbonate. | ||
Unlike all of the polyesters mentioned previously, PTMC degrades by surface erosion, retaining its structural form until late onset degradation, ideal for applications in regenerative medicine.26 Furthermore, unlike PLA or PGA-based polymers, PTMC does not produce acidic moieties upon degradation responsible tissue necrosis and autocatalytic degradation. Interestingly, it was found that the hydrolytic degradation of PTMC was extremely slow (>4 years), however samples exhibited rapid degradation in vivo. Feijen et al. hypothesised that changes in the pH of the implants environment, subsequent to biological responses, may contribute to increased hydrolytic degradation, however studies have concluded that pH has no effect and that enzymatic interactions are important to the rapid surface erosion in vivo.26 Furthermore, extensive studies have shown that molecular weight has a dramatic effect on the in vivo degradation of PTMC.96 Contrary to intuition, HMW PTMC exhibited a greater rate of mass loss than the low molecular weight equivalent. This has been attributed to the ‘interfacial activation’ of lipases at the more hydrophobic HMW PTMC surface (Fig. 11).26
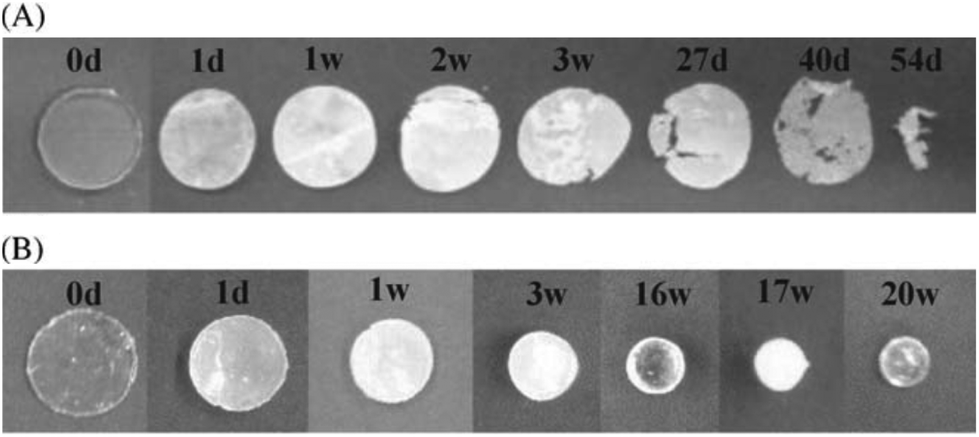 | ||
| Fig. 11 Degradation specimens of high molecular weight PMTC (A) and low molecular weight PMTC (B) at different time points.26 | ||
Another application of PTMC is the synthesis of thermoplastic elastomeric materials based on phase-separable block copolymers. Kim et al. first reported the synthesis of PLLA/PTMC ABA triblock copolymers and subsequent use as thermoplastic elastomers (TPEs).97,98 Consequently, PTMC has been extensively reported as effective ‘soft’ segments in triblock block copolymers in conjunction with high Tg or highly crystalline copolymers.99–102 Like PLA, PGA and PCL, PTMC has been effectively utilised in the synthesis of highly elastic degradable thermoplastic polyester-urethanes for applications ranging from textiles to biomedical devices.
Application of polyesters in the synthesis of thermoplastic polyester-urethanes and polyester-urethane-urea elastomers
Thermoplastic polyester-urethanes (TPEUs) and thermoplastic polyester-urethane-ureas (TPEUUs) are a class of segmented degradable multiblock copolymers, traditionally synthesised by the step-growth polymerisation of a telechelic polyester macrodiols, diisocyanates and small diol (urethane) or diamine (urea) extenders via the nucleophilic addition of the terminal hydroxyl/amine groups to the diisocyanate (Schemes 8 and 9).103,104 Characterised by their vast range of mechanical, thermal and degradation properties, determined by polymer composition, block lengths and ratios, TPEUs and TPEUUs have been extensively used in the development of devices for use in medical applications such as tissue fixation, wound dressings, regenerative medicine etc.105 Recently, as a consequence of hazardous starting materials used during TPEU/TPEUU synthesis, there has been extensive research into polyester-based elastomeric A–B–A triblock copolymers.106,107 Typically, these degradable TPEs consist of crystalline high Tg A blocks such as PLLA with an amorphous low Tg central B block such as PTMC, in which the thermo-mechanical properties being modulated by the ratio of each block.108–110 Although there has been advances in the development of polyester-based TPEs, these materials tend to lack the mechanical versatility of traditional TPEU/TPEUUs.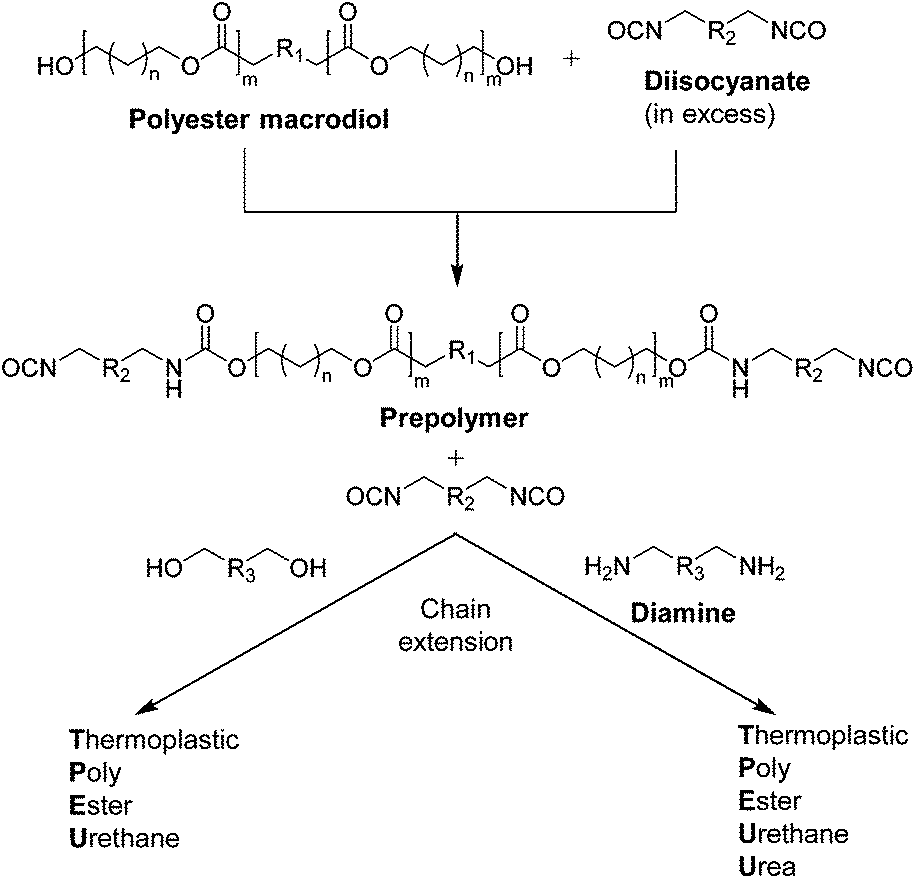 | ||
| Scheme 8 Schematic for the prepolymer formation and chain extension for the synthesis of TPEU/TPEUUs. | ||
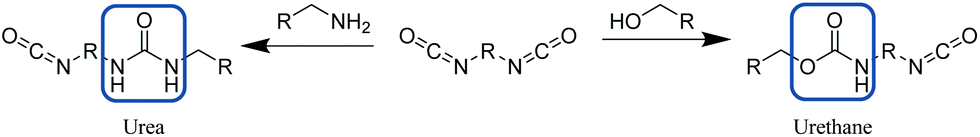 | ||
| Scheme 9 Synthesis of urea (left) or urethane (right) bond from reaction of a diisocyanate with an amine or alcohol respectively. | ||
TPEU/TPEUUs derive their mechanical versatility as a consequence of their phase separated and highly segmented nature (Fig. 12).44 While the implementation of flexible ‘soft’ blocks provide excellent elongations, highly ordered ‘hard’ blocks provide rigidity and strength through physical crosslinks.45
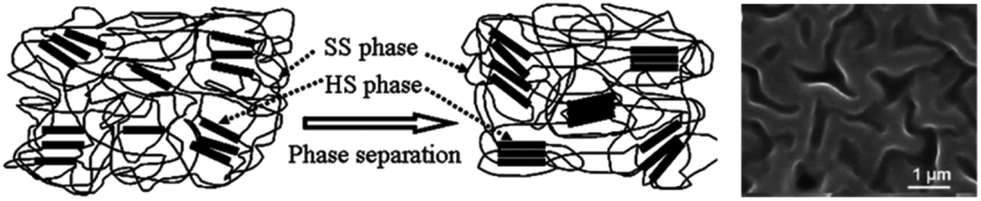 | ||
| Fig. 12 Schematic of phase separation in TPEU/TPEUUs between hard segments (HS) and soft segments (SS) and an SEM image of a phase separated TPEU. Reproduced from ref. 111 with permission from The Royal Society of Chemistry. | ||
This combination allows for the development and modulation of viscoelastic properties of the resultant materials.112 Although the phase separation of segmented polyurethanes allows access to a wide range of mechanical properties, the compartmentalisation of phases can lead to heterogeneous degradation profiles throughout the polymer matrix leading to severe loss of mechanical integrity.44,93 This occurs as a consequence of differences in diffusivity of degradation media and autocatalytic inducing degradation products, hydrolysis rates of esters/carbonates vs. urethanes/ureas, and topology of the ‘hard-soft’ phase separated polymer network.45 Although a range of polyurethane and urea-based materials have been developed since their inception, only the synthesis of PLGA, PCL and PTMC-based TPEU/TPEUUs and their applications will be discussed herein.
PLA and PLGA TPEUs
Owing to the rapid degradation of PGA and the brittle nature of PLLA, the use of glycolides and lactides as macrodiols in the synthesis of TPEU/TPEUUs has been relatively limited. However, both PLA and PLA/PGA copolymers (PLGA) have been investigated owed there improved mechanical properties and tunable degradation times (Lubrizol Estane® 58277 TPU (UTS = 62.1 MPa, εbreak = 450%), Huntsman IROGRAN® A 60E 4612N TPU-polyester (UTS = 25.5 MPa, εbreak = 900%)).In agreement with the degradation of the homopolyesters previously discussed, Storey and Hickey demonstrated the vastly increased degradation of PGA-based TPEUs (8% weight loss after 13 days) over PLA-based TPEUs (1.3% weight loss after 13 days). Furthermore it was observed that PLA-based TPEUs exhibited a superior Young's modulus (767 MPa) and UTS (41 MPa) than the PGA-based materials (E = 2.14 MPa, UTS = 9.22 MPa), coinciding with the trends observed in the mechanical analysis of the homopolyesters.113 In a separate study conducted by Storey and coworkers, PLGA (50:50 mol% PLA:PGA statistical copolymer) was incorporated into a poly(butylene adipate) (PBA) thermoplastic polyurethane (TPU) in order to introduce enhanced degradability.114 As predicted, PLGA-based TPEUs exhibited enhanced degradation however, interestingly, PLGA containing TPEUs also exhibited an increase in E (27 MPa) in comparison to PBA TPEUs (13 MPa) with no significant variance in UTS (∼20 MPa) or percentage strain at break (∼600%). Furthermore, the degradation of the PLGA-containing TPEUs was further enhanced by the utilisation of a more hydrophilic initiator (2,2-bis-(hydroxymethyl)butanoic acid) for the ROP of lactide/glycolide, consequently increasing the water uptake capability of the material.
Unlike the previous examples, Adhikari et al. designed injectable degradable polyurethane systems for orthopaedic applications utilising a mix of both PGA and PDLLA macrodiols in the same TPEU.115 It was found, as expected, that PDLLA-based TPEUs exhibited a slight reduction in mechanical strength when copolymerised with PGA macrodiols, with an inversely proportional change in degradation times. Furthermore, it was found that the onset of degradation was retarded by the addition of a β-tricalcium phosphate, owing to its quenching of acidic degradation products.
PCL TPEU/TPEUUs
For reasons analogous to the properties of the homopolyester (i.e. increased flexibility, biocompatibility, reduced autocatalytic degradation etc.), PCL-based TPEU/TPEUUs have been the most extensively examined and commercially utilised macrodiols in the synthesis of degradable polyurethane materials (Lubrizol Pearlthane® D95N85P (UTS = 27.0 MPa, εbreak = 850%), Huntsman IROGRAN® A 95 K 4640 (UTS = 37.23 MPa, εbreak = 570%), SK Chemicals SKYTHANE® L194A (UTS = 49.0 MPa, εbreak = 500%)).116 Furthermore, as has been previously mentioned in the synthesis of polyesters for biomedical applications, Kylmä and Seppälä employed PCL in order to reduce hydrolytic susceptibility and improve viscoelasticity of PLA-containing TPEUUs.103 Although considered a semi-crystalline material, PCL's low Tg and mechanical flexibility in comparison with PLA and PGA-based polymers makes it an excellent candidate as a ‘soft’ block for TPEU/TPEUU synthesis.117In order to investigate the effect of molecular weight of macrodiols on the mechanical properties and morphology of resultant TPEUs, Schouten et al. used telechelic PCL initiated from 1,4-butanediol (1,4-BDO) with molecular weights ranging from 750 to 2800 g mol−1.118 The PCL macrodiols were end-capped with excess 1,4-butanediisocyanate (1,4-BDI) and chain extended with 1,4-BDO to yield high molecular (100–160 kg mol−1) weight TPEUs. Schouten et al. noted at higher molecular weight PCL, TPEUs exhibited an enhanced elastic behaviour with a reduced plastic deformation and an enhanced percentage strain at break (>1200%). This was owed to the decrease in ‘hard’ block crystallinity as a consequence of reduced ‘soft’ block mobility at higher molecular weights, retarding ‘hard’ block association. The reduced mobility of the ‘soft’ block was characterised by an increase in the glass transition temperature of the incorporated PCL macrodiols, indicative of free volume in accordance with the Flory–Fox equation.119 In addition to this, Watanabe and co-workers demonstrated the reduction in biodegradability of TPEUs with increasing molecular weight PCL macrodiols.120 Parallel to previous results, this was attributed to poor diffusivity of degradation media through the polymer matrix as a consequence of low chain mobility at higher molecular weights. Furthermore, it was noted that the degradation was also retarded by the recrystallisation of low molecular weight PCL degradants further reducing diffusivity.120
In order to improve the mechanical rigidity and biocompatibility of PCL-based polyurethane materials, Guan et al. employed the use of the diamine chain extenders putrescine and lysine ethyl ester in the synthesis of TPEUUs (Fig. 13).121 It was found that upon biodegradation of the TPEUU ‘hard’ blocks, the biocompatible and excretable lysine ethyl ester, an amino acid derivative, or putrescine, a diamine essential for cell growth and differentiation, were released.
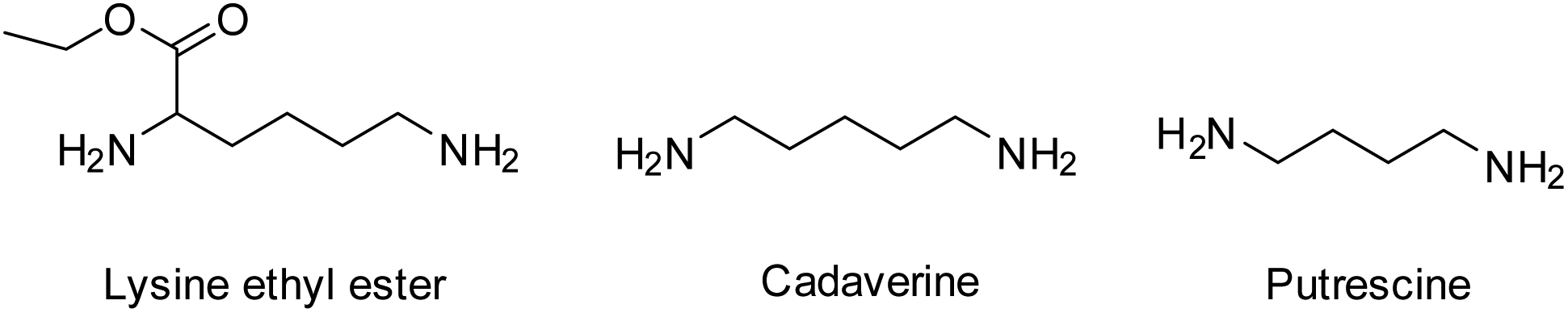 | ||
| Fig. 13 Natural diamine chain extenders for the synthesis of TPEUUs. | ||
In addition to this, Guan et al. demonstrated the improved viability and proliferation of human umbilical vein endothelial cells (HUVECs) when seeded on the putrescine-containing TPEUUs. Furthermore, the choice of chain extender was also found to significantly affect the mechanical properties of the PCL-based TPEUUs. It was found that putrescine-containing TPEUUs exhibited a higher E (78 MPa), UTS (11.9 MPa) and a reduced plastic deformation over its lysine counterpart (E = 38 MPa, UTS = 4.7 MPa). This was attributed to the enhanced hydrogen bonding of the lysine-containing TPEUUs through the ester side group causing rigid plastic-like properties.
PTMC TPEU/TPEUUs
Although homo-PTMC exhibits an enhanced elasticity in comparison with other polyesters, it has been relatively under-utilised exclusively of other macrodiols in the synthesis of TPEU/TPEUU elastomers.122–124 To date, and to our knowledge, no commercially available TPEU/TPEUUs utilising PTMC macrodiols are available. Similar to studies discussed previously, Wagner et al. synthesised a series of PTMC macrodiols with varying molecular weights (1.5–5.4 kg mol−1) and utilised them in the synthesis TPEUUs employing 1,4-BDI and putrescine as the diisocyanate and extender respectively.122 Although each TPEUU exhibited comparable strains at break (∼750%), it was noted at low strains (50%), TPEUUs comprised of higher molecular weight PTMC macrodiols exhibited a reduction in unrecoverable deformation through hysteresis (<5%). This trend continued at higher strains (400%), however the degree of deformation increased by an order of magnitude (50–80%). In conjunction to this, it was found that PTMC-based TPEUUs exhibited excellent cell viability and proliferation with very low susceptibility to hydrolysis (>80% mass remaining after 28 days), similar to the PTMC homopolymer discussed earlier (Fig. 14). Furthermore, utilising an extender free methodology, Hilborn et al. reported the synthesis of highly elastic TPEUUs which exhibited high strains at break, in excess of 1900%, UTS (>30 MPa) and reasonable Young's moduli (∼4.5 MPa).123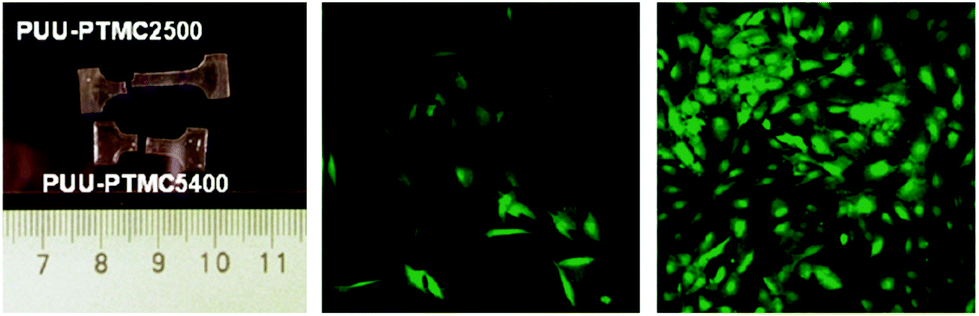 | ||
| Fig. 14 PTMC-based TPEUUs with improved elasticity with higher molecular weight diols (right), good cell viability (middle) and excelent cell proliferation (right). Reprinted with permission from ref. 122. Copyright 2011, American Chemical Society. | ||
In a comprehensive analysis of hydrolytic stability, cytotoxicity and mechanical robustness after partial degradation of PTMC-based TPEUs, Sobczak et al.124 reported that after 5 weeks under accelerated degradation conditions (10% aq. HCl at 70 °C), PTMC-TPEUs retained greater than 85% of their original mass by gravimetric analysis. Furthermore, these materials were reported to retain between 78–80% of their initial strain at break and UTS. In addition to this Sobczak and co-workers found that PTMC-based TPEUs exhibited no significant cytotoxic effect when evaluated with luminescent bacteria (V. fischeri) or ciliated protozoans (S. ambiguum and T. thermophila).
Conclusion and outlook
In conclusion, the development and production of synthetic hydrolytically degradable polymers for applications as biomaterials is of upmost importance for the advancement of effective biomedical therapeutics such as tissue regeneration, sustained API release, degradable implants etc. As is evident, even from the small selection of polymers discussed, aliphatic polyester and polycarbonates offer a facile route to a plethora of materials with a variety of thermal, degradation and mechanical properties. Furthermore, these properties can be defined through stereochemistry, copolymerisation, initiating species chosen, targeted molecular weight and morphology.Although synthetic hydrolytically degradable polymers offer a facile route to medically relevant biomaterials, there remain a number of challenges to be overcome and advancements made. While much technically excellent work has focussed on developing an understanding of these materials, ultimately more advanced properties such as more ready access to surface erodible materials, materials with more ready functional group access and those with better mechanical properties are required to truly advance the field. Most importantly, there is a need to overcome premature degradation-induced mechanical failure. Current methodologies to realise such materials seem to be reaching their limit and only through investigation of a wider range of materials will these be overcome. In itself, this presents the challenge of translating from the chemistry laboratory to biomedical studies.
Beyond this, an additional challenge that is clearly evident from the published results, is the development of standardised in vitro degradation analysis, ideally that can replicate in vivo behaviour of the materials. The wide range of tests used creates a confusing picture by which to compare materials. To this end, standardisation would allow for efficient and accurate selection of purpose-matched degradable biomaterials. With such a vast and ever expanding selection of monomers and improvements in polymerisation techniques, the arena of degradable polymers proposes a great deal of exciting prospects and challenges.
Acknowledgements
The Royal Society are acknowledged for funding an Industrial Fellowship to A. P. D.References
- M. Okada, Prog. Polym. Sci., 2002, 27, 87–113 CrossRef CAS.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273–1335 CrossRef CAS.
- C. H. Lee, A. Singla and Y. Lee, Int. J. Pharm., 2001, 221, 1–22 CrossRef CAS PubMed.
- L. Buttafoco, N. G. Kolkman, P. Engbers-Buijtenhuijs, A. A. Poot, P. J. Dijkstra, I. Vermes and J. Feijen, Biomaterials, 2006, 27, 724–734 CrossRef CAS PubMed.
- B. Kundu, R. Rajkhowa, S. C. Kundu and X. Wang, Adv. Drug Delivery Rev., 2013, 65, 457–470 CrossRef CAS PubMed.
- H. W. T. Matthew and J. K. F. Suh, Biomaterials, 2000, 21, 2589–2598 CrossRef.
- Y. Luo, K. R. Kirker and G. D. Prestwich, J. Controlled Release, 2000, 69, 169–184 CrossRef CAS PubMed.
- G. Q. Chen and Q. Wu, Biomaterials, 2005, 26, 6565–6578 CrossRef CAS PubMed.
- T. Freiera, C. Kunzea, C. Nischana, S. Kramera, K. Sternberga, M. Sab, U. T. Hoptb and K.-P. Schmitza, Biomaterials, 2002, 23, 2649–2657 CrossRef.
- G. T. Köse, H. Kenar, N. Hasırcı and V. Hasırcı, Biomaterials, 2003, 24, 1949–1958 CrossRef.
- J. A. Hubbell, Nat. Biotechnol., 1995, 13, 565–576 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- J. R. Lentino, Clin. Infect. Dis., 2003, 36, 1157 CrossRef PubMed.
- A. van Ooij, S. M. Kurtz, F. Stessels, H. Noten and L. van Rhijn, Spine, 2007, 32, 223–229 CrossRef PubMed.
- N. Goonoo, A. Bhaw-Luximon, G. L. Bowlin and D. Jhurry, Polym. Int., 2013, 62, 523–533 CrossRef CAS.
- G. H. Altman, F. Diaz, C. Jakuba, T. Calabro, R. L. Horan, J. Chen, H. Lu, J. Richmond and D. L. Kaplan, Biomaterials, 2003, 24, 401–416 CrossRef CAS PubMed.
- H. S. Azevedo, F. M. Gama and R. L. Reis, Biomacromolecules, 2003, 4, 1703–1712 CrossRef CAS PubMed.
- D. S. Katti, S. Lakshmi, R. Langer and C. T. Laurencin, Adv. Drug Delivery Rev., 2002, 54, 933–961 CrossRef CAS PubMed.
- A. Göpferich, Biomaterials, 1996, 17, 103–114 CrossRef.
- S. Lyu and D. Untereker, Int. J. Mol. Sci., 2009, 10, 4033–4065 CrossRef CAS PubMed.
- R. A. Larson and E. J. Weber, Reaction Mechanisims in Environmental Chemistry, CRC Press, 1994 Search PubMed.
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181–3198 CrossRef CAS PubMed.
- C. G. Pitt, M. M. Gratzl, G. L. Kimmel, J. Surles and A. Schindler, Biomaterials, 1981, 2, 215–220 CrossRef CAS PubMed.
- F. von Burkersrodaa, L. Schedlb and A. Gopferich, Biomaterials, 2002, 23, 4221–4231 CrossRef.
- M. Oak, R. Mandke, S. Lakkadwala, L. Lipp and J. Singh, Polymer, 2015, 7, 1510–1521 CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741–1748 CrossRef CAS PubMed.
- J. Heller, J. Barr, S. Y. Ng, K. S. Abdellauoi and R. Gurny, Adv. Drug Delivery Rev., 2002, 54, 1015–1039 CrossRef CAS PubMed.
- D. S. Muggli, A. K. Burkoth and K. S. Anseth, J. Biomed. Mater. Res., Part A, 1999, 46, 271–278 CrossRef CAS.
- H. S. Azevedo and R. L. Reis, Understanding the Enzymatic Degradation of Biodegradable Polymers and Strategies to Control Their Degradation Rate, CRC Press, 2005 Search PubMed.
- A. Ghaffar, P. J. Schoenmakers and S. van der Wal, Crit. Rev. Anal. Chem., 2014, 44, 23–40 CrossRef CAS PubMed.
- H. Shirahama, Y. Kawaguchi, M. S. Aludin and H. Yasuda, J. Appl. Polym. Sci., 2001, 80, 340–347 CrossRef CAS.
- H. Shirahama, A. Kanetani and H. Yasuda, Polym. J., 2000, 32, 280–286 CrossRef CAS.
- H. Pranamuda, Y. Tokiwa and H. Tanaka, Appl. Environ. Microbiol., 1995, 61, 1828–1832 CAS.
- J. Casas, Q. Zhao, M. Donovan, P. Schroeder, K. Stokes and D. Untereker, J. Biomed. Mater. Res. Symp., 1999, 46, 475–484 CrossRef CAS.
- M. C. Sobieraj and C. M. Rimnac, J. Mech. Behav. Biomed. Mater., 2009, 2, 433–443 CrossRef CAS PubMed.
- W. L. Hawkins, Polym. Eng. Sci., 1964, 4, 187–192 CAS.
- O. Chiantore, L. Trossarelli and M. Lazzari, Polymer, 2000, 41, 1657–1668 CrossRef CAS.
- T. Seguchi, K. Arakawa, M. Ito, N. Hayakawa and S. Machi, Radiat. Phys. Chem., 1983, 21, 495–501 CrossRef CAS.
- J. F. Rabek, in Degradation of Polymers, ed. R. G. Compton, C. H. Bamford and C. F. H. Tipper, Elsevier, 1975, ch. 4, pp. 425–539 Search PubMed.
- M. C. Celina, Polym. Degrad. Stab., 2013, 98, 2419–2429 CrossRef CAS.
- C. Booth, Polymer, 1963, 4, 471–478 CrossRef CAS.
- F. Bueche, J. Appl. Polym. Sci., 1960, 4, 101–106 CrossRef CAS.
- A. A. Hamouda, C. Eliassen, C. Idsoe and T. Jacobsen, Annu. Trans. - Nord. Rheol. Soc., 2005, 13, 65–71 Search PubMed.
- K. Stokes, R. McVenes and J. M. Anderson, J. Biomater. Appl., 1995, 9, 321–353 CAS.
- P. Krol, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef CAS.
- D. J. King and M. J. Matava, Oper. Techn. Sport Med., 2004, 12, 161–169 CrossRef.
- O. Bostman, E. Hirvensalo, J. Makinen and P. Rokkanen, J. Bone Joint Surg. Br., 1990, 72, 592–596 CAS.
- M. Vert, Prog. Polym. Sci., 2007, 32, 755–761 CrossRef CAS.
- B. K. Ambati, M. Nozaki, N. Singh, A. Takeda, P. D. Jani, T. Suthar, R. J. Albuquerque, E. Richter, E. Sakurai, M. T. Newcomb, M. E. Kleinman, R. B. Caldwell, Q. Lin, Y. Ogura, A. Orecchia, D. A. Samuelson, D. W. Agnew, J. St Leger, W. R. Green, P. J. Mahasreshti, D. T. Curiel, D. Kwan, H. Marsh, S. Ikeda, L. J. Leiper, J. M. Collinson, S. Bogdanovich, T. S. Khurana, M. Shibuya, M. E. Baldwin, N. Ferrara, H. P. Gerber, S. De Falco, J. Witta, J. Z. Baffi, B. J. Raisler and J. Ambati, Nature, 2006, 443, 993–997 CrossRef CAS PubMed.
- E. B. Hunziker, Osteoarthr. Cartil., 2002, 10, 432–463 CrossRef CAS PubMed.
- M. G. Freshney and I. Freshney, Culture of Epithelial Cells, Wiley, 2nd edn, 2002 Search PubMed.
- J. E. Bergsma, W. C. de Bruiju, F. R. Rozema, R. R. M. Bos and G. Boering, Biomaterials, 1995, 16, 25–31 CrossRef CAS PubMed.
- P.-J. Roumanet, F. Laflèche, N. Jarroux, Y. Raoul, S. Claude and P. Guégan, Eur. Polym. J., 2013, 49, 813–822 CrossRef CAS.
- H. W. Gibson, S. Liu, C. Gong, Q. Ji and E. Joseph, Macromolecules, 1997, 30, 3711–3726 CrossRef CAS.
- M. Kolitz, N. Cohen-Arazi, I. Hagag, J. Katzhendler and A. J. Domb, Macromolecules, 2009, 42, 4520–4530 CrossRef CAS.
- Z. Zhang, X. Lou, Y. Lu and D. Ma, Eur. Polym. J., 2001, 37, 99–104 CrossRef CAS.
- D. M. Hunsicker, B. C. Dauphinais, S. P. Mc Ilrath and N. J. Robertson, Macromol. Rapid Commun., 2012, 33, 232–236 CrossRef CAS PubMed.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- R. J. Pounder and A. P. Dove, Biomacromolecules, 2010, 11, 1930–1939 CrossRef CAS PubMed.
- S. M. Guillaume, Eur. Polym. J., 2013, 49, 768–779 CrossRef CAS.
- I. van der Meulen, M. de Geus, H. Antheunis, R. Deumens, E. A. J. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2008, 9, 3404–3410 CrossRef CAS PubMed.
- I. van der Meulen, Y. Li, R. Deumens, E. A. Joosten, C. E. Koning and A. Heise, Biomacromolecules, 2011, 12, 837–843 CrossRef CAS PubMed.
- K. S. Bisht, L. A. Henderson, R. Gross, D. L. Kaplan and G. Swift, Macromolecules, 1997, 30, 2705–2711 CrossRef CAS.
- E. J. Frazza and E. E. Schmitt, J. Biomed. Mater. Res. Symp., 1971, 1, 43–58 CrossRef PubMed.
- D. K. Gilding and A. M. Reed, Polymer, 1980, 22, 494–498 Search PubMed.
- I. Engelberg and J. Kohn, Biomaterials, 1990, 12, 292–304 CrossRef.
- A. Södergård and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123–1163 CrossRef.
- C. C. Chu, Polymer, 1985, 26, 591–594 CrossRef CAS.
- R. A. Kenley, M. Ott Lee, R. T. Mahoney and L. M. Sanders, Macromolecules, 1987, 20, 2398–2403 CrossRef CAS.
- C. A. P. Joziasse, D. W. Grijpma, J. E. Bergsma, F. W. Cordewener and A. J. Pennings, Colloid Polym. Sci., 1998, 276, 968–975 CAS.
- S. Li and S. McCarthy, Biomaterials, 1999, 20, 35–44 CrossRef CAS PubMed.
- H. Tsuji, Macromol. Biosci., 2005, 5, 569–597 CrossRef CAS PubMed.
- S. Li and M. Vert, Polym. Int., 1994, 33, 37–41 CrossRef CAS.
- R. A. Jain, Biomaterials, 2000, 21, 2475–2490 CrossRef CAS PubMed.
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- M. H. Sheridan, L. D. Shea, M. C. Peters and D. J. Mooney, J. Controlled Release, 2000, 64, 91–102 CrossRef CAS PubMed.
- E. Vey, C. Rodger, L. Meehan, J. Booth, M. Claybourn, A. F. Miller and A. Saiani, Polym. Degrad. Stab., 2012, 97, 358–365 CrossRef CAS.
- R. A. Miller, J. M. Brady and D. E. Cutright, J. Biomed. Mater. Res., 1977, 11, 711–719 CrossRef CAS PubMed.
- F. J. Natta, J. W. Hill and W. H. Carothers, J. Am. Chem. Soc., 1934, 56, 455–457 CrossRef.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef CAS.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- A. L. Luis, J. M. Rodrigues, J. V. Lobato, M. A. Lopes, S. Amado, A. P. Veloso, P. A. S. Armada-da Silva, S. Raimondo, S. Geuna, A. J. Ferreira, A. S. P. Varejao, J. D. Santos and A. C. Maurício, Bio-Med. Mater. Eng., 2007, 17, 39–52 CAS.
- A. Elzubair, C. N. Elias, J. C. Suarez, H. P. Lopes and M. V. Vieira, J. Dent., 2006, 34, 784–789 CrossRef CAS PubMed.
- K. Gissefalt, B. Edberg and P. Flodin, Biomacromolecules, 2002, 3, 951–958 CrossRef.
- L. S. Klopp, B. J. Simon, J. M. Bush, M. R. Enns and S. A. Turner, Spine, 2008, 33, 1518–1526 CrossRef PubMed.
- K. W. Ng, H. N. Achuth, S. Moochhala, T. C. Lim and D. W. Hutmacher, J. Biomater. Sci., Polym. Ed., 2007, 18, 925–938 CrossRef CAS PubMed.
- D. S. Jones, J. Djokic, C. P. McCoy and S. P. Gorman, Biomaterials, 2002, 23, 4449–4458 CrossRef CAS PubMed.
- W. H. Carothers and F. J. Van Natta, J. Am. Chem. Soc., 1930, 52, 314–325 CrossRef CAS.
- B. Amecke, D. Bendix and G. Entenmann, in Encyclopedia handbook of biomaterials and bioengineering, Part A: Materials, ed. D. L. Wise, D. J. Trantolo, D. E. Altobelli, M. J. Yaszemski, J. D. Gresser and E. R. Schwartz, Marcel Dekker Inc., 1995, vol. 1 and 2, pp. 97–1007 Search PubMed.
- E. Bat, B. H. Kothman, G. A. Higuera, C. A. van Blitterswijk, J. Feijen and D. W. Grijpma, Biomaterials, 2010, 31, 8696–8705 CrossRef CAS PubMed.
- A. P. Pêgo, A. A. Poot, D. W. Grijpma and J. Feijen, J. Controlled Release, 2003, 87, 69–79 CrossRef.
- Z. Zhang, D. W. Grijpma and J. Feijen, J. Controlled Release, 2006, 111, 263–270 CrossRef CAS PubMed.
- A. Albertsson and M. Eklund, J. Appl. Polym. Sci., 1995, 57, 87–103 CrossRef CAS.
- J. H. Kim, S. Y. Lee and D. J. Chung, Polym. J., 2000, 32, 1056–1059 CrossRef CAS.
- J. H. Kim and J. H. Lee, Polym. J., 2002, 34, 203–208 CrossRef CAS.
- S. Dai, L. Xue and Z. Li, ACS Catal., 2011, 1, 1421–1429 CrossRef CAS.
- Z. Zhang, D. W. Grijpma and J. Feijen, Macromol. Chem. Phys., 2004, 205, 867–875 CrossRef CAS.
- L. K. Widjaja, J. F. Kong, S. Chattopadhyay, V. T. Lipik, S. S. Liow, M. J. Abadie and S. S. Venkatraman, J. Mech. Behav. Biomed. Mater., 2012, 6, 80–88 CrossRef CAS PubMed.
- R. Shi, D. Chen, Q. Liu, Y. Wu, X. Xu, L. Zhang and W. Tian, Int. J. Mol. Sci., 2009, 10, 4223–4256 CrossRef CAS PubMed.
- J. Kylmä and J. V. Seppälä, Macromolecules, 1997, 30, 2876–2882 CrossRef.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS.
- N. M. K. Lamba, K. A. Woodhouse and S. L. Cooper, Polyurethanes in Biomedical Applications, CRC Press LLC, 1997 Search PubMed.
- R. J. Spontak and N. P. Patel, Curr. Opin. Colloid Interface Sci., 2000, 5, 334–341 CrossRef CAS.
- M. A. Hillmyer and W. B. Tolman, Acc. Chem. Res., 2014, 47, 2390–2396 CrossRef CAS PubMed.
- A. Harrane, A. Leroy, H. Nouailhas, X. Garric, J. Coudane and B. Nottelet, Biomed. Mater., 2011, 6, 065006 CrossRef PubMed.
- Z. Zhang, D. W. Grijpma and J. Feijen, J. Controlled Release, 2006, 116, 29–31 CrossRef PubMed.
- C. L. Wanamaker, L. E. O'Leary, N. A. Lynd, M. A. Hillmyer and W. B. Tolman, Biomacromolecules, 2007, 8, 3634–3640 CrossRef CAS PubMed.
- Z. Wu, H. Wang, X. Tian, P. Cui, X. Ding and X. Ye, Phys. Chem. Chem. Phys., 2014, 16, 6787–6794 RSC.
- T. O. Ahn, S. U. Jung, H. M. Jeong and S. W. Lee, J. Appl. Polym. Sci., 2003, 51, 43–49 CrossRef.
- R. F. Storey and T. P. Hickey, Polymer, 1994, 35, 830–838 CrossRef CAS.
- S. J. Moravek, M. K. Hassan, D. J. Drake, T. R. Cooper, J. S. Wiggins, K. A. Mauritz and R. F. Storey, J. Appl. Polym. Sci., 2010, 115, 1873–1880 CrossRef CAS.
- R. Adhikari, P. A. Gunatillake, I. Griffiths, L. Tatai, M. Wickramaratna, S. Houshyar, T. Moore, R. T. Mayadunne, J. Field, M. McGee and T. Carbone, Biomaterials, 2008, 29, 3762–3770 CrossRef CAS PubMed.
- C. J. Bettinger, Macromol. Biosci., 2011, 11, 467–482 CrossRef CAS PubMed.
- I. Vroman and L. Tighzert, Materials, 2009, 2, 307–344 CrossRef CAS.
- R. G. Heijkants, R. V. van Calck, T. G. van Tienen, J. H. de Groot, P. Buma, A. J. Pennings, R. P. Veth and A. J. Schouten, Biomaterials, 2005, 26, 4219–4228 CrossRef CAS PubMed.
- G. Odian, Principles of Polymerization, John Wiley & Sons, Inc., 4th edn, 2004 Search PubMed.
- A. Watanabe, Y. Takebayashi, T. Ohtsubo and M. Furukawa, J. Appl. Polym. Sci., 2009, 114, 246–253 CrossRef CAS.
- J. Guan, M. S. Sacks, E. J. Beckman and W. R. Wagner, J. Biomed. Mater. Res., Part A, 2002, 61, 493–503 CrossRef CAS PubMed.
- Z. Ma, Y. Hong, D. M. Nelson, J. E. Pichamuthu, C. E. Leeson and W. R. Wagner, Biomacromolecules, 2011, 12, 3265–3274 CrossRef CAS PubMed.
- B. Asplund, T. Bowden, T. Mathisen and J. Hilborn, Biomacromolecules, 2007, 8, 905–911 CrossRef PubMed.
- M. Sobczak, C. Dębek, E. Olędzka, G. Nałęcz-Jawecki, W. L. Kołodziejski and M. Rajkiewicz, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3904–3913 CrossRef CAS.
- S. Clarke, W. Hagberg, R. A. Kaufmann, A. Grand and R. Wollstein, Hand, 2011, 6, 282–286 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |